

Abstract
The mitotic kinesin Eg5 is critical for the assembly of the mitotic spindle and is a promising chemotherapy target. Previously, we identified S-trityl-l-cysteine as a selective inhibitor of Eg5 and developed triphenylbutanamine analogues with improved potency, favorable drug-like properties, but moderate in vivo activity. We report here their further optimization to produce extremely potent inhibitors of Eg5 (Kiapp < 10 nM) with broad-spectrum activity against cancer cell lines comparable to the Phase II drug candidates ispinesib and SB-743921. They have good oral bioavailability and pharmacokinetics and induced complete tumor regression in nude mice explanted with lung cancer patient xenografts. Furthermore, they display fewer liabilities with CYP-metabolizing enzymes and hERG compared with ispinesib and SB-743921, which is important given the likely application of Eg5 inhibitors in combination therapies. We present the case for this preclinical series to be investigated in single and combination chemotherapies, especially targeting hematological malignancies.
Introduction
Mitotic kinesins represent an exciting class of oncology targets.1 The members of this structurally divergent superfamily of microtubule (MT)-based molecular motor proteins have extensive involvement throughout mitosis, with functions ranging from the organization of the mitotic architecture to the transport of regulatory proteins.2,3 One of the most studied is the kinesin spindle protein (KSP, also known as Eg5, Kif11, KNSL1; kinesin-5 family), whose primary role is to establish the bipolar spindle in early prometaphase.4,5 Inhibition of Eg5 by small molecule inhibitors results in monopolar spindles and mitotic arrest which can lead to cell death.6 This has proved highly effective across a variety of in vitro and in vivo models of cancer, and total regression has been demonstrated in a number of xenograft models.7−9 A great variety of inhibitor scaffolds have been described in the literature,3,10,11 the majority of which are allosteric inhibitors that bind to an induced fit pocket formed by loop L5, approximately 10 Å from the nucleotide binding site.12 These allosteric inhibitors exhibit excellent selectivity for Eg5 among kinesins due to the unique length of loop L5 among the kinesin superfamily.13 A number of compounds have progressed to clinical trials, with the most advanced in phase II (e.g., 1–3, Figure 1); however, to date the best outcome in the majority of studies against advanced solid tumors has been limited to disease stabilization.1,11 Better responses were achieved targeting hematological malignancies with Eg5 inhibitors.14,15 In a phase II study with the 1,3,4,-thiadiazole-based candidate ARRY-520 (3),8 a 19% objective response rate was recorded in patients with relapsed/refractory multiple myelomas.15 Additive responses were reported on treatment with 3 and the proteasome inhibitor bortezomib in xenograft models of multiple myeloma, and combination clinical trials are now planned.15,16 Targeting hematological malignancies with Eg5 inhibitors as single agents or in combination with existing therapies therefore represents an exciting area of investigation which may offer clinical advantages over current regimens.
Figure 1.

Selected Eg5 inhibitors ispinesib (1), SB-743921 (2), and ARRY-520 (3) in clinical development.
Our group previously identified S-trityl-l-cysteine (STLC, 4) as a tight-binding and selective allosteric inhibitor of Eg5 (Figure 2).17−19 In common with other loop L5 Eg5 inhibitors, 4 blocks bipolar spindle formation causing mitotic arrest, which ultimately leads to apoptotic cell death across a variety of tumor cell lines.18 This primarily occurs through activation of the spindle checkpoint and the intrinsic apoptotic pathway.20 Antitumor activity has been reported for 4 and the p-methoxyphenyl analogue 7 across leukemia, bladder, and prostate xenograft models.21−23 In initial SAR investigations, analogues with improved cellular potency were identified which incorporated a phenyl containing lipophilic para substituents (e.g., 5–7, Figure 2).24−26 The subsequent elucidation of the crystal structure of 4 in complex with Eg527,28 enabled us to pursue the rational SAR-based optimization of this scaffold, which led to the development of butanamine analogues with improved activity against Eg5 (11–14, Figure 2).29 The lead triphenylbutanamine from our previous study (11) had improved potency, favorable drug-like properties, and in vivo antitumor activity in a lung cancer xenograft model. We report here on the next phase of development, which has produced analogues that significantly improve on both the potency and in vivo efficacy of the previous leads. Across multiple cancer cell lines, these new leads demonstrate comparable in vitro activity to the phase II candidate ispinesib (1), and its more potent second generation analogue SB-743921 (3) which is also undergoing clinical studies (Figure 1).1,9,30 Favorable drug-like properties are evident across a selection of the most potent analogues, in addition to compelling in vivo pharmacokinetics. Finally, the new lead analogue induces complete tumor regression in a lung cancer xenograft model. In addition to surpassing the activity of all previously described S-trityl-l-cysteine analogues, this in vivo efficacy is comparable with that of the drug candidates 1–3 and strongly warrants further development.
Figure 2.

The structure of S-trityl-l-cysteine (4) and selected previously reported analogues. The crystal structures of Eg5 in complex with 4, 5, and 11 have previously been determined and used for structure-based drug design.26,27,29
Results and Discussion
Synthesis
S-Tritylthioethanamine and cysteine analogues were prepared by dehydration of trityl alcohols in trifluoroacetic acid and thioetherification with l-cysteine or cysteamine hydrochloride (Scheme 1).31 Intermediate trityl alcohols were synthesized by the reduction of benzophenone analogues with lithiated aryl bromides; key thioethanamines were therefore accessible in two steps. The trityl alcohol 59 containing a disubstituted m-ethyl,p-methylphenyl was prepared from the 5-bromobenzoic acid derivative 55 by converting to the Weinreb amide 56 to enable controlled access to ketone 57,32 which was subsequently reduced with hydrazine hydrate to afford the dialkyl aryl bromide 58 (Scheme 2). Subsequently, lithium bromine exchange and reduction of benzophenone and thioetherification of 59 afforded the target thioether 36.
Scheme 1. General Route for Synthesis of Thioethers.

Reagents and conditions: (i) n-BuLi, −78 °C, 1 h; (ii) benzophenone or 3-hydroxybenzophenone, THF, −78 °C, 4–6 h, then rt overnight; (iii) cysteamine hydrochloride or l-cysteine, TFA, rt, 3 h.
Scheme 2.

Triphenylbutanamine (CH2-trityl) analogues with the carboxylic acid were prepared by the route reported previously from common trityl alcohol intermediates, as illustrated for rac-42 (Scheme 3).29 Allylation of trityl alcohol 60 with iron trichloride mediating carbon–oxygen bond cleavage afforded the carbon framework,33 prior to successive oxidations by hydroboration–oxidation to the primary alcohol 62 and then Dess–Martin periodinane (DMP) to aldehyde 63.34 This was converted to α-aminonitrile rac-64 by a modified version of the Strecker synthesis employing Montmorillonite KSF clay.35 Hydrolysis of rac-64 and subsequent hydrogenation of benzylamine rac-65 using ammonium formate gave the racemic amino acid 45.36 Attempts to resolve rac-42 by chiral HPLC using a ChiralPak IC column as described previously were not successful.29 Triphenylbutanamines without the carboxylic acid were prepared by functional group interconversion from primary alcohol intermediates (e.g., 62) to the primary amine via the mesylate and azide (Supporting Information, Schemes S6–S8).29
Scheme 3.

The β-fluorinated triphenylbutanamine rac-52 was prepared by the enamine-based organocatalyzed α-fluorination of aldehyde 66 with l-proline and N-fluorobenzenesulfonimide, followed by immediate reduction37 to afford β-fluorinated alcohol rac-67 (Scheme 4).38 Expedient access to rac-28 was then achieved by stepwise conversion via the tosylate and azide. The β,β-difluorinated amine 53 was synthesized by a comparable methodology with SelectFluor as the electrophilic source of fluorine.39 Direct access to azide 69 was possible from β,β-difluorinated alcohol 68 under microwave conditions, and subsequent reduction of 69 yielded the β,β-difluorinated-amine 53. The α-trifluoromethyl amine rac-54 was prepared from aldehyde 66 by trifluoromethylation with TMS-CF3;40 subsequent functional group interconversion of trifluoromethyl alcohol rac-70 via the triflate and azide gave the desired product rac-54.41
Scheme 4.

Structure–Activity Relationship (SAR) Studies: Monosubstituted Phenyl Rings
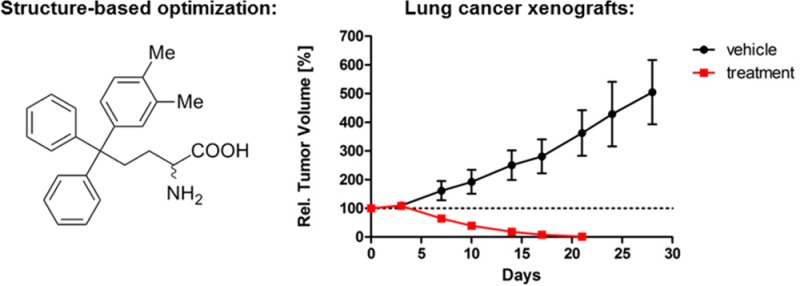
The most potent analogues identified previously contained a lipophilic substituent in the meta or para position of one phenyl ring.24,29 Crystallographic studies with p-chlorophenyl- and m-tolyl-containing analogues 5 and 11 revealed these substituents to be preferentially positioned in the predominantly hydrophobic environment of the P3 pocket (Figure 3B).24,27 In the meta position, the optimal substituents were typically small alkyl groups that were electron-donating and hydrophobic,29 which we rationalized as being due to strengthening the C–H···π interaction between the isopropyl side chain of Leu 214 and the phenyl ring (Figure 3A).27 A small number of thioethanamines was prepared to determine whether this was also applicable for substituents in the para position (15–21, Table 1). The p-methyl and p-methoxy analogues 17 and 19 demonstrated similar basal inhibition activity, that was comparable to 16 and improved ∼4-fold over 4, and while a drop in potency was apparent upon increasing from methyl to ethyl (17 and 18), this was not seen with the ether analogues 19–21. We evaluated all inhibitors of Eg5 in the basal assay for growth inhibition against K562 human leukemia cells. This cell line has previously been shown to be susceptible to Eg5 inhibition and to overexpress Eg5.42,43 Improved activity compared with 4 was exhibited in the cellular assay by 17, 18, and 19 containing p-alkyl and p-methoxy substituents, with 18 and 19 matching that of the potent meta-substituted analogue 16.29 Ethoxy and triofluoromethoxy derivatives 20 and 21 proved less active, which may be due to less appropriate physicochemical properties such as reduced aqueous solubility (e.g., 20: turbidimetric solubility at pH 7.4 = 3.75 μM). To determine whether the optimal thioethanamine trityl substituents were transferrable to the butanamine-based inhibitors, several triphenylbutanamine analogues containing a single substituent in the meta or para position were prepared (22–27, Table 1). Triphenylbutanamines 22 and 23 incorporating m-ethyl and m-isopropyl substituents, respectively, proved very potent in both assays, with 22 ∼3-fold more active against cells than the comparable thioether 16. The p-methoxyphenyl analogues rac-25 and 26 with and without the α-carboxylate both displayed GI50 < 100 nM, which improved significantly over the equivalent S-trityl pair 7 and 19. While these findings confirmed the systematically improved activity of the butanamines versus analogous thioethanamines, in general these new compounds exhibited equivalent in vitro activity to the previously described leads (e.g., rac-11 and 13). The activity of the p-methoxy analogue 26 was however improved (∼2-fold) versus other analogues without the carboxylate and now equivalent in the cell-based assay to the zwitterionic rac-11. Interestingly, while rac-25 and 26 exhibited comparably strong activity across both assays, the zwitterionic thioether 7 was clearly superior in the cellular assay to 19.
Figure 3.

(A) The Eg5–4 allosteric binding site, illustrating interactions with proximal (4 Å) protein residues.29 Hydrogen bonds are represented as dashed lines. (B) The Eg5–4 inhibitor-binding pocket with a solid surface illustrating the nomenclature for subpockets P1, P2, and P3. (C) Stereoplot of (R)-46 in the allosteric binding site. Hydrogen bonds between inhibitor (blue) and Eg5 residues (beige) are depicted by black broken lines. Coordinate and structure factor files for the Eg5–46 complex (PDB ID: 4BBG) were deposited at the PDB.
Table 1. Analogues with Monosubstituted Phenyl Ringsa.
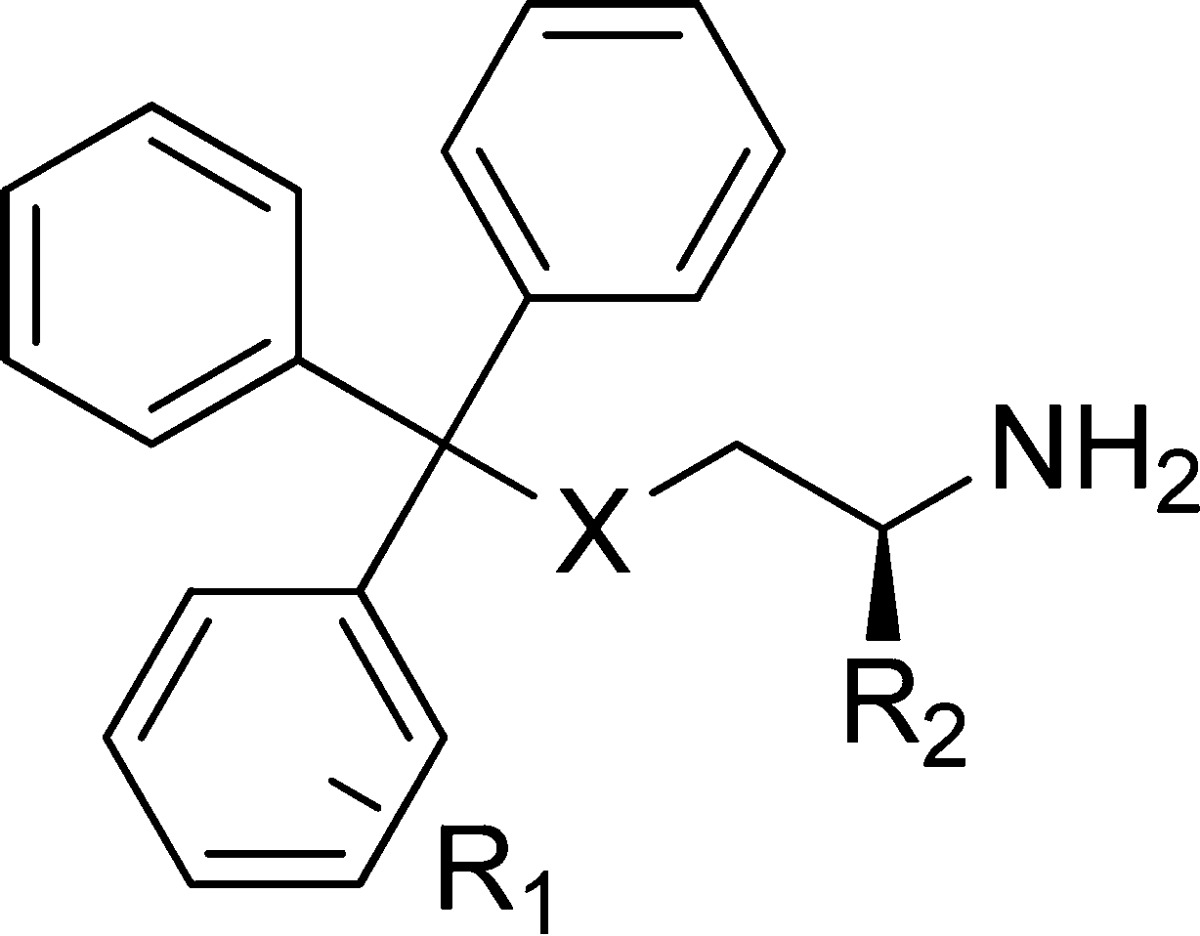
| compd | X | R1 | R2 | inhibition of basal ATPase activity Kiapp (nM) | LEb | K562 cells GI50 (nM) |
|---|---|---|---|---|---|---|
| 4 | S | H | (R)-CO2H | 107.3 ± 2.6 | 0.37 | 1452 ± 76 |
| 15 | S | H | H | 245.0 ± 1.8 | 0.39 | 1791 ± 166 |
| 16a | S | 3-Et | H | 23.4 ± 5.2 | 0.42 | 680 ± 84 |
| 17 | S | 4-Me | H | 27.4 ± 0.7 | 0.43 | 731 ± 36 |
| 18 | S | 4-Et | H | 57.5 ± 9.1 | 0.39 | 871 ± 59 |
| 7 | S | 4-OMe | (R)-CO2H | 15.7 ± 0.9 | 0.38 | 240 ± 17 |
| 19 | S | 4-OMe | H | 21.5 ± 2.8 | 0.42 | 700 ± 27 |
| 20 | S | 4-OEt | H | 17.3 ± 2.5 | 0.41 | 1901 ± 212 |
| 21 | S | 4-OCF3 | H | 29.7 ± 3.8 | 0.37 | 2218 ± 198 |
| rac-11a | C | 3-Me | CO2H | 12.2 ± 3.8 | 0.40 | 73 ± 3 |
| 13a | C | 3-Me | H | 8.8 ± 1.8 | 0.46 | 200 ± 16 |
| 22 | C | 3-Et | H | 6.4 ± 0.9 | 0.45 | 253 ± 13 |
| 23 | C | 3-i-Pr | H | 6.7 ± 1.3 | 0.43 | 305 ± 30 |
| 14a | C | 4-Me | H | 16.4 ± 1.9 | 0.44 | 219 ± 21 |
| 24 | C | 4-Et | H | 9.7 ± 3.2 | 0.44 | 750 ± 34 |
| rac-25 | C | 4-OMe | CO2H | 9.0 ± 1.7 | 0.39 | 94 ± 8 |
| 26 | C | 4-OMe | H | 15.5 ± 5.3 | 0.43 | 83 ± 4 |
| rac-27 | C | 4-OH | CO2H | 128.6 ± 12.6 | 0.35 | 1111 ± 68 |
Values provided for reference from our previous study.29
LE = ligand efficiency.
