

Abstract
Prion diseases are a group of neurodegenerative disorders characterized by accumulation of abnormal prion proteins in the central nervous system. The prions resist conventional sterilization procedures especially when infected tissue becomes dried onto metal or glass surfaces. This article, a review of literature collected using Pubmed as search engine, describes the oral manifestations of prion diseases in addition to studying the possibility of cross contamination in the dental office. The article emphasizes the importance for dentists to be aware of these diseases, to identify the high-risk patients by obtaining adequate medical history and to know the appropriate deactivation procedures to be followed.
Keywords: Contamination, Deactivation, Oral, Prions
Introduction
Prion diseases also known as transmissible spongiform encephalopathies (TSEs), are a group of fatal neurodegenerative diseases occurring in both humans and animals. Stanley B. Prusiner was the first person to purify the infectious agent of prion disease and won the 1997 Nobel Prize in Physiology/Medicine. Prusiner defined prions as infectious, transmissible proteinaceous particles that lack nucleic acid. The normal cellular prion protein (PrPc) is encoded by PrPc gene, which is located on the short arm of chromosome 20. PrPc has predominantly alpha helical structure, is soluble and proteinase sensitive. The normal function of PrPc is not well-known, but the suggested functions are signal transduction, cell adhesion, regulation and distribution of acetylcholine receptors. [1]
PrPc is transformed into abnormal isoform of the protein (PrPSc) due to post translational modification or mutation in the PrPc gene. PrPSc has predominantly beta structure, is insoluble and partially proteinase resistant. This mutated PrPSc gives rise to TSEs, including bovine spongiform encephalopathy (BSE) in cattle, scrapie in sheep and goats and Creutzfeldt-Jakob disease (CJD) in humans. These diseases are characterized by vacuolization of the gray matter and these vacuoles are located in the neuropils between the nerve cell bodies.[2,3]
Although the risk of transmission of CJD through dental procedures is still unclear, the theoretical possibility of transmission through contaminated dental instruments should be kept in mind. This article aims to give a brief overview of the clinical characteristics, risk of transmission and infection control methods of prion diseases for dentists using data obtained from literature search in Pubmed search engine.
Etiopathogenesis
The infectious agents for prior disease are composed of a 35-kD brain sialoglycoprotein called PrPSc that is essential for the transmission and pathogenesis of several neurodegenerative diseases. PrPSc is able to propagate itself in the host by stimulating the conversion PrPc to PrPSc, leading to its accumulation.[4]
Accumulation of PrPSc can result either from exposure to infectious prions iatrogenically or through ingestion, or because of mutations in the PrP gene. Sporadic CJD has an unknown cause; that is, thus far no apparent infections or mutations of the PrP gene have been found in association with such cases, although the brain in these patients also accumulates PrPSc.[2]
Human Transmissible Spongiform Encephalopathies
Based on the etiopathogenesis, different types of human TSEs have been recognized. Table 1 describes the transmission routes and clinical features of TSEs in humans.
Table 1.
Transmissible spongiform encephalopathies in humans

Sporadic Creutzfeldt-Jakob disease
This is the most common type of CJD accounting for 85% of all CJD cases, occurring in middle or older age group. The disease is characterized by progressive dementia, ataxia, myoclonus, cortical blindness, akinesia and speech loss, followed by death within 4 months.[5]
Iatrogenic Creutzfeldt-Jakob disease
The disease occurs following neurosurgery, duramater transplantation, corneal grafting, and injection of pituitary hormones obtained from human cadavers. This type of prion disease is important to dentists due to the risk of cross contamination after the use of infected dental instruments. The incubation period is variable ranging from 2 to 35 years. The clinical features are similar to sporadic form, but cerebellar motor symptoms are predominant in this type.[5]
Variant Creutzfeldt-Jakob disease
vCJD is associated with the intake of BSE-contaminated beef and beef products. The disease is characterized by depression, delirium, hallucinations, paresthesia and dysesthesia in hands, feet and mouth followed by dementia and akinesia. Deposition of amyloid plaques in the lymphatic tissues throughout the body is a prominent feature.[5]
