

Abstract
The cyanuric acid hydrolase, AtzD, is the founding member of a newly identified family of ring-opening amidases. We report the first X-ray structure for this family, which is a novel fold (termed the ‘Toblerone’ fold) that likely evolved via the concatenation of monomers of the trimeric YjgF superfamily and the acquisition of a metal binding site. Structures of AtzD with bound substrate (cyanuric acid) and inhibitors (phosphate, barbituric acid and melamine), along with mutagenesis studies, allowed the identification of the active site. The AtzD monomer, active site and substrate all possess threefold rotational symmetry, to the extent that the active site possesses three potential Ser–Lys catalytic dyads. A single catalytic dyad (Ser85–Lys42) is hypothesized, based on biochemical evidence and crystallographic data. A plausible catalytic mechanism based on these observations is also presented. A comparison with a homology model of the related barbiturase, Bar, was used to infer the active-site residues responsible for substrate specificity, and the phylogeny of the 68 AtzD-like enzymes in the database were analysed in light of this structure–function relationship.
Introduction
The deployment of the s-triazines, such as atrazine (1-chloro-3-ethylamino-5-isopropylamino-2,4,6-triazine), into the environment has resulted in the evolution of new atrazine catabolic pathways in bacteria (Wackett, 2009). These pathways, and the enzymes that comprise them, have garnered considerable attention in light of their potential application in the bioremediation of atrazine (Udikovic-Kolic et al., 2012), which is a potential human carcinogen, endocrine disrupter and teratogen (Wiegand et al., 2001; MacLennan et al., 2002; Hayes et al., 2003). s-Triazine degrading systems are also model systems for the study of evolving enzymes and pathways (Wackett, 2009; Noor et al., 2012; Udikovic-Kolic et al., 2012).
Atrazine is mineralized in bacteria by a series of hydrolases, with the first three hydrolytic steps resulting in the dechlorination and deamination of atrazine to produce cyanuric acid (CA; 2,4,6-trihydroxy-1,3,5 triazine; Seffernick et al., 2012), which is then hydrolysed by the ring-opening amide hydrolase, cyanuric acid hydrolase (AtzD; E.C. 3.5.2.15). AtzD catalyses the hydrolytic ring opening of CA to yield 1-carboxybiuret which then spontaneously decomposes to biuret and carbon dioxide (Fig. 1A; Seffernick et al., 2012). Although CA is a naturally occurring compound, occasionally formed during oxidative damage of DNA (Wackett, 2009), its environmental abundance has increased markedly since the introduction of synthetic s-triazine compounds.
Fig. 1.
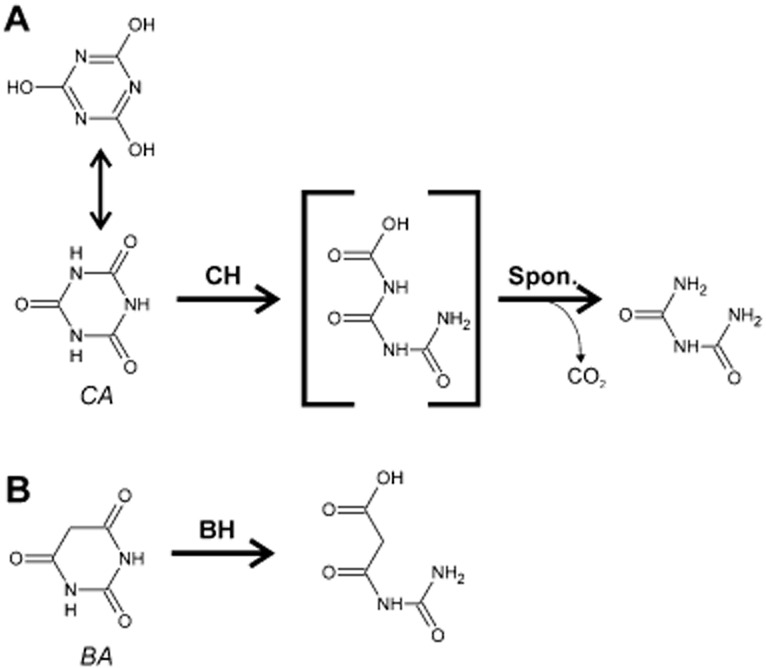
Enzymatic hydrolysis of cyanuric acid and barbituric acid.
A. Cyanuric acid (CA) tautomerizes between its trihydro- and triketo-forms, the latter acting as the substrate for cyanuric acid hydrolase (CH). The product of hydrolysis is 1-carboxybiuret, which spontaneously decomposes to biuret (liberating CO2).
B. Barbituric acid (BA) is hydrolysed by barbiturate hydrolase (BH) to form 3-oxo-3-uridopropionate.
AtzD, is the archetype for a family of ring-opening amidases (Seffernick et al., 2012) that includes a related CA hydrolase (TrzD; 56% identical to AtzD; Karns, 1999) and barbiturase (Bar; E.C. 3.5.2.1; Soong et al., 2002; Seffernick et al., 2012). Barbituric acid (2,4,6-trihydroxy-1,3-pyrimidine; BA), is an intermediate in the oxidative degradation of pyrimidines (Soong et al., 2002). Pyrimidines are degraded reductively in eukaryotes and most prokaryotes (Soong et al., 2001); however, an oxidative pathway was described from a small number of eubacteria in the 1950s in which pyrimidines are first oxidized to BA, which is then hydrolysed by Bar and ureidomalonase to yield malonic acid and urea (Soong et al., 2001). This metabolic pathway is rare, as AtzD/Bar homologues were described in only 3% of 6423 surveyed genomes in a recent study (Seffernick et al., 2012).
Bar catalyses an equivalent ring-opening reaction to that of AtzD, using BA as substrate and yielding the stable product 3-oxo-3-ureidopropanoate (Soong et al., 2002; Fig. 1B). BA differs from CA at just a single position in the ring (C5 in BA and N5 in CA; Fig. 1A and B). It is therefore perhaps surprising that BA is an inhibitor, rather than a substrate, for AtzD (Seffernick et al., 2012) and that CA is an inhibitor of Bar (Soong et al., 2002). With no available structure for AtzD or Bar, the determinants of specificity were not identified.
Early studies of this family suggested that these enzymes were members of the metal-dependent amidohydrolase family; Soong and co-workers were able to inactivate Bar by treatment with a chelator and the activity was restored upon incubation with Zn2+ (Soong et al., 2002). However, more recent work from Seffernick et al. (2012) demonstrated that this family is unrelated to amidohydrolases, forming a discrete evolutionary class of their own. Moreover, it was suggested that the newly defined family was metal-independent.
Herein, we describe the first X-ray structure for this family, an unusual concatenated knot fold (which we term the ‘Toblerone’ fold), that possesses a threefold rotational symmetry that extends to the active-site architecture. Structures of AtzD with the substrate and inhibitors, and kinetic and mutagenesis analysis of these with wild-type and mutant proteins are presented. We identify Lys42 and Ser85 as a plausible catalytic dyad and hypothesize a catalytic mechanism consistent with that assignation.
Results
Structure of AtzD
The AtzD monomer is a unique arrangement of a core of 12 β-strands with six helices on the outside (Fig. 2A). This structure is formed from three ‘repeating units’ (RUs) that share the same structure as one another; they have been designated RU A (residues 3–104), B (residues 113–250) and C (residues 256–364). Each RU is comprised of a four strand antiparallel β-sheet and two helices. This arrangement of RUs confers threefold rotational symmetry in the AtzD monomer reminiscent of a ‘Toblerone’ Bar (viewed end on; Fig. 2A), and it is proposed that this new fold be termed the Toblerone fold. The three RUs of the monomer align with the following root mean square deviation (rmsd) over the Cα atoms and sequence identities: B to A = 3.0 Å/9.3%, C to A = 2.3 Å/16.2%, C to B = 2.2 Å/17.8% (Fig. 2B).
Fig. 2.
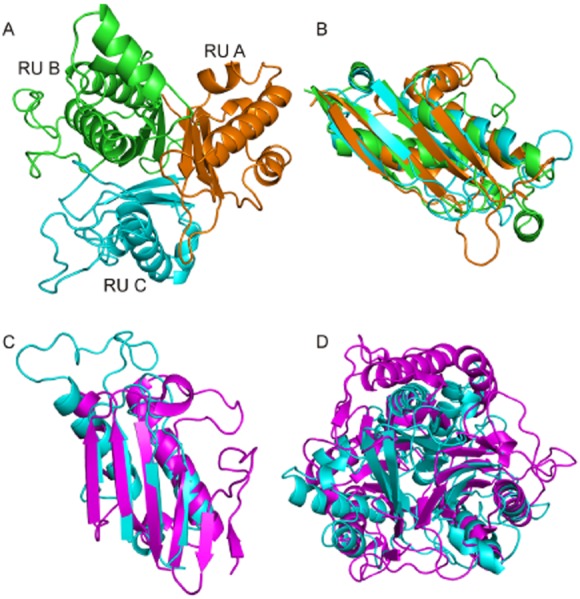
Structure of AtzD monomer and comparison to the perchloric acid soluble protein (PSP).