Disubstituted Phenyl Rings
Having identified the optimal substituents to improve affinity, we next investigated disubstituted phenyl rings. By broadening the scope of our SAR data, we hoped to utilize this knowledge to allow for more permutations to modulate the metabolic and pharmacokinetic properties of the ultimate lead candidates by unearthing multiple modifications capable of imbuing Eg5 affinity. A 2,5-difluorophenyl ring in the P3 pocket produced impressive increases in Eg5 inhibitory activity during optimization of the clinical candidate MK-0731.44 In our own series, although a m-fluoro substituent had proved detrimental to Eg5 affinity,29 we had not examined the influence of fluoro substituents in alternative positions in combination with the optimal alkyl and methoxy substituents. An o-fluoro substituent diminished inhibitory activity against Eg5 substantially when combined with a methyl substituent in either meta or para (28 and 29, Table 2). However, when combined with a p-methoxy substituent (31), the o-fluorine produced a ∼2-fold improvement in potency against Eg5 compared with the p-methoxyphenyl 19. Retaining the p-methoxy substituent and repositioning the fluoro substituent in the meta position was not tolerated (32). In the cellular assay, 28, 29, 32, and 33 proved weak inhibitors, but the improvements evident in the basal assay for 31 translated to a modest improvement in growth inhibition compared with the p-methoxyphenyl containing 19. On introduction of the carboxylic acid (30), a further 3-fold improvement in growth inhibitory activity was realized compared with 7, equivalent to the butanamine lead rac-11. Interestingly the butanamine analogue of 30, compound 40 proved less potent in both the enzymatic and cellular assays.
Table 2. Analogues Containing Disubstituted Phenyl Rings.
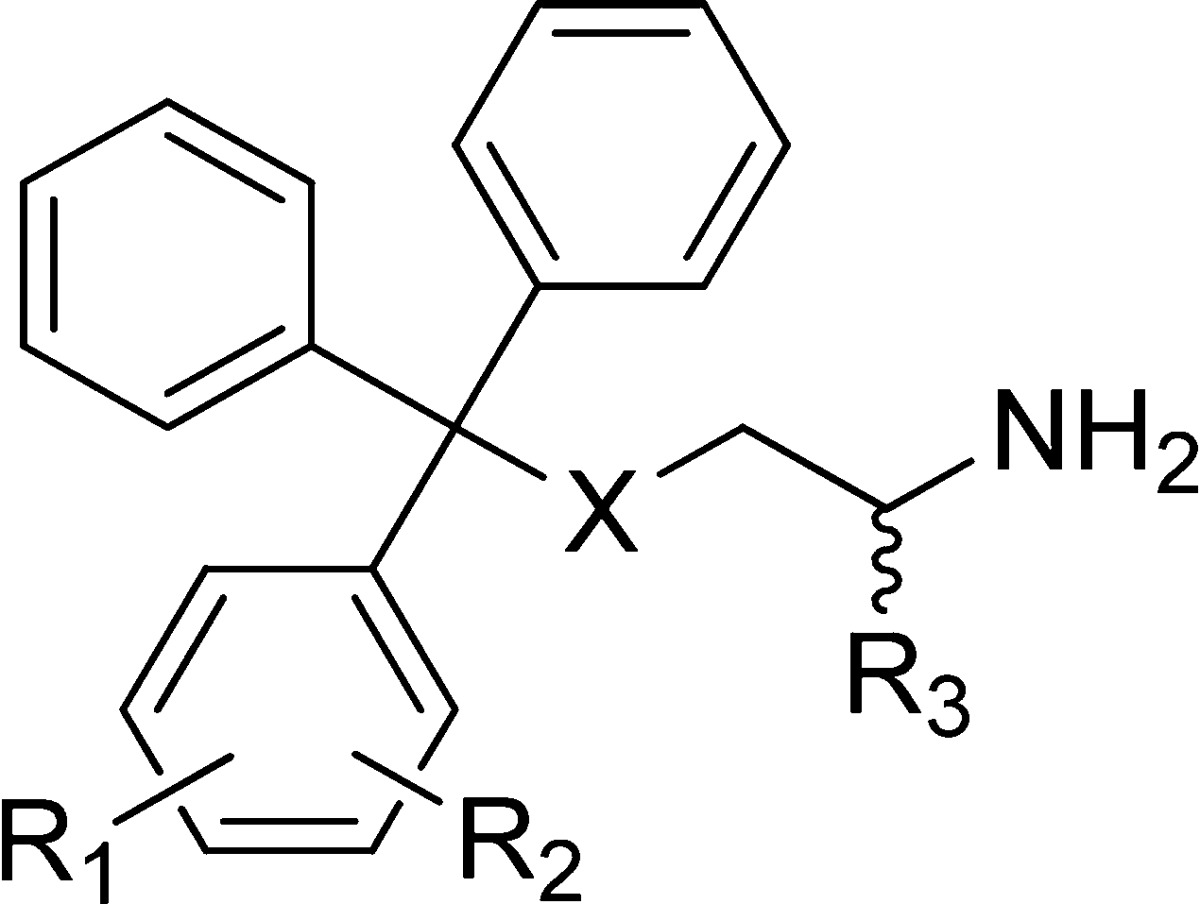
| compd | X | R1 | R2 | R3 | inhibition of basal ATPase activity Kiapp (nM) | LE | K562 cells GI50 (nM) |
|---|---|---|---|---|---|---|---|
| 28 | S | 2-F | 3-Me | H | 293.6 ± 23.2 | 0.36 | 2547 ± 141 |
| 29 | S | 2-F | 4-Me | H | 201.3 ± 18.7 | 0.37 | 2084 ± 109 |
| 30 | S | 2-F | 4-OMe | (R)-CO2H | 10.4 ± 4.5 | 0.38 | 82 ± 3 |
| 31 | S | 2-F | 4-OMe | H | 11.6 ± 3.7 | 0.42 | 489 ± 26 |
| 32 | S | 3-F | 4-OMe | H | 162.2 ± 15.6 | 0.36 | 1892 ± 134 |
| 33 | S | 3-Cl | 4-Cl | H | 35.2 ± 4.9 | 0.41 | 1993 ± 343 |
| 34 | S | 3-Me | 4-Me | (R)-CO2H | 1.2 ± 0.1 | 0.43 | 72 ± 8 |
| 35 | S | 3-Me | 4-Me | H | 25.7 ± 6.3 | 0.41 | 729 ± 43 |
| 36 | S | 3-Et | 4-Me | (R)-CO2H | 4.6 ± 1.7 | 0.39 | 34 ± 2 |
| 37 | S | 3-Et | 4-Me | H | 7.8 ± 3.9 | 0.43 | 1045 ± 42 |
| 38 | S | 3,4-(CH2)4 | (R)-CO2H | 2.1 ± 0.5 | 0.40 | 56 ± 2 | |
| 39 | S | 3,4-(CH2)4 | H | 10.6 ± 3.2 | 0.40 | 934 ± 127 | |
| 40 | C | 2-F | 4-OMe | H | 37.9 ± 4.7 | 0.39 | 764 ± 42 |
| 41 | C | 3-Cl | 4-Cl | H | 31.4 ± 6.4 | 0.41 | 633 ± 126 |
| rac-42 | C | 3-Me | 4-Me | CO2H | 12.4 ± 4.4 | 0.38 | 23.4 ± 1.8 |
Next, we investigated combinations of dialkyl substitutuents following from the observation that a β-naphthyl analogue of 4 demonstrated good potency against both Eg5, in vitro and in HeLa cells, with docking studies suggesting that the bicyclic ring occupied the P3 pocket.24 Compounds 34–39 were prepared to investigate whether more metabolically amenable and physiochemically favorable combinations of dialkyl substituents could be implemented in place of the naphthyl moiety (Table 2).45 The dialkylphenyl and tetralene analogues (35, 37, and 39) all proved comparably effective inhibitors of Eg5 basal activity to singly substituted analogues (e.g., 16–27). When the terminal carboxylic acid was introduced, incremental improvements in activity were apparent: the dimethylphenyl compound 34 displayed the highest affinity for Eg5 of any S-trityl-l-cysteine analogue, with an estimated Kiapp ≈ 1.2 nM. However, in the cell-based assay, 35, 37, and 39 lacking the carboxylate were disappointingly weak inhibitors, with poor solubility probably the principal cause of their reduced cellular efficacy (e.g., turbidimetric solubility at pH 7.4: 35 = 3.75 μM, Supporting Information, Table S3). The presence of the terminal α-carboxylic acid in the tail ameliorated this: in addition to Kiapp < 5 nM against Eg5 in the basal assay, the l-cysteine analogues 34, 36, and 38 all exhibited improved activity over the butanamine lead rac-11 in the cell-based assay. The most potent growth inhibition activity was exhibited by 36 (GI50 ≈ 34 nM), which was ∼30-fold better than 37 without the α-carboxylate, highlighting the importance of achieving the correct physicochemical balance. S-Alkylated derivatives of cysteine were first designed as anticancer agents following the observation that radiolabeled cysteine was incorporated by leukemic white blood cells,46,47 so active uptake of the amino acid zwitterion also cannot be discounted as contributing to the improved activity of 36 compared with 37. Next, we transferred the optimal dialkyl substituent pattern to the butanamine scaffold. The poor aqueous solubility of tetralene 38 even when the carboxylic acid was present precluded continuing with this modification (turbidimetric solubility at pH 7.4 = 20 μM). Although 36 was more potent in the cellular assay, the superior Eg5 affinity and physicochemical properties of the dimethylphenyl analogue 34 marked this as the optimal modification from this selection. The prepared compound rac-42 was less potent in the basal assay but importantly proved 3-fold more active in the cellular growth inhibition assay (GI50 ≈ 23 nM), thereby improving on the activity of both the previously reported butanamine leads 11 and 12 (Figure 2)29 and the l-cysteine analogues 34, 36, and 38 in this assay.
Analogues with Modifications on Two Phenyl Rings
All the modifications to the trityl group described so far increased its hydrophobicity, which in certain cases compromised cellular efficacy due to poor aqueous solubility (e.g., 38). In addition to this immediately apparent penalty, excessively lipophilic (i.e., log P/log D7.4 > 3) compounds are more likely to possess unfavorable ADME properties and bind more promiscuously, which is likely to result in greater side effects and general toxicity.48 We were interested in developing hydrophilic modifications to the trityl group to produce a more physicochemically balanced scaffold to serve as a backup to the lead amino acid-based compounds. One way to achieve this was by replacing a phenyl ring with a heterocycle; however, none of the investigated analogues displayed noteworthy activity (S1–S5, Supporting Information, Table S1), in agreement with other reports on thiophene and pyridyl analogues.26,49 Previously we identified certain hydrophilic phenyl substituents that reduced the lipophilicity of the trityl headgroup and improved cellular efficacy (e.g., 8 and 9, Figure 2).29 The most effective of these modifications was the m-hydroxy 8, designed to form hydrogen-bonding interactions with the peptide backbone of the P2 pocket (Figure 3A).12,29 Modest improvements in cellular efficacy were also noted in analogues containing primary and secondary amide phenyl substituents (e.g., 9, Figure 2). These amides were proposed to occupy the solvent-exposed P1 region because of the steric restrictions in the P2 pocket observed in other SAR studies (Figure 3B).44,50,51 We combined each of these modifications in turn, with a lipophilic substituent on a different phenyl in the trityl group, to investigate if synergistic increments in activity could be achieved (44–51, Table 3). While no improvement was apparent for thioethanamine rac-45 containing a m-phenol and m-chlorophenyl, the related m-phenol analogues rac-46 and rac-47 with m-ethyl- and p-methylphenyl substituents were 5-fold and 4-fold more potent inhibitors of Eg5 basal activity, respectively, than comparable analogues 16 and 17. These analogues also exhibited improved inhibition over their respective benchmarks in the K562 assay: e.g., rac-46 improved ≥2-fold over the benchmarks 8 and 16, and rac-47 improved ≥5-fold over 8 and 17. These data implied a synergistic binding motif was being realized, with hydrogen bonding interactions from the m-hydroxyl in the P2 pocket augmented by hydrophobic interactions with the alkyl-substituted phenyl rings situated in the P3 pocket. While no increase in potency was evident in the basal assay when a p-tolyl was combined with a m-(primary amide)-containing phenyl, this combination afforded a ∼2-fold improvement in the cellular assay (rac-49 compared with 9 and 17) which also supported the proposed binding conformation. With GI50 ≈ 100 nM, rac-47 is the most active thioethanamine without the terminal α-carboxylic acid in the cellular assay reported to date and comparable to the lead butanamine 11. These modifications did not prove advantageous when applied in the comparable butanamine rac-51, which was less active in both assays than rac-47 or 11. Better activity was evident on addition of the terminal carboxylate in the basal assay but not in the cellular assay (dia-50). We previously noted that passive cellular diffusion was adversely affected by the presence of both the α-carboxylate in the tail and amide or hydroxyl phenyl substituents, which may offer a partial explanation for the comparatively weak cellular activity of the zwitterionic butanamine dia-50.
Table 3. Analogues with Modifications on Two Phenyl Rings.