Kuru
Kuru is endemic in Papua New Guinea and is transmitted by intake of infected nervous tissue in cannibalistic practices. The disease is characterized by ataxia, tremors, dysarthria and death. Though cannibalism is banned in 1950, the incubation period is more than 40 years and the chance of appearance of new cases is still possible.[4]
Fatal familial insomnia
The disease presents with progressive insomnia, dysautonomy in the form of hyperthermia, myosis and loss of sphincter control, followed by dysarthria, tremors, motor dysfunction and cognitive deterioration. Death occurs within 7-18 months.[6]
Gerstmann-Straussler-Scheinker syndrome
Gerstmann-Straussler-Scheinker syndrome gives rise to lack of coordination leading to ataxia, dysarthria and nystagmus. Death occurs after 1-10 years.[6]
Oral Manifestations
Oral manifestations are rarely seen in prion diseases. Dysphagia (difficulty in swallowing) and dysarthria (poor articulation of speech) are noticed in all forms of human TSEs. Dysphagia and dysarthria could be early symptoms of the disease and occur as a consequence of pseudobulbar paralysis.[7] In vCJD, parasthesia (tingling, pricking or numbness), orofacial dysesthesia (abnormal sensations in the absence of stimulation) and one case of loss of taste and smell have been reported in the literature.[8,9]
Infectivity of human oral tissues
Studies on human oral tissues for the presence of PrPSc showed positivity in limited number of oral tissues. Various human tissues like tonsil, tongue, submandibular and parotid salivary glands, trigeminal ganglia, inferior alveolar nerve, dental pulp and gingiva taken from different post mortem cases of vCJD were analyzed for the presence of PrPSc. Majority of the cases showed positive PrPSc in tonsils and trigeminal ganglia, while the other human tissues were negative for PrPSc. Western blot, paraffin-embedded tissue blot, and immunohistochemical techniques were used for the study and the sensitivity of these assay indicated that PrPSc must have been at the level of less than 1% of that found in the brain tissue.[10]
Presence of PrPSc in trigeminal ganglia may raise concerns about the extent of deposition of PrPSc along the cranial nerves and possible extension into oral and nasal cavities which are innervated by the ganglia. Guiroy et al.,[11] have noted positive PrP immunostaining of axons in the nerve root and around the degenerating ganglion cells of trigeminal ganglion, suggesting centripetal or centrifugal extension of the infectious agent along the axons.
Blanquet-Grossard et al.,[12] have investigated the presence of protease-resistant PrPSc in pulp tissues from eight patients with sCJD using specific monoclonal antibody by Western blotting. Though the authors were unable to detect protease-resistant PrPSc in pulpal tissues, they suggested that the negative results could be due to low sensitivity of the technique used and the potential for transmission of CJD via dental procedures could not be dismissed. The authors have calculated that 1 g of sCJD infected pulp would be expected to contain 40 log10 LD50 /g of infectivity, compared to 108-9 LD50 /g of infectivity in brain tissue.
Prions and Dentistry
There is no evidence to show that TSE is transmissible from one person to other by normal social contact, sexual contact or airborne droplets. Studies have failed to show evidence of transmission of sCJD by blood components or plasma products. On the contrary, there have been four reports of probable transmission of vCJD via blood transfusion, where the donors were at preclinical phase of the disease at the time of blood donation [Table 1].[13]
Occupational exposure
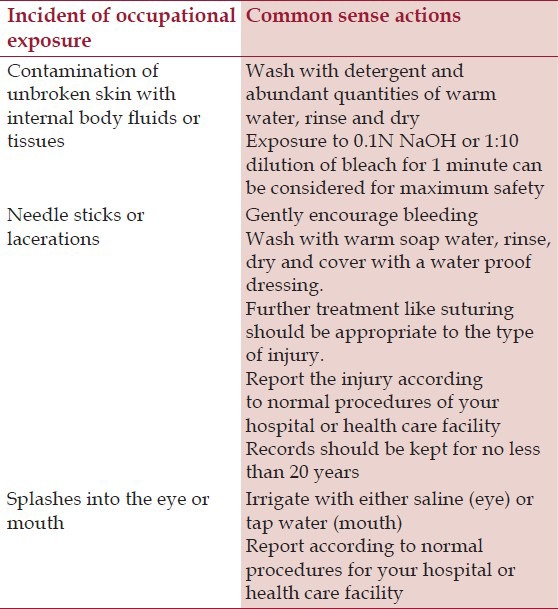
There is no risk of transmission of TSE to health care workers including medical doctors and dentists through clinical contact or noninvasive clinical investigative procedure. A total of 24 cases of sCJD have been reported in health care workers as of 2005.[14] Theoretically, it is possible that the health care workers may acquire prion diseases from affected patients through needle stick injuries. However, there is no epidemiological evidence to prove an association between occupational exposure and sCJD. The health care personnel should be informed about the nature of the hazard as well as the relevant safety procedures. The World Health Organization (WHO) has recommended “common-sense” actions in case of an occupational exposure while performing dental procedures on TSE patients [Table 2].[15]
Table 2.