A. Structure and substructure of an AtzD monomer. The RUs are coloured as follows: RU A (residues 2–104) is orange, RU B (residues 113–250) is green and RU C (residues 256–364) is cyan.
B. Overlay between RUs A, B and C (coloured as Fig. 2A).
C. PSP (PDB: 3K0T) monomer (magenta) superposed with RU C of AtzD (cyan).
D. A trimer of PSP (magenta) superposed with a monomer of AtzD (cyan).
There were no homologues to the full-length AtzD in the PDBe (using SSM). However, a single RU has homology with proteins of the YgjF superfamily: perchloric acid-soluble protein (PSP) from Pseudomonas syringae (PDB: 3K0T), YGJF from Streptococcus pyogenes (PDB: 3EWC) and RutC from Escherichia coli (PDB: 3V4D). PSP overlays an AtzD RU with a rmsd of ∼ 1.3 Å over 109 residues (sequence identity ∼ 16.5%; Fig. 2C), notwithstanding that the YgjF proteins have a six strand antiparallel β-sheet (rather than four). YgjF proteins form homo-trimers that align poorly with the structure of an AtzD monomer (Fig. 2D), due to the two additional β-strands per monomer/RU.
AtzD possesses a single metal binding site per monomer, in RU C, for which there is no equivalent in the YgjF family of proteins (Fig. 3A–C; Zhang et al., 2010; Knapik et al., 2012), but which is completely conserved in 90% of the AtzD homologues currently available (data not shown). The metal binding site consists of backbone carbonyls from residues Ala347, Gln350, Pro352, Gly355, the δ-carboxylate of Glu298 and a water molecule. The octahedral co-ordination, electron density and B factor associated with the cation suggest that the metal bound by AtzD is likely to be magnesium (Dudev et al., 1999; Fig. 3D). Anomalous scattering data show that the native metal ion is either magnesium or sodium (Supplemental Fig. S1). Unfortunately, it is not possible to distinguish between Na+ and Mg2+ by anomalous scattering as both cations have an identical number of electrons and the theoretical anomalous difference at accessible X-ray wavelengths is consequently near identical. However, it is certain from these data that the density found is not Zn2+, nor is it any other transition metal. The metal appears to stabilize an extended loop that contains residues that make polar contact with amino acids in the adjacent RU (Fig. 3B and C), which may stabilize the β-strand that holds Lys296 in the active site and contribute to the stability of the enzyme more generally. In addition, this highly ordered structure orients Glu348 in an interaction with Arg195, which sits in the active site (Fig. 3C).
Fig. 3.
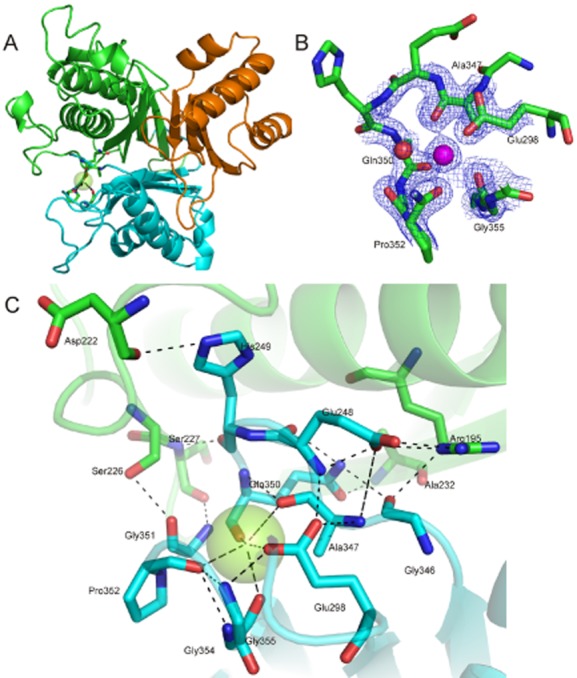
The metal binding site of AtzD.
A. The position of the bound metal (green sphere) in the AtzD monomer (coloured as per Fig. 2A).
B. The bound metal (magenta sphere), the metal-binding residues, the water ligand (red sphere) and their electron densities are shown.
C. The interactions between the metal, metal-stabilized loop and the surrounding protein.
Gel filtration during purification suggested that the native enzyme was a dimer (data not shown); however, Small Angle X-ray Scattering (SAXS; Supplemental Fig. S2) shows that AtzD forms a compact tetramer with D2 symmetry in solution (Fig. 4A and B). The diameter of the compact tetramer is near identical to that of the dimer, explaining the underestimate in native molecular weight using a lower resolution method (gel filtration) during purification. The core of the D2 tetramer is highly ordered, as indicated by the low B-factors found in the crystal structure (Fig. 4C and D). The surface of the tetrameric protein assembly has two cavities per monomer that could provide access to each active site for substrate ingress/product egress (Fig. 4E). One is formed by residues Tyr188, Met191, Thr321 and Gly345. The second is formed by residues Thr58, Phe82, Glu237 and Val242, and between the two cavities sits Lys162 (hydrogen bonded to the Met84 carbonyl oxygen).
Fig. 4.
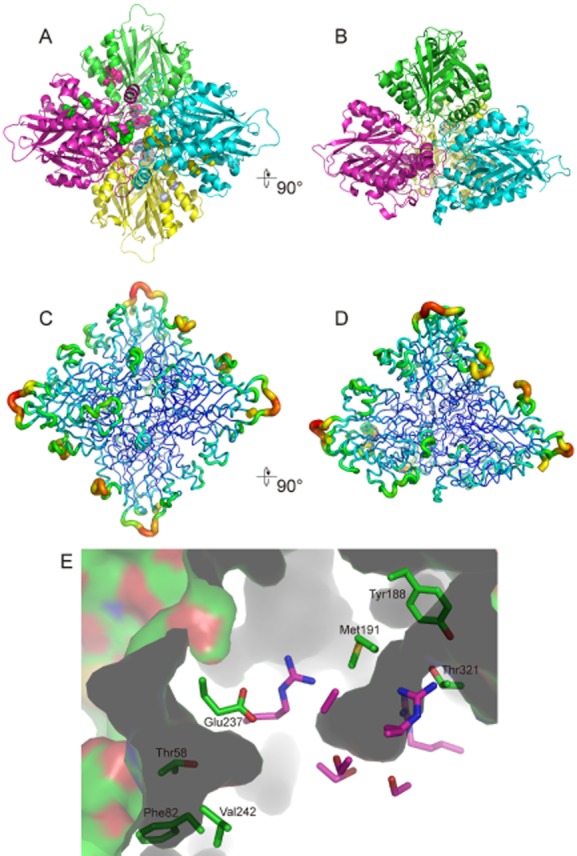
The AtzD tetramer.
A and B. Ribbon depictions of the tetrameric structure of AtzD and are approximate 90 degree rotations of each other. In (A) residues 58 and 81 (towards the periphery of the tetramer), and 188 and 191 (towards the centre of the tetramer) are shown as balls in colours that contrast with the main chain; these resides contribute to the two potential substrate channels.
C and D. The same rotations, but in this case the chains are coloured by B factors.
E. Two channels potentially provide access to the active site of each AtzD monomer. Active-site amino acid residues are shown in pink, but are otherwise unlabelled. The amino acid side-chains that form the channels are shown in green and their identities given.
Active site of AtzD
A phosphate ion was found in a polar, positively charged cavity in the AtzD monomer, making several hydrogen bonds with the protein. This cavity contains three lysine/serine dyads (Lys42, Ser85, Lys162, Ser233, Lys296 and Ser344) any, or all, of which could plausibly form the catalytic site (Botos and Wlodawer, 2007; Ekici et al., 2008).
Structures of AtzD were then obtained bound to CA or to melamine (1,3,5-triazine-2,4,6-triamine; Table 1, Fig. 5A–C). There was clear density for melamine and CA, displacing two well-ordered water molecules and the phosphate found in the ‘native’ structure (Fig. 5D). Melamine and CA bound in an identical fashion (0.47 Å rmsd) via an extensive hydrogen bonding network between CA/melamine and Arg54, Arg195, Arg325, Gly86, Ala234, Gly345, Ser85, Ser233 and Ser344 (Fig. 5B). BA-containing crystals were also obtained, but at lower resolution. The position of BA superposed well with the position of the melamine and CA.
Table 1.
