

Abstract
MbtA catalyzes the first committed step of mycobactin biosynthesis in Mycobacterium tuberculosis (Mtb) and is responsible for the incorporation of salicylic acid into the mycobactin siderophores. 5′-O-[N-(Salicyl)sulfamoyl]adenosine (Sal-AMS) is an extremely potent nucleoside inhibitor of MbtA that possesses excellent activity against whole-cell Mtb, but suffers from poor bioavailability. In an effort to improve the bioavailability, we have designed four conformationally constrained analogues of Sal-AMS that remove two rotatable bonds and the ionized sulfamate group based on computational and structural studies. Herein we describe the synthesis, biochemical, and microbiological evaluation of chromone-, quinolone-, and benzoxazinone-3-sulfonamide derivatives of Sal-AMS. We developed new chemistry to assemble these three heterocycles from common β-ketosulfonamide intermediates. The synthesis of the chromone- and quinolone-3-sulfonamide intermediates features formylation of a β-ketosulfonamide employing dimethylformamide dimethyl acetal to afford an enaminone that can react intramolecularly with a phenol or intermolecularly with a primary amine via addition-elimination reaction(s). The benzoxazinone-3-sulfonamide was prepared by nitrosation of a β-ketosulfonamide followed by intramolecular nucleophilic aromatic substitution. Mitsunobu coupling of these bicyclic sulfonamides with a protected adenosine derivative followed by global deprotection provides a concise synthesis of the respective inhibitors.
Introduction
Tuberculosis (TB) is caused primarily by the acid-fast bacillus Mycobacterium tuberculosis (Mtb) and is the leading cause of bacterial infectious disease mortality, responsible for 1.4 million deaths and 8.7 million new infections in 2011.1 It is estimated that one-third of the world’s population is infected with latent TB.1 The current standard of treatment for drug-susceptible TB, known as Directly Observed Treatment Short-course, requires six to nine months of combination chemotherapy of the four frontline TB agents: isoniazid, rifampin, pyrazinamide, and ethambutol. The emergence of multidrug resistant and extensively drug resistant TB strains demands the development of new drugs ideally with novel mechanisms of action.2
Iron is an essential micronutrient for almost all known organisms; its redox tuneability makes it an indispensable cofactor for life processes and biochemistry.3 However, the extreme insolubility of ferric hydroxide at physiological pH and the further sequestration of ferric iron in a mammalian host by iron transport proteins like lactoferrin and transferrin suppress its free concentration to an astonishing 10−24 M, which is far too low to support bacterial colonization and growth.3 To overcome this lack of readily available iron, pathogenic bacteria have evolved the ability to obtain iron from the serum and/or tissues of their host via the synthesis, secretion, and reuptake of small-molecule iron chelators known as siderophores.4 Mtb produces a suite of structurally related siderophores that are essential for iron acquisition.5 Disruption of genes involved in mycobactin biosynthesis results in Mtb strains unable to replicate in vitro unless chemically complemented with exogenous mycobactin, which in turns suggests inhibition of mycobactin biosynthesis may represent a novel strategy for the development of new antitubercular agents.6
Mycobactins (Figure 1A) are biosynthesized by a mixed nonribosomal peptide synthetatse– polyketide synthase (NRPS–PKS) pathway in Mtb.7 MbtA, an aryl acid adenylating enzyme (AAAE), is responsible for initiating mycobactin biosynthesis by priming the NRPS–PKS assembly line.7a MbtA does this by catalyzing a two-step adenylation–acylation reaction (Figure 1B). In the adenylation half-reaction, salicylate and ATP are condensed to form an acyl-adenylate intermediate that remains tightly bound to prevent adventitious hydrolysis of this labile mixed anhydride. MbtA then catalyzes the transfer of the acyl moiety onto MbtB, the second protein in the mycobactin pathway. 5′-O-[N-(salicyl)sulfamoyl]adenosine (Sal-AMS) is a rationally designed inhibitor of MbtA wherein the hydrolytically labile acyl-phosphate moiety of the acyl-adenylate intermediate is replaced by a stabile acyl-sulfamate linker (Figure 1C).8 The sulfamate linker is inspired by the natural product ascamycin isolated from an unknown Streptomyces species in Japan.9
Figure 1.

(A) Aryl-capped siderophores from Mtb, the mycobactins. (B) Enzymatic reactions catalyzed by MbtA, with hydrolytically labile acylphosphate outlined in blue. (C) Sal-AMS and natural product ascamycin. The hydrolytically stabile acylsulfamate bioisostere is outlined in blue.
Sal-AMS is a potent inhibitor of MbtA with an apparent Ki in a functional kinetic assay of 7 nM.10 Furthermore, Sal-AMS displays potent whole cell activity against Mtb H37Rv under iron-limiting conditions with a minimum inhibitory concentration (MIC) of 0.39 µM, rivaling the first-line clinical agent isoniazid.8c To date, our laboratory has conducted extensive structure– activity relationship (SAR) studies on the Sal-AMS scaffold, systematically exploring its aryl,11 linker,8c, 12 glycosyl,10 and nucleobase domains.13 These SAR findings, in conjunction with a quantum mechanical study in which Sal-AMS was docked in the binding site of an MbtA homology model, indicate that an internal hydrogen bond is formed between the phenol and sulfamate nitrogen atom (estimated pKa around 2) of Sal-AMS.14 This enforces a coplanar arrangement of the salicyl group when bound in the MbtA active site. Further evidence in support of this binding mode is observed in the co-crystal structures of homologous AAAEs from Bacillus subtilis (DhbE)15 and Acinetobacter baumannii (BasE)16 with the acyl-adenylate ligands 5′-O-[N-(2,3-dihydroxybenzoyl)sulfamoyl]adenosine (DHB-AMS) and 2,3-dihydroxybenzoyl adenosine monophosphate (DHB-AMP) (Figure 2).
Figure 2.

(A) (Left; center; right). Molecular structure of DHB-AMP; co-crystal structure of enzymatic intermediate DHB-AMP bound to AAAE DhbE from B. subtilis (PDB ID: 1MDB) with aryl ring in plane of paper; same co-crystal structure, rotated 90°, with aryl ring perpendicular to paper. (B) (Left; center; right). Molecular structure of DHB-AMS; co-crystal structure of bisubstrate mimic DHB-AMS bound to AAAE BasE from A. baumannii (PDB ID: 3O82) with aryl ring in plane of paper; same co-crystal structure, rotated 90°, with aryl ring perpendicular to paper.
Preliminary pharmacokinetic (PK) studies demonstrated that Sal-AMS has poor oral bioavailability.17 Veber and co-workers have shown that oral bioavailibility inversely correlates with two criteria: the number of rotatable bonds and polar surface area.18 In an attempt to improve the oral bioavailability of Sal-AMS, we designed conformationally constrained analogues 1–3, shown in Figure 3. These analogues mimic the hypothesized MbtA-bound conformation of Sal-AMS and could potentially improve its oral bioavailability through removal of two rotatable bonds and the charged sulfamate moiety, which reduces the total polar surface area (tPSA). The calculated tPSA and octanol-water coefficient (cLogP) for all compounds in shown in Table 2. Herein we report the synthesis, biochemical and microbiological evaluation of conformationally constrained analogues 1–4 of Sal-AMS and DHB-AMS.
Figure 3.

Conformationally constrained analogues of Sal-AMS (i.e. 1–3) and DHB-AMS (i.e. 4).
Table 2.
Combined Biochemical and Antitubercular Evaluation of 1–4.
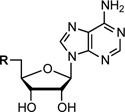 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R | ClogPb | tPSAb |
KD (µM) |
Kiapp (µM) |
MIC50 (µM) |
||||
| MbtA | BasE | EntE | VibE | MbtA | –Fe | +Fe | ||||
| Fl-Sal-AMSa | n.a. | n.a. | n.a. | 0.0093 | 0.093 | 0.23 | 0.13 | — | — | — |
| Sal-AMS | 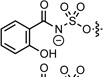 |
−1.79 | 210.8 | <0.01 | <0.1 | <0.2 | <0.1 | 0.0066 | 0.39c | 1.56c |
| 1 | 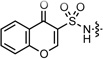 |
−0.65 | 188.5 | 3.6 ± 0.1 | 108 ± 8 | 280 ± 60 | 98 ± 7 | — | >50 | >50 |
| 2 | 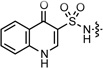 |
−0.57 | 193.4 | <0.01 | <0.1 | 0.32 ± 0.04 | <0.1 | 0.12 ± 0.02 | >50 | >50 |
| 3 | 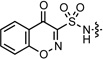 |
−1.26 | 200.9 | 0.37 ± 0.01 | 1.19 ± 0.07 | 12 ± 1 | 2.5 ± 0.7 | — | >50 | >50 |
| 4 | 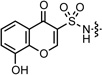 |
−1.10 | 208.7 | 290 ± 70 | >300 | >300 | >300 | — | >50 | >50 |
Structure of Fl-Sal-AMS: 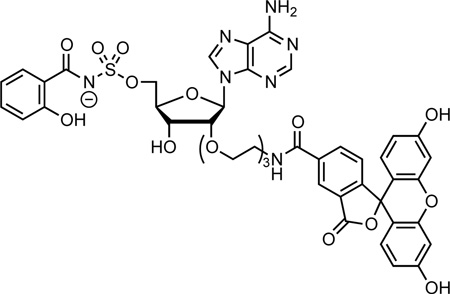
Calculated using ChemBioDraw Ultra Version 13.0.0.3015
Previously reported
Results and Discussion
Synthesis
The most concise synthesis we envisioned to the proposed bicyclic sulfonamide adenosine analogues involves disconnection of 1–4 by Mitsunobu reaction to bicyclic sulfonamides and an appropriately protected adenosine derivative (Scheme 1). Further retrosynthetic disconnection leads to acetophenone or benzoic acid derivatives.