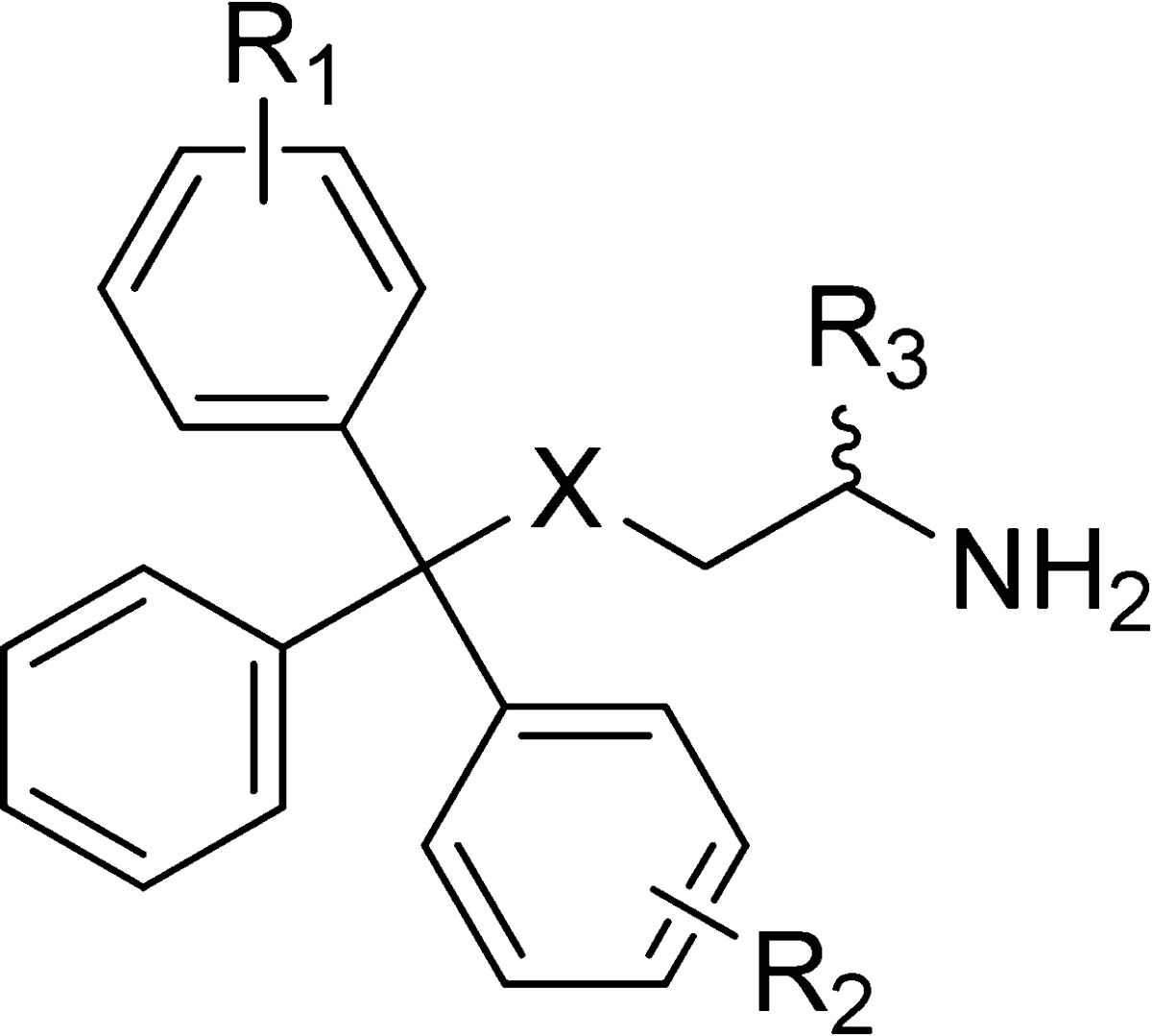
| compd | X | R1 | R2 | R3 | Inhibition of basal ATPase activity Kiapp (nM) | LE | K562 cells GI50 (nM) |
|---|---|---|---|---|---|---|---|
| 8a | S | 3-OH | H | H | 200.3 ± 51.9 | 0.38 | 555 ± 121 |
| 43a | S | 3-CN | H | H | 542.4 ± 41.3 | 0.34 | 2128 ± 108 |
| 9a | S | 3-CONH2 | H | H | 419.7 ± 38.8 | 0.33 | 802 ± 51 |
| 44 | S | 4-Me | 4-Me | H | 56.0 ± 5.0 | 0.40 | 2174 ± 119 |
| rac-45 | S | 3-OH | 3-Cl | H | 200.3 ± 35.3 | 0.37 | 1662 ± 77 |
| rac-46 | S | 3-OH | 3-Et | H | 4.8 ± 1.2 | 0.44 | 260 ± 19 |
| rac-47 | S | 3-OH | 4-Me | H | 7.1 ± 1.8 | 0.44 | 98 ± 6 |
| rac-48 | S | 3-CN | 4-Me | H | 64.2 ± 5.2 | 0.38 | 1186 ± 34 |
| rac-49 | S | 3-CONH2 | 4-Me | H | 55.6 ± 8.0 | 0.37 | 308 ± 16 |
| dia-50 | C | 3-OH | 4-Me | CO2H | 7.8 ± 2.0 | 0.39 | 813 ± 20 |
| rac-51 | C | 3-OH | 4-Me | H | 33.6 ± 6.4 | 0.41 | 232 ± 24 |
Values provided for reference from previous paper.29
Crystal Structure of the Eg5–46 Complex
The binding mode proposed for analogues with two orthogonal phenyl substituents was confirmed when the crystal structure of 46 in complex with Eg5 was solved to a resolution of 2.65 Å. This showed the m-phenol in P2 and the m-ethyl in P3 (Figure 3C). Data collection and refinement statistics are presented in Table 4. The Eg5–46 complex crystallized in space group I213 with one molecule per asymmetric unit. Although the racemic mixture of 46 was used for crystallization, only the R-enantiomer was observed in the inhibitor-binding pocket. The three phenyl rings of (R)-46 are positioned in the same overall conformation as 4, and consequently the hydrophobic and aromatic interactions between the three phenyl rings and the residues in the inhibitor-binding pocket are conserved.27,28 In the thioethanamine tail, the primary amine maintains the crucial hydrogen bonds exhibited by all analogues of 4 with the carboxylate of Glu116 and the main chain amide carbonyl of Gly117.26,27,29 The well-defined electron density affords accurate positioning of the two phenyl substituents and provides a clear structural rationale for the improvements exhibited in binding affinity. The m-ethylphenyl is situated in the predominantly hydrophobic P3 pocket bounded by Leu160, Gly217, Ala218, Arg221, mimicking the m-tolyl and p-chlorophenyl rings in previous crystal structures.26,29 However, the terminal CH3 of the ethyl extends into previously unutilized space between the methyl of Ala218 and the aliphatic chain of Arg221. The m-phenol of 46 occupies the P2 pocket formed by Glu118, Arg119, Trp127, Asp130, Leu132, Ala133, and Pro137, with the hydroxyl pointing toward the core of the protein instead of outward toward the solvent. Hydrogen-bonding interactions are evident in this position from the hydroxyl to the main chain carbonyl of Glu118, the main chain nitrogen of Ala133 and additional weak electrostatic interactions with the main chain nitrogen of Arg119, one of the side chain oxygens of Asp130, and the carbonyl oxygen of Leu132 (distance cutoff = 4.0 Å), similar to those observed in the Eg5–monastrol complex and structurally related dihydropyrimidines, thereby stabilizing the inhibitor in the allosteric site and increasing the affinity for the protein.12,52
Table 4. Data Collection and Refinement Statistics for the Eg5–46 Complexa.
| Eg5–46 | |
|---|---|
| unit cell dimensions a, b, c, γ (Å, deg) | 158.25, 158.25, 158.25, 90 |
| space group | I213 |
| beamline/detector | I23-2/MAR 225 |
| molecules per asymmetric unit | 1 |
| resolution range (Å) | 30–2.65 |
| no. of unique reflections | 19305 (2826)b |
| completeness (%) | 99.9 (100.0) |
| multiplicity | 7.5 (7.6) |
| Rsym (%) | 8.7 (58.5) |
| I/σ(I) | 15.4 (3.9) |
| Wilson B (Å2)/DPIc (Å) | 66.89 |
| refinement statistics | |
| Rwork/Rfree (%) | 18.5/22.8 |
| average B factors | |
| overall | 60.5 |
| main chain/side chain | 58.9/62.0 |
| no. of ADP/inhibitor/water | 1/1/110 |
| r.m.s.d. in bond length (Å)d | 0.012 |
| r.m.s.d. in bond angle (deg) | 1.68 |
The racemic mixture was used for crystallization.
Values in parentheses pertain to the highest resolution shell.
DPI: Diffraction-component precision index;76
r.m.s.d. is the root-mean-square deviation from ideal geometry.`
Inhibition of MT-Stimulated ATPase Activity and Kinesin Selectivity
The inhibitory activity of the most potent analogues from the initial in vitro assays was next examined in a MT-stimulated Eg5 ATPase assay, together with selected control compounds (4, 7, rac-11, and 13) and external benchmarks (1 and 2, Table 5). In cells, Eg5 uses MT filaments as tracks to push antiparallel MTs apart, which makes this particular assay closer to physiological conditions. Three new analogues (36, rac-42, and rac-47) possessed Kiapp estimates of <10 nM. Recently, the inhibitory activity of monastrol against Eg5 was shown to be dependent on the ionic strength of the buffer.65 To investigate whether this was the case for our scaffold, we assessed MT-stimulated ATPase activity of compounds 1 and rac-42 at different salt concentrations. For both compounds, the IC50 values systematically decrease with increasing salt concentrations, indicating that activity is optimal under the more physiologically relevant conditions represented by 150 mM NaCl (Supporting Information, Figure S2).
Table 5. Testing of a Series of Representative Analogues in Human Colon (HCT116), Prostate (LNCaP and PC3), Leukemia (K562), Pancreas (BxPC-3), and Lung (NCI-H1299) Cancer Cell Linesa.
| tumor cell
line growth inhibition GI50 (nM) |
|||||||
|---|---|---|---|---|---|---|---|
| compd | MT-stimulated ATPase activity Kiapp (nM) | HCT116 | LNCaP | K562 | PC3 | BxPC-3 | NCI-H1299 |
| 1b | 2.3 ± 0.002 | 25 ± 3 | 22 ± 4 | 48 ± 4 | 50 ± 5 | 80 ± 15 | 82 ± 10 |
| 2 | 0.5 ± 0.002 | 9.7 ± 0.8 | 15 ± 3.5 | 24 ± 1 | 21.2 ± 2.4 | 25 ± 4 | 35 ± 7 |
| 4 | 129.0 ± 8.9 | 553 ± 57 | 811 ± 116 | 1452 ± 76 | 1371 ± 96 | 1563 ± 155 | 1549 ± 111 |
| 7b | 21.6 ± 0.07 | 130 ± 7 | 131 ± 10 | 240 ± 17 | 226 ± 19 | 291 ± 54 | 294 ± 24 |
| (R)-11 | 7.1 ± 0.04 | 40 ± 2 | 32 ± 2 | 82 ± 4 | 58 ± 4 | 85 ± 12 | 111 ± 6 |
| 13b | 13.7 ± 0.06 | 95 ± 5 | 104 ± 20 | 200 ± 16 | 98 ± 14 | 1211 ± 265 | 360 ± 73 |
| rac-25 | 15.4 ± 0.7 | 100 ± 7 | 80 ± 9 | 94 ± 8 | 140 ± 8 | 187 ± 40 | 271 ± 17 |
| 26 | 10.1 ± 1.2 | 33 ± 2 | 60 ± 6 | 83 ± 4 | 158 ± 11 | 168 ± 12 | 158 ± 10 |
| 30 | 10.1 ± 0.7 | 114 ± 8 | 93 ± 8 | 82 ± 3 | 94 ± 10 | 77 ± 15 | 325 ± 45 |
| 34 | 18.6 ± 0.6 | 63 ± 9 | 34 ± 6 | 72 ± 8 | 73 ± 8 | 124 ± 29 | 111 ± 10 |
| 36 | 4.9 ± 0.004 | 34 ± 3 | 9.2 ± 1.2 | 34 ± 2 | 27.3 ± 2.3 | 26 ± 6 | 39 ± 2 |
| 38 | 16.3 ± 0.001 | 39 ± 2 | 31 ± 2 | 56 ± 2 | 43 ± 5 | 53 ± 5 | 101 ± 9 |
| rac-42 | 9.9 ± 0.6 | 28 ± 2 | 11.4 ± 0.8 | 23.4 ± 1.8 | 21 ± 1 | 44 ± 10 | 41 ± 3 |
| rac-47 | 6.8 ± 1.0 | 91 ± 10 | 97 ± 10 | 98 ± 6 | 258 ± 24 | 427 ± 85 | 221 ± 16 |
| rac-51 | 38.1 ± 0.4 | 95 ± 6 | 103 ± 14 | 232 ± 24 | 220 ± 17 | 184 ± 70 | 1074 ± 244 |
Estimates of Kiapp values for the inhibition of the MT-stimulated ATPase activity are also included.
Data reproduced in part from our previous study.29
Analogues 4, 7, and 11 were all previously tested against multiple kinesins and found to specifically inhibit Eg5. Against a panel of seven human kinesins containing Kif3B (kinesin-2; organelle transport), neuron-specific Kif5A and Kif5B/conventional kinesin (both kinesin-1; cargo transport), Kif7 (kinesin-4, involved in Hedgehog signaling), Kif9 (kinesin-9; regulation of matrix degradation), and Kif20A/MKLP-2 and Kif20B/MPP1 (both kinesin-6; required for cytokinesis),2,3rac-42 displayed no activity at a maximum inhibitor concentration of 200 μM (Supporting Information, Table S4 and Figure S1), which confirms the proposed high selectivity of this class of inhibitors within the kinesin superfamily.17,18,25,29 Our analogues have not yet been tested on ATPases beyond the kinesins, although other S-trityl-l-cysteine analogues were recently shown to be weak inhibitors of hepatitis C virus NS5B polymerase.53
Evaluation of Inhibitors across Multiple Cell Lines
A selection of the new lead compounds was evaluated in five additional cell lines derived from colon (HCT116), pancreas (BxPC-3), prostate (LNCaP and PC3), and lung (NCI-H1299) tumors, to determine in vitro efficacy against a histologically broader panel of cancer cell lines (Table 5). The lead compounds from our previous study with and without the carboxylic acid (11 and 13, respectively) were included to assess progress,29 along with the original hit 4 and the phase II drug candidates 1 and 2. In general, HCT116 cells were the most responsive to the Eg5 inhibitors tested, whereas NCI-H1299 cells were the least affected. Three of the new analogues (34, 36, and rac-42) displayed activity higher than that of the original lead (rac-11);29 additionally both 36 and rac-42 were more potent than the clinical candidate 1 and equal to its more potent second generation analogue 2. For those compounds without the carboxylic acid group, three (26, rac-47, rac-51) were more active than the previous lead 13 but less active than rac-11. We have shown previously that for 4, 11, and 12, the absolute configuration of the stereocenter in the amino acid tail has very little effect on efficacy.18,29 The phenol compounds rac-47 and rac-51 were prepared racemically and contain a stereocenter at the trityl carbon more likely to affect activity, especially given that only the (R)-enantiomer was observed in the crystal structure of the Eg5–46 complex. Compounds prepared via the TFA-mediated dehydration of chiral tertiary alcohols form a 1:1 ratio of enantiomers (Scheme 1).49 However, we did not investigate the resolution of rac-47 and rac-51 further, as rac-47 displayed very high clearance in human hepatocytes (Clint = 50.1 ± 4.2 μL min–1; t1/2 = 27.7 min) and thus was not suitable for progression as a drug candidate.