Crystallographic statistics
| Data collection | ||||||
|---|---|---|---|---|---|---|
| PDB code | 3ZGR | 3ZGT | 3ZGU | 3ZGS | ||
| Ligand | Native | Melamine | Barbituric acid | Cyanuric acid | Xe-derivative | |
| Spacegroup | R32:H | R32:H | R32:H | R32:H | Spacegroup | R32:H |
| Cell (a = b × c) | 130.7 × 236.9 | 128.4 × 228.4 | 129.2 × 233.8 | 129.1 × 229.7 | Cell (a = b × c) | 130.7 × 233.1 |
| Resolution (Å) | 1.90 | 2.60 | 3.09 | 2.58 | Resolution (Å) | 2.55 |
| Completeness (%) | 99.8 (99.1) | 99.9 (99.9) | 99.5 (96.5) | 100 (100) | Completeness (%) | 99.8 (100) |
| Rmerge % | 0.056 (0.573) | 0.137 (0.784) | 0.145 (0.621) | 0.141 (0.799) | Rmerge % | 0.101 (0.739) |
| Mean I/sigI | 27.9 (4.4) | 12.8 (3.3) | 12.9 (3.6) | 12.5 (3.1) | Mean I/sigI | 21.9 (3.7) |
| # unique reflections | 61 745 | 22 546 | 13 929 | 23 460 | # unique reflections | 25 199 |
| Multiplicity | 11.1 | 11.1 | 9.7 | 11.1 | Multiplicity | 9.8 |
| Anomalous completeness | 99.8 (100) | |||||
| Anomalous multiplicity | 5.0 | |||||
| # Xe | 4 | |||||
| Refinement | Wavelength (Å) | 1.378 | ||||
| Resolution (Å) | 102.1–1.90 | 99.9–2.60 | 100.9–3.10 | 100.5–2.58 | ||
| No. Reflections | 58 623 | 21 401 | 13 210 | 22 272 | ||
| Rwork % | 17.5 (23.1) | 17.9 (25.7) | 15.5 (23.6) | 18.0 (24.8) | ||
| Rfree % | 19.9 (25.1) | 21.9 (29.3) | 22.3 (26.3) | 23.0 (29.0) | ||
| # atoms (total) | 5861 | 5376 | 5361 | 5440 | ||
| # waters | 264 | 20 | 5 | 37 | ||
| # metal ions | 2 | 2 | 2 | 2 | ||
| Mean B value overall (Å2) | 33.0 | 53.8 | 60.0 | 47.8 | ||
| Mean B value inhibitor (Å2) | NA | 28.2 | 40.4 | 37.9 | ||
| r.m.s.d. bond lengths (Å2) | 0.006 | 0.006 | 0.006 | 0.007 | ||
| r.m.s.d. bond angles (°) | 1.10 | 1.12 | 1.10 | 1.09 | ||
| Ramachandran analysis (%) preferred/allowed/outliers | 96.9/2.5/0.6 | 95.4/3.3/1.3 | 95.3/3.6/1.1 | 95.1/3.9/1.0 |
Values in parentheses are for the high-resolution bin.
Fig. 5.
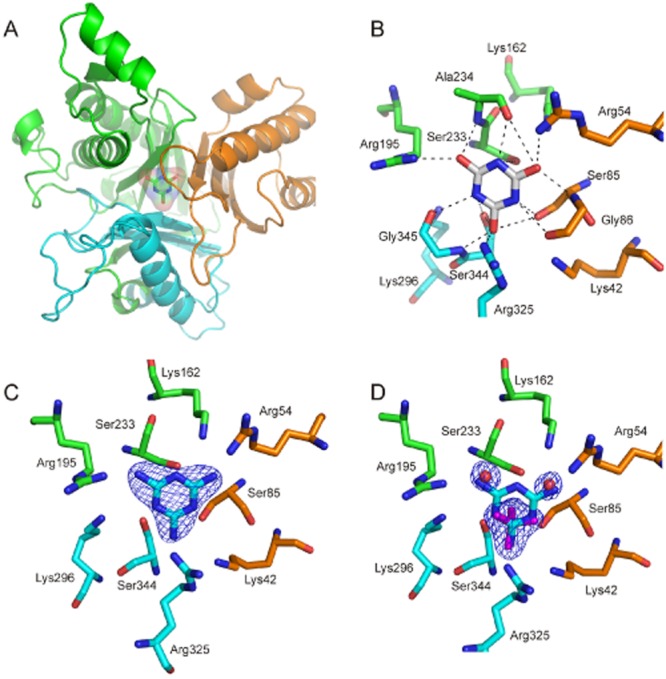
The active site of AtzD.
A. The cyanuric acid-bound monomer showing the Toblerone fold (cartoon with RUs coloured as in Fig. 2A) with the substrate bound in the active site (stick and surface).
B. Active site of AtzD with cyanuric acid bound. Hydrogen bonds are shown (dashed lines).
C. The AtzD active site with bound melamine.
D. Electron density for the inhibitor is shown.
Density for the phosphate (magenta) and two water molecules (red balls) in the ‘native’ data set with the melamine bound AtzD structure superposed.
The threefold symmetry of the Toblerone fold is also present in the active site (Fig. 5B and C), with each RU contributing an arginine, a serine, a lysine and a main-chain carbonyl (glycine/alanine) to the active site (Fig. 5B and C). Although all three serine/lysine pairs are found in the vicinity of the substrate, only the side-chain of Ser85 is within strong hydrogen bonding distance of the molecules (i.e. < 3 Å). In addition, we were able to detect significant polarization of the substrate's electron density that suggested that a covalent bond could form between the substrate and Ser85 (Fig. 6).
Fig. 6.
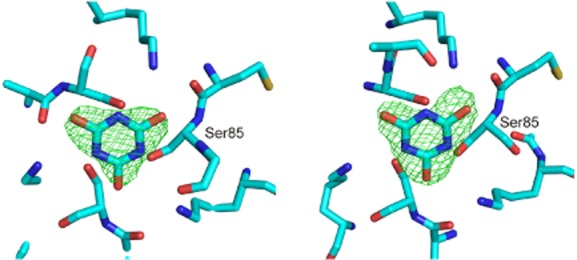
Electron density of cyanuric acid bound in the AtzD active site. Left structure obtained at pH 4.5. Right structure obtained at pH 6.6. In each case Ser85 has been labelled and the electron density obtained for the substrate is shown. In the crystal obtained at pH 4.5 (too low for hydrolysis) the density has a near symmetrical distribution. In the crystal obtained at pH 6.5 (i.e. with hydrolytically active enzyme) the density has become polarized in the vicinity of Ser85, suggesting that Ser85 is the catalytic serine.
Identification of catalytic amino acids
Consistent with earlier observations, the optimal pH for the reaction was 8.5–9.0 (Supplemental Fig. S3; Fruchey et al., 2003) and similar to other Lys–Ser containing hydrolases, such as the leader peptidase of E. coli, which uses a buried lysine with an apparent pKa of 8.7 as a general base to activate a nucleophilic serine (Paetzel et al., 1998). Inhibitor studies were also consistent with a Lys–Ser-dependent hydrolytic mechanism, as AtzD was inhibited by phenylmethanesulphonylfluoride (PMSF) and lysine methylation (Table 2).
Table 2.
Biochemical analysis of AtzD
| Inhibitor/treatment | Residual activity (%) |
|---|---|
| PMSF | 8.5 |
| o-Phenanthroline | 1.2 |
| EDTA | 100 |
| ApoAtzD plus Mg2+ (50 μM)a | 60 |
| Lysine alkylation | 7.5 |
ApoAtzD treated by the addition of 50 μM Zn2+, Na+, K+, Mn2+, Co2+, Cu2+, Ca2+ or Fe2+ failed to recover any catalytic activity.
Tryptic fingerprinting of AtzD that had been treated with PMSF was used in an attempt to determine which of the active-site serine residues participates in catalysis. In the untreated control, the tryptic fragments of AtzD containing all three active-site serine residues were observed (Supplemental Table S1). When the enzyme was treated with PMSF, the abundance of the Ser85-containing fragment was far lower than in the untreated control, while the other tryptic fragments (including those containing Ser233 and Ser344) do not vary substantively from the control (Supplemental Table S1). Although it was not possible to detect masses equivalent to those expected from the covalently modified Ser85-containing fragments, these observations imply that Ser85 may be the active-site serine, consistent with the crystallographic evidence observed (Fig. 6).
Unexpectedly, an unreported substrate inhibition was observed when substrate was supplied in molar excess (Supplemental Fig. S3; Fruchey et al., 2003; Seffernick et al., 2012). As it was not possible to delineate the effects of inhibition from the overall rate of the reaction, it was not possible to derive accurate steady-state estimates of kcat and KM. Reactions conducted in the presence of biuret were not inhibited (M. Wilding, unpubl. obs.), suggesting that the inhibition is mediated by either the substrate or 1-carboxybiuret.
