

Abstract
The Caenorhabditis elegans gene eat-4affects multiple glutamatergic neurotransmission pathways. We find thateat-4 encodes a protein similar in sequence to a mammalian brain-specific sodium-dependent inorganic phosphate cotransporter I (BNPI). Like BNPI in the rat CNS,eat-4 is expressed predominantly in a specific subset of neurons, including several proposed to be glutamatergic. Loss-of-function mutations in eat-4 cause defective glutamatergic chemical transmission but appear to have little effect on other functions of neurons. Our data suggest that phosphate ions imported into glutamatergic neurons through transporters such as EAT-4 and BNPI are required specifically for glutamatergic neurotransmission.
Keywords: C. elegans, behavior, genetics, glutamate, synaptic neurotransmission, sodium-dependent inorganic phosphate cotransporter
Glutamate is widely used as a neurotransmitter. For example, glutamate is a neurotransmitter at neuromuscular junctions in arthropods and at specific peripheral and central synapses in arthropods and molluscs (Gerschenfeld, 1973; Walker and Roberts, 1982; Bicker et al., 1988; Horseman et al., 1988; Quinlan and Murphy, 1991; Dale and Kandel, 1993; Trudeau and Castellucci, 1993). In the vertebrate CNS glutamate is also a major excitatory transmitter capable of exciting virtually all central neurons (Jahr and Lester, 1992). Although its normal function is important for animal behavior, glutamatergic transmission, when excessive, can contribute to neuronal degeneration after acute insults to the brain, e.g., in ischemia and epilepsy and possibly in chronic neurodegenerative diseases such as Alzheimer’s, Huntington’s, and Parkinson’s (Choi, 1988; Meldrum and Garthwaite, 1990; Whetsell, 1996).
Glutamatergic neurotransmission also occurs in the nematodeCaenorhabditis elegans. To date, two glutamate receptor genes, glr-1 and avr-15, have been characterized functionally in C. elegans. GLR-1 is most similar to the vertebrate AMPA-type receptors (Hart et al., 1995; Maricq et al., 1995), whereas AVR-15 is a member of the recently identified invertebrate glutamate-gated Cl− channel family (Dent et al., 1997).
In our studies of glutamatergic transmission in C. elegans, we have focused on the eat-4 (EATing defective) gene (Avery, 1993a). In eat-4 mutants, pharyngeal muscle relaxation is delayed because of dramatically reduced pharyngeal motor neuron M3 synaptic transmission (Avery, 1993a; Raizen and Avery, 1994). M3 neurotransmission is mediated by the AVR-15 glutamate receptor (Dent et al., 1997). Mutations in avr-15 cause a feeding-defective phenotype similar to that seen in eat-4 mutant animals, i.e., longer pharyngeal muscle contractions caused by a severe reduction or a complete block of M3-dependent inhibitory postsynaptic potentials (IPSPs) (Raizen and Avery, 1994; Dent et al., 1997). However, eat-4 mutant pharynxes, unlike those ofavr-15 animals, have a normal sensitivity to iontophoretically applied glutamate (Dent et al., 1997). This result indicates that eat-4 affects M3 transmission presynaptically.
In contrast to their strong effects on M3 glutamatergic neurotransmission, eat-4 mutations do not appear to affect the behaviors that involve neuronal pathways that are known to use other small neurotransmitters (Avery, 1993a). eat-4 mutants are apparently normal for behaviors such as locomotion [involving GABA, McIntire et al. (1993a,b) and ACh (acetylcholine), Chalfie and White (1988)], egg-laying [involving 5-HT (serotonin), Trent et al. (1983)], male-mating [involving 5-HT, Loer and Kenyon (1993)], and defecation [involving GABA, McIntire et al. (1993a,b)].
However, the effect of eat-4 apparently is not restricted to M3 neurotransmission. In eat-4 mutant animals the muscle contractions of the anterior pharynx (corpus) not only last longer, but they also are often less complete than those in normal animals (Avery, 1993a). This phenotype of feeble muscle contraction cannot be explained by the loss of M3 neurotransmission (Avery, 1993a,b). Furthermore, defects in several extrapharyngeal behaviors, such as thermotaxis and chemotaxis, also have been observed in eat-4 mutant animals (I. Mori and C. I. Bargmann, personal communications). Although the cellular basis for these eat-4 mutant defects is not known, it seems likely that eat-4 affects several specific neurotransmission pathways. Why does eat-4 affect some, but not all, neurotransmission pathways in C. elegans? Because M3 transmission is glutamatergic and because behaviors that are known to be mediated by neurotransmitters other than glutamate are not affected by eat-4, it is possible that eat-4 may function specifically in glutamatergic neurotransmission.
To help determine how eat-4 affects the function of specific neuronal pathways, we cloned and analyzed the eat-4gene.
MATERIALS AND METHODS
General methods. Nematodes were grown onEscherichia coli strain HB101 on nematode growth medium (NGM) plates at 20°C. Wild-type animals were C. elegansstrain N2. Electropharyngeograms (EPGs) were recorded as described byRaizen and Avery (1994). Germline transformation methods were as described by Mello et al. (1992). For general cloning methods we followed those described in Sambrook et al. (1989). We used the Genetics Computer Group (GCG) Wisconsin Package for sequence management and comparisons.
Molecular cloning. The ability of cosmid ZK512 and the various DNA constructs derived from it to rescue eat-4mutants was tested in transgenic animals. The DNA to be tested was coinjected into the gonads of eat-4 mutant animals with pRAK3 (Davis et al., 1995), which contains a dominantrol-6 marker (Mello et al., 1992). Independent lines of animals that transmitted the transgenes via the germline were established and scored for eat-4 phenotypes. We relied primarily on the pharyngeal phenotypes (Avery, 1993a), namely the extent of lumen opening of the anterior pharynx and M3 transmission, which was measured by EPG recordings (Raizen and Avery, 1994). We found that the cosmid ZK512 rescued eat-4(ad572) in three of three transgenic lines. We then found that ZK512 digested with any of the restriction endonucleases ApaLI (two of two lines), BsaHI (two of two lines), NaeI (two of two lines), and XhoI (two of three lines) was still able to rescue and that ZK512 restricted with either HindIII (one line) or ApaI (one line) was unable to rescue the mutant phenotype. Because the sequence of ZK512 had been determined (Sulston et al., 1992), the exact sites of restriction by each of these endonucleases were known, and we were able to deduce thateat-4 rescuing activity was located in a 6.9 kb region (23,392–30,260 in the reported sequence). This conclusion then was verified with plasmid subclones of ZK512. pRE4-4, which contains a 6.5 kb fragment (22339–28863) of ZK512, was not able to rescueeat-4(ad819) (four lines); neither was pRE4-8, containing 25834–30259 of ZK512, able to rescueeat-4(ky5) (one line) when injected individually. When mixed together, however, they were able to rescue (three of six lines) eat-4(ky5). This result suggested that the recombined products of these two plasmids contained eat-4rescuing activity.
A 2.2 kb cDNA clone, yk32h2, was isolated and mapped to the 6.9 kb region of ZK512 (Y. Kohara, personal communication). We determined and analyzed the sequence of yk32h2 and found that the largest open reading frame (ORF) within the 2.2 kb insert potentially encodes a polypeptide of 563 amino acids (see Fig. 2A). We tested the possibility that yk32h2 represents an eat-4 transcript by asking whether yk32h2 expressed in appropriate cells could rescueeat-4 mutants. We made a minigene (illustrated in Fig.1A) construct by fusing aSacI–PstI (22339–24738) genomic DNA fragment from ZK512 to the corresponding PstI site in yk32h2. By germline transformation experiments we found that the minigene was able to rescue eat-4(ky5) partially but significantly in seven of nine independent transgenic lines.