Profiling of Druglike Properties of Lead Compounds
We subsequently subjected the new lead compounds from this study (34, 36, and rac-42) to a series of in vitro and in vivo physicochemical and ADMET assays to evaluate if in addition to pharmacodynamics improvements, they exhibited favorable drug-like properties (Table 6). The phase II candidate 2 was included as an external benchmark; the profile for the first generation candidate 1 has been reported previously.29 The p-methoxy analogue 7 was also profiled, as this compound was reported to inhibit the rate of tumor growth in subcutaneous prostate (PC3Luc cells) and bladder xenograft tumor models.22,23 Analogue 7 has a balanced profile, with favorable physiochemical characteristics and reasonable metabolic stability in both human microsomes and hepatocytes. Moderate affinity is displayed for the 2C19 CYP isoform. In comparison to the other lead compounds, 7 is systematically less potent in vitro (Table 5), and so was not investigated further. The most potent analogue lacking the carboxylic acid 26, which also contains a p-methoxyphenyl, was profiled and interestingly proved metabolically more resilient in human microsomal and hepatocyte assays than 7. Several liabilities were however evident, including low turbidimetric solubility at physiological pH (>30 μM), hERG inhibition, and moderate to significant inhibition of CYP isoforms from four of the main families.54 This agreed with the trend previously observed that compounds lacking the carboxylic acid group have generally more liabilities than those possessing it.29 At this stage, we investigated whether modulating the basicity of the primary amine would be an effective strategy to ameliorate these.29 Although alkylation of primary amines can overcome hERG inhibition,55 the hydrogen bonds formed by the primary amine in 4 are crucial to maintaining activity (Figure 3A), with secondary amines only weakly active.26,27 We therefore applied the subtler strategy of modulating the pKa by β-fluorination to produce 52–54 (Table 7).56 In the basal assay, while inhibitory activity comparable to that of the unmodified 26 was displayed by β-fluorinated rac-52, the binding affinity was substantially reduced for β,β-difluorinated 53 and lowered to micromolar levels for α-trifluoromethyl-containing rac-54. Despite good potency against Eg5, in cells rac-52 proved ∼8-fold less active than the unmodified 26, and 53 only exhibited micromolar levels of growth inhibition. While these results reinforce the importance of the protonated state of the primary amine at physiological pH, the reasonable basal activity suggests that multiple factors contributed to the reduced cellular efficacy. As this strategy did not appear useful in this series, we did not investigate further.
Table 6. ADME Profiling of Selected S-Trityl-l-cysteine Analogues in Comparison with Clinical Candidate 2.
| assay/compound | 2 | 7 | 26 | 34 | 36 | rac-42 |
|---|---|---|---|---|---|---|
| MW (Da) | 517.06 | 393.50 | 331.45 | 391.53 | 405.55 | 373.49 |
| turb solubilitya (μM) | ||||||
| pH 2.0 | >100 | >100 | >100 | 65 | >100 | >100 |
| pH 6.0 | >100 | >100 | >100 | 65 | 65 | >100 |
| pH 7.4 | >100 | >100 | >30 | 65 | 65 | >100 |
| log P | 3.08 ± 0.03 | 1.77 ± 0.07 | 3.96 ± 0.04 | 2.94 ± 0.03 | 4.13 ± 0.03 | 1.96 ± 0.51 |
| pKa | 8.50 ± 0.07 | pKa 1: 9.43 ± 0.02 | 9.86 ± 0.05 | pKa 1: 8.21 ± 0.09 | pKa 1: 7.10 ± 0.10 | pKa 1: 9.36 ± 0.05 |
| pKa 2: 6.14 ± 0.01 | pKa 2: 2.51 ± 0.11 | pKa 2: 3.28 ± 0.07 | pKa 2: 1.69 ± 0.46 | |||
| log D7.4 | 3.09 ± 0.19 | 1.57 ± 0.20 | 1.99 ± 0.17 | 3.17 ± 0.2 | 3.08 ± 0.15 | 3.28 ± 0.37 |
| microsomal stability (μL/min/mg protein) | ||||||
| human | 4.5 ± 3.6 | 8.6 ± 1.4 | stable | stable | stable | stable |
| mouse | stable | 15.1 ± 2.1 | 26.4 ± 3.8 | 17.8 ± 3.7 | stable | stable |
| half life (min) | 309/– | 162/92.0 | –/52.6 | –/77.8 | –/– | –/– |
| human hepatocytes (μL/min/106 cells) | 12.1 ± 3.6 | 6.2 ± 2.5 | 0.5 ± 2.9 | 7.9 ± 3.4 | 24.4 ± 4.7 | 5.9 ± 1.9 |
| half life (min) | 114 | 225 | 2590 | 176 | 56.9 | 235 |
| human plasma protein binding (fu100%) | 0.223 ± 0.050 | 0.093 ± 0.004 | 0.093 ± 0.010 | 0.045 ± 0.011 | 0.016 ± 0.001 | 0.051 ± 0.012 |
| recovery (%) | 81.5 | 80.7 | 73.0 | 79.6 | 70.7 | 78.9 |
| hERG (μM) | 1.6 ± 0.2 | >25 | 6.6 ± 1.1 | >25 | >25 | >25 |
| CYP450 inhibition (μM) | ||||||
| 1A2 | >25 | >25 | >25 | >25 | >25 | >25 |
| 2C9 | >25 | >25 | 1.9 ± 0.2 | >25 | 14.0 ± 4.9 | >25 |
| 2C19 | >25 | 6.3 ± 1.0 | 2.8 ± 0.3 | 7.9 ± 1.3 | >25 | >25 |
| 2D6 | >25 | >25 | 12.8 ± 1.5 | >25 | >25 | >25 |
| 3A4 | 4.0 ± 0.4 | >25 | 7.3 ± 0.8 | >25 | >25 | >25 |
Turbidimetric solubility.
Table 7. β-Fluorinated Analogues of 4-Methoxytriphenylbutanamine 26.
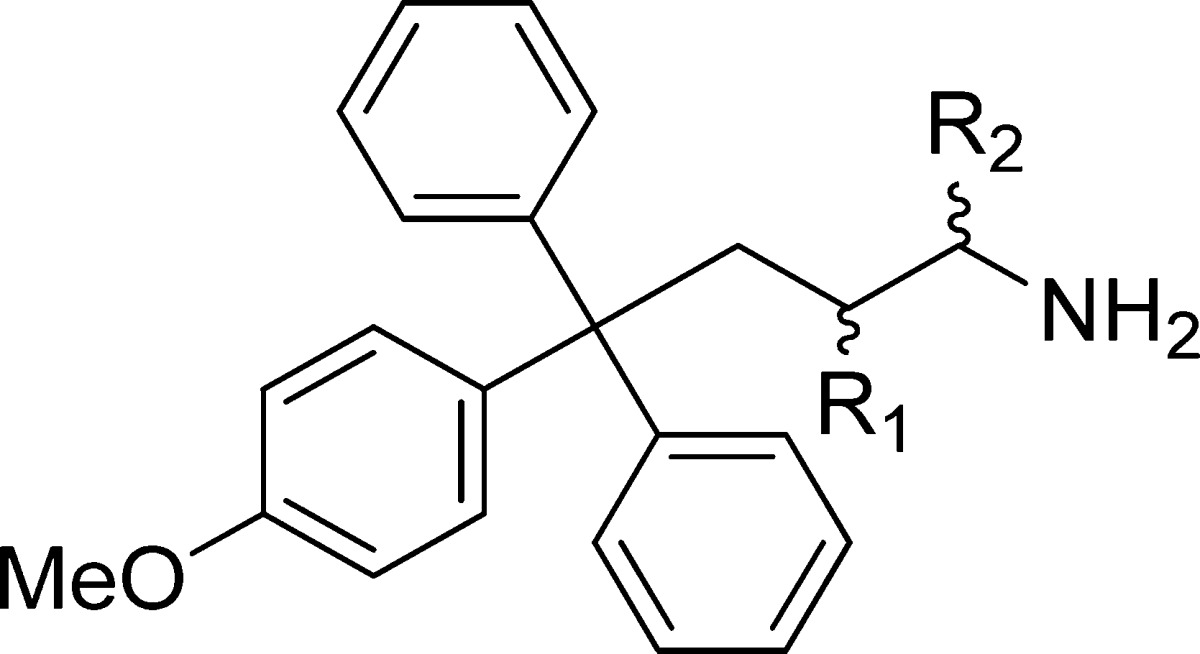
| compd | R1 | R2 | pKa | inhibition of basal ATPase activity Kiapp (nM) | LE | K562 cells GI50 (nM) |
|---|---|---|---|---|---|---|
| 26 | H | H | 9.86 ± 0.05 | 15.5 ± 5.3 | 0.43 | 83 ± 4 |
| rac-52 | F | H | 7.86 ± 0.07 | 27.3 ± 1.2 | 0.40 | 652 ± 61 |
| 53 | F2 | H | 7.11 ± 0.05 | 88.4 ± 1.9 | 0.36 | 2624 ± 379 |
| rac-54 | H | CF3 | 4.63 ± 0.10 | 2268 ± 39 | 0.27 | >100000 |
Consequently, we profiled our two most potent cysteine-based analogues, 34 and 36, which differ only by an extra methylene in the meta position that imparts 3- to 4-fold higher potency across cellular assays (Table 5). This gain in potency comes at a cost: 36 exhibits higher clearance in human hepatocytes (24.4 ± 4.7 versus 7.9 ± 3.4 μL/min/million cells) and is less available in human plasma (1.55% versus 4.51%, Table 6). A distinct profile is also evident toward CYP enzymes: like the p-methoxy analogue 7, 34 inhibits CYP 2C19 (IC50 = 7.9 ± 1.3 μM), whereas 36 does not but instead weakly inhibits CYP 2C9 (IC50 = 14.0 ± 4.9 μM). Compound 36 displays high bioavailability (63%) when compared to that determined previously for 1 (45%), 13 (4%), and rac-11 (51%) (Table 8).29 The pharmacokinetic data is in good agreement with that from the in vitro mouse metabolic profiling: 36 has a shorter half-life and higher clearance than the clinical candidate 1, with rac-11 displaying low clearance in comparison.29
Table 8. Bioavailability and Pharmacokinetic Parameters for rac-11, 13, and 36 Compared to Clinical Candidate 1.
| oral dosing (po) |
intravenous dosing (iv) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compd | F (%) | Cmax (μg/mL) | tmax (h) | AUClast (μg-h/mL) | C0 (μg/mL) | t1/2 (h) | VD(L/kg) | CL (mL/min/kg) | AUClast (μg-h/mL) | comments |
| 1 | 45 | 0.3 | 2.7 | 1.5 | 2.0 | 4.1 | 2.5 | 20.5 | 3.1 | moderate clearance, good po levels |
| rac-11 | 51 | 1.2 | 3.7 | 7.2 | 8.2 | 6.5 | 0.6 | 3.8 | 13.1 | low clearance, good po levels |
| 13 | a4 | 0.02 | 0.3 | 0.01 | 0.7 | 1.8 | 6.9 | 203.0 | 0.3 | high clearance, low po levels |
| 36 | 63 | 1.3 | 0.5 | 4.4 | 13.6 | 2.1 | 0.4 | 11.4 | 7.1 | high clearance, good po levels |
Possibly a consequence of the high clearance.
The butanamine analogue of 34 and most active from the in vitro profiling, rac-42, was investigated next. Direct comparison with 34 illustrates that rac-42 displays higher turbidimetric solubility (>100 μM versus 65 μM), increased metabolic resilience in human hepatocytes (5.9 ± 1.9 versus 7.9 ± 3.4 μL/min/million cells) and does not inhibit any of the common CYP isoforms examined. In common with the profiled zwitterionic S-trityl analogues, no affinity is evident toward the hERG channel. This attractive profile is analogous to that reported for our previous lead, rac-11.29
Examining clinical candidate 2 in the same assays, we observed improved solubility at the three pHs tested compared to 1 (>100 μM versus 65 μM).29 The reduced log P of 2 also translates to a higher proportion of unbound compound in human plasma (22% versus 8%). However, 2 retains the potentially physiologically significant hERG channel interactions and affinity for the CYP isoform 3A4 evident for 1.29 Therefore, the butanamines rac-11 and rac-42 possess a more balanced ADMET profile, which may offer distinct clinical advantages.
In Vivo Xenograft Studies
We advanced the three most active and balanced of the new analogues (34, 36, and rac-42) into in vivo xenograft experiments with lung cancer patient explants (LXFS 538) passaged as subcutaneous xenografts in nude mice. As controls, our previous lead analogue rac-11 and the clinical candidate 1 were selected. We adopted lung cancer xenograft models, as this is a key area for improving therapy due to the poor prognosis for most patients;57 the indications that Eg5 inhibitors may achieve better clinical outcomes in hematological malignancies rather than in solid tumors came after this work had commenced. We used explanted tumors, as these are a more realistic model than solid tumors derived from in vitro cell lines. The transplanted tumors retain the histological complexity from the patient, reflect prior treatment and have been shown to be better predictors of clinical response.58,59 Administering our previous lead, rac-11, by an improved dosing schedule resulted in complete tumor regression at day 44 (Figure 4A), in comparison to partial remission recorded previously.29 When treatment stopped, tumor growth relapsed around day 76 (data not shown). Xenograft experiments for 1 have previously been reported at a dose of 8–10 mg/kg in breast tumor models.60 In mice implanted with LXFS 538 tumors, treatment at this dose proved to be lethal (data not shown), and similar toxicity issues have been observed previously.7 When we repeated at a lower dose of 6 mg/kg, total tumor regression was evident around day 13 and the mice remained tumor free over the entire observation time (Figure 4A). For the cysteine containing 36, although significantly more potent than rac-11 in cell-based assays, only a reduced tumor growth rate was observed (Figure 4B), although the difference was still statistically significant (p = 0.0124). No tumor growth inhibition was evident for 34 (Figure 4C). Metabolic profiling of 34 and 36 explained this poor in vivo activity compared to the strong in vitro efficacy: although stable in mouse microsomes, 36 showed high clearance in mouse hepatocytes (58.7 ± 5.64 μL/min/million cells; t1/2 = 23.6 min). A similar explanation applied for 34: while stable in human microsomes with only low clearance in human hepatocytes (7.9 ± 3.4 μL/min/million cells), medium clearance was evident in mouse microsomes (17.8 ± 3.7 μL/min/mg protein) and extremely high clearance by mouse hepatocytes (170 ± 9.4 μL/min/million cells; t1/2 = 8.2 min). Therefore, nude mice xenografts are not an appropriate model for assessing the therapeutic potential of 34 and 36: in the future, alternative models such as nude rats will need to be employed, if these compounds prove metabolically more stable in rat hepatocytes. In contrast to the thioethanamines 34 and 36, the activity of the most potent butanamine rac-42 was translated effectively into the xenograft model, with total tumor regression by day 17, improving on the activity recorded for rac-11 and comparable to the response achieved on treatment with 1 (Figure 4C).
Figure 4.