Common sense actions in case of an occupational exposure (World health organization, 2000)[15]
Dental procedures
To date, there are no reported definite or suspected cases of human TSEs arising from dental procedures. Bourvis et al.,[16] had theoretically assessed the risk of iatrogenic transmission of sCJD during endodontic treatment. They estimated that the risk of being infected during endodontic treatment ranged from 3.4 to 13 per million procedures, if no effective prior deactivation procedures were used. However, the probability that more than one case was infected secondary to endodontic treatment ranged from 47% to 77% depending on the quantity of the infective material. The results of this study showed that the risk of sCJD transmission is higher because of the reuse of endodontic instruments in the absence of effective prion decontamination procedures.
Achieving appropriate decontamination of endodontic instruments intended for reuse is extremely difficult. Therefore, there is a possibility that these decontaminated instruments that were in contact with dental pulpal tissue may transfer the prion proteins from the infected patients to other patients.[17]
There are two possible mechanisms assessed for the transfer of CJD via dental instruments:
Accidental abrasion of lingual tonsil during dental procedures. Such a chance is extremely low (104-109 times less likely than tonsillectomy).
Contact of dental instruments with pulp tissue. As dental pulp originates from richly innervated neural crest cells, it is theoretically possible that the dental pulp of individuals infected with CJD may be infectious.[12]
General recommendations for dentists
The role of the dentist is to identify the patients with different forms of CJD and to take appropriate measures to reduce the possible risk of cross contamination. This can be achieved by obtaining: (a) complete medical history of the patient, (b) family history of prion diseases, (c) travel history to know about the possible exposure during visits to endemic areas like United Kingdom. Based on this information, patients can be classified as being at high risk or low risk for developing the disease.[2]
High-risk patients
Patients with diagnosis or suspicion of CJD; asymptomatic patients with a gene mutation; all members of the family with a case of hereditary CJD; all members of a family with a case of vCJD.
Low-risk patients
Patients with undiagnosed progressive neurological disease with or without dementia; members of a family with history of undiagnosed dementia or neurological disease; recipients of human pituitary hormone or duramater; patients undergoing transdural surgery between 1972 and 1989 as infectious agent could be transmitted during those procedures.
Prion inactivation methods
The routine physical and chemical sterilizing procedures are ineffective against prion agents, as they are heat resistant and bind tightly to surgical steel instruments. The prions also become more resistant to inactivation when dried and have shown to transmit disease experimentally and clinically, even after disinfection.[18]
It is advisable to use disposable instruments whenever possible and incinerate reusable instruments that are difficult to clean (endodontic files, broaches, carbide and diamond burs and dental matrix bands). Endodontic files used in the treatment of pulp cavity contain blood and peripheral nerves known to carry the prion proteins and their intricate surface topography enable to trap the proteins. Therefore, a recent communication in 2007 titled “Advise for dentists on re use of endodontic instruments and variant Creutzfeldt-Jakob Disease” issued by UK Department of Health, has advised dentists to ensure single use of endodontic reamers and files as a precaution to reduce any potential risk of transmission of vCJD. The nondisposable instruments should be mechanically cleaned and passed thorough stringent decontamination protocols before reuse, as recommended by WHO in 2000 [Table 3].[15]
Table 3.
Recommendations from world health organization for disinfection/sterilization approaches for preventing iatrogenic transmission of creutzfeldt Jakob disease (World Health Organization 2000)[15]

The handling of instruments depends on the risk of the patient being treated. When treating high-risk patients, all materials must be incinerated. The source of refrigeration and aspiration system should be external to the equipment due to the possibility that some residues might pass via internal systems and compromise sterilization. The patient should never use the normal spittoon but a disposable receptacle that is later incinerated. The histological samples of high-risk patients must be handled by specialized staffs that are aware of the risk. As routine formalin fixation does not inactivate prion proteins, the samples must be immediately immersed in 98% formic acid for 1 h prior to paraffin embedding and labelled as biohazardous.[5]