Fig. 2.
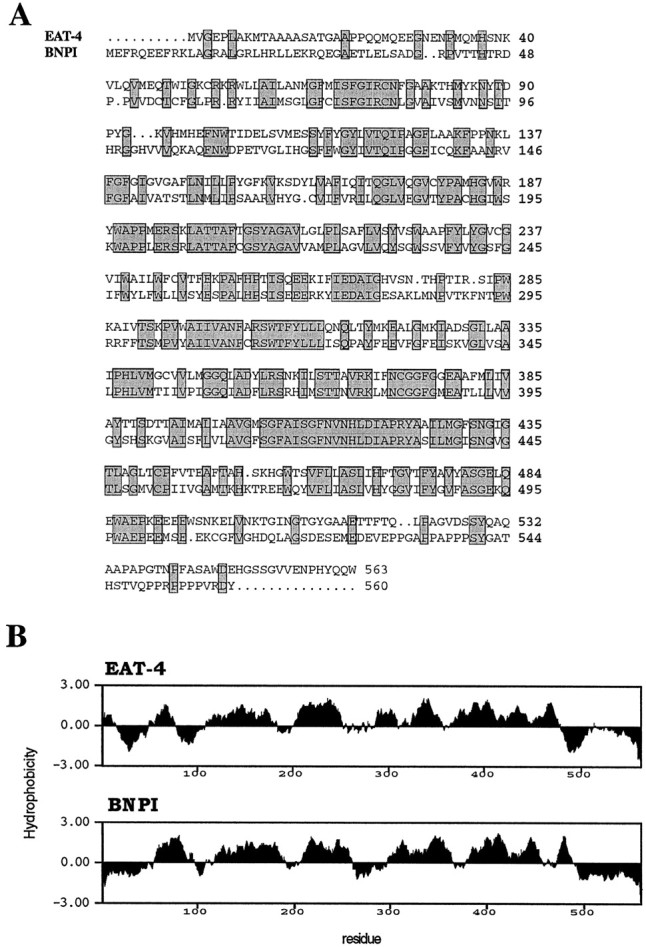
Comparisons between EAT-4 and rat brain-specific sodium inorganic phosphate cotransporter I (BNPI). A, Protein sequence alignment of EAT-4 and BNPI. Identical amino acid residues are highlighted by the gray boxes. The overall identity between these two proteins is 48%. B, Kyte and Doolittle (1982) hydrophobicity profiles of EAT-4 and BNPI. The calculation is based on a window size of 20 residues.
Fig. 1.

eat-4 cloning. This figure summarizes our strategy for and the results from cloningeat-4, starting with the cosmid ZK512. A, The abilities of ZK512 and its derivatives to rescueeat-4 mutants were tested by germline transformation rescue experiments (right column; for details, see Materials and Methods). A 2.4 kb region of ZK512 in which a restriction-fragment length polymorphism (RFLP) was found in eat-4(ad572) (seeC) is marked by ↔. Circles are used to mark the sites of restriction enzymes that did not interfere with the ability of ZK512 to rescue, whereas crosses are used to mark the sites of enzymes that did appear to interfere. pRE4-4 and pRE4-8 are two plasmid subclones of ZK512. The deduced extent (∼6.9 kb) and location of the minimal eat-4 rescuing activity are indicated. At the bottom, drawn to an expanded scale, the relationships among the cDNA, the minigene construct, theeat-4:: reporter constructs, and the cosmid are illustrated. The GenBank accession number for eat-4 cDNA is AF095787. B, Examples of M3 neurotransmission phenotypes, assayed by electropharyngeogram (EPG) recordings, observed in wild-type, eat-4 mutant, and transgene-carrying eat-4 mutant animals. Examples of M3 transients [one in each EPG record, except that of the eat-4(ky5) animal] are marked by asterisks. C, A Southern blot analysis of genomic DNA isolated from animals of three different genotypes: wild type, eat-4(ad572), and a spontaneous intragenic revertant eat-4(ad572 ad613), digested with the restriction enzymesPstI or HindIII and probed with labeled ZK512 cosmid. In the set of lanes for each restriction enzyme a band (marked by >), apparent in both the wild-type and the revertant lanes, disappears in the eat-4(ad572) lane where a novel, apparently 1.3 kb larger, band (marked by anasterisk) is visible. This apparent insertion found ineat-4(ad572) was deduced by restriction patterns to be in a 2.4 kb region of ZK512, as indicated by ↔ inA.
Because the dominant coinjection marker rol-6 we had used thus far caused transgenic animals to roll, we were unable to score the nose touch and foraging abnormal phenotype. To circumvent this problem, we did rescue experiments, using lin-15 as a coinjection marker (Clark et al., 1994; Huang et al., 1994). We transformedeat-4(ky5);lin-15(n765ts) mutant animals (which have a multivulva phenotype) with ZK512 along with DA#735 (which contains the wild-type lin-15 gene; Huang et al., 1994). Within each of six established lines there were transgenic animals that had apparently wild-type feeding and foraging behaviors, whereas the control transgenic animals (yk32h2 and DA#735 coinjection) did not show any rescue (five lines).
The eat-4(ad572) mutation reverts spontaneously at a low frequency (<1 in 6800 meiotic events) to an apparently wild-type phenotype (data not shown), suggesting the possibility thatad572 may be caused by a chromosomal rearrangement. We tested this possibility by Southern blot analysis, using ZK512 as a probe.
Analysis of expression patterns. We usedeat-4:: lacZ(nls) andeat-4:: gfp fusion reporter constructs to identify the cells in which the presumptive eat-4 promoter is active (Fire et al., 1990; Chalfie et al., 1994). Theeat-4:: lacZ(nls) construct was made by fusing the 2.4 kb SacI–PstI fragment from ZK512 to the lacZ vector pPD22.11 (Fire et al., 1990). The resulting construct (pRE4–lacZ) has the potential to express a fusion protein that contains the first 10 amino acids of EAT-4, a nuclear localization signal (NLS), and the E. coli β-galactosidase (β-gal). We used pRE4–lacZ to transform wild-type animals. We found strong expression of the transgene in many extrapharyngeal neurons in a pattern similar to the one we saw in eat-4:: gfptransgenic animals (see below and Fig. 3B,C). We also found consistent transgene expression in the nuclei of pharyngeal neurons M3 and neurosecretory motor neurons (NSM), although the signal was lower than in the extrapharyngeal cells. For clarity of presentation we selected a pharynx that had little staining in extrapharyngeal neurons for Figure 3A. The failure of staining in the extrapharyngeal neurons in this animal probably was caused by genetic mosaicism, which is expected from extrachromosomal transgenes (Mello et al., 1992).
Fig. 3.