Scheme 1.

Retrosynthetic analysis
A review of the reported methods for synthesizing chromone and other derivatives caused our attention to focus on a potentially short route to chromone-3-sulfonamides. Enaminone 5 from Föhlisch’s chromone synthesis19 was successfully used by Löwe and Matzanke in a tandem sulfamoylation–cyclization with chlorosulfonylureas to produce chromone-3-sulfonylureas in modest yields.20 We pursued this route by screening a few sulfamoylating reagents to effect the desired transformation (Table 1). After extensive experimentation with sulfamoyl chloride 6, we successfully isolated the desired chromone-3-sulfonamide 9 in 5% yield (Table 1, entry 1). The use of recrystallized sulfamoyl chloride21 was essential to obtain this meager yield. The remainder of material was cyclized, non-sulfamoylated chromone (32%), recovered enaminone 5 (15%), and a highly insoluble material that could not be characterized, potentially polymerized product and/or multi-sulfamoylated material. The less reactive DMAP-stabilized and Boc-protected sulfamoylating reagent 722 was then tried. We considered it an attractive reagent because the sulfonamide functionality is already protected and unlike sulfamoyl chloride, it is stable for prolonged periods at room temperature and is not air-senstive. To our dismay, 7 was unable to effect the desired transformation even with heating, which resulted in cyclization to non-sulfamoylated chromone (Table 1, entry 2). Based on the presumed over-reactivity of 6 and the non-reactivity of 7 we investigated tert-butyl chlorosulfonylcarbamate 8 that we hypothesized would possess intermediate reactivity. This was prepared by addition of chlorosulfonyl isocyanate to a stirring solution of t-BuOH in dichloromethane and successfully purified by recrystallization. Tandem sulfamoylation–cyclization of enaminone 5 with 8 afforded 10 in a final optimized yield of 21% (Table 1, entry 3), which represent a 4-fold improvement over sulfamoyl chloride 6. This low yield was primarily the result of competitive cyclization of the enaminone to chromone, but was commensurate with the yields achieved by Löwe and Matzanke.20
Table 1.
Optimization of tandem sulfamoylation-cyclization of enaminone 5
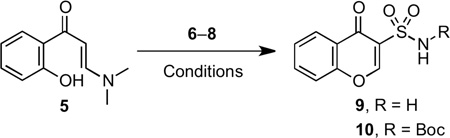 | |||
|---|---|---|---|
| Entry | Reagent | Conditions | Results |
| 1 | DCM, 0 °C | 5% of 9 32% of chromone 15% of 5 recovered |
|
| 2 | 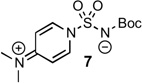 |
1,4-dioxane, 80 °C |
Total conversion to chromone |
| 3 | 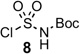 |
DCM, 22 °C | 21% of 10 |
The modest yield of chromone-3-sulfonamide 10 is due to competitive direct cyclization of enaminone 5 to chromone 12 (Scheme 2, pathway A). Since we were unable to convert chromone 12 to chromone-3-sulfonamide 10 under any reaction conditions, we propose that sulfamoylation of enaminone 5 must occur first to afford an α-formyl-β-ketosulfonamide intermediate 13 followed by rapid cyclization to furnish 10 and Me2NH·HCl (Scheme 2, pathway B), the latter of which could serve to catalyze either pathway. Addition of triethylamine or other bases suppresses both pathways. HCl can also be formed through decomposition of the sulfamoylating reagents 6 and 8.
Scheme 2.

Proposed mechanism for tandem sulfamoylation-cyclization of enaminone 5
Based on the proposed mechanism shown in Scheme 2, we hypothesized that 10 could be obtained through α-formylation of a β-ketosulfonamide followed by cyclization (Scheme 3). The necessary β-ketosulfonamide was synthesized by TBS protection of methyl salicylate 15 followed by Claisen-like condensation12 with the LDA-generated dianion of N-Boc-methanesulfonamide 16 (Scheme 4).23 The intermediate 18 was not isolated, but directly treated with TBAF to furnsih β-ketosulfonamide 19 in 86% overall yield. Formylation was initially attempted with triethyl orthoformate in acetic anhydride as described by Chu and coworkers24 in their synthesis of quinolone-3-carboxylic acids; however, its sluggish reactivity required elevated temperatures (~100 °C) that led to Boc-deprotection and imidate formation at the sulfonamide in conjunction with the desired α-formylation. We next investigated the use of dimethylformamide dimethyl acetal (DMF–DMA, 25) since this has been shown to formylate active methylenes under mild conditions.25 Treatment of β-ketosulfonamide 19 with DMF-DMA at ambient temperature resulted in full conversion to enaminone 20 as monitored by TLC and indicated by 1H NMR and MS of the crude material. Addition of saturated aqueous NH4Cl to crude 20 in THF induced rapid cyclization (~5 minutes) to chromone 10. This second generation synthesis of chromone-3-sulfonamide 10 requires 3 steps from methyl salicylate 15 and proceeds in 50% overall yield. The corresponding 8-hydroxychromone-3-sulfonamide 27 was prepared analogously from methyl 2,3-dihydroxybenzoate 22 in 3 steps in 64% overall yield. The free phenol in 27 was converted to MOM ether 28 in order to avoid complication with the subsequent Mitsunobu reaction.
Scheme 3.

Revised retrosynthetic analysis of chromone-3-sulfonamide
Scheme 4.

Second generation chromone synthesis
Mitsunobu coupling of chromone 10 with bis-Boc-adenosine 2926 afforded chromone nucleoside 30 in 80% yield (Scheme 5).12 Mitsunobu couplings of purine nucleosides at the 5′ position is notoriously problematic as noted in Mitsunobu’s seminal review27 due to cyclonucleoside formation between purine N-3 and ribose C-5′. We avoided this competitive reaction through bis-Boc protection of the exocyclic N-6 amino group of adenosine, which served to attenuate the nucleophilicity at N-3. Global deprotection of the Boc and acetonide groups in 30 with 80% aqueous TFA provided the desired chromone analogue 1. The 8-hydroxychromone analogue 4 was prepared analogously from 28.
Scheme 5.

Mitsunobu coupling to chromone 1
We developed a new synthesis of quinolone-3-sulfonamides from β-ketosulfonamides by adapting the classic Grohe-Heitzer route used for the preparation of the related quinolone-3-carboxylic acids, which involves formylation of a β-ketoester followed by introduction of the quinolone nitrogen through a tandem addition-elimination reaction of a primary amine (Scheme 6).28 The requisite β-ketosulfonamide 33 was synthesized from methyl 2-fluorobenzoate 32 through Claisen-like condensation with the dianion of N-Boc-methanesulfonamide 16 (Scheme 7).12 Reaction of β-ketosulfonamide 33 with DMF–DMA at room temperature for 18 h resulted in total conversion to enaminone 34, which was not isolated but directly treated with excess benzylamine. Transamination of 34 via an addition–elimination pathway was complete in 10 minutes and furnished N-benzyl enaminone 35 as an inseperable 2:1 mixture of geometric isomers in 85% overall yield. Cyclization of enaminone 35 to N-benzyl quinolone 36 was achieved by treatment with sodium hydride at ambient temperature in 1 hour. Mitsunobu coupling of 36 with adenosine derivative 29 afforded quinolone nucleoside 37. Debenzylation under standard hydrogenolysis conditions yielded 38 and subsequent deprotection of the acetonide and Boc groups with aqueous TFA provided the desired quinolone 2. Overall, this new route to quinolone-3-sulfonamides is notable for the facile installation of the nitrogen at N-1, which occures at 0 °C to ambient temperature as well as the use of DMF-DMA for formylation of the β-ketosulfonamide.
Scheme 6.
Grohe-Heitzer quinolone synthesis
Scheme 7.