AtzD activity was significantly reduced by incubation with o-phenanthroline (Table 2), suggesting that the bound metal is required for activity, albeit it appears to be too distant from the substrate-binding pocket to be involved directly in catalysis. Removal of the metal also resulted in a destabilization of the protein (> 80% precipitated at room temperature), which hampered our efforts to obtain a crystal of metal-free AtzD or accurately determine the Kd value for the bound metal. The activity of apoAtzD could be recovered by providing magnesium (MgCl2), but not Zn2+ as reported by Soong et al. for Bar (Soong et al., 2002) or other metal ions (Na+, K+, Mn2+, Co2+, Cu2+, Ca2+ and Fe2+). Along with the crystallographic and anomalous scattering data, these observations suggest that the native metal for AtzD is magnesium.
The three potential catalytic dyads were investigated by site-directed mutagenesis. Amino acid substitutions at Lys42 (Ala), Ser85 (Ala), Lys162 (Ala, Arg), Ser233 (Ala), Lys296 (Ala, Arg) and Ser344 (Ala) yielded soluble, but inactive, protein. The unfolding profiles for the variants, as judged by differential scanning fluorimetry (data not shown), were consistent with correctly folded protein. While this provides no direct evidence for the identities of the catalytic residues of AtzD, it does suggest that these residues are critical for catalysis.
Determinants of substrate specificity
AtzD is inhibited by BA and Bar is inhibited by CA, despite the near identical structures of the compounds (Fig. 1A and B). To understand the differences in substrate specificity between AtzD and Bar, the structure of Bar was modelled using the AtzD structure as a template (∼ 44% identical in sequence; Supplemental Fig. S4). The model shows a similar active site with changes in active-site residues being (AtzD to Bar): Arg195/Asn194, Gly345/Val348, Arg325/Lys328, Thr321/His324 and Gly346/Ser349 (Fig. 7). The Gly345Val change is of particular interest as it impinges sterically on the active-site space and introduces a hydrophobic residue in a predominantly hydrophilic pocket.
Fig. 7.
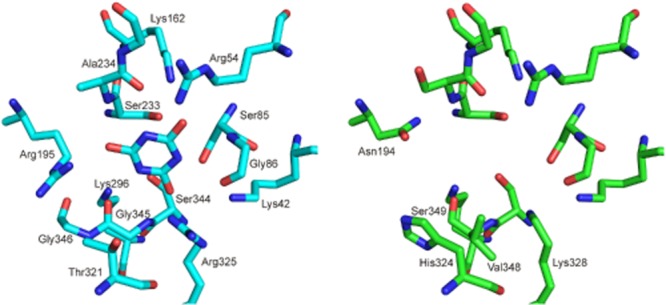
Comparison of the active sites of AtzD and a homology model of Bar. The AtzD active site is shown (left, cyan) with cyanuric acid bound. The modelled Bar active site (right, green) is shown without cyanuric acid docked, only the amino acid residues that differ between AtzD and Bar are indicated on the Bar structure.
The major effect of these differences is that the Bar active site does not possess the almost perfect rotational symmetry that AtzD has in the active site, instead Bar has a single axis of bilateral symmetry (Fig. 7). The differences in the active site mirror those of the substrates, in that CA possesses rotational symmetry, while BA does not due to the atomic composition at position 5 in the ring (Fig. 1A and B). The introduction of the hydrophobic surface formed by Val348 in Bar and the loss of positive charge associated with the Arg195-Asn194 substitution in Bar when compared with AtzD also dramatically changes the electronic structure of the active site. It is speculated that these steric and electronic differences underpin the substrate specificity for AtzD and Bar, albeit further data are required to support this hypothesis.
As residues 195, 321, 325, 345 and 346 in AtzD appear to play a role in determining substrate specificity, the identities of the amino acids at equivalent positions in all 68 known AtzD homologues were analysed and compared with a phylogenetic analysis of these sequences (Fig. 8). Six major phylogenetic groups were discovered. Groups I, II and III possess AtzD-like signatures at positions equivalent to 195, 321, 325, 345 and 346 in AtzD (i.e. Arg195, Thr321, Arg325, Gly345 and Gly346). Indeed, seven characterized CA hydrolases (AtzD and homologues from Pseudomonas sp. ADP, Moorella thermoacetica, Pseudomonas sp. NRRLB-12227, Bradyrhizobium japonicum USDA 110, Rhizobium leguminosarum bv. viciae 3841, Methylobacterium sp. 4-46 and locus AZC_3892 from Azorhizobium caulinodans ORS 571; Karns, 1999; Seffernick et al., 2009; 2012) are located within Groups II and Group III (Fig. 8).
Fig. 8.
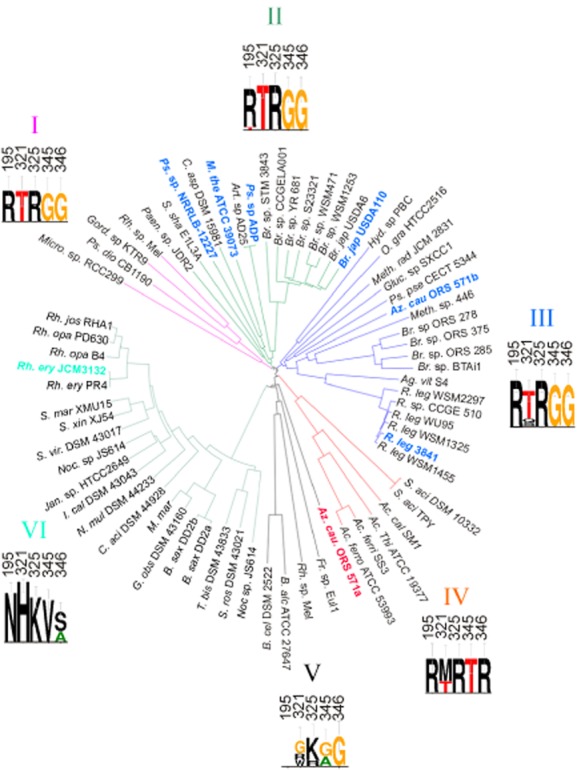
Phylogeny of the AtzD/Bar family. The sequences for the AtzD/Bar homologues in the six groups were sourced from the following organisms (listed in order, clockwise): Group I (purple); Micromonas sp. RC299, Pseudomonas dioxanivorans CB1190, Gordonia sp. KTR8, Rhodococcus sp. Mel. Group II (green); Paenibacillus sp. JDR2, Salinisphaera shabanensis E1L3A, Pseudomonas sp. NRRLB-12227, Clostridium asparagiforme DSM 15981, Moorellia thermoacetica ATCC 39073, Arthrobacter sp. AD25, Pseudomonas sp. ADP, Bradyrhizobium sp. STM3843, Bradyrhizobium sp. CCGELA001, Bradyrhizobium sp. YR681, Bradyrhizobium sp. S23321, Bradyrhizobium sp. WSM471, Bradyrhizobium sp. WSM1253 Bradyrhizobium japonicum USDA6, Bradyrhizobium japonicum USDA110. Group III (blue); Hydrogenophaga sp. PBC, Oceanicoia granulosus HTCC2516, Methylobacterium radiotolerans JCM 2831, Pseudomonas pseudoalcaligenes CECT 5344, Azorhizobium caulinodans ORS 571, Methylobacterium sp. 446, Bradyrhizobium sp. ORS 278, Bradyrhizobium sp. ORS 375, Bradyrhizobium sp. ORS 285, Bradyrhizobium sp. BTAi1, Agrobacterium vitis S4, Rhizobium leguminosarum bv. trifolii WSM2297, Rhizobium sp. CCGE 510, Rhizobium leguminosarum bv. trifolii WU95, Rhizobium leguminosarum bv. trifolii WSM1325, Rhizobium leguminosarum bv. viciae 3841, Rhizobium leguminosarum bv. viciae WSM1455. Group IV (red): Sulfobacillus acidophilus DSM 10332, Sulfobacillus acidophilus TPY, Acidithiobacillus caldus SM1, Acidithiobacillus thiooxidans ATCC 19377, Acidithiobacillus ferrivorans SS3, Acidithiobacillus ferrooxidans ATCC 27647. Group V (black): Azorhizobium caulinodans ORS571, Frankia sp. Eul1c, Rhodococcus sp. Mel, Bacillus alcalophilus ATC 27647, Bacillus cellulolytitcus DSM 2552. Group VI (cyan): Nocardioides sp. JS614, Streptosporangium roseum DSM 43021, Thermobispora bispora DSM 43833, Blastococcus saxobsidens DD2, Blastococcus saxobsidens DD2, Geodermatophilus obscures DSM 44928, Modestobacter marinus, Catenulispora acidophia DSM 44928, Nakamurella multipartita DSM 43233, Intrasporangium calvum DSM 43043, Janibacter sp. HTCC2649, Nocardioides sp. JS614, Saccharomonospora viridis DSM 43017, Saccharomonospora xinjiangensis XJ54, Saccharomonospora marina XMU15, Rhodococcus erythropolis PR4, Rhodococcus erythropolis JCM3132, Rhodococcus opacus B4, Rhodococcus opacus PD630, Rhodococcus jostii RHA1. The identities and conservation of amino acids found in each at positions equivalent to AtzD 195, 321, 325, 345 and 346 are shown as logos. The names of bacterial species from which Toblerone fold enzymes have had their catalytic activities characterized are emboldened and coloured to indicate their substrate specificity: blue for cyanuric acid hydrolase, cyan for barbituric acid hydrolase and red for the absence of hydrolytic activity with either cyanuric acid or barbituric acid.