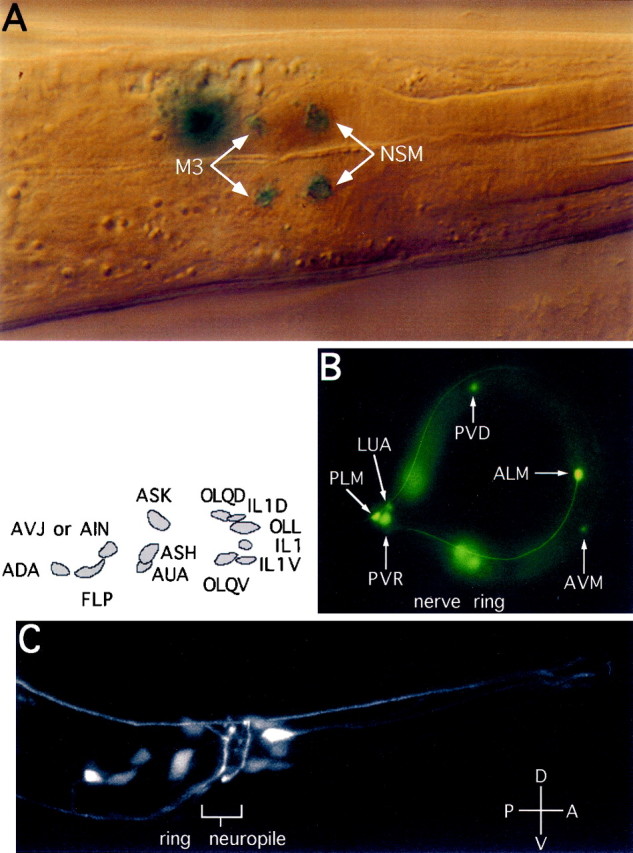
Expression ofeat-4:: reporters. The reporter constructs schematically shown in Figure 1A were used to generate transgenic animals. A, Expression ofeat-4:: lacZ(nls) in the pharynx, observed with Nomarski optics. β-Galactosidase activity was found in the nuclei of two types (each consists of a bilaterally symmetric pair) of pharyngeal neurons, M3 andNSM. Also seen in this panel is the strong signal in the nucleus of an extrapharyngeal cell posterior to M3. This extrapharyngeal cell is probably a neuron in the IL1 class.B, C, Expression ofeat-4:: gfp in extrapharyngeal neurons. The identity of these cells is marked either in the figure (B) or above the photograph (C). The identity of one cell (marked asAVJ or AIN) is uncertain. InB, the entire animal is shown, whereas inC only the head is shown. Only the right half of the animal is shown in C, because these classes of cells are bilaterally symmetric. C, Shown is an overlay (with Adobe Photoshop software) of nine serial confocal images with an intersection spacing of 0.9 μm.
The eat-4:: gfp fusion gene construct was made by inserting the 2.4 kb SacI–PstI ZK512 fragment into a green fluorescent protein (GFP) construct pPD95.77 (A. Fire, J. Ahnn, G. Seydoux, and S. Xu, personal communication). The construct (pRE4–sGFP) mixed with DA#735 was used to transform MT1642 (lin-15 mutant) animals. Eleven transgenic lines were analyzed and all had similar expression patterns, although the levels of expression were quite variable. To circumvent the problem of mosaicism, we integrated the transgene in one selected line by γ-ray irradiation (Mello and Fire, 1995). One integrated line was obtained. To see whether mutations in eat-4 have an effect on the expression of the transgene, we crossed the transgene, which was mapped to chromosome X, into an eat-4(ky5) mutant background. The expression patterns of the transgene were identical in eat-4 wild-type and mutant genetic backgrounds, except that the level of expression appeared slightly higher in the mutant than in the wild-type background. We did most of our cell identification analysis with an eat-4(ky5) mutant transgenic strain because of its higher level of GFP expression (see Fig. 3B,C). In ∼50% of the transgeniceat-4(ky5) mutant animals the pharyngeal interneuron I5 showed weak GFP staining (data not shown). Because of the interference by the GFP staining in extrapharyngeal neurons in the head, it was difficult to observe GFP expression in the pharynx in the integrated transgenic line. Therefore, pharyngeal reporter expression was analyzed in mosaic animals carrying theeat-4:: lacZ transgene as extrachromosomal arrays. In the pharynx of these animals we found consistent β-gal staining in the pharyngeal neurons M3 and NSM (see Fig. 3A).
The identification of the cells expressing β-gal or GFP was based on their stereotypic locations and the morphologies of the cell bodies and, when possible, the neuronal process morphologies (Albertson and Thomson, 1976; Sulston et al., 1983; White et al., 1986).
Laser ablation of neurons. Neurons were killed by a laser microbeam as previously described (Avery and Horvitz, 1989; Bargmann and Horvitz, 1991). PVC and NSM were killed in the first larval stage. All other neurons were killed in the second larval stage. Mock-treated animals were transferred to pads and anesthetized in parallel to the animals that underwent laser ablation.
Behavioral assays. The pharyngeal pumping rate was assayed for young adult animals as previously described (Raizen et al., 1995). Anterior touch behavioral assays on young adult animals were performed 2 d after laser ablation. The anterior touch response was scored in young adult animals and was scored blind to the genotype and laser ablation treatment of the animal. Each animal was touched gently in the anterior body region with an eyelash attached to a Pasteur pipette. An animal that backed immediately in response to the touch was scored as touch-sensitive. An animal that backed only after a delay was scored as partially sensitive, and an animal that did not back within ∼2 sec was scored as touch-insensitive. The touch sensitivity trials were spaced by 45–60 min to prevent habituation to gentle touch.
RESULTS
eat-4 encodes a putative plasma membrane Na-Pi cotransporter
Previous genetic mapping experiments placed eat-4between glp-1 and emb-9 on the third chromosome (LGIII) (Avery, 1993a). A cosmid, ZK512, in this ∼240 kb region restored the wild-type feeding phenotype when it was introduced as a transgene into eat-4 mutant animals (Fig.1A,B). Aided by the fact that the ZK512 sequence had been determined (Sulston et al., 1992), we further mapped the rescuing activity to a 6.9 kb region by transformation rescue experiments, using restriction-digested fragments and plasmid subclones of ZK512. A cDNA clone yk32h2 was found to map within this 6.9 kb region (Fig. 1A) (Y. Kohara, personal communication). We found that yk32h2, when fused to the presumptive 5′ regulatory sequence of eat-4, also significantly, albeit incompletely, restored the pharyngeal muscle relaxation defect of eat-4 mutants in germline transformation experiments. For example, the yk32h2 minigene caused incomplete but significant restoration of M3 neural activity (Fig.1B). This result indicates that the yk32h2 cDNA represents an eat-4 transcript.
The eat-4(ad572) mutation reverted spontaneously (see Materials and Methods), raising the possibility that it was a chromosomal rearrangement. By Southern blot analysis with ZK512 as the probe, we compared the restriction patterns of genomic DNA isolated from wild-type, eat-4(ad572), and a revertanteat-4(ad572 ad613) strain (Fig. 1C). We found that eat-4(ad572) is associated with an ∼1.3 kb insertion, which is not present in either the wild-type or the revertant genomes. We localized the insertion in a 2.4 kb region in the ZK512 sequence on the basis of the cosmid restriction map (data not shown). The 2.4 kb region is within the 6.9 kb of DNA that containseat-4 rescuing activity (Fig. 1A). Therefore, the genomic analysis of eat-4(ad572) independently supports the conclusion that the cDNA yk32h2 is encoded by the eat-4 gene.
The largest ORF in eat-4 cDNA potentially encodes a polypeptide (EAT-4) of 563 amino acids. By searching sequence databases, we found that EAT-4 is most similar (48% identical; see Fig. 2A) to brain-specific sodium-dependent inorganic phosphate cotransporter I (BNPI; Ni et al., 1994). EAT-4 has significant sequence similarity (∼29% identity) to known sodium-inorganic phosphate (Na-Pi) cotransporters found in rabbit, mouse, and human kidney cortex (Werner et al., 1991; Chong et al., 1993, 1995; Miyamoto et al., 1995). EAT-4 is also similar in sequence to at least three predicted C. elegans genes: C38C10.2 (32% identity), K10G9.1 (42% identity), and T07A5.3 (44% identity), revealed by the Genome Sequencing Project (Sulston et al., 1992). The sequence identity between BNPI and EAT-4 is higher by at least 9% than that between BNPI and any other putativeC. elegans Na-Pi cotransporter.