Synthesis of quinolone 2
Given the facile cyclization of enaminones 35, it was hypothesized that an oxime derivative (i.e. α-hydroxyimino-β-ketosulfonamide) could also easily cyclize to afford the 1,2-benzoxazin-4-one heterocycle of 3. The key oxime precursor was prepared using the classic Meyer nitrosation reaction (Scheme 8).29 Thus, treatment of β-ketosulfonamide 33 with sodium nitrite in a mixed AcOH–H2O–THF solvent system30 yielded a nitroso intermediate that tautomerized to oxime 39 as a single undefined geometric isomer that was moderately stabile and was directly utilized in the next step following purification. We explored several methods to induce cyclization of 39 to the desired 1,2-benzoxazin-4-one 40 and ultimately identified cesium carbonate in DMF as the optimal conditions, which provided 40 in 58% yield at room temperature in 1 hour. Under these conditions total consumption of the substrate was observed. We speculate that the modest yield is due to decomposition of the labile oxime under the basic reaction conditions. The only other precedence for this transformation (i.e. 39→40) is found in a patent from Pharmacia & Upjohn that discloses a single example of an analogous α-hydroxyimino-β-ketoester.31 However, the reported cyclization in this patent did not employ a base and required reflux in toluene for 48 hours. The synthesis of target molecule 3 was completed by Mitsunobu coupling of 40 with adenosine derivative 29 to provide 41 followed by deprotection with aqueous TFA. Overall, this concise route provides 1,2-benzoxazin-4-one 3 in only 4 steps from β-ketosulfonamide intermediate 33 and employs extremely mild reaction conditions to assemble this rare heterocycle.32
Scheme 8.

Synthesis of benzoxazinone 3
Biochemical Evaluation
The compounds were evaluated for their binding affinity to MbtA using a previously described fluorescence polarization (FP) assay.33 FP assays are competitive binding assays in which a fluorescently labeled probe molecule is displaced from its receptor (i.e. MbtA) by a competitive ligand (i.e. 1–4) and they allow direct determination of ligand dissociation constants. The binding affinity of chromone 1 toward MbtA using the FP assay is 3.6 µM. Replacement of the ‘CH’ at C-2 in chromone 1 with a ‘N’ atom in benzoxazinone 3 results in a nearly 10-fold increase in potency to 0.37 µM. The higher binding affinity of 3 versus 1 is most likely due to the more isosteric design of 3 when compared to the proposed bound conformation of Sal-AMS in the MbtA active site. The 8-hydroxychromone 4 shows very low affinity to MbtA with a KD of 290 µM, which represents an 80-fold loss of potency relative to chromone 1. This result is readily reconciled since MbtA binds salicylic acid preferably over 2,3-dihydroxybenzoic acid.7a The KD of quinolone 2 is below the detection limit of the assay and is <0.01 µM. The acidity of the quinolone nitrogen (estimated pKa ~ 7.3)34 is low enough that it may be ionized when bound to MbtA, which might more closely mimic the electrostatics of the native acyl-adenylate and Sal-AMS, which are both negatively charged. Bicyclic sulfonamides 1–4 were also evaluated against other AAAE homologues including BasE, EntE, and VibE from Acinetobacter baumannii, Escherichia coli, and Vibrio cholerae respectively. The native substrate of these AAAE’s is 2,3-dihydroxybenzoic acid, hence we expected 8-hydroxychromone 4 to have enhanced potency toward these enzymes. Surprisingly, 4 was inactive against all three AAAE’s (KD > 300 µM). The relative potency trends of 1–3 toward these other AAAE’s are the same as observed with MbtA. Thus, quinolone 2 displays the highest potency with KD’s generally below the lower limit of the assay (<0.1 to 0.32 µM). Benzoxazinone 3 is the next most potent with KD ranging from 1–12 µM while chromone 1 binds the weakest with KD’s ranging from 98–280 µM, which represents more than a 1000-fold loss of potency relative to quinolone 2.
FP assays are limited in the accuracy of binding constants they provide when an inhibitor’s affinity is greater than that of the probe.35 We were unable to discriminate the potency of quinolone 2 versus Sal-AMS using our FP assay since both compounds bound more tightly than the fluorescent probe Fl-Sal-AMS. In order to address this shortcoming, we used a functional [32P]PPi-ATP steady-state kinetic exchange assay10 employing super-saturating concentrations of substrates to determine apparent Ki values for Sal-AMS and quinolone 2 with respect to MbtA under identical assay conditions. This analysis afforded a Kiapp of 120 nM for 2 against MbtA, which is approximately 18-fold less active than Sal-AMS (Kiapp of 6.6 nM). The compromise in activity of 2 is offset by the substantially improved physicochemical properties of quinolone 2.
Antitubercular Evaluation
All compounds were tested against Mtb under iron-deficient and iron-replete conditions as previously described (Table 2).8c Unexpectedly, compounds 1–4 are inactive with minimum inhibitory concentrations greater than 50 µM, the highest concentration evaluated. The lack of activity of quinolone 2 is disappointing and suggests it may not accumulate at sufficient concentrations intracellularly, despite improved ClogP and tPSA values relative to SalAMS, due to poor penetration or active efflux. Cellular accumulation studies will be required to address this possibility.
Conclusion
We have designed a divergent strategy for the synthesis of chromone-, quinolone-, and benzoxazinone-3-sulfonamides from common β-ketosulfonamide intermediates, which were assembled via a Claisen-like condensation between an appropriate benzoate ester derivative and the dianion of N-Boc-methanesulfonamide. Formylation of the active methylene of the β-ketosulfonamide was found to optimially proceed with dimethylformamide dimethyl acetal at room temperature to afford an enaminone intermediate, which underwent intramolecular cyclization with a phenol to provide the chromone-3-sulfonamides. The quinolone-3-sulfonamide was accessed from a similar enaminone intermediate by transamination with benzylamine followed by intramolecular cyclization via nucleophilic aromatic substitution onto an ortho-fluorophenyl moiety. The versatility of the β-ketosulfonamide intermediate was futher demonstrated by the electrophilic nitrosation of the active methylene to yield an intermediate oxime. Subsequent intramolecular cyclization onto an ortho-fluorophenyl group via nucleophilic aromatic substitution provided the benzoxazinone-3-sulfonamide. Each of the bicyclic-3-sulfonamides was efficiently coupled to a protected adenosine via a Mitsunobu reaction to furnish the desired inhibitors 1–4 following global deprotection. These compounds were designed as conformationally restricted analogues of 5′-O-[N-(salicyl)sulfamoyl]adenosine (Sal-AMS) to improve oral bioavailability by removal of two rotatable bonds and the charged sulfamate moiety. Biochemical studies with MbtA showed that the negative charge of Sal-AMS appears critical to maintain potent activity as chromone 1 and benzoxazine 3 analogues that lack an ionizable function in the heterocycle display substantially reduced potency while quinolone 2, which contains an ionizable NH moiety at N-1 is only 18-fold less active than Sal-AMS toward MbtA as measured in a functional steady-state kinetic assay. This is likely a general phenomenon of adenylating enzymes and has been noted previously.12, 26 We hypothesized that the compromise in activity of 2 would be offset by the substantially improved physicochemical properties of quinolone 2. Unfortunately, quinolone 2 is inactive against Mtb in a whole-cell assay with a minimum inhibitor concentration of greater than 50 µM, which represents more than a 128-fold loss of activity relative to Sal-AMS. Quinolone 2 shows significant biochemical potency, thus the inactivity against Mtb may be due to reduced cellular accumulation. While our primary goal was to remove the ionizable and negatively charged sulfamate group, it may be necessary to lower the pKa of quinolone 2 to ensure this is fully ionized. Additional studies will be required to understand the loss of biological activity, which is critical to further optimize this scaffold.
Experimental Section
General Synthetic Methods
All commercial reagents were used as provided unless otherwise indicated. An anhydrous solvent dispensing system using two packed columns of neutral alumina was used for drying THF and CH2Cl2, while two packed columns of molecular sieves were used to dry DMF, and the solvents were dispensed under Argon. Anhydrous grade MeOH was purchased from Aldrich. All reactions were performed under an inert atmosphere of dry Ar in oven-dried (150 °C) glassware. TLC analyses were performed on TLC silica gel plates and were visualized with UV light. Purification by flash chromatography was performed using a medium-pressure flash chromatograpy system equipped with flash column silica cartridges with the indicated solvent system. Reversed-phase HPLC (RP-HPLC) purification was performed on a Phenomenex Gemini 10 micron C18 250 × 10.00 mm column operating at 5.0 mL/min with detection at 254 nm with the indicated solvent system. 1H, 13C, and 19F NMR spectra were recorded on a 600 MHz spectrometer. Proton chemical shifts are reported in ppm from an internal standard of residual chloroform (7.26), methanol (3.31), dichloromethane (5.32), dimethyl sulfoxide (2.50), or mono-deuterated water (HDO, 4.79); carbon chemical shifts are reported in ppm from an internal standard of residual chloroform (77.16), methanol (49.00), dichloromethane (54.00), or dimethyl sulfoxide (39.52); and fluorine chemical shifts are reported in ppm from an internal standard of 2-fluorobenzoic acid (−112.05).36 Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, ap = apparent, br = broad, ovlp = overlapping), coupling constant(s), integration. High-resolution mass spectra were obtained on an TOF/MS instrument equipped with either an ESI or APCI interface. Compounds 5,19 6,21 7,22 15, 16,23 21,40 29,26 32 were prepared as described.