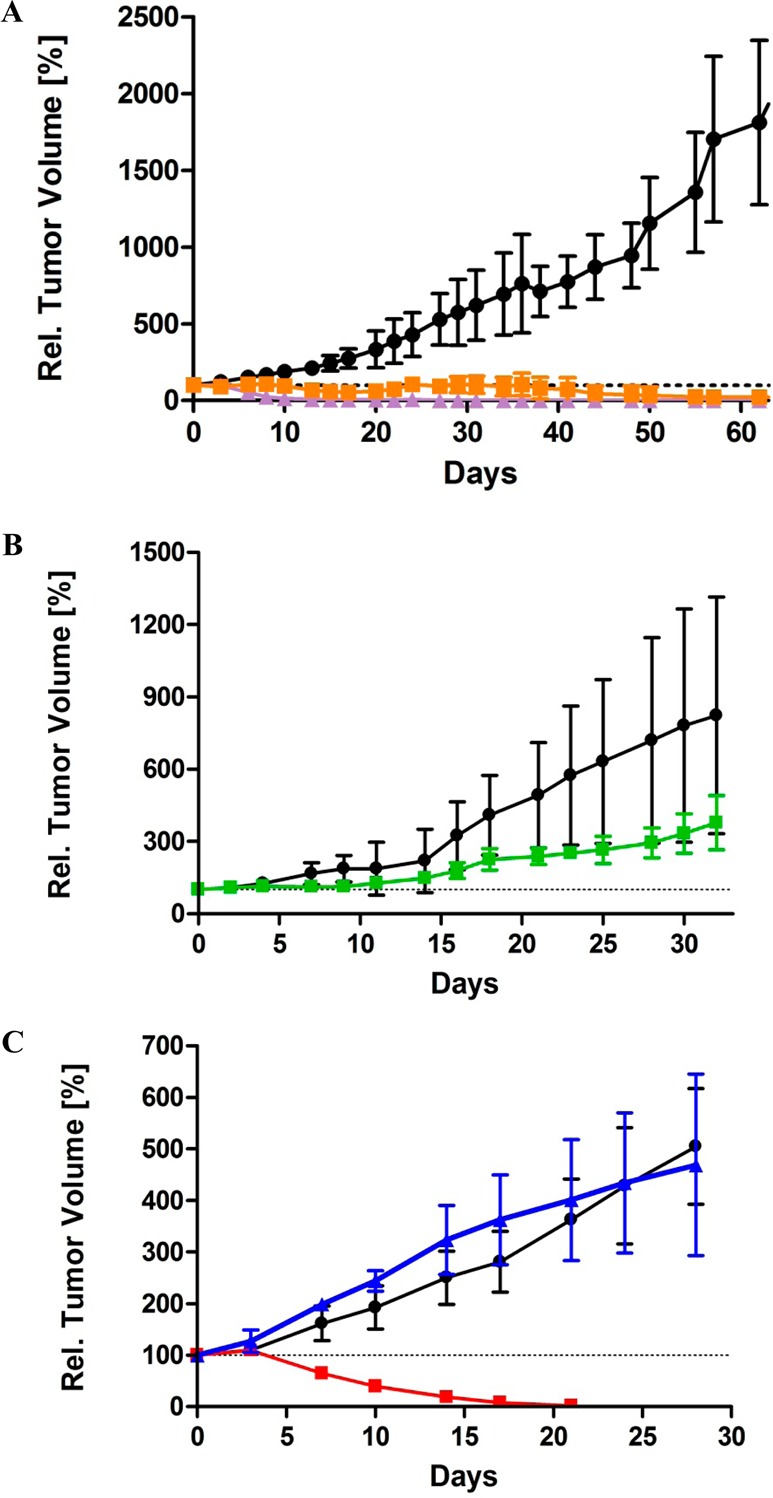
Antitumor efficacy of rac-11, 34, 36, and rac-42 compared to the clinical candidate 1 in a subcutaneous tumor xenograft model with LXFS 538. The control groups (black ●) received only vehicle on the same days as treatment with inhibitors. The data are plotted as the mean of the RTV ± standard deviation. (A) Tumor xenografts for 1 (purple ▲) and rac-11 (orange ■) showing total regression after 13 days of treatment for 1 and at day 44 for rac-11. The difference between the treated groups and vehicle are statistically significant (p < 0.0001 for both 1 and rac-11). (B) Reduced tumor growth rate by 36. The difference between the treated group (36, green ■) and vehicle is statistically significant (p = 0.0124). (C) Differing responses of xenografts to 34 (blue ▲) and rac-42 (red ■). Treatment with rac-42 results in complete tumor regression within 17 days. No response was recorded for 34. The difference between the treated group (rac-42, red ■) and vehicle is statistically significant (p = 0.0003).
Conclusions
Optimization of 4 has led to structurally relatively simple yet extremely effective Eg5 inhibitors which display in vitro and in vivo antitumor activity comparable to benchmark inhibitors further advanced in clinical trials. The only other Eg5 inhibitors reported to induce complete tumor regression in mice xenograft models, to the best of our knowledge, are the three clinical candidates 1–3 in phase II.7−9 We propose that the favorable drug-like properties, as highlighted by in vitro profiling, strengthens the case for the progression of triphenylbutanamine analogues based on rac-42 for further evaluation. In general, the butanamines proved systematically more potent than the equivalent thioethanamines, but most notably their increased metabolic stability imparted improved efficacy in xenograft studies with nude mice. Both the profiled butanamine and thioethanamine leads rac-11 and 36 show good oral bioavailability and pharmacokinetics; however, for an accurate assessment of the potential of these S-trityl analogues, alternative in vivo models will need to be examined. In this and our preceding study, we have developed a range of modifications to the core scaffold which impart excellent affinity for the target protein Eg5. The potential of clinical candidates based on rac-42 will be determined by selecting those which provide optimal bioavailability and robust pharmacokinetics, a process we have initiated through the profiling described here. The wider challenge facing Eg5 inhibitors is translating the efficacy evident in preclinical studies into the clinic. The responses to Eg5 inhibitors in clinical trials have been limited to date, and one explanation for this is that tumor doubling times in xenograft models are significantly faster than those in human patients and thus not sufficiently realistic models for developing chemotherapeutics.61 This hypothesis is corroborated by the improved responses observed against faster growing hematological cancers and the principal dose-limiting toxicities for most clinical candidates of neutropenia and myelosuppression.1 Early clinical indications suggest a possible application for Eg5 inhibitors in advanced multiple myeloma, which will most likely be in combination with existing therapies.15 In combined chemotherapies, the potential for inducing drug–drug interactions and toxicities becomes increasingly important. For example, the hepatic metabolism of the synthetic glucocorticoid dexamethasone currently used in the treatment of multiple myeloma is mediated primarily by CYP 3A4.62 Both of the trial Eg5 drugs we profiled, 1 and 2, are CYP 3A4 inhibitors, so careful consideration of the physiological relevance of these interactions would be required prior to undertaking combination-based therapy. Limited information is available on the relevant properties of other candidates under investigation.1 We have demonstrated that in comparison to the clinical candidates 1 and 2, the lead S-trityl-l-cysteine-derived analogues rac-11 and rac-42 exhibit comparable in vivo efficacy by inducing complete tumor regression in an advanced tumor explant xenograft model but display fewer liabilities toward CYP enzymes and the hERG channel. Furthermore, unlike other highly advanced Eg5 inhibitors, the described compounds are not patented and therefore are freely available for investigation in single or combined cancer chemotherapies by the scientific and medical community, which we propose, based on the recent results for ARRY-520, should focus on targeting hematological malignancies.
Material and Methods
Chemistry
General
Compounds 4 and 2 were purchased from Nova Biochem, Sigma Aldrich, and Selleck Chemicals, respectively, and used without further purification. Compound 1 was a gift from Sanofi-Aventis. Compounds 8, 9, 11–16, and 43 were prepared as reported previously.29 Compound 7 (NSC123528) was obtained from the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov) of the National Cancer Institute. All reagents and solvents were of commercial quality and used without further purification. Anhydrous reactions were carried out in oven-dried glassware under a nitrogen atmosphere unless otherwise noted. Microwave reactions were performed using a Biotage Initiator-8 microwave synthesizer (operating at 2.45 GHz). Thin-layer chromatography (TLC) was carried out on aluminum-backed SiO2 plates (silica gel 60, F254), and spots were visualized using ultraviolet light (254 nm) or by staining with phosphomolybdic acid (alcohols) or ninhydrin (amines). Flash column chromatography was performed on silica gel (SNAP KP-Sil, 60 Å, 40–63 μm cartridges) using a Biotage SP4 automated chromatography system (detection wavelength, 254 nm; monitoring, 280 nm). Melting points were determined using a Stuart Scientific SMP1 melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a JEOL ECX-400 (400 MHz), Avance DPX400 (400 MHz), or Avance DPX500 (500 MHz) spectrometer. 19F NMR spectra were recorded on an Avance AV400 (400 MHz) instrument equipped with a multinuclear probe. 1H chemical shifts (δ) are reported in ppm relative to the residual signal of the deuterated solvent (7.26 in CDCl3, 3.31 in CD3OD, and 2.50 in DMSO-d6). Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), m (unresolved multiplet), and br (broad signal). 13C chemical shifts (δ) are reported in ppm relative to the carbon resonance of the deuterated solvent (77.16 in CDCl3, 49.00 in CD3OD, and 39.52 in DMSO-d6). 19F spectra are referenced relative to CFCl3. High-resolution mass spectra were recorded on a Thermo Electron LTQ ORBITRAP mass spectrometer using electrospray ionization. Gas chromatography mass spectra (GC-MS) using electron ionization (EI) were recorded on a Thermo Scientific Focus GC with DSQ2 single quadrupole mass spectrometer. GC-MS using chemical ionization (CI) were recorded on an Agilent Technologies 7890A GC system and an Agilent 5975C Inert XL EI/CI MSD with a DSQ2 single quadrupole mass spectrometer, equipped with an Agilent Technologies DB5-MS column (30 m × 0.25 mm × 0.25 μm). Helium was the carrier gas (flow rate = 1 mL/min–1). Elemental analysis data were recorded on a Perkin-Elmer 2400 series 2 CHN analyzer. LC-MS analyzes were performed with an Agilent Quaternary 1200 series pump and an Agilent 6130 dual source mass spectrometer with UV detection at 254 nM. Retention times (tR) were in minutes, and purity was calculated as percentage of total area. The method for determining purity consisted of the following: Zorbax Eclipse XDB-C18 reverse phase column (15 cm × 4.3 μm, particle size 5 μm); column temperature 40 °C; solvent A: H2O (5 mM ammonium acetate); solvent B: MeCN (5 mM ammonium acetate); gradient of A:B, 95:5 (0–3 min), A:B, 95:5 → B, 100% (3–17 min), B, 100% (17–27 min), B, 100% → A:B, 95:5 (27–33 min), A:B, 95:5 (33–36 min); flow rate 1 mL min–1. All tested compounds were ≥95% pure by elemental or LCMS analyses. New compounds were named according to IUPAC nomenclature by ACD ChemSketch 12.01 (Windows, Advanced Chemistry Development, Toronto, Canada).
Procedures
5-Bromo-N-methoxy-N,2-dimethylbenzamide (56)
Oxalyl chloride (3.43 mL, 40 mmol) was added to a solution of 5-bromo-2-methylbenzoic acid 55 (4.30 g, 20 mmol) in anhydrous CH2Cl2 (20 mL), with a catalytic amount of DMF (1 drop), and stirred at room temperature for 2 h. The reaction mixture was concentrated in vacuo, and the residual oil was added by slow dropwise addition over 5 min to a cooled (0 °C) solution of N,O-dimethylhydroxylamine hydrochloride (2.24 g, 23 mmol) and triethylamine (3.67 mL, 50 mmol) in anhydrous CH2Cl2 (20 mL). After being stirred for 1 h, the reaction mixture was warmed to room temperature, stirred for 20.5 h, quenched with water followed by aqueous HCl (0.5 M, 25 mL), and extracted with CH2Cl2 (3 × 25 mL). The combined organic extracts were dried (MgSO4), concentrated in vacuo, and purified by flash chromatography (SiO2; 0–50% EtOAc in hexane) to afford 56 as a colorless oil (3.63 g, 70%). 1H NMR (CDCl3, 500 MHz) δ 2.26 (s, 3H, CH3), 3.30 (br s, 3H, CH3), 3.48 (br s, 3H, CH3), 7.07 (d, J = 8.6 Hz, 2H), 7.38–7.40 (m, 2H). 13C NMR (CDCl3, 125 MHz) δ 18.77, 61.33, 118.95, 129.13, 131.95, 132.23, 134.06, 137.20. HRMS (ESI+) calcd for C10H13BrNO2 (M + H)+: 258.01242; found: 258.01244. Anal. Calcd for C10H12BrNO2: C, 46.53; H, 4.69; N, 5.43. Found: C, 45.03; H, 4.78; N, 5.60.
1-(5-Bromo-2-methylphenyl)ethanone (57)
The title compound was prepared using an adaptation of the procedure reported by Hirashima et al.63 MeMgCl (3.0 M in THF, 8.27 mL, 24.8 mmol) was added by slow dropwise addition over 10 min to a cooled (0 °C) solution of 5-bromo-N-methoxy-N,2-dimethylbenzamide 56 (3.27 g, 12.4 mmol) in THF (12.4 mL) and stirred for 2 h. The reaction mixture was warmed to room temperature and stirred for 2 h, before quenching with saturated aqueous NH4Cl solution (20 mL) and extracting with EtOAc (3 × 25 mL). The combined organic extracts were washed successively with water and brine (75 mL each), dried (MgSO4), and concentrated in vacuo to give 57 as a clear, pale brown oil (2.40 g, 91%), which was used without further purification. 1H NMR (CDCl3, 500 MHz) δ 2.45 (s, 3H, CH3), 2.56 (s, 3H, CH3), 7.11 (d, J = 8.2 Hz, 1H), 7.48 (dd, J = 2.1, 8.2 Hz, 1H), 7.77 (d, J = 2.1 Hz, 1H). 13C NMR (CDCl3, 125 MHz) δ 22.10, 29.62, 119.24, 132.11, 133.77, 134.39, 137.29, 139.46. GC-MS (EI, 70 eV) tR = 4.68 min (m/z = 211.9, M+). Anal. Calcd for C9H9BrO C, 50.73; H, 4.26. Found: C, 50.44; H, 4.30.
4-Bromo-2-ethyl-1-methylbenzene (58)
The title compound was prepared using an adaptation of the procedure reported by Chackal-Catoen et al.64 Hydrazine hydrate monohydrate (1.46 mL, 30 mmol) was added to a solution of 1-(5-bromo-2-methylphenyl)ethanone 57 (2.13 g, 10 mmol) and powdered KOH (1.68 g, 30 mmol) in anhydrous ethylene glycol (10 mL) and refluxed for 4 h. After being cooled to room temperature, the reaction was quenched was aqueous HCl (1.0 M, 30 mL) and extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed successively with water and brine (75 mL each), dried (MgSO4), and concentrated in vacuo, and the residue was purified by flash chromatography (SiO2; hexane) to afford alkane 58 as a colorless oil (1.10 g, 55%). 1H NMR (CDCl3, 400 MHz) δ 1.20 (t, J = 7.5 Hz, 3H, CH3), 2.24 (s, 3H, CH3), 2.59 (q, J = 7.5 Hz, 2H, CH2), 7.00 (d, J = 8.0 Hz, 1H), 7.22 (dd, J = 2.0, 8.0 Hz, 1H), 7.28 (d, J = 1.9 Hz, 1H). 13C NMR (CDCl3, 100 MHz) δ 14.21, 18.84, 26.18, 119.63, 128.72, 130.80, 131.71, 134.83, 144.68. GC-MS (EI, 70 eV) tR = 4.90 min (m/z = 199.8, M+).