BNPI was cloned from rat cerebellar granule cells, and BNPI message is found predominantly in rat brain, in neurons of the cerebral cortex, hippocampus, and cerebellum (Ni et al., 1994, 1995). BNPI expressed inXenopus oocytes has been demonstrated to transport Pi across the plasma membrane in a Na+-dependent manner (Ni et al., 1994). In addition to their similarity at the primary sequence level (Fig. 2A), EAT-4 and BNPI also may have similar secondary structures, as indicated by the striking similarity of theirKyte and Doolittle (1982) hydrophobicity profiles (Fig.2B). As predicted for BNPI (Ni et al., 1994), EAT-4 may form six to eight membrane-spanning domains (data not shown). On the basis of the sequence similarity to BNPI and other known Na-Pi cotransporters, it seems likely that eat-4 encodes aC. elegans sodium-dependent inorganic phosphate cotransporter.
eat-4 expresses and functions in known glutamatergic neurons
That pharyngeal muscles in eat-4 mutant animals are sensitive to exogenously applied glutamate, the M3 neurotransmitter, suggests that eat-4 acts presynaptically, i.e., in M3 neurons (Dent et al., 1997; Li et al., 1997). To see if indeedeat-4 is expressed in M3 and possibly other glutamatergic neurons, we assayed the expression pattern ofeat-4:: lacZ and eat-4:: gfpreporters in transgenic animals. The reporter genes, lacZand GFP, respectively, were fused to an eat-4 5′ fragment (the same one that was found to be sufficient to driveeat-4 cDNA expression to rescue the pharyngeal pumping defect of eat-4 mutants) in the same translational frame aseat-4 (see Fig. 1A). As summarized in Figure 3, we foundeat-4:: reporter gene expression in a subset of neurons in the pharynx and in the extrapharyngeal nervous system. However, transgene expression was weak in pharyngeal neurons. We also noticed expression in intestine cells (int1–int9; data not shown). The expression pattern is the same in eat-4(ky5) mutant as in wild-type animals (see Materials and Methods), indicating that the eat-4 mutation does not prevent the development of the cells that express this gene.
In the pharynx eat-4 was found to be expressed in the M3, NSM (Fig. 3A), and possibly I5 (data not shown) (see Materials and Methods) neurons, but not in muscle cells. The expression in M3 neurons suggests that eat-4 functions in the M3 neurons to affect their glutamatergic transmission.
The NSM neurons are serotonergic (Horvitz et al., 1982; Avery and Horvitz, 1990) neurosecretory motor neurons (Albertson and Thomson, 1976) that can stimulate pharyngeal pumping (Avery and Horvitz, 1990). We addressed the role of eat-4 in the NSM neurons by asking whether NSM neurons are functional in eat-4 mutant animals. If eat-4 is necessary for NSM function, we would expect that laser-ablating NSM neurons in eat-4 mutant animals should have little effect, whereas the same operation should reduce the pharyngeal pumping rate in the wild type. We found that laser-ablating the NSM neurons caused a comparable reduction in pharyngeal pumping rate in either the presence (11.6%) or the absence (11.4%) of a functional eat-4 gene (Table1). This result suggests that the serotonergic function of NSM neurons is unaffected by theeat-4(ky5) mutation.
Table 1.
Effects of eat-4 on NSM function
| eat-4 genotype | Pharyngeal pumps/min mean ± SEM (n) | Reduction of pumping rate by operation | |
|---|---|---|---|
| Control | NSM− | ||
| Wild-type | 276 ± 3.7 (20) | 244 ± 4.8 (8)* | 11.6% |
| ky5 | 202 ± 4.4 (18) | 179 ± 5.0 (12)** | 11.4% |
The unc-31 (e928) mutation was present in all animals used to achieve consistently rapid pumping (Avery et al., 1993b).
*NSM− pumping rate is significantly different from operated control pumping rate; p < 0.001.
**NSM− pumping rate is significantly different from unoperated control pumping rate; p < 0.002.
Clear and consistent eat-4 reporter expression was found in 15 different anatomical types (ADA, ALM, ASH, ASK, AUA, and AVJ or AIN, AVM, FLP, IL1, LUA, OLL, OLQ, PLM, PVD, and PVR; 34 cells total) of extrapharyngeal neurons (Fig. 3B,C) (White et al., 1986). The neurotransmitter(s) used by most of these neurons is not known. The exceptions are the four types ASH, IL1V, OLQV, and PVD, which are thought to be glutamatergic because their functions are mediated by the GLR-1 glutamate receptor, which is expressed in the respective postsynaptic cells (Hart et al., 1995; Maricq et al., 1995). Becauseeat-4 mutant animals respond poorly to nose touch and forage abnormally (A. C. Hart and J. M. Kaplan, personal communication), behaviors that require the normal function of ASH, and IL1V and OLQV, respectively (Kaplan and Horvitz, 1993; Hart et al., 1995; Maricq et al., 1995), eat-4 is probably necessary for the function of at least these three extrapharyngeal neuron types. We have not tested the effect of eat-4 on PVD function.
We conclude, on the basis of the analysis of the expression pattern and its mutant phenotypes, that eat-4 expresses and functions in glutamatergic neurons.
eat-4 affects specifically the glutamatergic transmission function of anterior touch cells
The eat-4:: gfp fusion also is expressed in the mechanosensory neurons ALM, AVM, and PLM. We addressed the functional role of eat-4 in the anterior touch response because the neural circuit involved has been well characterized (Chalfie et al., 1985).
In adult hermaphrodites, touch to the anterior body is detected by the two ALM neurons and the single AVM neuron (together referred to as the anterior touch cells; Chalfie et al., 1985). These mechanosensory neurons synapse onto the AVB, AVD, and PVC command interneurons of the locomotory circuitry via both chemical synapses (AVB, PVC) and gap junctions (AVD; Chalfie et al., 1985) (Fig.4A). We tested the response of eat-4 mutant animals to gentle anterior touch, the stimulus that is detected by the anterior touch cells (Chalfie et al., 1985). We found that eat-4 animals that are mutant for either of two independent alleles showed a significantly, albeit slightly, decreased fidelity of response (Fig. 4B). Wild-type animals backed in response to 94.0 ± 1.4% of touches, whereas eat-4(ky5) animals backed in response to 76.3 ± 3.1% of touches, andeat-4(n2474) animals backed in response to 87.4 ± 4.1% of touches.
Fig. 4.

eat-4 function in the anterior touch cells. A, Schematic circuit diagram of the anterior touch response. The diagram is modified from Chalfie et al. (1985). The arrows represent chemical synapses, and the bars represent gap junctions. Dashed lines represent the chemical synapses affected byeat-4, as suggested by the data in B.B, Quantitation of the anterior touch response. For each gentle touch to the anterior body the animal’s response was scored as sensitive if backing occurred immediately, partially sensitive if backing occurred with a delay, and insensitive if backing did not occur within several seconds. The response of each animal was tested at least seven times, and the bars represent the average percentage of trials in which the animals responded as sensitive (black portion of the bar) or partially sensitive (gray portion of thebar). Error bars represent the SEM for the percentages of responses that were either sensitive or partially sensitive. Cases in which the behavior of the operated animals was significantly different from that of the mock-operated animals of the same genotype are indicated by an asterisk(p < 0.05); n, number of animals tested.