Experimental for compounds from Table 1
tert-Butyl chlorosulfonylcarbamate (8)
To a stirred solution of tert-butanol (1.9 mL, 20 mmol, 1.3 equiv) in CH2Cl2 (12 mL) at 0 °C was added chlorosulfonyl isocyanate (1.4 mL, 15 mmol, 1.0 equiv) dropwise over the course of 10 min. The reaction mixture was removed from the 0 °C bath after 5 min of additional stirring. After warming to 22 °C, stirring was stopped and the reaction mixture was concentrated in vacuo to one-third volume. The flask was placed back into the 0 °C bath and the product crystallized out of solution. After 50 min, the product was filtered and washed with hexanes yielding the title compound (1.5 g, 46%) as a colorless solid. Additional product (1.2 g, 37%) was obtained by crystallizing the concentrated mother liquor in CH2Cl2 at 0 °C. Mp 60–68 °C; 1H NMR (600 MHz, CD2Cl2) δ 1.56 (s, 9H), 8.50 (s, 1H); 13C NMR (150 MHz, CD2Cl2) δ 28.1, 87.4, 147.8; HRMS (ESI–) calcd for C5H10NO5S− (hydrolysis product) [M – H]− 196.0285, found 196.0274 (error 5.6 ppm).
Chromone-3-sulfonamide (9)
To a stirred solution of sulfamoyl chloride 6 (347 mg, 3.00 mmol, 1.00 equiv) in CH2Cl2 (6 mL) at 0 °C was added enaminone 5 (574 mg, 3.00 mmol, 1.00 equiv) in one portion. After stirring 5 h, the reaction was quenched with addition of saturated aqueous NaHCO3 (50 mL). The layers were separated and the pH of the aqueous layer was adjusted to neutral (~7 by pH paper). The aqueous layer was then extracted with EtOAc (3 × 75 mL). The combined organic layer was dried (MgSO4), concentrated, and chromatographed (20:80 to 40:60 EtOAc–hexanes gradient) yielding chromone 12 (140 mg, 32%; characterization data matched that of authentic commercially obtained sample) and recovered enaminone 5 (86 mg, 15%). MS showed the possibility of product remaining in the aqueous layer (major peak of m/z = 224 in negative mode), so the aqueous layer was further extracted with n-BuOH (3 × 75 mL). The combined n-BuOH layers were dried (MgSO4) and concentrated. The resultant residue was taken up in MeOH–MeCN (20 mL, 1:1) and insoluble solids filtered away. Upon sitting overnight, the product had crystallized to afford the title compound (34 mg, 5%) as off-white crystals: 1H NMR (600 MHz, DMSO-d6) δ 7.34 (s, 2H), 7.60 (t, J = 7.6 Hz, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.90 (td, J = 7.6, 1.5 Hz, 1H), 8.14 (dd, J = 7.6, 1.5 Hz, 1H), 8.97 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 118.9, 123.8, 125.2, 126.7, 127.3, 135.3, 155.7, 158.6, 171.7; HRMS (ESI–) calcd for C9H6NO4S– [M – H]– 224.0016, found 224.0023 (error 3.1 ppm).
tert-Butyl (chromon-3-yl)sulfonylcarbamate (10). Method A – Sulfamoylation of 5
To a solution of 8 (1.5 g, 7.0 mmol, 1.0 equiv) in CH2Cl2 (14 mL) at 22 °C was added enaminone 5 (1.3 g, 7.0 mmol, 1.0 equiv). The reaction was stirred 13 h then concentrated in vacuo. Purification by flash chromatography (30:70 to 100:0 CH2Cl2–hexanes, linear gradient) afforded the title compound (377 mg, 21%) as a yellow amorphous solid: Rf 0.29 (1:5:95 Et3N–MeOH– CH2Cl2); 1H NMR (600 MHz, CDCl3) δ 1.40 (s, 9H), 7.54 (t, J = 7.8 Hz, 1H), 7.59 (d, J = 8.3 Hz, 1H), 7.80 (t, J = 8.3 Hz, 1H), 8.27 (d, J = 7.8 Hz, 1H), 8.82 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 28.0, 84.4, 118.9, 123.6, 124.4, 126.4, 127.1, 135.4, 149.2, 156.3, 162.0, 171.8; HRMS (ESI–) calcd for C14H14NO6S– [M – H]– 324.0547, found 324.0559 (error 3.7 ppm).
Experimental for compounds from Scheme 4
Methyl 2-(tert-butyldimethylsilyloxy)benzoate (17)
TBSCl (3.01 g, 20.0 mmol, 3.00 equiv) was added to a solution of 15 (862 µL, 6.65 mmol, 1.00 equiv), imidazole (1.81 g, 26.6 mmol, 4.00 equiv), and DMAP (8 mg, 0.07 mmol, 0.01 equiv) in DMF (10 mL) at 0 °C. After stirring 1 h, the homogeneous solution had become a thick suspension. The ice bath was removed and the reaction stirred a further 24 h at room temperature. A 5% aqueous NaHCO3 (100 mL) was added to the reaction mixture and the resulting aqueous solution was extracted with Et2O (3 × 100 mL). The combined organic layers were dried (MgSO4) and concentrated yielding a colorless oil which was purified by flash chromatography (1:99 to 10:90 Et2O–hexanes) to afford the title compound (1.41 g, 79%) as a colorless oil: Rf 0.19 (1:99 EtOAc–hexanes); 1H NMR (600 MHz, CDCl3) δ 0.21 (s, 6H), 1.01 (s, 9H), 3.86 (s, 3H), 6.87 (dd, J = 8.2, 1.2 Hz, 1H), 6.98 (td, J = 7.6, 1.2 Hz, 1H), 7.35 (td, J = 8.2, 1.8 Hz, 1H), 7.75 (dd, J = 7.6, 1.8 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ −4.2, 18.4, 25.8, 52.0, 121.0, 121.3, 123.0, 131.7, 133.1, 155.2, 167.5; HRMS (ESI+) calcd for C14H23O3Si+ [M + H]+ 267.1411, found 267.1424 (error 4.9 ppm).
tert-Butyl [2-(2-hydroxyphenyl)-2-oxoethyl]sulfonylcarbamate (19)
Freshly titrated n-BuLi (2.1 M in hexane, 5.0 mL, 11 mmol, 3.1 equiv) was added dropwise to freshly distilled (i-Pr)2NH (1.6 mL, 11 mmol, 3.3 equiv) in THF (10 mL) at 0 °C. The mixture was stirred for 30 min, then sulfonamide 16 (666 mg, 3.41 mmol, 1.00 equiv) in THF (10 mL) was added and the reaction was stirred for a further 1 h at 0 °C. Next, methyl ester 17 (1.00 g, 3.75 mmol, 1.10 equiv) in THF (2 mL) was added and the reaction was stirred for 3 d at 0 °C. The reaction mixture was quenched with saturated aqueous NaCl (10 mL) and 0.5 M aqueous NaH2PO4 (10 mL), then diluted with EtOAc (10 mL). The layers were separated and the aqueous layer acidified to pH ~5–6 (pH paper) with 6 N aqueous HCl. The aqueous layer was extracted with EtOAc (2 × 100 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure yielding a golden oily residue (2.19 g). The residue was dissolved in THF (10 mL) and cooled to 0 °C. TBAF (1.0 M in THF, 8.2 mL, 8.2 mmol, 2.4 equiv) was added and the solution stirred at 0 °C for 17 h. The reaction mixture was quenched with satdurated aqueous NH4Cl (25 mL) and was diluted with Et2O (25 mL). The layers were separated and the aqueous layer acidified to pH ~5–6 (pH paper) with 6 N aqueous HCl. The aqueous layer was then extracted with Et2O (25 mL) and EtOAc (50 mL). The organic layers were combined, dried (MgSO4), and concentrated under reduced pressure yielding a golden oil (2.40 g). Purification by flash chromatography (0.2:0.2:19.6:80 HCO2H–MeOH–EtOAc–hexanes) afforded the title compound (930 mg, 86%) as an off-white solid: Rf 0.83 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, CDCl3) δ 1.53 (s, 9H), 4.99 (s, 2H), 6.98 (t, J = 8.2 Hz, 1H), 7.03 (d, J = 8.2 Hz, 1H), 7.57 (t, J = 8.2 Hz, 1H), 7.76 (d, J = 8.2 Hz, 1H), 11.67 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 28.1, 57.9, 85.3, 119.0, 119.1, 119.9, 131.1, 138.4, 149.5, 163.5, 193.2; HRMS (APCI– ) calcd for C13H16NO6S− [M – H]− 314.0704, found 314.0732 (error 8.9 ppm).
tert-Butyl (chromon-3-yl)sulfonylcarbamate (10). Method B – Formylation–cyclization of 19
N,N-Dimethylformamide dimethylacetal (638 µL, 4.80 mmol, 2.40 equiv) was added to a solution of β-ketosulfonamide 19 (631 mg, 2.00 mmol, 1.00 equiv) in THF (20 mL) at 22 °C. The solution was stirred for 18 h, then was acidified with satdurated aqueous NH4Cl (25 mL) and diluted with EtOAc (25 mL). The layers were separated and the aqueous layer acidified to pH ~2−3 (pH paper) with aqueous 6 N HCl. The aqueous layer was then extracted with EtOAc (2 × 25 mL). The organic layers were combined, dried (MgSO4), and concentrated under reduced pressure yielding a golden foamy residue. Purification by flash chromatography (0.2:0.2:19.6:80 HCO2H–MeOH–EtOAc–hexanes) afforded the title compound (484 mg, 74%) as an off-white amorphous solid: Characterization data matched that as given above for Method A.