Group VI contains the characterized Bar enzyme and there is strong conservation of a Bar-like signature within this group (i.e. Asn195, His321, Lys325, Val345 and Ser/Ala346). Groups IV and V possess signatures unlike either AtzD or Bar; Group IV has an Arg195, Met/Thr321, Arg325, Thr345 and Arg346 motif, while Group V has low conservation at positions 321 (a Gly, Arg or Trp), 325 (Lys/Arg) and 345 (Gly/Ala) and a conserved Gly at position 346. The equivalent of position 195 is absent in Group V. A single enzyme in Group V has been examined for CA and BA hydrolase activity (locus AZC_3203 from A. caulinodans ORS 571; Seffernick et al., 2012) and no such activity could be detected. The remaining AtzD active-site residues (Lys42, Lys162, Lys296, Arg54, Arg325, Gly86, Ala234, Ser85, Ser233 and Ser344) were conserved throughout the Toblerone fold proteins (not shown), suggesting that they are not involved in substrate specificity.
Discussion
Reaction mechanism
Structural, mutagenesis and biochemical studies suggest that AtzD, and the other members of the Toblerone fold family (TrzD and Bar), are lysine-serine hydrolases. The Michaelis complex is formed when CA binds to AtzD via an extensive hydrogen bonding network (Fig. 5B). Binding to this highly positively charged pocket promotes the formation of the triketide, eliminating the aromaticity of the substrate, with the hydrogen bonding interactions provided by Arg54, Arg195 and Arg325 possibly polarizing the substrate, making the carbonyl carbons more electrophilic and the lactones more scissile. Hydrolysis and formation of the enzyme:acyl intermediate then occurs via nucleophilic attack by a lysine-activated serine (evidenced by inhibitor and pH studies).
A number of potential catalytic mechanisms involving serine and lysine residues are possible, from a simple Ser–Lys dyad to more complex catalytic mechanisms involving multiple serine and lysine residues, with such configurations as Ser–cisSer–Lys, Ser–Xxx–Xxx–Lys and Lys–Ser–Ser–Lys (McKinney and Cravatt, 2003; Ekici et al., 2008; Pratt and McLeish, 2010). Although there are three pseudo-equivalent Lys–Ser dyads, it is presumed that a single serine acts as the nucleophile during a single catalytic event, as the product (carboxybiuret; Fig. 1A) is produced via hydrolysis of a single amide bond. It is not possible to definitively attribute the nucleophilic activity to any one of the three active-site serines, and it is possible that any or all three may act as nucleophile (albeit only one can do so per catalytic cycle).
The position of the substrate in the active site (Fig. 5A and B) places Ser85 such that it is closer to a scissile bond than are the other serine residues. Additionally, pH-dependent polarization of the substrate's electron density and a reduction in abundance of the Ser85 containing polypeptide in mass spectra of PMSF-treated tryptic digests of AtzD are suggestive that Ser85 is the dominant (if not sole) nucleophile in the hydrolytic mechanism. A hypothetical reaction mechanism consistent with a Ser85 nucleophile is therefore proposed: Lys42 is a general base, activating Ser85 and promoting formation of a tetrahedral intermediate between Ser85 and the closest substrate carbonyl carbon, this then resolves into the acyl:enzyme intermediate following ring opening (Fig. 9). Thereafter, a solvent water molecule is required to hydrolyse the acyl intermediate and regenerate the serine, liberating carboxybiuret (which then spontaneously decarboxylates to form biuret; Seffernick et al., 2012).
Fig. 9.

Plausible catalytic mechanism for cyanuric acid hydrolysis by AtzD. Ser85 is activated by Lys42 and acts as nucleophile with bound CA (1), allowing nucleophilic attack at one of the three ketone positions of CA. The tetrahedral intermediate formed undergoes ring opening to form an acyl intermediate. The acylated serine is subsequently hydrolysed by a solvent water molecule, again activated by Lys42, via a tetrahedral intermediate to form carboxybiuret (2) which spontaneously decarboxylates to form biuret (3) and the active site is regenerated by intramolecular proton transfer.
Evolution of the Toblerone fold
To the authors' knowledge AtzD possesses a unique fold, related to, but distinct from, the YgjF-family of trimeric proteins. At some point in their evolutionary history, the subunits of the trimeric ancestor of AtzD and Bar concatenated, possibly following successive gene duplications. On the other hand, the RUs may have been ‘recruited’ from other members of the YgjF protein family. An alternative hypothesis is that an ancestral Toblerone fold protein was truncated to one-third its original length (i.e. a single RU) that was capable of forming stable trimers. However, YgjF-family members are found in all domains of life, catalysing a wide range of reactions and participating in diverse physiological processes (Kim et al., 2001; Schaap et al., 2002; Burman et al., 2007). Until the introduction of s-triazine herbicides, the AtzD/Bar enzymes were only known to participate in a single physiological process (BA hydrolysis) in a limited number of species (Seffernick et al., 2012). The concatenation scenario is therefore the more likely of the two possible evolutionary histories.
The AtzD active site is located in the central ‘barrel’ of the protein (Fig. 5). In contrast, the three binding pockets of each PSP/RutC trimer are formed by the interactions between adjoining monomers and are located on the outside of the protein (Zhang et al., 2010; Knapik et al., 2012). In both PSP and RutC, the central barrel provides the core interactions that maintain the trimer (Zhang et al., 2010; Knapik et al., 2012). In the AtzD family, the strength of these core interactions may have been reduced in an evolutionary trade-off with the optimization of the catalytic function, which could have resulted in a destabilization of the protein. It is plausible that concatenation of the three RUs of AtzD compensates for the loss of the stabilizing core interactions, although it is unclear if concatenation was a prerequisite or a consequence of this trade off.
Removal of the metal ion from AtzD greatly reduced its stability, resulting in protein precipitation at room temperature, suggesting that formation of the metal binding site is also an evolutionary innovation that provides enhanced stability. It is notable that none of the other cations tested appear to substitute for Mg2+. Bar has been reported to be Zn2+-dependent (Soong et al., 2002), despite the fact that the metal binding sites of AtzD and Bar are completely conserved (as it is in 90% of the known AtzD/Bar homologues). Moreover, the metal binding site does not appear well adapted for binding Zn2+, albeit the structure of apoAtzD was not determined due to its inherent instability, and it is conceivable that the structure of the metal-binding pockets in holoAtzD/Bar are, in part, determined by the identity of the bound cation. It is also possible that there is a second, unidentified, Zn2+ binding site in Bar. However, there are no canonical Zn2+-binding motifs in the primary sequence of Bar (Seffernick et al., 2012), and so such a site would necessarily be another novel metal binding site.
Distribution of CA and BA hydrolase activities
It appears that there are six phylogenetically distinct groups within the AtzD/Bar family. The six groups are also separated by the identities of the amino acids that are likely to distinguish substrate specificities of AtzD and Bar. Phylogenetic Group II contains AtzD and TrzD and the predicted determinants of substrate specificity for CA are highly conserved throughout this group (Fig. 8), as they are in Groups I and III. This may suggest that Groups I–III contain CA hydrolases. Group VI contains Bar and the Bar-like specificity signature is highly conserved within this group, suggesting that Group VI contains BA hydrolases. The high level of conservation for the ‘specificity signature’ residues within these groups is consistent with their inferred roles in determining substrate specificity. Moreover, a number of enzymes in Groups II, III and VI have been characterized elsewhere and found to possess activities consistent with the proposed CA or BA hydrolases (Fig. 8; Seffernick et al., 2012).
Groups IV and V are unlike the CA hydrolases, BA hydrolases or each other, and may fulfil different physiological roles. Indeed, a single Group V enzyme from A. caulinodans ORS571 has been characterized (Seffernick et al., 2012), and found to possess neither AtzD-like nor Bar-like substrate specificities. Group V is composed of proteins from five bacterial species (from the genera Rhizobium, Azorhizobium, Frankia and Bacillus). There is far lower conservation in the amino acids identified in this study as specificity determinants, suggesting that these enzymes may not share substrate specificities.