Because eat-4 affects glutamatergic M3 transmission in the pharynx, we reasoned that the slight deficit in the anterior touch response observed in eat-4 mutants might reflect a defect in chemical, but not electrical, neurotransmission. If this were the case, then sensory information from the touch cells in eat-4mutants would be transmitted exclusively through the gap junctions between ALM and AVM and the AVD interneurons (Fig.4A). If this model is accurate, then the anterior touch response in eat-4 mutants should be more dependent on the function of the AVD neurons than the anterior touch response in wild-type animals, because wild-type, but not eat-4 mutant, animals would retain the chemical synapse-mediated transmission even in the absence of the AVD interneurons.
We tested this hypothesis by comparing the effect of laser ablation of the AVD interneurons in wild-type and eat-4 mutant animals (Fig. 4B) and found that, whereas the loss of AVD in the wild type resulted in a slight decrease in the fidelity of the response (animals responded in 65.3 ± 7.0% of trials), the loss of AVD resulted in a more pronounced defect in the anterior touch response in eat-4 mutants (the animals responded in 23.9 ± 4.3% and 19.8 ± 7.4% of the trials forky5 and n2474, respectively). These results suggest that the AVD interneurons, and thus the gap junction-mediated pathway of the anterior touch circuit, play a relatively more important role in eat-4 animals than in the wild type. This finding is consistent with the hypothesis that the chemical synapse-mediated component of the response is not functioning efficiently ineat-4 mutants.
We further tested this possibility by killing the AVB and PVC interneurons, which receive chemical synapses from ALM and AVM (Fig.4B). We found that ablation of these interneurons slightly reduced the fidelity of the response in wild-type animals (animals responded in 80.0 ± 4.8% of trials) but did not decrease the fidelity of the response ofeat-4(ky5) animals (74.2 ± 3.2%). This result is again consistent with the hypothesis that the chemical synapse-mediated pathway is defective in eat-4 mutants (Fig.4A).
The neurotransmitter of the touch cells has not been identified. However, because our results showed that eat-4 function is required for chemical neurotransmission from the touch cells, it seemed possible that the touch cells, like the M3 cell, might be glutamatergic and that glutamate signaling might be involved in transmitting information from the touch cells to the interneurons. We therefore examined the anterior touch response of avr-15 animals, which lack a class of glutamate receptor (Dent et al., 1997). We found that, as was the case for eat-4 mutants,avr-15(ad1051) animals exhibited a dramatic decrease in the fidelity of the anterior touch response after AVD laser ablation (animals responded in 30.8 ± 7.2% of the trials). This result supports the model that glutamate is the chemical neurotransmitter of the touch cells. Furthermore, thatavr-15 encodes a glutamate-gated Cl−channel and that the AVR-15 receptor mediates inhibitory signaling in the pharynx (Dent et al., 1997) argue that some or all of the chemical synapses between the touch cells and the command interneurons might be inhibitory, a possibility previously suggested on the basis of an analysis of the connectivity of the circuitry for the touch response (Chalfie et al., 1985; Wicks et al., 1996).
The fact that in eat-4 mutants the ALM and AVM neurons appear to be inefficient at signaling via chemical synapses but able to participate in the touch reflex via gap junctions indicates that these neurons are differentiated, able to detect stimuli, and electrically active in the eat-4 mutant background and that the loss ofeat-4 function appears to affect only chemical transmission by the anterior touch cells.
DISCUSSION
eat-4 mutant animals are defective in behaviors known to be mediated by glutamatergic neurotransmission but normal in behaviors known to be mediated by other neurotransmitters such as GABA, ACh, and 5-HT (Avery, 1993a) (A. C. Hart and J. M. Kaplan, personal communication). Because of this correlation we hypothesized that in the nervous system eat-4 functions exclusively in glutamatergic pathways. The results of our present study are consistent with this hypothesis.
eat-4 is expressed and functions in known glutamatergic neurons
Reporter genes fused to an eat-4 5′ regulatory element were expressed predominantly in a subset of neurons in the pharyngeal and extrapharyngeal nervous systems and in cells of the intestine (see Fig. 3; data not shown). We do not know the significance ofeat-4 expression in intestinal cells. eat-4mutants have pale intestine pigmentation, which could be explained by malnutrition caused by feeding defects (Avery, 1993a). However, it remains possible that food absorption in the intestine is compromised also in eat-4 mutant animals.
Among eat-4-expressing neuron types, glutamate appears to be the neurotransmitter used by M3 in the pharynx and ASH, IL1V, OLQV, and PVD in the somatic nervous system (Hart et al., 1995; Maricq et al., 1995; Dent et al., 1997). Presently, the neurotransmitter or transmitters used by all (except ALM, AVM, and NSM, discussed below) of the other eat-4-expressing neurons are not known.
The functional role of eat-4 in M3 transmission has been well characterized. In eat-4 mutants the pharyngeal motor neuron M3-dependent inhibitory synaptic transmission is diminished, and consequently the duration of muscle contraction is prolonged (see Fig.1B) (Avery, 1993b; Raizen and Avery, 1994). It has been shown that the M3 neurotransmitter is glutamate and thateat-4 mutants have a presynaptic defect (Dent et al., 1997). These results, together with the fact thateat-4:: reporter genes are expressed in M3 (see Fig. 2A), argue that eat-4 functions in the M3 neurons to affect their glutamatergic synaptic transmission.
We demonstrated a similar presynaptic role of eat-4 in the synaptic transmission of the anterior touch cells ALM and AVM. We found that eat-4 is expressed in ALM and AVM cells (see Fig.2B) and that eat-4 is required for the fidelity of chemical transmission from these sensory neurons to their interneuron targets (see Fig. 4). That avr-15, which encodes a subunit of a glutamate receptor, affects the transmission of ALM and AVM suggests that ALM and AVM also are glutamatergic.
It appears that the expression of eat-4 in neurons ASH, IL1V, and OLQV is also functionally significant. The neurotransmission of these three types of sensory neurons is mediated by a glutamate receptor encoded by the glr-1 gene. glr-1 mutants show defects in their response to a touch to the nose, caused by a reduced ASH transmission, and they are abnormal for IL1V- and OLQV-dependent head withdrawal and foraging behaviors (Hart et al., 1995; Maricq et al., 1995). It has been found by A. C. Hart and J. M. Kaplan (personal communication) that eat-4mutants are defective in these ASH-, and IL1V- and OLQV-mediated behaviors.
Our reporter assay suggested that eat-4 may be expressed in the pharyngeal serotonergic NSM neurons (see Fig. 3A). However, our analysis showed that eat-4 has little effect on the serotonergic function of the NSM neurons in stimulating pharyngeal pumping (Table 1). It remains possible that eat-4 affects an NSM function independent from the one mediated by serotonergic transmission, the only function that we were able to assay. It is interesting to note that there are two types of vesicles (large and small) in the NSM neurons, suggesting that these neurons may use more than one neurotransmitter (Albertson and Thomson, 1976). Perhaps a second neurotransmitter is glutamate.
eat-4 is involved specifically in the synaptic function of glutamatergic neurons
How does eat-4 affect the function of the presynaptic neurons in glutamatergic transmission pathways? Our results are most consistent with the possibility that eat-4 affects a cellular function specific to glutamatergic synaptic transmission. The expression pattern of eat-4:: gfp ineat-4 mutants was similar to that in wild-type animals (see Fig. 3; data not shown), indicating that eat-4 does not prevent the development of eat-4-expressing cells. Our analysis of the effect of eat-4 mutations on the function of the anterior touch cells, ALM and AVM, showed that, although the chemical transmission of these cells was greatly affected, their other capacities necessary for mechanosensory neuron function, such as their ability to sense environmental stimuli and to transduce electrical signals through gap junction connections, were not affected byeat-4 (see Fig. 4A). Together, these observations suggest that the loss of eat-4 function specifically leads to a reduced capacity of glutamatergic neurons to release glutamate into synaptic junctions. Thus, eat-4affects either the synthesis or release of the neurotransmitter glutamate.