Methyl 2,3-bis(tert-butyldimethylsilyloxy)benzoate (22)
To a solution of methyl 2,3-dihydroxybenzoate 21 (2.95 g, 17.5 mmol, 1.00 equiv) in DMF (35 mL) was added imidazole (6.20 g, 91.1 mmol, 5.20 equiv) and TBSCl (7.92 g, 52.6 mmol, 3.00 equiv). The solution was heated at 65 °C. After 19 h, the solution was cooled to room temperature and diluted with 5% aqueous NaHCO3 (100 mL), extracted with hexanes (3 × 100 mL), dried (MgSO4) and concentrated yielding a colorless oil (8.77 g). Purification by flash chromatography (2:98 EtOAc–hexanes) afforded the title compound (6.93 g, 99%) as a colorless oil: Rf 0.37 (0.025:0.025:2.45:97.5 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, DMSO-d6) δ 0.03 (s, 6H), 0.22 (s, 6H), 0.94 (s, 9H), 0.95 (s, 9H), 3.76 (s, 3H), 6.93 (t, J = 7.9 Hz, 1H), 7.07 (d, J = 8.2 Hz, 1H), 7.19 (d, J = 7.6 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ −4.4, −3.9, 17.9, 18.3, 25.7, 25.9, 51.8, 121.4, 122.9, 123.9, 125.3, 145.1, 147.6, 166.7; HRMS (ESI+) calcd for C20H37O4Si2+ [M + H]+ 397. 2225, found 397.2226 (error 0.3 ppm)
tert-Butyl [2-(2,3-dihydroxyphenyl)-2-oxoethyl]sulfonylcarbamate (24)
Freshly titrated n-BuLi (2.1 M in hexane, 11.0 mL, 23.2 mmol, 3.10 equiv) was added dropwise to freshly distilled (i-Pr)2NH (3.5 mL, 24.8 mmol, 3.30 equiv) in THF (24 mL) at 0 °C. The mixture was stirred for 1.5 h, then 16 (1.46 g, 7.50 mmol, 1.00 equiv) in THF (24 mL) was added and the reaction stirred for a further 1.5 h at 0 °C. Next, methyl ester 22 (3.25 g, 8.20 mmol, 1.10 equiv) was added and the reaction was stirred for 1.5 h at 0 °C. The reaction mixture was quenched with saturated aqueous NaCl (25 mL) and 0.5 M aqueous NaH2PO4 (25 mL) and was diluted with EtOAc (25 mL). The layers were separated and the aqueous layer acidified to pH ~3–4 (pH paper) with aqueous 6 N HCl. The aqueous layer was then extracted with EtOAc (3 × 100 mL). The organic layers were combined, dried (MgSO4), and concentrated under reduced pressure yielding a golden foamy residue (4.73 g). The residue was dissolved in THF (24 mL) and cooled to 0 °C. TBAF (1.0 M in THF, 18.0 mL, 18.0 mmol, 2.40 equiv) was added and the solution stirred at 0 °C for 1 h. The reaction mixture was quenched with saturated aqueous NH4Cl (25 mL). The layers were separated and the aqueous layer acidified to pH ~3–4 (pH paper) with aqueous 6 N HCl. The aqueous was then extracted with EtOAc (3 × 40 mL). The organic layers were combined, dried (MgSO4), and concentrated under reduced pressure yielding a dark oily residue (7.25 g). Purification by flash chromatography (0.3:0.3:29.4:70 HCO2H–MeOH–EtOAc– hexanes) afforded the title compound (2.05 g, 82%) as a yellow solid: Rf 0.69 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, DMSO-d6) δ 1.43 (s, 9H), 5.16 (s, 2H), 6.79 (t, J = 7.9 Hz, 1H), 7.09 (d, J = 7.6 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 9.69 (br s, 1H), 10.70–11.90 (br s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 27.8, 59.7, 82.5, 119.1, 121.5, 121.6, 121.7, 146.3, 150.0, 150.7, 193.0; HRMS (ESI–) calcd for C13H16NO7S– [M – H]– 330.0653, found 330.0662 (error 2.7 ppm).
tert-Butyl (8-hydroxychromon-3-yl)sulfonylcarbamate (27)
N,N-Dimethylformamide dimethyl acetal (638 µL, 4.80 mmol, 2.40 equiv) was added to a solution of β-ketosulfonamide 24 (663 mg, 2.00 mmol, 1.00 equiv) in THF (20 mL) at 22 °C. The solution was stirred for 18 h, then was acidified with saturated aqueous NH4Cl (25 mL) and diluted with EtOAc (25 mL). The layers were separated and the aqueous layer acidified to pH ~5–6 (pH paper) with aqueous 6 N HCl. The aqueous layer was then extracted with EtOAc (2 × 25 mL). The organic layers were combined, dried (MgSO4), and concentrated under reduced pressure yielding a golden foamy residue. Purification by flash chromatography (0.2:0.2:19.6:80 HCO2H–MeOH–EtOAc– hexanes) afforded the title compound (536 mg, 79%) as an off-white amorphous solid: Rf 0.48 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, DMSO-d6) δ 1.29 (s, 9H), 7.35 (dd, J = 7.6, 1.2 Hz, 1H), 7.39 (t, J = 7.6 Hz, 1H), 7.52 (dd, J = 7.6, 1.2 Hz, 1H), 9.07 (s, 1H), 10.91 (s, 1H), 11.94 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 27.5, 82.2, 114.3, 120.6, 122.8, 125.0, 127.0, 145.1, 147.2, 149.7 162.6, 170.7; HRMS (ESI–) calcd for C14H14NO7S− [M – H]− 340.0496, found 340.0529 (error 9.7 ppm).
tert-Butyl [8-(methoxymethoxy)chromon-3-yl]sulfonylcarbamate (28)
To a solution of 27 (465 mg, 1.36 mmol, 1.00 equiv) and DIPEA (0.30 mL, 1.7 mmol, 1.3 equiv) in DMF (10 mL) at 0 °C was added MOMCl (124 µL, 1.63 mmol, 1.20 equiv). The reaction was stirred 25 h then diluted with CH2Cl2 (100 mL). The solution was washed with H2O (2 × 100 mL) and saturated aqueous NaCl (100 mL). The organic layer was dried (MgSO4) and concentrated yielding a yellow-orange solid (480 mg). Purification by flash chromatography (0.3:0.3:29.4:70 HCO2H– MeOH–EtOAc–hexanes) afforded the title compound (310 mg, 58%) as an off-white amorphous solid: Rf 0.51 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, DMSO-d6) δ 1.30 (s, 9H), 3.35 (s, 3H), 5.22 (s, 2H), 7.37 (dd, J = 7.6, 1.2 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 7.50 (dd, J = 7.6, 1.2 Hz, 1H), 9.21 (s, 1H), 10.99 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 27.3, 56.1, 78.8, 84.6, 114.2, 120.8, 123.2, 124.8, 127.2, 145.2, 147.3, 150.0, 162.8, 171.0; HRMS (ESI–) calcd for C16H18NO8S– [M – H]– 384.0759, found 384.0790 (error 8.1 ppm).
Experimental for compounds from Scheme 5
N6,N6-bis(tert-Butoxycarbonyl)-5′-amino-5′-N-(tert-butoxycarbonyl)-5′-N-[(chromon-3-yl)sulfonyl]-5′-deoxy-2′,3′-O-isopropylideneadenosine (30)
To a stirred solution of 10 (270 mg, 0.83 mmol, 1.1 equiv), 29 (381 mg, 0.75 mmol, 1.0 equiv) and PPh3 (218 mg, 0.83 mmol, 1.1 equiv) in THF (20 mL) at 0 °C was added DIAD (0.16 mL, 0.83 mmol, 1.1 equiv) dropwise over the course of 10 min. The reaction was stirred 2.5 h at 0 °C and was then allowed to warm to 22 °C. The reaction was stirred another 1.5 h then concentrated in vacuo. Purification by flash chromatography (40:60 EtOAc–hexanes) afforded the title compound (491 mg, 80%) as a colorless oil: Rf 0.63 (3:1 EtOAc–hexanes); 1H NMR (600 MHz, CDCl3) δ 1.25 (s, 3H), 1.34 (s, 9H), 1.41 (s, 18H), 1.65 (s, 3H), 4.25 (dd, J = 15.3, 6.5 Hz, 1H), 4.36 (dd, J = 15.3, 6.5 Hz, 1H), 4.67 (td, J = 6.5, 3.5 Hz, 1H), 5.21 (dd, J = 6.2, 3.5 Hz, 1H), 5.42 (dd, J = 6.2, 2.3 Hz, 1H), 6.23 (d, J = 2.3 Hz, 1H), 7.51 (t, J = 7.9 Hz, 1H), 7.56 (d, J = 8.5 Hz, 1H), 7.77 (td, J = 8.5, 1.5 Hz, 1H), 8.19 (dd, J = 7.9, 1.5 Hz, 1H), 8.28 (s, 1H), 8.69 (s, 1H), 8.91 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 25.6, 27.4, 27.9, 28.0, 49.5, 82.5, 83.8, 84.6, 85.2, 85.5, 90.8, 114.9, 118.7, 124.4, 124.8, 125.4, 126.3, 127.1, 129.1, 135.2, 144.1, 150.4, 150.6, 152.4, 152.7, 156.0, 161.8, 171.3; HRMS (ESI+) calcd for C37H47N6O13S+ [M + H]+ 815.2916, found 815.2926 (error 1.2 ppm).