It is interesting to note that the Group IV enzymes belong to the genera Acidothiobacillus (Gram-negative, γ-proteobacter) and Sulfobacillus (Gram-positive, Clostridiales). Although phylogenetically distinct, Acidothiobacillus and Sulfobacillus are physiologically similar (Waksman and Joffe, 1922; Temple and Colmer, 1951; Golovacheva and Karavaiko, 1978; Kovalenko and Malakhova, 1983; Karavajko et al., 1990; Narayan and Sahana, 2009); both are acidophiles that oxidize iron and elemental sulphur, indeed both are exploited in biomining/bioleaching applications (Narayan and Sahana, 2009; Tang et al., 2009). It may be that the Toblerone fold enzymes of these two genera fulfil a physiological role that is unique to their unusual physiologies.
Conclusion
AtzD is the archetype for the Toblerone fold, which has likely evolved by the concatenation of three genes encoding proteins of the YgjF superfamily. The concatenation, along with the evolution of a stabilizing metal binding site (that may also help organize the active site), has allowed the repurposing of the stabilizing central barrel of the monomer for catalysis, which has resulted in a unique family of ring-opening hydrolases. Active-site residues have been identified that differ between AtzD and Bar, and are therefore implicated in determining substrate specificity. The inferred ‘specificity signature’ varies between the six phylogenetic groups of the Toblerone fold family, and is well conserved within Groups I–IV and VI, suggesting that these residues may indeed confer substrate specificities to this family of enzymes.
Experimental procedures
DNA manipulation
The atzD gene (Accession No. U66917) was provided by GenScript, as an NdeI/BamHI insert in pUC57. The atzD gene was subcloned into the NdeI and BamHI sites of pETCC2. The pETCC2 expression vector was a gift from Dr Christopher Coppin (CSIRO Ecosystem Sciences) and is derived from pET14b (Novagen). An in-frame N-terminal hexahis-tag along with thrombin cleavage site (MGSSHHHHHHSSGLVPRGSH) was added to the encoded enzyme as a result of the subcloning.
Mutagenesis was by the over-lapping PCR method of Ho et al. (1989) The oligonucleotide primers used are detailed in Supplemental Table S2. Amplicons were resolved on a 0.8% agarose gel and purified using NucleoSpin® Gel kits (Machery Nagel). Purified BamHI and NdeI digested amplicons were cloned into the pETCC2 vector using T4 DNA ligase (NEB). Sequence verification was done by Macrogen (Korea).
Protein expression and purification
Electrocompetent E. coli BL21 λ(DE3) (Invitrogen) were transformed with appropriate plasmids (Suppl Table 2), then grown in Lennox medium (LB; Lennox, 1955) or on LB agar (15 mg ml−1 agar, Merck) supplemented with 100 μg ml−1 ampicillin (Sigma Aldrich). Starter cultures (50 ml) were grown from single colonies at 37°C for 18 h while being shaken at 200 r.p.m., used to inoculate 950 ml of LB and incubated at 37°C to an OD600 of 0.6–0.8. Cultures were induced by addition of 100 μM isopropyl-beta-d-thiogalactopyranoside (IPTG; Astral) and incubated at 37°C overnight while shaking at 200 r.p.m. Cultures were then harvested by centrifugation (4000 g, 10 min) in an Avanti J-E centrifuge (Beckman Coulter) and resuspended in lysis buffer (50 mM sodium phosphate pH 7.5, 50 mM NaCl) and lysed by passing through Avestin C3 homogenizer three times at 18 000 p.s.i. Insoluble material was removed by centrifugation (18 000 g) using an Avanti J-E centrifuge.
Metal ion affinity chromatography (Ni-NTA Superflow Cartridge; Qiagen) was used as per the manufacturer's instructions. AtzD eluted at 160 mM imidazole. AtzD was further purified by size exclusion chromatography using a 130 ml column packed with Superdex 200 prep grade resin (GE Healthcare Life Sciences) using lysis buffer. AtzD was estimated to be > 98% pure by Coomassie (Sigma Aldrich) stained precast SDS-PAGE gel (4–20% Tris-HEPES-SDS, Thermo Scientific), and typical yields were 7.5–8.5 mg from 1 l of culture.
Crystallization
Crystals of native AtzD were prepared as follows: concentrated protein (10.4 mg ml−1 in lysis buffer) was set up in crystallization droplets consisting of 200 nl protein solution, 250 nl crystallization cocktail and 50 nl additive solution [0.1 M sodium HEPES pH 6.9, 0.2 M NH4Cl, 20% (w/v) polyethylene glycol (PEG) 6000]. The crystallization cocktail contained a low-molecular-weight PEG (either PEG 300, PEG monomethyl ether 350 or PEG 400) between 35% and 50% (v/v) in sodium phosphate (50–100 mM; pH 6.5–8) buffer, with or without NaCl (100–200 mM). All chemicals were obtained from Sigma Chemicals (St. Louis, USA). Droplets were set up with a Phoenix nanodispensing robot (ARI, Sunnyvale, CA) into SD-2 plates (Molecular Dimensions, UK) and were stored at 20°C. Triangular prism-shaped crystals appeared within 24 h, and grew large enough to obtain X-ray data within a week (Supplemental Fig. S5).
Substrate/inhibitor bound AtzD crystals were prepared as follows: AtzD protein in 50 mM sodium phosphate pH 7.5, 50 mM NaCl was dialysed against HEPES buffer (50 mM, pH 7.5) containing NaCl (50 mM), DTT (5 mM) and melamine (10 mM), barbituric acid (10 mM) or cyanuric acid (0.1 mM) and then set up in crystallization experiments as described above, against crystallization conditions without phosphate.
Crystal data collection
Crystals were taken to the MX-2 beamline of the Australian Synchrotron and cryo-cooled to 100 K directly in the nitrogen stream. A xenon derivative was prepared by exposing a crystal to xenon gas at 20 p.s.i. for 2 min, and flash-cooled in liquid nitrogen. A total of 720 frames of 0.5° oscillation were used to collect 360 degrees of data for the Xe derivative data set. All further data sets consisted of 360 frames of 0.5° oscillation for a total of 180 degrees of data. Each was processed as described below. Melamine, barbituic acid and cyanuric acid were all placed in clear density using Afitt (OpenEye Scientific Software, USA) for those data sets collected after co-crystallization with these compounds.
Anomalous data were collected at 1.7149 Å (7230 eV) on 17 isomorphous AtzD native crystals and the best data were merged to give a data set with 140-fold multiplicity which gave exquisite anomalous difference maps, showing every sulphur atom, the phosphate ions as well as the two metal binding sites (Supplemental Fig. S1).
Structural determination
Data sets were indexed with XDS (Kabsch, 2010) and processed with SCALA (Evans, 2006), and entered into the SIRAS protocol in Auto-Rickshaw (Panjikar et al., 2005) to locate the four xenon atoms. Phases from this derivative were sufficient to allow Buccaneer (Cowtan, 2006) to autobuild a partial model, which was used in Phaser (McCoy et al., 2007) as a molecular replacement model (crystallographic statistics are found in Table 1). These data allowed the complete tracing of the chain from residue 2 to residue 364, using the program coot (Emsley et al., 2010). The model was refined using Refmac (Murshudov et al., 1997) to a final resolution of 1.90 Å, and showed clean density for the entire chain excepting weak density for the residues of the loop from residues 105 to 109. This model was subsequently used for molecular replacement using Phaser (McCoy et al., 2007) for subsequent data sets with inhibitors bound.
Small-angle X-ray scattering (SAXS)
AtzD was dialysed overnight into a 50 mM phosphate buffer, 50 mM NaCl, 1 mM DTT (pH 7.5). The same buffer was used as the buffer standard during data collection. A dilution series of AtzD (from 0.19 to 3 mg ml−1) was prepared, and scattering data were collected for 1 s using a Pilatus 1 M photon counting detector (Dektris) with a sample to detector distance of 1.6 m. Ten replicate images were collected for each sample and averaged, with outlier rejection, to control for radiation damage. Data were measured in a Q range from 0.01 to 0.5 Å−1 and at the highest protein concentration the scattering remained above the noise threshold to the edge of the detector.
AtzD (75 μl, 6 mg ml−1) was injected on to a size exclusion column (Wyatt Silica Bead column, 300 Å pore size) that had been pre-equilibrated with the PO4/NaCl buffer. The column was developed at 0.2 ml min−1, and a single peak eluted. The SAXS scattering showed no change over the peak (data not shown). A CRYSOL (Svergun et al., 1995) fit of various quaternary structures for the protein to the scattering data was performed (Supplemental Fig. S2).