(3-Ethyl-4-methylphenyl)(diphenyl)methanol (59)
n-Butyllithium (2.5 M in hexane, 2.21 mL, 5.53 mmol) was added by slow dropwise addition over 2 min to a cooled (−78 °C) solution of 4-bromo-2-ethyl-1-methylbenzene 58 (918 mg, 4.61 mmol) in anhydrous THF (4.61 mL) and stirred for 1 h at ≤−70 °C. A solution of benzophenone (966 mg, 5.30 mmol) in anhydrous THF (5.30 mL) was added by slow dropwise addition over 5 min, and the reaction mixture was stirred with the temperature maintained at ≤−70 °C for 5 h, before allowing the reaction to warm to room temperature and stirring for a further 20 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic extracts were then washed successively with H2O and brine (30 mL each), dried (MgSO4), and concentrated in vacuo. Purification by flash chromatography (SiO2; 0–10% EtOAc in hexane) afforded trityl alcohol 59 as a yellow oil (1.263 g, 91%). 1H NMR (CDCl3, 500 MHz) 1.14 (t, J = 7.6 Hz, 3H, CH3), 2.31 (s, 3H, CH3), 2.59 (q, J = 7.6 Hz, 2H, CH2), 2.77 (s, 1H, OH), 6.93 (dd, J = 2.0, 7.9 Hz, 1H), 7.07 (d, J = 7.9 Hz, 1H), 7.12 (d, J = 1.9 Hz, 1H), 7.26–7.32 (m, 10H). 13C NMR (CDCl3, 125 MHz) δ 14.60, 18.92, 82.11, 125.60, 127.24, 127.66, 127.98, 128.06, 129.67, 134.96, 142.11, 144.77, 147.26. HRMS (ESI+) calcd for C21H21 (M – OH)+: 285.16378; found: 285.16348. Anal. Calcd for C22H22O: C, 87.38; H, 7.33. Found: C, 87.23; H, 6.81.
(2R)-2-Amino-3-(((3-ethyl-4-methylphenyl)(diphenyl)methyl)sulfanyl)propanoic Acid (36)
A solution of the tertiary alcohol 59 (254 mg, 0.84 mmol) with l-cysteine (112 mg, 0.93 mmol) in TFA (1.0 mL) was stirred for 3 h at room temperature. The volatiles were removed in vacuo, and the crude basified (ca. pH 10) with saturated aqueous sodium carbonate solution. The aqueous mixture was extracted with CH2Cl2 (3 × 10 mL) and the organic layer dried (MgSO4) and concentrated in vacuo. Purification by flash chromatography (SiO2; 0–25% MeOH in CH2Cl2) afforded the thioether 36 as a white solid (242 mg, 71%): mp 149–152 °C. 1H NMR (500 MHz, MeOD) δ = 1.10 (t, 3H, J = 7.6 Hz), 2.27 (s, 3H), 2.56 (q, 2H, J = 7.6 Hz), 2.69 (dd, 1H, J = 9.2, 13.4 Hz), 2.82 (dd, 1H, J = 4.2, 13.4 Hz), 3.05 (dd, 1H, J = 4.2, 9.2 Hz), 7.06 (d, 1H, J = 8.0 Hz), 7.14 (dd, 1H, J = 2.1, 8.0 Hz), 7.18–7.24 (m, 3H), 7.27–7.32 (m, 4H), 7.42–7.46 (m, 4H). 13C NMR (125 MHz, MeOD) δ = 14.94, 18.73, 27.28, 34.36, 55.13, 68.05, 127.92, 128.07, 129.04, 130.47, 130.70, 130.74, 135.60, 143.16, 143.29, 145.91, 145.95, 172.53. HRMS (ESI+) Calcd for C25H28NO2S (M + H)+: 406.1835; found 406.1843. Anal. Calcd for C25H27NO2S·1/2H2O: C, 72.43; H, 6.81; N, 3.38. Found: C, 72.18; H, 6.37; N, 3.14.
4-(1,1-Diphenylbut-3-en-1-yl)-1,2-dimethylbenzene (61)
The title compound was prepared using an adaptation of the method reported by Kabalka et al.33n-Butyllithium (2.5 M in hexane, 6.12 mL, 15.3 mmol) was added cautiously by dropwise addition to a cooled (0 °C) solution of (3,4-dimethylphenyl)(diphenyl)methanol 61 (4.00 g, 13.9 mmol) in anhydrous CH2Cl2 (85 mL), and the reaction mixture was allowed to warm to room temperature and stirred for 30 min. Allyltrimethylsilane (6.02 mL, 36.9 mmol) and iron trichloride (5.19 g, 32.0 mmol) were then added, and the reaction was stirred at room temperature for 6 h. The reaction was quenched with H2O (100 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organic extracts were washed successively with saturated aqueous NaHCO3 solution (150 mL) and brine (200 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash chromatography (SiO2; 0–10% EtOAc in hexane) afforded alkene 61 as a brown oil [3.119 g, 91% (based on 79% conversion)] and recovered starting material 60 (834 mg). 1H NMR (400 MHz, CDCl3) δ = 2.22 (s, 3H, CH3), 2.26 (s, 3H, CH3), 3.43–3.46 (m, 2H), 4.97 (ddd, J = 1.4, 3.4, 10.3 Hz, 1H), 5.06 (ddd, J = 1.5, 3.5, 17.0 Hz, 1H), 5.70 (ddd, J = 6.6, 10.4, 17.1 Hz, 1H), 6.94–6.98 (m, 1H), 7.03–7.08 (m, 2H), 7.19–7.34 (m, 10H). 13C NMR (100 MHz, CDCl3) δ = 19.40, 20.24, 45.67, 56.01, 117.18, 125.97, 126.97, 127.79, 129.14, 129.58, 130.62, 134.23, 135.87, 136.35, 144.92, 147.65. GC-MS (CI, methane) tR = 16.44 min (m/z = 311.2, [M – H]+). Anal. Calcd for C24H24: C, 92.26; H, 7.74. Found: C, 91.51; H, 7.28.
4-(3,4-Dimethylphenyl)-4,4-diphenylbutan-1-ol (62)
A solution of BH3·THF (1.0 M in THF, 20.0 mL, 20.00 mmol) was added to a cooled (0 °C) solution of alkene 61 (3.11 g, 9.95 mmol) in THF (20 mL), and stirred at room temperature for 19 h. The reaction was cooled to 0 °C and quenched cautiously with H2O (5 mL) and aqueous NaOH (3.0 M, 6.8 mL, 20.40 mmol), followed by slow dropwise addition of hydrogen peroxide (30% in H2O, 5.11 mL, 50.01 mmol) over 5 min. The mixture was maintained at 0 °C for 30 min and then allowed to warm to room temperature and stirred for a further 3.5 h, diluted with H2O (50 mL), and extracted with Et2O (3 × 50 mL). The combined organic extracts were washed successively with saturated aqueous NaHCO3 solution (200 mL) and brine (200 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash chromatography (SiO2; 0–22% EtOAc in hexane) afforded the primary alcohol 62 as a colorless oil (1.90 g, 58%). 1H NMR (400 MHz, CDCl3) δ = 1.31–1.40 (m, 2H, CH2), 2.19 (s, 3H, CH3), 2.21 (s, 3H, CH3), 2.59–2.66 (m, 2H, CH2), 3.62 (t, J = 6.4 Hz, 2H, CH2), 6.97–7.03 (m, 2H), 7.04–7.09 (m, 1H), 7.14–7.19 (m, 2H), 7.22–7.31 (m, 8H). 13C NMR (100 MHz, CDCl3) δ = 19.36, 20.25, 29.36, 36.61, 56.06, 63.54, 125.85, 126.75, 127.89, 129.19, 129.36, 130.44, 134.11, 135.90, 144.94, 147.67. GC-MS (CI, methane) tR = 17.91 min (m/z = 371.1, [M + C3H5]+). Anal. Calcd for C24H26O·1/4EtOAc: C, 85.19; H, 8.01. Found: C, 85.11; H, 7.81.
4-(3,4-Dimethylphenyl)-4,4-diphenylbutanal (63)
The title compound was prepared using an adaptation of the method developed by Dess et al. and the procedure reported by Wang et al.29,34 Dess–Martin periodinane (3.069 g, 7.24 mmol) was added to a solution 4-(3,4-dimethylphenyl)-4,4-diphenylbutan-1-ol 62 (1.993 g, 6.03 mmol) in anhydrous CH2Cl2 (24 mL). The reaction was stirred at room temperature for 4 h and then quenched cautiously with sodium thiosulfate solution (0.26 M in saturated aqueous NaHCO3 solution, 100 mL) and extracted with CH2Cl2 (3 × 50 mL). The combined organic extracts were washed successively with saturated aqueous NaHCO3 solution, H2O, and brine (150 mL each), dried (MgSO4), and concentrated in vacuo. Purification by flash chromatography (SiO2; 0–16% EtOAc in hexane) afforded aldehyde 63 as a colorless oil (830 mg, 42%). 1H (400 MHz, CDCl3) δ = 2.20 (s, 3H, CH3), 2.22 (s, 3H, CH3), 2.29–2.35 (m, 2H, CH2), 2.88–2.94 (m, 2H, CH2), 6.97–7.06 (m, 3H), 7.15–7.22 (m, 2H), 7.24–7.30 (m, 8H). 13C NMR (100 MHz, CDCl3) δ = 19.36, 20.24, 31.99, 41.30, 55.63, 126.17, 126.60, 128.13, 129.18, 129.40, 130.26, 134.48, 136.21, 144.11, 146.96, 202.01. GC-MS (CI, methane) tR = 17.57 min (m/z = 329.2 [M + H]+).
2-(Benzylamino)-5-(3,4-dimethylphenyl)-5,5-diphenylpentanenitrile (rac-64)
The title compound was prepared by an adaptation of the method reported by Yadav et al.35 Montmorillonite KSF clay (4.5 g) and 1-phenylmethanamine (549 μL, 5.02 mmol) were added to a solution of 4-(3,4-dimethylphenyl)-4,4-diphenylbutanal 63 (300 mg, 1.0 mmol) in anhydrous CH2Cl2 (45 mL) at room temperature. Trimethylsilyl cyanide (686 μL, 5.48 mmol) was then added, the reaction mixture was stirred at room temperature for 2.5 h and filtered, and the clay was rinsed with CH2Cl2. The filtrate was concentrated in vacuo and purification of the residue by flash chromatography (SiO2; 0–16% EtOAc in hexane) afforded the racemic α-aminonitrile 64 as a pale yellow oil [931 mg, 68% (based on 79% conversion)] and unreacted 4-(3,4-dimethylphenyl)-4,4-diphenylbutanal 63 (485 mg). 1H (500 MHz, CDCl3) δ = 1.56–1.62 (m, 2H, CH2), 2.20 (s, 3H, CH3), 2.23 (s, 3H, CH3), 2.71–2.81 (m, 2H, CH2), 3.36–3.40 (m, 1H, CH), 3.78 (d, J = 12.8 Hz, 1H, CHaHbBn), 4.02 (d, J = 12.8 Hz, 1H, CHaHbBn), 6.98–7.06 (m, 3H), 7.17–7.21 (m, 2H), 7.25–7.31 (m, 9H), 7.33–7.36 (m, 4H). 13C NMR (100 MHz, CDCl3) δ = 19.34, 20.22, 30.12, 36.31, 50.17, 51.78, 55.95, 120.33, 126.14, 126.64, 127.72, 128.10, 128.49, 128.74, 129.18, 129.39, 130.22, 134.43, 136.17, 138.42, 144.13, 147.03, 147.06. HRMS (ESI+) calcd for C32H33N2 (M + H)+: 445.2638; found 445.2640.
2-(Benzylamino)-5-(3,4-dimethylphenyl)-5,5-diphenylpentanoic Acid (rac-65)
The title compound was prepared using an adaptation of the procedure reported by Wang et al.29 Concentrated HCl (12 M, 25 mL) was added to a solution of the nitrile rac-64 (900 mg, 2.02 mmol) in dioxane (25 mL) and the mixture heated at reflux for 48 h. After cooling, the mixture was concentrated in vacuo. The crude residue was basified (ca. pH 9.5) with saturated aqueous sodium carbonate solution and extracted with CH2Cl2 (3 × 100 mL). The combined organic extracts were dried (MgSO4) and concentrated in vacuo. Purification by flash chromatography (SiO2; 0–20% MeOH in CH2Cl2 with 1% NH4OH) afforded the protected amino acid rac-65 as a white solid (538 mg, 57%): mp 176–178 °C. 1H (400 MHz, MeOD) δ = 1.51–1.70 (m, 2H, CH2), 2.16 (s, 3H, CH3), 2.19 (s, 3H, CH3), 2.64–2.86 (m, 2H, CH2), 2.64–2.85 (m, 2H, CH2), 3.40–3.44 (m, 1H, CH), 3.92 (d, J = 12.8 Hz, 1H, CHaHbPh), 4.02 (d, J = 12.8 Hz, 1H, CHaHbPh), 6.95–7.05 (m, 2H), 7.11–7.17 (m, 2H), 7.20–7.31 (m, 8H), 7.34–7.40 (m, 5H). 13C NMR (100 MHz, MeOD) δ = 19.23, 20.09, 27.89, 36.66, 51.22, 57.06, 63.35, 126.93, 127.81, 128.87, 130.09, 130.30, 130.41, 131.08, 131.50, 132.78, 135.20, 136.91, 145.69, 148.55, 173.40. HRMS (ESI+) calcd for C32H34NO2 (M + H)+: 464.2584; found 464.2585.
2-Amino-5-(3,4-dimethylphenyl)-5,5-diphenylpentanoic Acid (rac-42)
The title compound was prepared using an adaptation of the method reported by Ram et al.36 A solution of benzylamine rac-65 (520 mg, 1.12 mmol), 10% Pd/C (250 mg), and HCOONH4 (354 mg, 5.61 mmol) in anhydrous MeOH (7.5 mL) was heated at reflux for 2 h. After being cooled to room temperature, the mixture was filtered through a thick pad of Celite, the residue washed with MeOH (25 mL), and the filtrate concentrated in vacuo. Purification by flash chromatography (SiO2; 0–25% MeOH in CH2Cl2) afforded the racemic amino acid 42 as a white solid (346 mg, 83%): mp 258–259 °C. 1H (500 MHz, MeOD) δ = 1.57–1.65 (m, 2H, CH2), 2.17 (s, 3H, CH3), 2.20 (s, 3H, CH3), 2.65–2.86 (m, 2H, CH2), 3.53 (t, 1H, J = 6.0 Hz, CH), 6.97–7.05 (m, 2H), 7.13–7.17 (m, 2H), 7.22–7.31 (m, 8H). 13C NMR (125 MHz, MeOD) δ = 19.22, 20.08, 28.93, 36.75, 56.33, 57.04, 126.92, 127.80, 128.85, 130.06, 130.33, 131.52, 135.18, 136.89, 145.72, 148.55, 148.59, 166.57. HRMS (ESI+) calcd for C25H28NO2 (M + H)+: 374.2122; found: 374.2115. Anal. Calcd for C25H27NO2·H2O: C, 76.70; H, 7.47; N, 3.58. Found: C, 76.74; H, 7.27; N, 3.45.
Biology
Inhibition of Basal and MT-Stimulated Eg5 ATPase Activities
The inhibition of the basal and MT-stimulated ATPase activities was measured as previously described using a TECAN Sunrise photometer.29 The experiments were performed at 25 °C. In short, the inhibition of the basal ATPase activity was measured at a concentration of 80–100 nM Eg5 in the presence of 150 mM NaCl. The inhibition of the MT-stimulated ATPase activity was determined at an Eg5 concentration of ∼5 nM in the presence of 150 mM NaCl. For this Eg5 construct, it has been noted that the Kiapp estimates are dependent on the salt concentration of the buffer.65S-Trityl-l-cysteine and related inhibitors are tight-binding inhibitors;18 estimates for Kiapp values were therefore calculated using the Morrison equation.66
Calculation of Ligand Efficiencies
Ligand efficiencies (LE) were calculated from the equation: LE = −ΔG/HAC ≈ −RT ln(Kiapp)/HAC, where ΔG is the change in Gibbs free energy, T is the absolute temperature, R represents the gas constant, and HAC is the heavy atom count for non-hydrogen atoms.67 Ligand efficiencies were calculated in Pipeline Pilot 7.4 for Windows (Accelerys Software, San Diego, CA).