EAT-4 may function as a phosphate transporter regulating the synthesis of neurotransmitter glutamate
eat-4 potentially encodes a protein (EAT-4) that is highly similar in sequence to several mammalian Na-Pi cotransporters, suggesting that EAT-4 may function as a Na-Pi cotransporter in C. elegans. EAT-4 is most similar (48% identical) to BNPI, a brain-specific Na-Pi cotransporter cloned from rat cerebellar granule cells (Ni et al., 1994). BNPI mRNA appears to be expressed exclusively in discrete populations of neurons in rat brain (Ni et al., 1994,1995). Prominent BNPI RNA expression is found (Ni et al., 1995) in several regions of the brain that have concentrated perikarya of presumed glutamatergic neurons, such as cerebral cortical layers II–VI, the pyramidal cell layer of the hippocampus, the granule cell layers of the dentate gyrus and the cerebellum, the entorhinal cortex, and the piriform cortex (Storm-Mathisen and Ottersen, 1986; Knowles, 1992; Conti and Minelli, 1994). The general expression pattern of BNPI is suggestive of a preferential localization in glutamatergic neurons. The similarities of their protein sequences, their predicted secondary protein structures (see Fig. 2B), and their expression in glutamatergic neurons suggest that EAT-4 and BNPI may be functional homologs, although we do not have direct evidence supporting this notion.
In the mammalian CNS glutamine is a major precursor for the transmitter pool of glutamate. Results from metabolic labeling experiments showed that glutamine is used preferentially over glucose as the precursor for depolarization-releasable glutamate in brain slice and synaptosomal preparations (Bradford et al., 1978; Hamberger et al., 1979a,b; Ward et al., 1983). A phosphate-activated glutaminase (PAG; EC 3.5.1.2) hydrolyzes glutamine to glutamate and ammonia in the mitochondria of glutamatergic neurons (Kvamme, 1983; Erecinska and Silver, 1990;Fonnum, 1993). Biochemical analysis showed that mammalian PAG is activated by inorganic phosphate; increasing [Pi] lowers theKm for glutamine (Sayre and Roberts, 1958), probably as a result of the oligomerization of PAG homomers (Godfrey et al., 1977). The maximal activity of purified PAG is achieved when the [Pi] is in the range of 100 mm (Sayre and Roberts, 1958; Kovacevic and McGivan, 1983). However, the [Pi] in cerebrospinal fluid is estimated to be only 1–2 mm(Erecinska and Silver, 1990). Therefore, for neurons to have a substantially activated PAG, an active transport of phosphate across the plasma membrane may be required. One possible mechanism would be to couple phosphate transport with that of sodium ions that are driven to flow into a neuron by a positive concentration gradient. Indeed, it has been shown that such a Na+-dependent inorganic phosphate transport system (possibly encoded by BNPI) does exist in cultured rat cortical neurons (Glinn et al., 1995) and in rabbit brain synaptosomes (Salamin et al., 1981). We propose that in C. elegans glutamatergic neurons a similar Na-Pi cotransporter encoded by eat-4 is required for activating a neuronal PAG; a reduction in eat-4 function would lead to a reduced intracellular [Pi] and consequently to an insufficient supply of the neurotransmitter glutamate.
The amino acid glutamate is not only a neurotransmitter but also an essential building block for proteins and a component of general cellular metabolism. A reduction in overall glutamate synthesis would be expected to impair many cellular functions. If eat-4indeed affects glutamate synthesis as we propose, our results suggest that the presumed reduction in the level of neuronal glutamate preferentially drains the neurotransmitter pool but spares the metabolic pool in eat-4 mutants. In the mammalian CNS, PAG is concentrated in presynaptic nerve terminals. PAG thus has been postulated to function preferentially in the supply of the neurotransmitter pool as opposed to the metabolic pool of glutamate (for review, see Erecinska and Silver, 1990). The apparent chemical neurotransmission-specific defects we observed in anterior touch cells are consistent with this view.
Recently, Bellocchio and coworkers showed by immunohistochemistry that BNPI protein is localized in axon terminals of presumptive glutamatergic neurons in rat brain (Bellocchio et al., 1998). This exquisite protein localization suggests that, similar to EAT-4 inC. elegans, BNPI may have a glutamatergic neurotransmission-specific function in the rat CNS.
Ni et al. (1995) have suggested a contrasting model: that BNPI-mediated Pi transport may be important for the maintenance of high phosphorylation potentials in neurons. Indeed, the Pi transported into cultured rat cortical neurons can be incorporated into high-energy compounds such as ATP and ADP (Glinn et al., 1995). Our data indicate that if EAT-4 is involved in a general aspect of cellular energy metabolism there must be substantially different energy requirements between chemical neurotransmission and other functions of glutamatergic neurons.
We suggest that eat-4 mutations might cause a defect in the synthesis of the transmitter glutamate. Alternatively, the effect ofeat-4 mutations on glutamatergic neurotransmission could be explained by a reduction in the loading or the exocytosis of synaptic vesicles. A defect in any of these three processes could result in reduced neurotransmitter release. Future biochemical analyses are required to distinguish among these three possibilities.
In conclusion, we have identified eat-4 as a crucial component in glutamatergic neuron function in C. elegans. The molecular similarities between EAT-4 and BNPI suggest a phylogenetically conserved function of Na-Pi cotransporters in glutamatergic neurotransmission.
Footnotes
This research was supported by United States Public Health Service Research Grants HL46154 to L.A. and GM24663 to H.R.H. H.R.H. is an Investigator of the Howard Hughes Medical Institute. We thank theC. elegans Genome Consortium for cosmid sequences, A. Coulson for cosmids, Y. Kohara for the cDNA clone ofeat-4, A. Fire for GFP and lacZ expression vectors, C. I. Bargmann for the eat-4(ky5) mutant strain, and J. M. Kaplan for identifying ky5and n2474 to be alleles of eat-4. We also thank C. I. Bargmann, A. C. Hart, J. M. Kaplan, and I. Mori for communicating unpublished observations on eat-4mutant phenotypes and E. E. Bellocchio, H. Hu, A. Pohorille, J. Chan, V. M. Pickel, and R. H. Edwards for communicating unpublished observations on BNPI. Finally, we thank R. H. Edwards for pointing out the fact that the transmitter pool of glutamate is made by PAG in mammals and J. M. Kaplan for the critical reading of and suggestions on this manuscript.
Correspondence should be addressed to Dr. Raymond Y. N. Lee at his present address: Department of Molecular Biology, Massachusetts General Hospital, Wellman 8, Blossom Street, Boston, MA 02114.