5′-Amino-5′-N-[(chromon-3-yl)sulfonyl]-5′-deoxy-adenosine (1)
To solid 30 (474 mg, 0.582 mmol) at 0 °C was added ice-cold 80% aqueous TFA (5 mL). The reaction was stirred 1.5 h at 0 °C, then warmed to 22 °C and stirred an additional 3 h. The reaction was concentrated in vacuo and subsequent purification by flash chromatography (10:90 MeOH–CHCl3) afforded the title compound (210 mg, 76%) as a colorless amorphous solid: Rf 0.33 (1:9 MeOH–CHCl3); 1H NMR (600 MHz, 1:10 D2O–DMSO-d6) δ 3.20 (dd, J = 14.1, 3.5 Hz, 1H), 3.27 (dd, J = 14.1, 4.7 Hz, 1H), 4.04–4.06 (m, 1H), 4.07 (dd, J = 5.0, 1.6 Hz, 1H), 4.68 (td, J = 6.4, 1.6 Hz, 1H), 5.74 (d, J = 6.4 Hz, 1H), 7.53 (t, J = 7.6 Hz, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.84 (td, J = 7.6, 1.2 Hz, 1H), 8.05 (dd, J = 7.6, 1.2 Hz, 1H), 8.22 (s, 1H), 8.26 (s, 1H), 8.83 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 45.0, 71.3, 72.3, 84.0, 88.3, 118.7, 119.6, 123.7, 124.3, 125.2, 126.5, 135.1, 140.5, 148.6, 152.5, 155.6, 156.2, 160.3, 171.6; HRMS (ESI+) calcd for C19H19N6O7S+ [M + H]+ 475.1030, found 475.1025 (error 1.1 ppm).
N6,N6-bis(tert-Butoxycarbonyl)-5′-amino-5′-N-(tert-butoxycarbonyl)-5′-deoxy-5′-N-{[(8-(methoxymethoxy)chromon-3-yl]sulfonyl}-2′,3′-O-isopropylideneadenosine (31)
To a solution of 28 (17 mg, 0.044 mmol, 1.1 equiv), 29 (20 mg, 0.040 mmol, 1.0 equiv), and PPh3 (12 mg, 0.044 mmol, 1.1 equiv) in THF (2 mL) at 22 °C was added DIAD (0.44 mL [100 mM in THF], 0.044 mmol, 1.1 equiv) dropwise. After 2 h stirring, TLC monitoring of the reaction suggested the limiting reagent 29 remained; therefore additional PPh3 (12 mg, 0.044 mmol, 1.1 equiv) and DIAD (0.44 mL [100 mM in THF], 0.044 mmol, 1.1 equiv) were added. After another 2.5 h stirring, the reaction appeared complete by TLC. The reaction mixture was concentrated to an off-white residue. Purification by flash chromatography (40:60 EtOAc– hexanes) afforded the title compound (36 mg, 45%) as an off-white amorphous solid: Rf 0.40 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, CDCl3) δ 1.26 (s, 3H), 1.41 (s, 9H), 1.48 (s, 18H), 1.68 (s, 3H), 3.51 (s, 3H), 4.33 (dd, J = 9.0, 5.2 Hz, 1H), 4.47 (dd, J = 9.0, 5.2 Hz, 1H), 4.77 (dd, J = 9.0, 3.6 Hz, 1H), 5.26 (dd, J = 5.8, 3.6 Hz, 1H), 5.38 (s, 2H), 5.45 (dd, J = 5.8, 2.0 Hz, 1H), 6.32 (d, J = 2.0 Hz, 1H), 7.19 (d, J = 8.2 Hz, 1H), 7.37 (t, J = 8.2 Hz, 1H), 7.77 (d, J = 8.2 Hz, 1H), 8.37 (br s, 1H), 8.77 (s, 1H), 8.87 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 25.6, 27.4, 28.0, 28.1, 53.2, 57.0, 77.5, 78.2, 79.9, 81.7, 84.2, 85.2, 91.0, 115.2, 117.4, 117.8, 124.8, 125.8, 126.9, 130.9, 134.9, 146.5, 147.8, 150.6, 150.7, 151.0, 152.4, 152.5, 161.6, 171.4; HRMS (ESI+) calcd for C39H51N6O15S+ [M + H]+ 875.3128, found 875.3136 (error 0.9 ppm).
5′-Amino-5′-deoxy-5′-N-[(8-hydroxychromon-3-yl)sulfonyl]adenosine (4)
To solid 31 (24 mg, 0.027 mmol) at 0 °C was added ice-cold 80% aqueous TFA (2.5 mL). The reaction was stirred 18 h at 0 °C. The reaction was concentrated in vacuo and subsequent purification by flash chromatography (1:1:98 to 1:4:95 HCO2H–MeOH–EtOAc, linear gradient) afforded the title compound (8.9 mg, 66%) as a colorless amorphous solid. Further purification by RP-HPLC (12.5:87.5 MeCN–H2O) and lyophilization of the appropriate fractions afforded the title compound (7.7 mg) as a fluffy colorless solid: Rf 0.22 (1:20:79 HCO2H–MeOH–EtOAc); 1H NMR (600 MHz, DMSO-d6) δ 4.33 (td, J = 5.3, 2.9 Hz, 1H), 4.41 (q, J = 5.3 Hz, 1H), 4.45 (dd, J = 11.2, 5.3 Hz, 1H), 4.49 (dd, J = 11.2, 2.9 Hz, 1H), 4.75 (q, J = 5.3 Hz, 1H), 5.44 (d, J = 5.3 Hz, 1H, D2O-exchangeable), 5.63 (d, J = 5.3 Hz, 1H, D2O-exchangeable), 6.01 (d, J = 5.3 Hz, 1H), 7.27 (br s, 2H, D2O-exchangeable), 7.38 (br s, 2H, D2O-exchangeable), 7.49 (t, J = 7.6 Hz, 1H), 7.59 (d, J = 7.6 Hz, 1H), 7.67 (d, J = 7.6 Hz, 1H), 8.15 (s, 1H), 8.38 (s, 1H), 8.97 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 69.2, 70.4, 73.3, 82.0, 87.5, 115.9, 117.4, 124.9, 126.5, 127.4, 132.7, 145.9, 147.7, 149.5, 152.7, 156.0, 158.0, 171.2, 171.6; HRMS (ESI+) calcd for C19H19N6O8S+ [M + H]+ 491.0980, found 491.0995 (error 3.1 ppm).
Experimental for compounds from Scheme 7
tert-Butyl [2-(2-fluorophenyl)-2-oxoethyl]sulfonylcarbamate (33)
Freshly titrated n-BuLi (2.1 M in hexane, 12.4 mL, 26.1 mmol, 3.10 equiv) was added dropwise to freshly distilled (i-Pr)2NH (3.9 mL, 27.8 mmol, 3.30 equiv) in THF (24 mL) at 0 °C. The mixture was stirred for 1 h, then 16 (1.65 g, 8.43 mmol, 1.00 equiv) in THF (24 mL) was added and the reaction stirred for a further 1 h at 0 °C. Next, methyl ester 32 (1.43 g, 9.28 mmol, 1.10 equiv) in THF (5 mL) was added and the reaction was stirred for 1.5 h at 0 °C. The reaction mixture was quenched with saturated aqueous NaCl (25 mL) and 0.5 M aqueous NaH2PO4 (25 mL). The layers were separated and the aqueous layer acidified to pH ~5–6 (pH paper) with aqueous 6 N HCl. The aqueous layer was then extracted with EtOAc (3 × 75 mL). The organic layers were combined, dried (MgSO4), and concentrated under reduced pressure. Purification by flash chromatography (0.2:0.2:19.6:80 HCO2H–MeOH–EtOAc–hexanes) afforded the title compound (1.92 g, 72%) as a yellow solid: Rf 0.74 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, CDCl3) δ 1.50 (s, 9H), 5.03 (s, 2H), 7.18–7.21 (m, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.62–7.64 (m, 1H), 7.91 (t, J = 7.6 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 28.1, 61.6 (d, 2JC–F = 9.2 Hz), 85.0, 117.2 (d, 2JC–F = 23.1 Hz), 124.3, 125.1 (d, 3JC–F = 3.5 Hz), 131.2, 136.6 (d, 3JC–F = 9.2 Hz), 149.6, 162.2 (d, 1JC–F = 256.6 Hz), 186.2 (d, 3JC–F = 3.5 Hz); 19F NMR (564 MHz, CDCl3) δ −112.7; HRMS (ESI−) calcd for C13H15FNO5S− [M – H]− 316.0660, found 316.0654 (error 1.9 ppm).
tert-Butyl [3-(2-fluorophenyl)-1-((benzylamino)-3-oxoprop-1-en-2-yl]sulfonylcarbamate (35)
Dimethylformamide dimethyl acetal (399 µL, 3.00 mmol, 1.50 equiv) was added to a solution of β-ketosulfonamide 33 (635 mg, 2.00 mmol, 1.00 equiv) in THF (10 mL) at 22 °C. The solution was stirred for 1.5 h, then was concentrated under reduced pressure yielding enaminone 34 as a yellow oily residue used directly without purification: Rf 0.25 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); LRMS (ESI−) calcd for C16H20FN2O5S− [M – H]− 371, found 371.