In patients with suspicion of CJD, all the instruments must be stored separately in a rigid container labelled with data of the patient, type of treatment provided and details of the attending clinician until a definitive diagnosis is arrived. The instruments are incinerated if the diagnosis is confirmed or sterilized by conventional methods like autoclaving if diagnosis is ruled out.[5]
Conclusion
TSEs are a group of fatal neurodegenerative disorders with no approved cure. The prion proteins resist conventional sterilization methods used in dental clinics and laboratories. Although there appears to be very low risk of CJD transmission during dental procedures, account must be taken of this possibility. As a general rule, appropriate medical and family history should be taken from all the patients before dental procedures. The dental professionals should have up to date knowledge about transmission, diagnosis, infection control and decontamination procedures regarding prion diseases.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Azarpazhooh A, Leake JL. Prions in dentistry-what are they, should we be concerned and what can we do? J Can Den Assoc. 2006;72:53–60. [PubMed] [Google Scholar]
- 2.Bebermeyer RD, Powell JF, Hobdell MH, Durban EM. Dental practice implications in prion diseases. Quintesence Int. 2003;34:38–44. [PubMed] [Google Scholar]
- 3.Azarpazhooh A, Fillery ED. Prion disease: The implications for dentistry. J Endod. 2008;34:1158–66. doi: 10.1016/j.joen.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Pruisner SB, Miller B. Prion Diseases. In: Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, editors. Harrison's Principles of Internal Medicine. New York: McGraw Hill; 2005. pp. 2495–8. [Google Scholar]
- 5.Scully C, Smith AJ, Bagg J. Prions and the human transmissible spongiform encephalopathies. Dent Clin North Am. 2003;47:493–516. doi: 10.1016/s0011-8532(03)00017-x. [DOI] [PubMed] [Google Scholar]
- 6.Palacios-Sanchez B, Esparza-Gomez GC, Campo-Trapero J, Cerero-Lapiedra R. Implications of prion disease for dentistry: An update. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;105:316–20. doi: 10.1016/j.tripleo.2007.09.033. [DOI] [PubMed] [Google Scholar]
- 7.Porter SR. Prion disease: Possible implications for oral health care. J Am Dent Assoc. 2003;134:1486–91. doi: 10.14219/jada.archive.2003.0079. [DOI] [PubMed] [Google Scholar]
- 8.Zeidler M, Johnstone EC, Bamber RK, Dicken CM, Fisher CJ, Francis AF, et al. New variant Creutzfeldt-Jakob disease: Psychiatric features. Lancet. 1997;350:908–10. doi: 10.1016/s0140-6736(97)03148-6. [DOI] [PubMed] [Google Scholar]
- 9.Reuber M, Al-Din AS, Baborie A, Chakrabraty A. New variant Creutzfeldt-Jakob disease presenting with loss of taste and smell. J Neurol Neurosurg Psychiatry. 2001;71:412–8. doi: 10.1136/jnnp.71.3.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Head MW, Ritchie D, McLoughlin V, Ironside JN. Investigation of PrPres in dental tissues in variant CJD. Br Dent J. 2003;195:339–43. doi: 10.1038/sj.bdj.4810536. [DOI] [PubMed] [Google Scholar]
- 11.Guiroy DC, Shankar SK, Gibbs CJ, Jr, Messenheimer JA, Das S, Gajdusek DC. Neuronal degeneration and neurofilament accumulation in the trigeminal ganglia in Creutzfeldt-Jakob disease. Ann Neurol. 1989;25:102–6. doi: 10.1002/ana.410250119. [DOI] [PubMed] [Google Scholar]
- 12.Blanquet-Grossard F, Sazdovitch V, Jean A, Deslys JP, Formant D, Hauw JJ, et al. Prion protein is not detectable in dental pulp from patients with Creutzfeldt-Jakob disease. J Dent Res. 2000;79:700. doi: 10.1177/00220345000790020101. [DOI] [PubMed] [Google Scholar]
- 13.Ludlam CA, Turner ML. Managing the risk of transmission of variant Creutzfeldt-Jakob disease by blood products. Br J Hematol. 2006;132:13–24. doi: 10.1111/j.1365-2141.2005.05796.x. [DOI] [PubMed] [Google Scholar]
- 14.Ena J. Prions: Who should worry about them? Arch Med Res. 2005;36:622–7. doi: 10.1016/j.arcmed.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Geneva, Switzerland: World Health Organization Communicable Disease Surveillance and Control; 2000. WHO Consultation: WHO infection control guidelines for transmissible spongiform encephalopathy. Report No: WHO/CDS/CSR/APH/2000. [Google Scholar]
- 16.Bourvis N, Boelle PY, Cesbron JY, Valleron AJ. Risk assessment of transmission of sporadic Creutzfeldt-Jakob disease in endodontic practice in absence of adequate prion inactivation. PLoS One. 2007;2:e1330. doi: 10.1371/journal.pone.0001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Letters S, Smith AJ, McHugh S, Bagg J. A study of visual and blood contamination on reprocessed endodontic files from general dental practice. Br Dent J. 2005;199:522–5. doi: 10.1038/sj.bdj.4812811. [DOI] [PubMed] [Google Scholar]
- 18.Dello Russo N. Understanding prions. J Am Dent Assoc. 2004;135:278. doi: 10.14219/jada.archive.2004.0165. [DOI] [PubMed] [Google Scholar]