REFERENCES
- 1.Albertson DG, Thomson JN. The pharynx of Caenorhabditis elegans. Philos Trans R Soc Lond [Biol] 1976;275:299–325. doi: 10.1098/rstb.1976.0085. [DOI] [PubMed] [Google Scholar]
- 2.Avery L. The genetics of feeding in Caenorhabditis elegans. Genetics. 1993a;133:897–917. doi: 10.1093/genetics/133.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avery L. Motor-neuron M3 controls pharyngeal muscle relaxation timing in Caenorhabditis elegans. J Exp Biol. 1993b;175:283–297. doi: 10.1242/jeb.175.1.283. [DOI] [PubMed] [Google Scholar]
- 4.Avery L, Horvitz HR. Pharyngeal pumping continues after laser killing of the pharyngeal nervous system of C. elegans. Neuron. 1989;3:473–485. doi: 10.1016/0896-6273(89)90206-7. [DOI] [PubMed] [Google Scholar]
- 5.Avery L, Horvitz HR. Effects of starvation and neuroactive drugs on feeding in Caenorhabditis elegans. J Exp Zool. 1990;253:263–270. doi: 10.1002/jez.1402530305. [DOI] [PubMed] [Google Scholar]
- 6.Bargmann CI, Horvitz HR. Chemosensory neurons with overlapping functions direct chemotaxis to multiple chemicals in C. elegans. Cell. 1991;7:729–742. doi: 10.1016/0896-6273(91)90276-6. [DOI] [PubMed] [Google Scholar]
- 7.Bellocchio EE, Hu H, Pohorille A, Chan J, Pickel VM, Edwards RH. The localization of the brain-specific inorganic phosphate transporter suggests a specific presynaptic role in glutamatergic transmission. J Neurosci. 1998;18:8648–8659. doi: 10.1523/JNEUROSCI.18-21-08648.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bicker G, Schäfer S, Ottersen OP, Storm-Mathisen J. Glutamate-like immunoreactivity in identified neuronal populations of insect nervous systems. J Neurosci. 1988;8:2108–2122. doi: 10.1523/JNEUROSCI.08-06-02108.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradford HF, Ward HK, Thomas AJ. Glutamine—a major substrate for nerve endings. J Neurochem. 1978;30:1453–1459. doi: 10.1111/j.1471-4159.1978.tb10477.x. [DOI] [PubMed] [Google Scholar]
- 10.Chalfie M, White J. The nervous system. In: Wood WB, the Community of C. elegans Researchers, editor. The nematode Caenorhabditis elegans. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1988. pp. 337–391. [Google Scholar]
- 11.Chalfie M, Sulston JE, White JG, Southgate E, Thomson JN, Brenner S. The neural circuit for touch sensitivity in Caenorhabditis elegans. J Neurosci. 1985;5:956–964. doi: 10.1523/JNEUROSCI.05-04-00956.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 13.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 14.Chong SS, Kristjansson K, Zoghbi HY, Hughes MR. Molecular cloning of the cDNA encoding a human renal sodium phosphate transport protein and its assignment to chromosome 6p21.3-p23. Genomics. 1993;18:355–359. doi: 10.1006/geno.1993.1476. [DOI] [PubMed] [Google Scholar]
- 15.Chong SS, Kozak CA, Liu L, Bordeau JE, Hughes MR, Kristjansson K, Dunn ST. Cloning, genetic mapping, and expression analysis of a mouse renal sodium-dependent phosphate cotransporter. Am J Physiol. 1995;268:1038–1045. doi: 10.1152/ajprenal.1995.268.6.F1038. [DOI] [PubMed] [Google Scholar]
- 16.Clark SG, Lu WX, Horvitz HR. The Caenorhabditis elegans locus lin-15, a negative regulator of a tyrosine kinase signaling pathway, encodes two different proteins. Genetics. 1994;137:987–997. doi: 10.1093/genetics/137.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conti F, Minelli A. Glutamate immunoreactivity in rat cerebral cortex is reversibly abolished by 6-diazo-5-oxo-l-norleucine (DON), an inhibitor of phosphate-activated glutaminase. J Histochem Cytochem. 1994;42:717–726. doi: 10.1177/42.6.7910617. [DOI] [PubMed] [Google Scholar]
- 18.Dale N, Kandel ER. l-Glutamate may be the fast excitatory transmitter of Aplysia sensory neurons. Proc Natl Acad Sci USA. 1993;90:7163–7167. doi: 10.1073/pnas.90.15.7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis MW, Somerville D, Lee RYN, Lockery S, Avery L, Fambrough DM. Mutations in the Caenorhabditis elegans Na, K-ATPase α-subunit gene, eat-6, disrupt excitable cell function. J Neurosci. 1995;15:8408–8418. doi: 10.1523/JNEUROSCI.15-12-08408.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dent JA, Davis MW, Avery L. avr-15 encodes a chloride channel subunit that mediates inhibitory glutamatergic neurotransmission and ivermectin sensitivity in Caenorhabditis elegans. EMBO J. 1997;16:5867–5879. doi: 10.1093/emboj/16.19.5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erecinska M, Silver IA. Metabolism and role of glutamate in mammalian brain. Prog Neurobiol. 1990;35:245–296. doi: 10.1016/0301-0082(90)90013-7. [DOI] [PubMed] [Google Scholar]
- 22.Fire A, Harrison SW, Dixon D. A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans. Gene. 1990;93:189–198. doi: 10.1016/0378-1119(90)90224-f. [DOI] [PubMed] [Google Scholar]
- 23.Fonnum F. Regulation of the synthesis of the transmitter glutamate pool. Prog Biophys Mol Biol. 1993;60:47–57. doi: 10.1016/0079-6107(93)90012-9. [DOI] [PubMed] [Google Scholar]
- 24.Gerschenfeld HM. Chemical transmission in invertebrate central nervous systems and neuromuscular junctions. Physiol Rev. 1973;53:1–119. doi: 10.1152/physrev.1973.53.1.1. [DOI] [PubMed] [Google Scholar]
- 25.Glinn M, Ni B, Paul SM. Characterization of Na+-dependent phosphate uptake in cultured fetal rat cortical neurons. J Neurochem. 1995;65:2358–2365. doi: 10.1046/j.1471-4159.1995.65052358.x. [DOI] [PubMed] [Google Scholar]
- 26.Godfrey S, Kuhlenschmidt T, Curthoys NP. Correlation between activation and dimer formation of rat renal phosphate-dependent glutaminase. J Biol Chem. 1977;252:1927–1931. [PubMed] [Google Scholar]
- 27.Hamberger AC, Chiang GH, Nylen ES, Scheff SW, Cotman CW. Glutamate as a CNS transmitter. I. Evaluation of glucose and glutamine as precursors for the synthesis of preferentially released glutamate. Brain Res. 1979a;168:513–530. doi: 10.1016/0006-8993(79)90306-8. [DOI] [PubMed] [Google Scholar]
- 28.Hamberger A, Chiang GH, Sandoval E, Cotman CW. Glutamate as a CNS transmitter. II. Regulation of synthesis in the releasable pool. Brain Res. 1979b;168:531–541. doi: 10.1016/0006-8993(79)90307-x. [DOI] [PubMed] [Google Scholar]
- 29.Hart AC, Sims S, Kaplan JM. Synaptic code for sensory modalities revealed by C. elegans GLR-1 glutamate receptor. Nature. 1995;378:82–85. doi: 10.1038/378082a0. [DOI] [PubMed] [Google Scholar]
- 30.Horseman BG, Seymour C, Bermudez I, Beadle DJ. The effects of l-glutamate on cultured insect neurons. Neurosci Lett. 1988;85:65–70. doi: 10.1016/0304-3940(88)90430-2. [DOI] [PubMed] [Google Scholar]
- 31.Horvitz HR, Chalfie M, Trent C, Sulston JE, Evans PD. Serotonin and octopamine in the nematode C. elegans. Science. 1982;216:1012–1014. doi: 10.1126/science.6805073. [DOI] [PubMed] [Google Scholar]