To a stirred solution of enaminone 34 prepared above in THF (6 mL) at 22 °C was added BnNH2 (328 µL, 3.00 mmol, 1.50 equiv). After stirring 10 min the reaction was concentrated under reduced pressure yielding a golden foamy oil. Purification by flash chromatography (0.2:0.2:19.6:80 HCO2H–MeOH–EtOAc-hexanes) afforded a mixture of isomers (~2:1) of the title compound (739 mg, 85% over two steps) as a golden foamy oil: Rf 0.13 (0.2:0.2:19.6:80 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, CDCl3) δ 1.46 (s, 3H, minor), 1.47 (s, 6H, major), 4.47 (d, J = 5.3 Hz, 0.67H, minor), 4.66 (d, J = 5.9 Hz, 1.33H, major), 6.64–6.67 (m, 0.67H, major), 7.10–7.25 (ovlp m, 1H), 7.14–7.19 (ovlp m, 1H), 7.24 (d, J = 7.0 Hz, 0.67H, major), 7.28 (t, J = 7.0 Hz, 0.67H, major), 7.31–7.43 (ovlp m, 5H), 7.62–7.64 (m, 0.33H, minor), 8.31 (d, J = 14.1 Hz, 0.67H, major); 8.70 (br s, 0.33H, minor), 11.03 (br s, 0.67H, major); 13C NMR (150 MHz, CDCl3) δ 28.08, 28.14, 54.0, 54.7, 83.4, 83.6, 108.4, 115.5, 115.6, 116.1, 116.3, 123.77, 123.80, 124.68, 124.70, 127.7, 127.8, 128.51, 128.54, 128.64, 128.65, 128.77, 128.80, 129.2, 129.3, 129.85, 129.88, 131.2, 131.3, 132.15, 132.20, 135.0, 135.2, 149.5, 150.3, 157.5, 157.9, 158.25, 158.26, 159.2, 159.6, 161.9, 185.2, 188.8; 19F NMR (564 MHz, CDCl3) δ − 120.1 (major), −117.4 (minor); HRMS (APCI−) calcd for C21H22FN2O5S− [M – H]− 433.1239, found 433.1265 (error 6.0 ppm).
tert-Butyl (1-benzylquinol-4-on-3-yl)sulfonylcarbamate (36)
To a stirred solution of enaminone 35 (434 mg, 1.00 mmol, 1.00 equiv) in THF (4 mL) at 22 °C was added NaH (60% dispersion in mineral oil, 88 mg, 2.2 mmol, 2.2 equiv) portionwise (~10 mg portions) such that noticeable gas evolution had ceased before next addition. After stirring 1 h, the reaction was quenched by addition of saturated aqueous NH4Cl (25 mL). The aqueous mixture was acidified to pH ~4 (pH paper) with 6 N aqueous HCl and extracted with EtOAc (3 × 25 mL). The organic layers were combined, dried (MgSO4), and concentrated under reduced pressure yielding an off-white solid. Purification by flash chromatography (0.4:0.4:39.2:60 to 1:10:89:0 HCO2H–MeOH– EtOAc–hexanes, linear gradient) afforded the title compound (336 mg, 81%) as an amorphous off-white solid: Rf 0.44 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, DMSO-d6) δ 1.29 (s, 9H), 5.80 (s, 2H), 7.25–7.35 (ovlp m, 5H), 7.48–7.50 (m, 1H), 7.74–7.76 (m, 2H), 8.26 (d, J = 7.6 Hz, 1H), 8.99 (s, 1H), 11.60 (br s, 1H, D2O-exchangeable); 13C NMR (150 MHz, 1:9 D2O–DMSO-d6) δ 27.9, 56.2, 82.2, 118.6, 126.2, 126.4, 126.8, 126.9, 128.0, 128.5, 129.2, 129.3, 133.9, 135.9, 139.5, 150.3, 171.3; HRMS (ESI−) calcd for C21H21N2O5S− [M – H]− 413.1177, found 413.1174 (error 0.7 ppm).
N6, N6-bis(tert-Butoxycarbonyl)-5’-amino-5’-N-(tert-butoxycarbonyl)-5’-N-[(1-benzylquinol-4-on-3-yl)sulfonyl]-5’-deoxy-2’,3’-O-isopropylideneadenosine (37)
To a stirred solution of quinolone 36 (228 mg, 0.550 mmol, 1.10 equiv), 29 (254 mg, 0.500 mmol, 1.00 equiv), and PPh3 (144 mg, 0.550 mmol, 1.10 equiv) in THF (50 mL) at 22 °C was added DIAD (108 µL, 0.550 mmol, 1.10) dropwise over 5 min. After stirring 1 h, the reaction was concentrated in vacuo yielding an off-white foamy residue (816 mg). Purification by flash chromatography (40:60 EtOAc–hexanes) afforded the title compound (385 mg, 85%) as an off-white foamy residue: Rf 0.27 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, CDCl3) δ 1.32 (s, 9H), 1.41 (s, 18H), 1.47 (s, 3H), 1.66 (s, 3H), 4.34 (dd, J = 15.0, 6.2 Hz, 1H), 4.46 (dd, J = 15.0, 6.2 Hz, 1H), 4.67 (td, J = 6.2, 3.5 Hz, 1H), 5.21 (dd, J = 6.5, 3.5 Hz, 1H), 5.36 (dd, J = 6.5, 2.6 Hz, 1H), 5.43 (s, 2H), 6.25 (d, J = 2.6 Hz, 1H), 7.19 (d, J = 7.0 Hz, 2H), 7.32–7.35 (ovlp m, 3H), 7.40 (d, J = 8.2 Hz, 1H), 7.45 (t, J = 8.2 Hz, 1H), 7.61 (t, J = 7.0 Hz, 1H), 8.36 (s, 1H), 8.42 (d, J = 8.2 Hz, 1H), 8.61 (s, 1H), 8.92 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 25.7, 27.5, 27.9, 28.0, 49.5, 58.0, 82.4, 83.8, 84.3, 84.6, 85.2, 90.6, 115.0, 117.2, 118.6, 126.0, 126.3, 127.5, 128.5, 129.0, 129.5, 129.6, 133.6, 133.8, 139.4, 143.9, 149.4, 150.4, 150.6, 151.1, 152.4, 152.8, 172.2; HRMS (ESI+) calcd for C44H54N7O12S+ [M + H]+ 904.3546, found 904.3537 (error 1.0 ppm).
N6, N6-bis(tert-Butoxycarbonyl)-5’-amino-5’-N-(tert-butoxycarbonyl)-5’-deoxy-5’-N-[(quinol-4-on-3-yl)sulfonyl]-2’,3’-O-isopropylideneadenosine (38)
To a Parr flask flushed with Ar was added Pd/C (10% by weight, 436 mg, 0.410 mmol, 1.00 equiv), a solution of 37 (371 mg, 0.410 mmol, 1.00 equiv) in anhydrous MeOH (10 mL), and AcOH (23 µL, 0.41 mmol, 1.0 equiv) respectively. The reaction vessel was evacuated, then backfilled with H2 to 40 psi, and the mixture was shaken at 22 °C for 4 h. The reaction mixture was filtered through celite and concentrated to an off-white amorphous solid (454 mg). Purification by flash chromatography (65:35 EtOAc–hexanes) afforded the title compound (195 mg, 58%) as an off-white amorphous solid: Rf 0.54 (0.75:0.75:73.5:25 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, DMSO-d6) δ 1.12 (s, 9H), 1.35 (s, 18H), 1.37 (s, 3H), 1.57 (s, 3H), 4.09 (dd, J = 15.0, 7.6 Hz, 1H), 4.14 (dd, J = 15.0, 6.8 Hz, 1H), 4.48 (td, J = 7.2, 3.4 Hz, 1H), 5.24 (dd, J = 6.2, 3.4 Hz, 1H), 5.60 (dd, J = 6.2, 1.6 Hz, 1H), 6.40 (d, J = 1.6 Hz, 1H), 7.47 (t, J = 8.2 Hz, 1H), 7.71 (d, J = 8.2 Hz, 1H), 7.77 (t, J = 8.2 Hz, 1H), 8.12 (d, J = 8.2 Hz, 1H), 8.30 (s, 1H), 8.58 (d, J = 6.2 Hz, 1H, D2O-exchangeable [collapses to singlet]), 8.81 (s, 1H), 8.88 (s, 1H), 12.79 (d, J = 6.2 Hz, 1H, D2O-exchangeable); 13C NMR (150 MHz, 1:10 D2O–DMSO-d6) δ 25.5, 27.2, 27.6, 49.4, 79.3, 82.2, 83.8, 83.90, 83.93, 86.3, 89.5, 113.9, 117.8, 119.5, 125.5, 126.1, 126.5, 128.6, 133.9, 139.2, 144.8, 146.0, 149.6, 150.3, 150.8, 152.0, 152.8, 171.8; HRMS (ESI+) calcd for C37H48N7O12S+ [M + H]+ 814.3076, found 814.3071 (error 0.6 ppm).