Kinetic analysis
Hydrolysis of CA and BA was assayed by UV-visible spectroscopy as described elsewhere (Seffernick et al., 2012), using a SpectraMax M2 spectrophotometer (Molecular Devices). Reactions [200 μl in 1 mM TAPS (N-Tris(hydroxymethyl)methyl-3-aminopropanesulphonic acid; pH 8.5); 1 μM AtzD] with 10–1000 μM substrate were assayed in UV-transparent 96-well microtitre plates (Greiner Bio-one). Absorbances were measured at 15 s intervals for 10 min. The pH-dependence of AtzD was determined in MOPS [3-(N-morpholino)propanesulphonic acid; 1 mM, pH 6.5, 6.9 and 7.5], TAPS (1 mM pH 7.5, 7.9 and 8.5) and CHES (N-Cyclohexyl-2-aminoethanesulphonic acid; 1 mM pH 9).
Enzyme inhibition
Alkylation of lysines in AtzD was carried out using JBS Methylation Kit (Jena Bioscience) following manufacturer's guidelines. A protein concentration of 6 mg ml−1 in sodium phosphate (50 mM, pH 7.5)/NaCl buffer (50 mM) was used at the time of treatment. Phenylmethylsulphonyl fluoride (PMSF) was prepared at 100 mM in isopropanol. Enzyme solutions were prepared at 4 μM concentrations in MOPS buffer (1 mM, pH 6.5) and treated twice with PMSF (1 mM final concentration) for 30 min. Trypsin was used to as a positive control using Nα-Benzoyl-l-arginine ethyl ester hydrochloride (BAEE) as a control substrate. Trypsin activity was monitored by UV spectrophotometrically at 253 nm.
Ethylenediaminetetraacetic acid (EDTA; 20 mM) and o-phenanthroline (75 mM) in 25 mM MOPS buffer (pH 8.5) were used to dialyse AtzD (117 μM) overnight at 4°C. Chelators were removed by centrifugation in a 30 kDa spin column (Amicon Ultra, Millipore). The zinc amidohydrolase TrzN (E.C. 3.8.1.8) was used as a positive control. Metals (Mg2+ Na+, K+, Mn2+, Co2+, Cu2+, Ca2, Zn2+ and Fe2+; 50 μM) were added to o-phenanthroline treated ApoAtzD and incubated at 4°C for 16 h.
Mass spectrometry
Peptides generated from tryptic digestions of AtzD were acidified with 0.5% formic acid, filtered [Millex-LG, low protein binding hydrophilic LCR (PTFE), 0.2 μm], and analysed by nanoflow, reversed phase, liquid chromatography-tandem mass spectrometry (MS) using an Agilent Chip Cube system coupled to an Agilent XCD ion trap mass spectrometer. MSMS spectra were assigned to sequences by several rounds searching and ‘autovalidation’ using SpectrumMill software (Agilent Rev A.03.03.084 SR4). First a small database of common contaminant sequences such as trypsin and keratins was searched assuming tryptic cleavages. Remaining unassigned spectra were used to search just the AtzD sequence allowing trypsin- and chymotrypsin-specific cleavages. The trypsin (Sigma-Aldrich) had not been treated to prevent chymotrypsin-specific cleavages and this generated some mixed tryptic/chymotryptic cleavages that usefully extended the sequence coverage. Finally, remaining unassigned spectra were used to search the AtzD sequence in ‘homology’ mode allowing single unassigned mass gaps in sequences that were otherwise consistent with MS/MS spectra. Spectra assigned in this last mode were inspected individually to verify putative modifications of amino acid residues.
Modelling of Bar
Bar was modelled using the Swiss-Model software as found on the web server (Arnold et al., 2006); the AtzD structure was used as the starting point (44% sequence identity over 364 residues). There are some minor differences between the structures in various loops: residues 74–78 (AtzD) has an extra residue in the AtzD loop; residues 106–111 (AtzD) this loop of weak density in the AtzD structure is built slightly differently in the Bar structure; residues 210–215 (AtzD) has two extra residues in the AtzD loop; residues 280–283 (AtzD) has six extra residues in the Bar modelled loop; the AtzD structure has a single amino acid residue extension at the C-terminus of the protein as well.
In silico sequence analysis
AtzD homologues were identified via a BlastP search of non-redundant databases. The following sequences were returned with E-values of less than 10−30 and identity scores of greater than 40%, there were no other hits with E-values of less than 101: NP862537 (Pseudomonas sp. ADP), ABK41866 (Arthrobacter sp. AD25), YP430955 (M. thermoacetica ATCC 39073), ZP10581004 (Bradyrhizobium sp. YR681), ZP09650932 (Bradyrhizobium sp. WSM471), P0A3V4 (Pseudomonas sp. NRRLB-12227), ZP10083023 (Bradyrhizobium sp. WSM1253), YP005453208 (Bradyrhizobium sp. S23321), YP005606973 (B. japonicum USDA 6), ZP09433530 (Bradyrhizobium sp. STM 3843), EJZ29306 (Bradyrhizobium sp. CCGE-LA001), NP773921 (B. japonicum USDA 110), ZP03758143 (Clostridium asparagiforme DSM 15981), YP003013624 (Paenibacillus sp. JDR-2), YP001757420 (Methylobacterium radiotolerans JCM 2831), ZP10760424 (Pseudomonas pseudoalcaligenes CECT 5344), ZP08551100 (Salinisphaera shabanensis E1L3A), ZP08318418 (Gluconacetobacter sp. SXCC-1), YP004783181 (Acidithiobacillus ferrivorans SS3), YP002219377 (Acidithiobacillus ferrooxidans ATCC 53993), YP001526119 (A. caulinodans ORS 571), YP001208170 (Bradyrhizobium sp. ORS 278), YP002547456 (Agrobacterium vitis S4), YP004750255 (Acidithiobacillus caldus SM-1), EIW44662 (R. leguminosarum bv. trifolii WU95), YP770629 (R. leguminosarum bv. viciae 3841), EJC71551 (R. leguminosarum bv. viciae WSM1455), YP002979447 (R. leguminosarum bv. trifolii WSM1325), ZP09420969 (Bradyrhizobium sp. ORS 375), YP001526808 (A. caulinodans ORS 571), ZP10837886 (Rhizobium sp. CCGE 510), EJC83804 (R. leguminosarum bv. trifolii WSM2297), ZP09473782 (Bradyrhizobium sp. ORS 285), YP001237458 (Bradyrhizobium sp. BTAi1), ZP01155857 (Oceanicola granulosus HTCC2516), ZP09997547 (Acidithiobacillus thiooxidans ATCC 19377), ZP10152173 (Hydrogenophaga sp. PBC), YP006671162 (Gordonia sp. KTR9), YP005257446 (Sulfobacillus acidophilus DSM 10332), YP004719285 (S. acidophilus TPY), YP001770627 (Methylobacterium sp. 4-46), AEX65082 (Rhodococcus sp. Mel), YP00433323 (Pseudonocardia dioxanivorans CB1190), XP002503480 (Micromonas sp. RCC299), CAC86669 (Rhodococcus erythropolis), YP004017027 (Frankia sp. EuI1c), YP004094229 (Bacillus cellulosilyticus DSM 2522), YP003112640 (Catenulispora acidiphila DSM 44928), YP002769329 (R. erythropolis PR4), YP922706 (Nocardioides sp. JS614), YP002779991 (Rhodococcus opacus B4), YP005331388 (Blastococcus saxobsidens DD2), ZP00996765 (Janibacter sp. HTCC2649), YP003133937 (Saccharomonospora viridis DSM 43017), YP005327200 (B. saxobsidens DD2), YP006364153 (Modestobacter marinus), ZP09986349 (Saccharomonospora xinjiangensis XJ-54), YP003204583 (Nakamurella multipartita DSM 44233), ZP09740740 (Saccharomonospora marina XMU15), EHI43897 (R. opacus PD630), YP004098318 (Intrasporangium calvum DSM 43043), YP003407358 (Geodermatophilus obscurus DSM 43160), YP003341997 (Streptosporangium roseum DSM 43021), YP705235 (Rhodococcus jostii RHA1), YP925454 (Nocardioides sp. JS614), YP003652816 (Thermobispora bispora DSM 43833), ZP10819722 (Bacillus alcalophilus ATCC 27647), AEX65050 (Rhodococcus sp. Mel).
Alignments and bootstrapping (1000×) of the AtzD superfamily sequences were carried out using clustalw (http://www.bioinformatics.nl/tools/clustalw.html). A phylogenetic tree was constructed from this alignment using the Interactive Tree of Life (iTol; Letunic and Bork, 2007;2011) and consensus sequences were created using Weblogo sequence generator (Crooks et al., 2004).
Accession codes
The molecular models for AtzD and AtzD bound to CA, melamine and BA were deposited in the Protein Data Bank, with PDB codes 3ZGR, 3ZGS, 3ZGT and 3ZGT.