- 32.Huang LS, Tzou P, Sternberg PW. The lin-15 locus encodes two negative regulators of Caenorhabditis elegans vulval development. Mol Biol Cell. 1994;5:395–411. doi: 10.1091/mbc.5.4.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jahr CE, Lester AJ. Synaptic excitation mediated by glutamate-gated ion channels. Curr Opin Neurobiol. 1992;2:270–274. doi: 10.1016/0959-4388(92)90114-z. [DOI] [PubMed] [Google Scholar]
- 34.Kaplan JM, Horvitz HR. A dual mechanosensory and chemosensory neuron in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1993;90:2227–2231. doi: 10.1073/pnas.90.6.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knowles WD. Normal anatomy and neurophysiology of the hippocampal formation. J Clin Neurophysiol. 1992;9:252–263. [PubMed] [Google Scholar]
- 36.Kovacevic Z, McGivan JD. Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol Rev. 1983;63:547–605. doi: 10.1152/physrev.1983.63.2.547. [DOI] [PubMed] [Google Scholar]
- 37.Kvamme E. Glutaminase (PAG). Neurol Neurobiol. 1983;7:51–67. [Google Scholar]
- 38.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Avery L, Denk W, Hess GP. Identification of chemical synapses in the pharynx of Caenorhabditis elegans. Proc Natl Acad Sci USA. 1997;94:5912–5916. doi: 10.1073/pnas.94.11.5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loer CM, Kenyon CJ. Serotonin-deficient mutants and male mating behavior in the nematode Caenorhabditis elegans. J Neurosci. 1993;13:5407–5417. doi: 10.1523/JNEUROSCI.13-12-05407.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maricq AV, Peckol E, Driscoll M, Bargmann CI. Mechanosensory signaling in C. elegans mediated by the GLR-1 glutamate receptor. Nature. 1995;378:78–81. doi: 10.1038/378078a0. [DOI] [PubMed] [Google Scholar]
- 42.McIntire SL, Jorgensen E, Horvitz HR. Genes required for GABA function in Caenorhabditis elegans. Nature. 1993a;364:334–337. doi: 10.1038/364334a0. [DOI] [PubMed] [Google Scholar]
- 43.McIntire SL, Jorgensen E, Kaplan J, Horvitz HR. The GABAergic nervous system of Caenorhabditis elegans. Nature. 1993b;364:337–341. doi: 10.1038/364337a0. [DOI] [PubMed] [Google Scholar]
- 44.Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- 45.Mello C, Fire A. DNA transformation. In: Epstein HF, Shakes DC, editors. Methods in cell biology, Vol 48, Caenorhabditis elegans: modern biological analysis of an organism. Academic; San Diego: 1995. pp. 451–482. [PubMed] [Google Scholar]
- 46.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1992;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyamoto K, Tatsumi S, Sonoda T, Yamamoto H, Minami H, Taketani Y, Takeda E. Cloning and functional expression of a Na+-dependent phosphate cotransporter from human kidney: cDNA cloning and functional expression. Biochem J. 1995;305:81–85. doi: 10.1042/bj3050081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ni B, Rosteck PR, Jr, Nadi NS, Paul SM. Cloning and expression of a cDNA encoding a brain-specific Na+-dependent inorganic phosphate cotransporter. Proc Natl Acad Sci USA. 1994;91:5607–5611. doi: 10.1073/pnas.91.12.5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ni B, Wu X, Yan G-M, Wang J, Paul SM. Regional expression and cellular localization of the Na+-dependent inorganic phosphate cotransporter of rat brain. J Neurosci. 1995;15:5789–5799. doi: 10.1523/JNEUROSCI.15-08-05789.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quinlan EM, Murphy AD. Glutamate as a putative neurotransmitter in the buccal central pattern generator of Helisoma trivolvis. J Neurophysiol. 1991;66:1264–1271. doi: 10.1152/jn.1991.66.4.1264. [DOI] [PubMed] [Google Scholar]
- 51.Raizen DM, Avery L. Electrical activity and behavior in the pharynx of Caenorhabditis elegans. Neuron. 1994;12:483–495. doi: 10.1016/0896-6273(94)90207-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raizen DM, Lee RYN, Avery L. Interacting genes required for pharyngeal excitation by motor neuron MC in Caenorhabditis elegans. Genetics. 1995;141:1365–1382. doi: 10.1093/genetics/141.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salamin A, Deshusses J, Straub RW. Phosphate ion transport in rabbit brain synaptosomes. J Neurochem. 1981;37:1419–1424. doi: 10.1111/j.1471-4159.1981.tb06310.x. [DOI] [PubMed] [Google Scholar]
- 54.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning, Ed 2. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 55.Sayre FW, Roberts E. Preparation and some properties of a phosphate-activated glutaminase from kidneys. J Biol Chem. 1958;233:1128–1134. [PubMed] [Google Scholar]
- 56.Storm-Mathisen J, Ottersen OP. Anatomy of putative glutamatergic neurons. In: Avoli M, Reader TA, Dykes RW, Gloor P, editors. Neurotransmitters and cortical function: from molecules to mind. Plenum; New York: 1986. pp. 39–70. [Google Scholar]
- 57.Sulston J, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode C. elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 58.Sulston J, Du Z, Thomas K, Wilson R, Hillier L, Staden R, Halloran N, Green P, Thierry-Mieg J, Qiu L, Drear S, Coulson A, Craxton M, Durbin R, Berks M, Metzstein M, Hawkins T, Ainscough R, Waterston R. The C. elegans genome sequencing project: a beginning. Nature. 1992;365:37–41. doi: 10.1038/356037a0. [DOI] [PubMed] [Google Scholar]
- 59.Trent C, Tsung N, Horvitz HR. Egg-laying defective mutants of the nematode C. elegans. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trudeau L-E, Castellucci VF. Excitatory amino acid neurotransmission at sensory-motor and interneuronal synapses of Aplysia californica. J Neurophysiol. 1993;70:1221–1230. doi: 10.1152/jn.1993.70.3.1221. [DOI] [PubMed] [Google Scholar]
- 61.Walker RJ, Roberts CJ. The pharmacology of Limulus central neurons. Comp Biochem Physiol [C] 1982;72:391–401. doi: 10.1016/0306-4492(82)90110-1. [DOI] [PubMed] [Google Scholar]
- 62.Ward HK, Thanki CM, Bradford HF. Glutamine and glucose as precursors of transmitter amino acids: ex vivo studies. J Neurochem. 1983;40:855–860. doi: 10.1111/j.1471-4159.1983.tb08058.x. [DOI] [PubMed] [Google Scholar]
- 63.Werner A, Moore ML, Mantei N, Biber J, Semenza G, Murer H. Cloning and expression of cDNA for a Na/Pi cotransport systems of kidney cortex. Proc Natl Acad Sci USA. 1991;88:9608–9612. doi: 10.1073/pnas.88.21.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whetsell WO., Jr Current concepts of excitotoxicity. J Neuropathol Exp Neurol. 1996;55:1–13. doi: 10.1097/00005072-199601000-00001. [DOI] [PubMed] [Google Scholar]
- 65.White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode C. elegans. Philos Trans R Soc Lond [Biol] 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 66.Wicks SR, Roehrig CJ, Rankin CH. A dynamic network simulation of the nematode tap withdrawal circuit: predictions concerning synaptic function using behavioral criteria. J Neurosci. 1996;16:4017–4031. doi: 10.1523/JNEUROSCI.16-12-04017.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]