Tissue Culture
HCT116 (ATCC CCL-247) cells were cultured in DMEM (Invitrogen, Paisley, U.K.), supplemented with 10% fetal bovine serum (PAA, Pasching, Austria). K562 (ATCC CCL-243), LNCaP (ATCC CRL-1740), PC-3 (ATCC CRL-1435), and NCI-H1299 (CRL-5803) cells were cultured in RPMI (Invitrogen, Paisley, U.K.), supplemented with 10% fetal bovine serum (PAA, Pasching, Austria). BxPC-3 (ATCC CRL-1687) cells were cultured in RPMI (Invitrogen, Paisley, U.K.), supplemented with 1% nonessential amino acids (Invitrogen, Paisley, U.K.), 1% sodium pyruvate (Invitrogen, Paisley, U.K.), 1% glutamine (Invitrogen, Paisley, U.K.), and 10% fetal bovine serum (PAA, Pasching, Austria). Cells were maintained at 37 °C, 95% humidity, and 5% carbon dioxide in a humidified incubator and used for experiments for 6–8 weeks before being replaced with fresh stocks that had been stored in liquid nitrogen.
Proliferation Assays
Cells were seeded in triplicate in 96-well assay plates at 1.250 cells (BxPC-3, HCT116), 1.500 cells (PC-3), 2.500 cells (NCI-H1299), 3.000 cells (LNCaP), or 5.000 cells (K562) per well in 100 μL of the respective growth medium. Medium blanks and cell blanks were also prepared for every cell line. On the next day, inhibitors were added with a starting concentration of 100 μM in a 3-fold serial dilution series. At 72 h after addition of inhibitors, 10% Alamar blue (Invitrogen, Paisley, U.K.) was added, and depending on the cell line, between 2 and 12 h later the absorbance was measured at 570 and 600 nm. All values were corrected for the absorbance of the medium blank, and the corrected cell blanks were set to 100%. Calculations for determining the relative proliferation were performed using the equations described in the manufacturer’s manual. The GI50 values were subsequently determined using a sigmoidal dose–response fitting (variable slope) with GraphPad Prism 5.03 for Windows (GraphPad Software, San Diego, CA).
Drug Profiling of Compounds
Profiling of drug-like properties of interesting compounds including physicochemical properties (turbidimetric solubility, permeability), metabolism and safety (human and mouse microsomal and hepatocyte stability, plasma protein binding, cytochrome P450 inhibition, hERG), and pharmacokinetics were determined at Cyprotex, as previously described.29
Tumor Xenografts
The animal experiments were performed at Oncotest GmbH with female NMRI nu/nu mice (Charles River, Sulzfeld, Germany). Tumor fragments were obtained from xenografts in serial passage in nude mice. Mice were randomized to the various groups, and dosing was started when the required number of mice carried a tumor of 50–250 mm3 volume, preferably 80–200 mm3. Vehicle for 1: 10% ethanol, 10% cremophor, 80% D5W (dextrose 5%); vehicle for all other compounds: 8% DMSO, 2% Tween 80, distilled water (pH 5). All treatments were given intraperitoneally. Vehicle control mice (group 1) were treated with 10 mL/kg vehicle on days 0, 3, 6, 8, 10, 13, 20, 22, 24, 29, 31, 34, 36, 38, 48, 51, 55, 58, 62, 65, and 69.
The rac-11 treatment group (group 2) received 20 mg/kg rac-11 on day 0; 14 mg/kg rac-11 on day 3; 18 mg/kg rac-11 on days 6 and 8; 14 mg/kg rac-11 on days 10 and 13; 16 mg/kg rac-11 on day 20; 18 mg/kg rac-11 on days 22, 24, 29, and 31; 20 mg/kg rac-11 on day 34; 16 mg/kg rac-11 on days 36 and 38; 20 mg/kg rac-11 on days 48, 51, 55, and 62; and 22.5 mg/kg rac-11 on days 65, 69, and 80. The treatment group for 1 (group 3) received 10 mg/kg of 1 on days 0 and 4, and 6 mg/kg of 1 on days 7, 20, and 24.
Vehicle control mice (group 1) were treated with 10 mL/kg vehicle on days 2, 4, 7, 11, 14, 16, 18, 21, 23, 25, 28, 30, and 32. The 36 treatment group (group 2) received 15 mg/kg 36 on days 0, 2, 4; 17 mg/kg 36 on day 7; 14 mg/kg 36 on days 11, 14, and 16; 17 mg/kg 36 on days 18, 21, and 23; 20 mg/kg 36 on days 25 and 28; and 22.5 mg/kg 36 on days 30 and 32.
Vehicle control mice (group 1) were treated with 10 mL/kg vehicle on days 0, 3, 7, 10, and 14. The rac-42 treatment group (group 2) received 15 mg/kg rac-42 on days 0, 3, 7, 10, and 14. The 34 treatment group (group 3) received 15 mg/kg 34 on days 0, 3, 7, 10, and 14 and up to 30 mg/kg during later treatment, without indications of toxicity at this dose.
Mortality checks were conducted at least daily during routine monitoring. Body weight was used as a means of determining toxicity. Mice were weighed twice a week. The tumor volume was determined by two-dimensional measurement with a caliper on the day of randomization (day 0) and then twice weekly (i.e., on the same days on which mice were weighed). Tumor volumes were calculated according to the formula (ab2) × 0.5 where a represents the largest and b the perpendicular tumor diameter. Tumor inhibition for a particular day (T/C in %) was calculated from the ratio of the median RTV (relative tumor volumes) values of test versus control groups multiplied by 100%. For the evaluation of the statistical significance of tumor inhibition, the Mann–Whitney U-test was performed. Individual RTVs were compared on days on which the minimum T/C was achieved, as long as sufficient animals were left for statistical analysis or otherwise on days as indicated. By convention, p ≤ 0.05 indicates significance of tumor inhibition. Statistical calculations were performed using GraphPad Prism 5.03 (GraphPad Software, San Diego, CA).
Crystallization of the Eg5–46 Complex
Purified Eg5 (20 mg/mL) in complex with 1 mM Mg2+ATP was incubated with 2 mM rac-46 (in DMSO) for 2 h on ice. Crystals of Eg5 with 46 appeared after 1 week in hanging drops by mixing 1 μL of protein–inhibitor complex with 1 μL of reservoir solution containing 22% PEG-3350, 0.3 M ammonium sulfate, 0.1 M MES pH 5.5, and 0.01 M trimethylamine hydrochloride in VDX plates (Hampton Research) at 4 °C. Dehydrating solution (32% PEG-3350, 0.36 M of ammonium sulfate, 0.12 M MES pH 5.5, 0.012 M trimethylamine hydrochloride, and 10% glycerol) was added slowly to the crystal droplet until the total volume of the drop was eight times the original. The drop was then equilibrated against air in 4 °C for 30 min. A cubic crystal with dimensions of approximately 0.1 mm on each side was then flash frozen in liquid nitrogen.
Data Collection and Processing
Diffraction data for the Eg5–inhibitor complexes Eg5–46 were recorded at the ESRF ID23-2. Data were processed using iMosflm68 and scaled using Scala69 from the CCP4 suite of programs.70 The calculation of Rfree used 5% of data. The Eg5–46 structure was solved by molecular replacement (Phaser)71 using one molecule of Eg5 motor domain from 3KEN.28 Refinement was carried out with PHENIX.72 Electron-density and difference density maps, all σA-weighted, were inspected, and the model was improved using Coot.73 Model geometry was analyzed using MolProbity.74 For the Eg5–46 complex, 97.0% (322) of the residues are in the preferred regions, 2.7% (9) are in the allowed regions, and 0.3% (1) are outliers. Figures were prepared using PyMOL.75
Acknowledgments
We thank Cancer Research UK for financial support. D.P. was funded by the Leonardo Da Vinci program. We are grateful to Pat Keating for helpful discussions on mass spectrometry and Denise Gilmore for microanalysis. We thank Dr. Gordon Leonard of the European Synchrotron Radiation Facility (ESRF) for assistance and support in using beamline ID23-2.
Glossary
Abbreviations Used
- dia-
diastereomeric mixture
- DMP
Dess–Martin periodinane
- GI50
the concentration required to achieve 50% growth inhibition
- Kiapp
apparent Ki value
- KSP
kinesin spindle protein
- LE
ligand efficiency
- MTs
microtubules
- rac-
racemate
- RTV
relative tumor volume
- STDC
S-trityl-d-cysteine
- STLC
S-trityl-l-cysteine
- n.d.
not determined
- n.i.
no inhibition
- T/C
relative test tumor versus control value
Supporting Information Available
Detailed experimental procedures and characterization data for all other compounds, supplementary results, and representative solubility data for a selection of analogues. This material is available free of charge via the Internet at http://pubs.acs.org. Coordinate and structure factor files for the Eg5–46 complex (PDB ID: 4BBG) was deposited at the Protein Data Bank.
The authors declare no competing financial interest.
Author Present Address
▽ Umeå University, Department of Chemistry, SE 901 87 Umeå, Sweden.
Author Present Address
⊥ Institute of Biophysics, Chinese Academy of Sciences, 15 Datun Rd., Chaoyang District, Beijing, 100101, China.
Author Present Address
§ Redx Oncology Ltd., Duncan Building, Royal Liverpool University Hospital, Liverpool, L69 3GA.
Author Present Address
# Institute of Molecular and Cell Biology, 61 Biopolis Dr., Proteos, Singapore 138673.
Author Present Address
¶ Wellcome Trust Centre for Cell Biology Michael Swann Building, Kings Building University of Edinburgh EH9 3JR
Author Contributions
∥ These authors contributed equally.
Supplementary Material
References
- Rath O.; Kozielski F. Kinesins and cancer. Nat. Rev. Cancer 2012, 12, 527–539. [DOI] [PubMed] [Google Scholar]
- Hirokawa N.; Noda Y.; Tanaka Y.; Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [DOI] [PubMed] [Google Scholar]
- Good J. A. D.; Skoufias D. A.; Kozielski F. Elucidating the functionality of kinesins: An overview of small molecule inhibitors. Semin. Cell Dev. Biol. 2011, 22, 935–945. [DOI] [PubMed] [Google Scholar]
- Sawin K. E.; LeGuellec K.; Philippe M.; Mitchison T. J. Mitotic spindle organization by a plus-end-directed microtubule motor. Nature 1992, 359, 540–543. [DOI] [PubMed] [Google Scholar]
- Blangy A.; Lane H. A.; d’Hérin P.; Harper M.; Kress M.; Nigg E. A. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 1995, 83, 1159–1169. [DOI] [PubMed] [Google Scholar]
- Mayer T. U.; Kapoor T. M.; Haggarty S. J.; King R. W.; Schreiber S. L.; Mitchison T. J. Small Molecule Inhibitor of Mitotic Spindle Bipolarity Identified in a Phenotype-Based Screen. Science 1999, 286, 971–974. [DOI] [PubMed] [Google Scholar]
- Carol H.; Lock R.; Houghton P. J.; Morton C. L.; Kolb E. A.; Gorlick R.; Reynolds C. P.; Maris J. M.; Keir S. T.; Billups C. A.; Smith M. A. Initial testing (stage 1) of the kinesin spindle protein inhibitor ispinesib by the pediatric preclinical testing program. Pediatr. Blood Cancer 2009, 53, 1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woessner R.; Tunquist B.; Lemieux C.; Chlipala E.; Jackinsky S.; Dewolf W.; Voegtli W.; Cox A.; Rana S.; Lee P.; Walker D. ARRY-520, a Novel KSP Inhibitor with Potent Activity in Hematological and Taxane-resistant Tumor Models. Anticancer Res. 2009, 29, 4373–4380. [PubMed] [Google Scholar]
- Jackson J. R.; Gilmartin A.; Dhanak D.; Knight S.; Parrish C.; Luo L.; Sutton D.; Caulder E.; Diamond M.; Giardiniere M.; Zhang S.; Huang P.; Bergnes G.; McDonald A.; Lee Y.; Sakowicz R.; Wood K. W. A second generation KSP inhibitor, SB-743921, is a highly potent and active therapeutic in preclinical models of cancer. AACR Meeting Abstracts 2006, 2006, B11. [Google Scholar]
- Knight S. D.; Parrish C. A. Recent Progress in the Identification and Clinical Evaluation of Inhibitors of the Mitotic Kinesin KSP. Curr. Topics Med. Chem. 2008, 8, 888–904. [DOI] [PubMed] [Google Scholar]
- Huszar D.; Theoclitou M.-E.; Skolnik J.; Herbst R. Kinesin motor proteins as targets for cancer therapy. Cancer Metastasis Rev. 2009, 28, 197–208. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Sardana V.; Xu B.; Homnick C.; Halczenko W.; Buser C. A.; Schaber M.; Hartman G. D.; Huber H. E.; Kuo L. C. Inhibition of a Mitotic Motor Protein: Where, How, and Conformational Consequences. J. Mol. Biol. 2004, 335, 547–554. [DOI] [PubMed] [Google Scholar]
- Turner J.; Anderson R.; Guo J.; Beraud C.; Fletterick R.; Sakowicz R. Crystal Structure of the Mitotic Spindle Kinesin Eg5 Reveals a Novel Conformation of the Neck-linker. J. Biol. Chem. 2001, 276, 25496–25502. [DOI] [PubMed] [Google Scholar]
- O’Connor O. A.; Gerecitano J.; Van Deventer H.; Afanasyev B.; Hainsworth J.; Chen M.; Saikali K.; Seroogy J.; Escandon R.; Wolff A.; Conlan M. G. A Phase I/II Trial of the Kinesin Spindle Protein (KSP) Inhibitor SB-743921 Dosed Q14D without and with Prophylactic G-CSF in Non-Hodgkin (NHL) or Hodgkin Lymphoma (HL). Blood 2009, 114, 667–667.19433855 [Google Scholar]
- Lonial S.; Cohen A.; Zonder J. A.; Benzinger W. I.; Kaufman J. L.; Orlowski R. Z.; Harvey R. D.; Alexanian R.; Thomas S. K.; Weber D.; Walker D.; Hilder B.; Ptazynski A.; Shah J. J. The Novel KSP Inhibitor ARRY-520 Demonstrates Single-Agent Activity in Refractory Myeloma: Results From a Phase 2 Trial in Patients with Relapsed/Refractory Multiple Myeloma (MM). Blood 2011, 118, 1266–1266. [Google Scholar]
- Woessner R.; Tunquist B.; Cox A.; Rana S.; Walker D.; Lee P. A. Combination of the KSP Inhibitor ARRY-520 with Bortezomib or Revlimid Causes Sustained Tumor Regressions and Significantly Increased Time to Regrowth in Models of Multiple Myeloma. Blood 2009, 114, 1115–1116. [Google Scholar]