Acknowledgments
We thank Dr Peter Campbell (CSIRO Ecosystem Sciences) for his assistance with mass-spectrometry, Drs Carol Hartley and Andrew Warden (CSIRO Ecosystem Sciences) for their constructive comments, and the beamline scientists at the Australian Synchrotron for their help in data collection. We also thank James Holton and Christine Gee for help with anomalous data collection. Crystals were grown in the CSIRO Collaborative Crystallisation Centre (http://www.csiro.au/C3) at CSIRO Materials Science and Engineering. This work was supported in part by an Office of the Chief Executive Julius Career Development Award (C.S.).
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
References
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Botos I, Wlodawer A. The expanding diversity of serine hydrolases. Curr Opin Struct Biol. 2007;17:683–690. doi: 10.1016/j.sbi.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman JD, Stevenson CEM, Sawers RG, Lawson DM. The crystal structure of Escherichia coli TdcF, a member of the highly conserved YjgF/YER057c/UK114 family. BMC Struct Biol. 2007;7:30. doi: 10.1186/1472-6807-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudev T, Cowan JA, Lim C. Competitive binding in magnesium coordination chemistry: water versus ligands of biological interest. J Am Chem Soc. 1999;121:7665–7673. [Google Scholar]
- Ekici OD, Paetzel M, Dalbey RE. Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008;17:2023–2037. doi: 10.1110/ps.035436.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Fruchey I, Shapir N, Sadowsky MJ, Wackett L. On the origins of cyanuric acid hydrolase: purification, substrates, and prevalence of AtzD from Pseudomonas sp strain ADP. Appl Environ Microbiol. 2003;69:3653–3657. doi: 10.1128/AEM.69.6.3653-3657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovacheva RS, Karavaiko GI. New genus of thermophilic spore-forming bacteria, Sulfobacillus. Microbiology. 1978;47:658–665. [PubMed] [Google Scholar]
- Hayes T, Haston K, Tsui M, Hoang A, Haeffele C, Vonk A. Atrazine-induced hermaphroditism at 0.1 ppb in American leopard frogs (Rana pipiens): laboratory and field evidence. Environ Health Perspect. 2003;111:568–575. doi: 10.1289/ehp.5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain-reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karavajko GI, Bulygina ES, Tsaplina IA, Bogdanova TI, Chumakov KM. Sulfobacillus thermonsulfoidooxidans – a new lineage of bacterial evolution. FEBS Lett. 1990;261:8–10. doi: 10.1016/0014-5793(90)80623-q. [DOI] [PubMed] [Google Scholar]
- Karns JS. Gene sequence and properties of an s-triazine ring-cleavage enzyme from Pseudomonas sp. strain NRRLB-12227. Appl Environ Microbiol. 1999;65:3512–3517. doi: 10.1128/aem.65.8.3512-3517.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Yoshikawa H, Shirahige K. A member of the YER057c/yjgf/Uk114 family links isoleucine biosynthesis and intact mitochondria maintenance in Saccharomyces cerevisiae. Genes Cells. 2001;6:507–517. doi: 10.1046/j.1365-2443.2001.00443.x. [DOI] [PubMed] [Google Scholar]
- Knapik AA, Petkowski JJ, Otwinowski Z, Cymborowski MT, Cooper DR, Chruszcz M, et al. Structure of Escherichia coli RutC, a member of the YjgF family and putative aminoacrylate peracid reductase of the rut operon. Acta Crystallograph Sect F Struct Biol Cryst Commun. 2012;68:1294–1299. doi: 10.1107/S1744309112041796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko EV, Malakhova PT. The spore-forming iron-oxidizing bacterium Sulfobacillus thermosulfidooxidans. Microbiology. 1983;52:760–763. [Google Scholar]
- Lennox ES. Transduction of linked genetic characters of the host by bacteriophage P1. Virology. 1955;1:190–206. doi: 10.1016/0042-6822(55)90016-7. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39:W475–W478. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney MK, Cravatt BF. Evidence for distinct roles in catalysis for residues of the serine-serine-lysine catalytic triad of fatty acid amide hydrolase. J Biol Chem. 2003;278:37393–37399. doi: 10.1074/jbc.M303922200. [DOI] [PubMed] [Google Scholar]
- MacLennan PA, Delzell E, Sathiakumar N, Myers SL, Cheng H, Grizzle W, et al. Cancer incidence among triazine herbicide manufacturing workers. J Occup Environ Med. 2002;44:1048–1058. doi: 10.1097/00043764-200211000-00011. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Narayan SJ, Sahana S. Bioleaching: a review. Res J Biotechnol. 2009;4:72–75. [Google Scholar]
- Noor S, Taylor MC, Russell RJ, Jermiin LS, Jackson CJ, Oakeshott JG, Scott C. Intramolecular epistasis and the evolution of a new enzymatic function. PLoS ONE. 2012;7:e39822. doi: 10.1371/journal.pone.0039822. doi: 10.1371/journal.pone.0039822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paetzel M, Dalbey RE, Strynadka NCJ. Crystal structure of a bacterial signal peptidase in complex with a beta-lactam inhibitor. Nature. 1998;396:186–190. doi: 10.1038/24196. [DOI] [PubMed] [Google Scholar]
- Panjikar S, Parthasarathy V, Lamzin VS, Weiss MS, Tucker PA. Auto-Rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr D Biol Crystallogr. 2005;61:449–457. doi: 10.1107/S0907444905001307. [DOI] [PubMed] [Google Scholar]
- Pratt RF, McLeish MJ. Structural relationship between the active sites of beta-lactam-recognizing and amidase signature enzymes: convergent evolution? Biochemistry. 2010;49:9688–9697. doi: 10.1021/bi1012222. [DOI] [PubMed] [Google Scholar]
- Schaap FG, van der Vusse GJ, Glatz JFC. Evolution of the family of intracellular lipid binding proteins in vertebrates. Mol Cell Biochem. 2002;239:69–77. [PubMed] [Google Scholar]
- Seffernick LJ, Sadowsky MJ, Wackett LP. Thermostable cyanuric acid hydrolase from Moorella thermoacetica ATCC 39073. Appl Environ Microbiol. 2009;75:6986–6991. doi: 10.1128/AEM.01605-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seffernick LJ, Erickson JS, Cameron SM, Cho S, Dodge AG, Richman JE, et al. Defining sequence space and reaction products within the cyanuric acid hydrolase (AtzD)/barbiturase protein family. J Bacteriol. 2012;194:4579–4588. doi: 10.1128/JB.00791-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong CL, Ogawa J, Shimizu S. Novel amidohydrolytic reactions in oxidative pyrimidine metabolism: analysis of the barbiturase reaction and discovery of a novel enzyme, ureidomalonase. Biochem Biophys Res Commun. 2001;286:222–226. doi: 10.1006/bbrc.2001.5356. [DOI] [PubMed] [Google Scholar]
- Soong CL, Ogawa J, Sakuradani E, Shimizu S. Barbiturase, a novel zinc-containing amidohydrolase involved in oxidative pyrimidine metabolism. J Biol Chem. 2002;277:7051–7058. doi: 10.1074/jbc.M110784200. [DOI] [PubMed] [Google Scholar]
- Svergun D, Barberato C, Koch MHJ. CRYSOL – a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- Tang K, Baskaran V, Nemati M. Bacteria of the sulphur cycle: an overview of microbiology, biokinetics and their role in petroleum and mining industries. Biochem Eng J. 2009;44:73–94. [Google Scholar]
- Temple KL, Colmer AR. The autotrophic oxidation of iron by a new bacterium Thiobacillus ferrooxidans. J Bacteriol. 1951;62:605–611. doi: 10.1128/jb.62.5.605-611.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udikovic-Kolic N, Scott C, Martin-Laurent F. Evolution of atrazine-degrading capabilities in the environment. Appl Microbiol Biotechnol. 2012;96:1175–1189. doi: 10.1007/s00253-012-4495-0. [DOI] [PubMed] [Google Scholar]
- Wackett LP. Questioning our perceptions about evolution of biodegradative enzymes. Curr Opin Microbiol. 2009;12:244–251. doi: 10.1016/j.mib.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Waksman SA, Joffe JS. Microorganisms concerned in the oxidation of sulfur in the soil II. Thiobacillus thiooxidans, a new sulfur-oxidizing organism isolated from the soil. J Bacteriol. 1922;7:239–256. doi: 10.1128/jb.7.2.239-256.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand C, Krause E, Steinberg C, Pflugmacher S. Toxicokinetics of atrazine in embryos of the zebrafish (Danio rerio. Ecotoxicol Environ Saf. 2001;49:199–205. doi: 10.1006/eesa.2001.2073. [DOI] [PubMed] [Google Scholar]
- Zhang HM, Gao Y, Li M, Chang WR. Crystal structure of the PSPTO-PSP protein from Pseudomonas syringae pv. tomato str. DC3000 in complex with d-glucose. Biochem Biophys Res Commun. 2010;397:82–86. doi: 10.1016/j.bbrc.2010.05.071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.