- DeBonis S.; Skoufias D. A.; Lebeau L.; Lopez R.; Robin G.; Margolis R. L.; Wade R. H.; Kozielski F. In vitro screening for inhibitors of the human mitotic kinesin Eg5 with antimitotic and antitumor activities. Mol. Cancer Ther. 2004, 3, 1079–1090. [PubMed] [Google Scholar]
- Skoufias D. A.; DeBonis S.; Saoudi Y.; Lebeau L.; Crevel I.; Cross R.; Wade R. H.; Hackney D.; Kozielski F. S-Trityl-l-cysteine Is a Reversible, Tight Binding Inhibitor of the Human Kinesin Eg5 That Specifically Blocks Mitotic Progression. J. Biol. Chem. 2006, 281, 17559–17569. [DOI] [PubMed] [Google Scholar]
- Brier S.; Lemaire D.; DeBonis S.; Forest E.; Kozielski F. Identification of the Protein Binding Region of S-Trityl-l-cysteine, a New Potent Inhibitor of the Mitotic Kinesin Eg5. Biochemistry 2004, 43, 13072–13082. [DOI] [PubMed] [Google Scholar]
- Kozielski F.; Skoufias D. A.; Indorato R.-L.; Saoudi Y.; Jungblut P. R.; Hustoft H. K.; Strozynski M.; Thiede B. Proteome analysis of apoptosis signaling by S-trityl-l-cysteine, a potent reversible inhibitor of human mitotic kinesin Eg5. Proteomics 2008, 8, 289–300. [DOI] [PubMed] [Google Scholar]
- Zee-Cheng K.-Y.; Cheng C.-C. Experimental antileukemic agents. Preparation and structure–activity study of S-tritylcysteine and related compounds. J. Med. Chem. 1970, 13, 414–418. [DOI] [PubMed] [Google Scholar]
- Ding S.; Nishizawa K.; Kobayashi T.; Oishi S.; Lv J.; Fujii N.; Ogawa O.; Nishiyama H. A Potent Chemotherapeutic Strategy for Bladder Cancer: (S)-Methoxy-Trityl-L-Cystein, a Novel Eg5 Inhibitor. J. Urol. 2010, 184, 1175–1181. [DOI] [PubMed] [Google Scholar]
- Xing N.-D.; Ding S.-T.; Saito R.; Nishizawa K.; Kobayashi T.; Inoue T.; Oishi S.; Fujii N.; Lv J.-J.; Ogawa O.; Nishiyama H. A potent chemotherapeutic strategy in prostate cancer: S-(methoxytrityl)-l-cysteine, a novel Eg5 inhibitor. Asian J. Androl. 2011, 13, 236–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBonis S.; Skoufias D. A.; Indorato R.-L.; Liger F.; Marquet B.; Laggner C.; Joseph B.; Kozielski F. Structure–Activity Relationship of S-Trityl-l-Cysteine Analogues as Inhibitors of the Human Mitotic Kinesin Eg5. J. Med. Chem. 2008, 51, 1115–1125. [DOI] [PubMed] [Google Scholar]
- Ogo N.; Oishi S.; Matsuno K.; Sawada J.-i.; Fujii N.; Asai A. Synthesis and biological evaluation of l-cysteine derivatives as mitotic kinesin Eg5 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3921–3924. [DOI] [PubMed] [Google Scholar]
- Kaan H. Y. K.; Weiss J.; Menger D.; Ulaganathan V.; Tkocz K.; Laggner C.; Popowycz F.; Joseph B.; Kozielski F. Structure–Activity Relationship and Multidrug Resistance Study of New S-trityl-l-Cysteine Derivatives As Inhibitors of Eg5. J. Med. Chem. 2011, 54, 1576–1586. [DOI] [PubMed] [Google Scholar]
- Kaan H. Y. K.; Ulaganathan V.; Hackney D. D.; Kozielski F. An allosteric transition trapped in an intermediate state of a new kinesin–inhibitor complex. Biochem. J. 2010, 425, 55–60. [DOI] [PubMed] [Google Scholar]
- Kim E. D.; Buckley R.; Learman S.; Richard J.; Parke C.; Worthylake D. K.; Wojcik E. J.; Walker R. A.; Kim S. Allosteric Drug Discrimination Is Coupled to Mechanochemical Changes in the Kinesin-5 Motor Core. J. Biol. Chem. 2010, 285, 18650–18661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Good J. A. D.; Rath O.; Kaan H. Y. K.; Sutcliffe O. B.; Mackay S. P.; Kozielski F. Triphenylbutanamines: Kinesin Spindle Protein Inhibitors with in Vivo Antitumor Activity. J. Med. Chem. 2012, 55, 1511–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. K.; McCabe F. L.; Caulder E.; Inlow-Porter L.; Whitacre M.; Winkler J. D.; Bergnes G.; Feng B.; Morgans D.; Wood K. W.; Jackson J. R. SB-715992, a potent and selective inhibitor of the mitotic kinesin KSP, demonstrates broad-spectrum activity in advanced murine tumors and human tumor xenografts. Proc. Am. Assoc. Cancer Res. Annu. Meet. 2002, 43, 269. [Google Scholar]
- Maltese M. Reductive Demercuration in Deprotection of Trityl Thioethers, Trityl Amines, and Trityl Ethers. J. Org. Chem. 2001, 66, 7615–7625. [DOI] [PubMed] [Google Scholar]
- Nahm S.; Weinreb S. M. N-methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar]
- Kabalka G. W.; Yao M.-L.; Borella S.; Goins L. K. Iron Trichloride Mediated Allylation of Lithium Alkoxides through an Unusual Carbon–Oxygen Bond Cleavage. Organometallics 2007, 26, 4112–4114. [Google Scholar]
- Dess D. B.; Martin J. C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983, 48, 4155–4156. [Google Scholar]
- Yadav J. S.; Reddy B. V. S.; Eeshwaraiah B.; Srinivas M. Montmorillonite KSF clay catalyzed one-pot synthesis of α-aminonitriles. Tetrahedron 2004, 60, 1767–1771. [Google Scholar]
- Ram S.; Spicer L. D. Debenzylation of N-Benzylamino Derivatives by Catalytic Transfer Hydrogenation With Ammonium Formate. Synth. Commun. 1987, 17, 415–418. [Google Scholar]
- Davis F. A.; Kasu P. V. N.; Sundarababu G.; Qi H. Nonracemic α-Fluoro Aldehydes: Asymmetric Synthesis of 4-Deoxy-4-fluoro-d-arabinopyranose. J. Org. Chem. 1997, 62, 7546–7547. [Google Scholar]
- Beeson T. D.; MacMillan D. W. C. Enantioselective Organocatalytic α-Fluorination of Aldehydes. J. Am. Chem. Soc. 2005, 127, 8826–8828. [DOI] [PubMed] [Google Scholar]
- Enders D.; Hüttl M. R. M. Direct Organocatalytic α-Fluorination of Aldehydes and Ketones. Synlett 2005, 2005, 0991–0993. [Google Scholar]
- Prakash G. K. S.; Krishnamurti R.; Olah G. A. Synthetic methods and reactions. 141. Fluoride-induced trifluoromethylation of carbonyl compounds with trifluoromethyltrimethylsilane (TMS-CF3). A trifluoromethide equivalent. J. Am. Chem. Soc. 1989, 111, 393–395. [Google Scholar]
- Jiang Z.-X.; Qing F.-L. Regioselective and Stereoselective Nucleophilic Ring Opening of Trifluoromethylated Cyclic Sulfates: Asymmetric Synthesis of Both Enantiomers of syn-(3-Trifluoromethyl)isoserine. J. Org. Chem. 2004, 69, 5486–5489. [DOI] [PubMed] [Google Scholar]
- Shimizu M.; Ishii H.; Ogo N.; Unno Y.; Matsuno K.; Sawada J.-i.; Akiyama Y.; Asai A. S-trityl-l-cysteine derivative induces caspase-independent cell death in K562 human chronic myeloid leukemia cell line. Cancer Lett. 2010, 298, 99–106. [DOI] [PubMed] [Google Scholar]
- Carter B. Z.; Mak D. H.; Shi Y.; Schober W. D.; Wang R.-Y.; Konopleva M.; Koller E.; Dean N. M.; Andreeff M. Regulation and Targeting of Eg5, a Mitotic Motor Protein in Blast Crisis CML: Overcoming Imatinib Resistance. Cell Cycle 2006, 5, 2223–2229. [DOI] [PubMed] [Google Scholar]
- Cox C. D.; Breslin M. J.; Mariano B. J.; Coleman P. J.; Buser C. A.; Walsh E. S.; Hamilton K.; Huber H. E.; Kohl N. E.; Torrent M.; Yan Y.; Kuo L. C.; Hartman G. D. Kinesin spindle protein (KSP) inhibitors. Part 1: The discovery of 3,5-diaryl-4,5-dihydropyrazoles as potent and selective inhibitors of the mitotic kinesin KSP. Bioorg. Med. Chem. Lett. 2005, 15, 2041–2045. [DOI] [PubMed] [Google Scholar]
- Wilson A. S.; Davis C. D.; Williams D. P.; Buckpitt A. R.; Pirmohamed M.; Park B. K. Characterisation of the toxic metabolite(s) of naphthalene. Toxicology 1996, 114, 233–242. [DOI] [PubMed] [Google Scholar]
- Weisberger A. S.; Levine B. Incorporation of Radioactive L-Cystine by Normal and Leukemic Leukocytes in Vivo. Blood 1954, 9, 1082–1094. [PubMed] [Google Scholar]
- Goodman L.; Ross L. O.; Baker B. R. Potential Anticancer Agents. V. Some Sulfur-Substituted Derivatives of Cysteine. J. Org. Chem. 1958, 23, 1251–1257. [Google Scholar]
- Waring M. J. Lipophilicity in drug discovery. Expert Opin. Drug Discovery 2010, 5, 235–248. [DOI] [PubMed] [Google Scholar]
- Abualhasan M. N.; Good J. A. D.; Wittayanarakul K.; Anthony N. G.; Berretta G.; Rath O.; Kozielski F.; Sutcliffe O. B.; Mackay S. P. Doing the methylene shuffle – Further insights into the inhibition of mitotic kinesin Eg5 with S-trityl l-cysteine. Eur. J. Med. Chem. 2012, 54, 483–498. [DOI] [PubMed] [Google Scholar]
- Barsanti P. A.; Wang W.; Ni Z.-J.; Duhl D.; Brammeier N.; Martin E.; Bussiere D.; Walter A. O. The discovery of tetrahydro-β-carbolines as inhibitors of the kinesin Eg5. Bioorg. Med. Chem. Lett. 2010, 20, 157–160. [DOI] [PubMed] [Google Scholar]
- Schiemann K.; Finsinger D.; Zenke F.; Amendt C.; Knöchel T.; Bruge D.; Buchstaller H.-P.; Emde U.; Stähle W.; Anzali S. The discovery and optimization of hexahydro-2H-pyrano[3,2-c]quinolines (HHPQs) as potent and selective inhibitors of the mitotic kinesin-5. Bioorg. Med. Chem. Lett. 2010, 20, 1491–1495. [DOI] [PubMed] [Google Scholar]
- Kaan H. Y. K.; Ulaganathan V.; Rath O.; Prokopcová H.; Dallinger D.; Kappe C. O.; Kozielski F. Structural Basis for Inhibition of Eg5 by Dihydropyrimidines: Stereoselectivity of Antimitotic Inhibitors Enastron, Dimethylenastron, and Fluorastrol. J. Med. Chem. 2010, 53, 5676–5683. [DOI] [PubMed] [Google Scholar]
- Nichols D. B.; Fournet G.; Gurukumar K. R.; Basu A.; Lee J.-C.; Sakamoto N.; Kozielski F.; Musmuca I.; Joseph B.; Ragno R.; Kaushik-Basu N. Inhibition of hepatitis C virus NS5B polymerase by S-trityl-l-cysteine derivatives. Eur. J. Med. Chem. 2012, 49, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienkers L. C.; Heath T. G. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discovery 2005, 4, 825–833. [DOI] [PubMed] [Google Scholar]
- Jamieson C.; Moir E. M.; Rankovic Z.; Wishart G. Medicinal Chemistry of hERG Optimizations: Highlights and Hang-Ups. J. Med. Chem. 2006, 49, 5029–5046. [DOI] [PubMed] [Google Scholar]
- Cox C. D.; Breslin M. J.; Whitman D. B.; Coleman P. J.; Garbaccio R. M.; Fraley M. E.; Zrada M. M.; Buser C. A.; Walsh E. S.; Hamilton K.; Lobell R. B.; Tao W.; Abrams M. T.; South V. J.; Huber H. E.; Kohl N. E.; Hartman G. D. Kinesin spindle protein (KSP) inhibitors. Part 5: Discovery of 2-propylamino-2,4-diaryl-2,5-dihydropyrroles as potent, water-soluble KSP inhibitors, and modulation of their basicity by β-fluorination to overcome cellular efflux by P-glycoprotein. Bioorg. Med. Chem. Lett. 2007, 17, 2697–2702. [DOI] [PubMed] [Google Scholar]
- Zee Y. K.; Eisen T. Survival from lung cancer in England and Wales up to 2001. Br. J. Cancer 2008, 99, S43–S46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebig H. H.; Dengler W. A.; Roth T.. Human tumor xenografts. Predictivity, characterization and discovery of new new anticancer agents. In Relevance of tumor models for anticancer drug development; Fiebig H. H.; Burger A. M., Eds.; Karger: Basel, 1999; Vol. 56, pp 29–50. [Google Scholar]
- Humphries M.; Woessner R.; Bouhana K.; Garrus J.; Napier C.; Lemieux C.; Walker D.; Winsk S. Human tumor explants are better predictors of clinical trial outcome than cell line xenografts for the KSP inhibitor ARRY-520. Presented at the AACR 103rd Annual Meeting, Chicago, IL, March 31–April 4, 2012; p 1782.
- Purcell J. W.; Davis J.; Reddy M.; Martin S.; Samayoa K.; Vo H.; Thomsen K.; Bean P.; Kuo W. L.; Ziyad S.; Billig J.; Feiler H. S.; Gray J. W.; Wood K. W.; Cases S. Activity of the Kinesin Spindle Protein Inhibitor Ispinesib (SB-715992) in Models of Breast Cancer. Clin. Cancer Res. 2010, 16, 566–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlodi-Pasztor E.; Sackett D. L.; Fojo A. T. Inhibitors Targeting Mitosis: Tales of How Great Drugs against a Promising Target Were Brought Down by a Flawed Rationale. Clin. Cancer Res. 2012, 18, 51–63. [DOI] [PubMed] [Google Scholar]
- Gentile D. M.; Tomlinson E. S.; Maggs J. L.; Park B. K.; Back D. J. Dexamethasone metabolism by human liver in vitro. Metabolite identification and inhibition of 6-hydroxylation. J. Pharmacol. Exp. Ther. 1996, 277, 105–112. [PubMed] [Google Scholar]
- Hirashima S.; Suzuki T.; Ishida T.; Noji S.; Yata S.; Ando I.; Komatsu M.; Ikeda S.; Hashimoto H. Benzimidazole Derivatives Bearing Substituted Biphenyls as Hepatitis C Virus NS5B RNA-Dependent RNA Polymerase Inhibitors: Structure–Activity Relationship Studies and Identification of a Potent and Highly Selective Inhibitor JTK-109. J. Med. Chem. 2006, 49, 4721–4736. [DOI] [PubMed] [Google Scholar]
- Chackal-Catoen S.; Miao Y.; Wilson W. D.; Wenzler T.; Brun R.; Boykin D. W. Dicationic DNA-targeted antiprotozoal agents: Naphthalene replacement of benzimidazole. Bioorg. Med. Chem. 2006, 14, 7434–7445. [DOI] [PubMed] [Google Scholar]
- Luo L.; Carson J. D.; Dhanak D.; Jackson J. R.; Huang P. S.; Lee Y.; Sakowicz R.; Copeland R. A. Mechanism of Inhibition of Human KSP by Monastrol: Insights from Kinetic Analysis and the Effect of Ionic Strength on KSP Inhibition. Biochemistry 2004, 43, 15258–15266. [DOI] [PubMed] [Google Scholar]
- Morrison J. F. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta, Enzymol. 1969, 185, 269–286. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Powell H. The Rossmann Fourier autoindexing algorithm in MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, 55, 1690–1695. [DOI] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- Collaborative. The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 56, 760-763. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. III; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.3r1, 2010.
- Cruickshank D. Remarks about protein structure precision. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, 55, 583–601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.