5′-Amino-5′-deoxy-5′-N-[(quinol-4-on-3-yl)sulfonyl]adenosine (2)
To solid 38 (97 mg, 0.12 mmol, 1.0 equiv) at 0 °C was added ice-cold 80% aqueous TFA (2 mL). The reaction was stirred 19 h at 0 °C. The reaction was concentrated in vacuo and subsequent purification by flash chromatography (1:10:98 HCO2H–MeOH–EtOAc) afforded the title compound (40 mg, 70%) as a colorless amorphous solid. Further purification of a portion (4.3 mg) by RP-HPLC (12.5:87.5 MeCN–H2O) and lyophilization of the appropriate fractions afforded the title compound (2 mg) as a fluffy colorless solid: Rf 0.07 (1:10:89 HCO2H–MeOH–EtOAc); 1H NMR (600 MHz, 1:10 D2O–DMSO-d6) δ 3.04 (dd, J = 13.8, 4.1 Hz, 1H), 3.10 (dd, J = 13.8, 4.1 Hz, 1H), 4.04–4.05 (m, 1H), 4.10–4.11 (m, 1H), 4.72 (t, J = 5.5 Hz, 1H), 5.28 (br s, 1H, D2O-exchangeable), 5.44 (br s, 1H, D2O-exchangeable), 5.79 (d, J = 6.4 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 7.62 (br s, 2H, D2O-exchangeable), 7.64 (d, J = 7.8 Hz, 1H), 7.76 (t, J = 7.6 1H), 8.13 (d, J = 7.6 Hz, 1H), 8.30 (s, 1H), 8.46 (s, 1H), 12.55 (d, J = 6.5 Hz, 1H, D2O-exchangeable); 13C NMR (150 MHz, DMSO-d6) δ 44.9, 71.3, 72.2, 84.0, 88.4, 118.2, 119.1, 119.6, 124.9, 125.2, 126.3, 132.9, 139.6, 140.6, 142.4, 148.8, 152.6, 156.2, 171.9; HRMS (ESI+) calcd for C19H20N7O6S+ [M + H]+ 474.1190, found 474.1181 (error 1.9 ppm).
Experimental for compounds from Scheme 8
tert-Butyl [2-(2-fluorophenyl)-1-(hydroxyimino)-2-oxoethyl]sulfonylcarbamate (39)
To a solution of β-ketosulfonamide 33 (635 mg, 2.00 mmol, 1.00 equiv) in AcOH–H2O–THF (1:1:2, 10 mL) at 0 °C was added NaNO2 (276 mg, 4.00 mmol, 2.00 equiv). After stirring 19 h, the reaction mixture was diluted with H2O (50 mL) and extracted with EtOAc (3 × 50 mL). The combined organic layer was dried (MgSO4) and concentrated, yielding a light yellow oil. Purification by flash chromatography (0.2:0.2:19.6:80 HCO2H–MeOH–EtOAc–hexanes) afforded the title compound (513 mg, 74%) as an off-white foamy residue: Rf 0.40 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, CD3OD) δ 1.48 (s, 9H), 7.25 (dd, J = 10.6, 8.8 Hz, 1H), 7.36 (t, J = 7.6 Hz, 1H), 7.71 (dddd, J = 8.8, 7.0, 5.3, 1.8 Hz, 1H), 7.92 (td, J = 7.6, 1.8 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 28.2, 84.6, 118.0 (d, 2JC–F = 22.0 Hz), 124.7 (d, 2JC–F = 8.1 Hz), 125.9 (d, 3JC–F = 3.5 Hz), 132.7, 138.1 (d, 3JC–F = 9.2 Hz), 151.5, 155.3, 163.5 (d, 1JC–F = 261.3 Hz), 184.9; 19F NMR (564 MHz, CDCl3) δ −115.3; HRMS (ESI−) calcd for C13H14FN2O6S− [M – H]− 345.0562, found 345.0569 (error 2.0 ppm).
tert-Butyl (4-oxo-4H-benzo[e][1,2]oxazin-3-yl)sulfonylcarbamate (40)
To a solution of oxime 39 (228 mg, 0.657 mmol, 1.00 equiv) in DMF (3.5 mL) at 22 °C was added Cs2CO3 (471 mg, 1.45 mmol, 2.20 equiv). After stirring 4.5 h the reaction mixture was quenched with saturated aqueous NH4Cl (20 mL). The aqueous solution was acidified to pH ~4–5 (pH paper) with 6 N aqueous HCl and extracted with EtOAc (3 × 30 mL). The organic layers were combined, dried (MgSO4), and concentrated under reduced pressure yielding a pale yellow residue. Purification by flash chromatography (0.2:0.2:19.6:80 HCO2H–MeOH–EtOAc– hexanes) afforded the title compound (124 mg, 58%) as a pale yellow residue: Rf 0.57 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, CDCl3) δ 1.42 (s, 9H), 7.54 (t, J = 7.6 Hz, 1H), 7.62 (d, J = 8.2 Hz, 1H), 7.90 (t, J = 7.7 Hz, 1H), 8.15 (d, J = 7.9 Hz, 1H), 8.58 (br s, 1H); 13C NMR (150 MHz, CDCl3) δ 27.8, 85.3, 116.9, 121.2, 125.4, 127.5, 137.3, 149.1, 156.2, 162.0, 163.6; HRMS (ESI−) calcd for C13H13N2O6S− [M – H]− 325.0500, found 325.0507 (error 2.2 ppm).
N6,N6-bis(tert-Butoxycarbonyl)-5′-amino-5′-N-(tert-butoxycarbonyl)-5′-deoxy-5′-N-[(4-oxo-4H-benzo[e][1,2]oxazin-3-yl)sulfonyl]-2′,3′-O-isopropylideneadenosine (41)
To a solution of 40 (45.4 mg, 0.139 mmol, 1.10 equiv), 29 (64 mg, 0.13 mmol, 1.0 equiv), and PPh3 (36 mg, 0.14 mmol, 1.1 equiv) in THF (5 mL) was added DIAD (27 µL, 0.14 mmol, 1.1 equiv). After 4 h, MS monitoring of the reaction suggested limiting reagent 29 remained, therefore additional PPh3 (36 mg, 0.14 mmol, 1.1 equiv) and DIAD (27 µL, 0.14 mmol, 1.1 equiv) were added. After a further 19 h stirring, MS monitoring of the reaction suggested limiting reagent 29 still remained, thus more PPh3 (36 mg, 0.14 mmol, 1.1 equiv) and DIAD (27 µL, 0.14 mmol, 1.1 equiv) were added. After a final 7.5 h stirring, MS monitoring suggested the total consumption of 29. The reaction was concentrated to an off-white residue. Purification by flash chromatography (30:70 EtOAc–hexanes) afforded the title compound (46 mg, 45%) as an off-white oily residue: Rf 0.53 (0.5:0.5:49:50 HCO2H–MeOH–EtOAc–hexanes); 1H NMR (600 MHz, CDCl3) δ 1.32 (s, 9H), 1.40 (s, 18H), 1.41 (s, 3H), 1.64 (s, 3H), 4.18 (dd, J = 14.7, 7.6 Hz, 1H), 4.25 (dd, J = 14.7, 5.3 Hz, 1H), 4.69–4.70 (m, 1H), 5.23 (dd, J = 6.2, 2.9 Hz, 1H), 5.49 (dd, J = 5.9, 1.8 Hz, 1H), 6.22 (d, J = 1.8 Hz, 1H), 7.53 (t, J = 7.9 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.88 (td, J = 8.2, 1.2 Hz, 1H), 8.11 (dd, J = 7.9, 1.2 Hz, 1H), 8.22 (s, 1H), 8.88 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 25.6, 27.3, 27.89, 27.93, 49.5, 82.6, 83.8, 84.6, 85.8, 86.3, 91.0, 114.8, 116.9, 121.2, 125.4, 127.4, 128.4, 129.7, 137.1, 144.3, 150.4, 150.6, 152.2, 152.7, 157.1, 161.9, 163.3; HRMS (ESI+) calcd for C36H46N7O13S+ [M + H]+ 816.2869, found 816.2851 (error 2.2 ppm).
5′-Amino-5′-deoxy-5′-N-[(4-oxo-4H-benzo[e][1,2]oxazin-3-yl)sulfonyl]adenosine (3)
To solid 41 (47 mg, 0.058 mmol, 1.0 equiv) at 0 °C was added ice-cold 80% aqueous TFA (2 mL). The reaction was stirred 19 h at 0 °C. The reaction was concentrated in vacuo and subsequent purification by flash chromatography (1:1:98 HCO2H–MeOH–EtOAc) afforded the title compound (22 mg, 81%) as a colorless amorphous solid. Further purification by RP-HPLC (17.5:82.5 MeCN–H2O) and lyophilization of the appropriate fractions afforded the title compound (12 mg) as a fluffy colorless solid: Rf 0.32 (1:10:89 HCO2H–MeOH–EtOAc); 1H NMR (600 MHz, 1:10 D2O/DMSO-d6) δ 3.42–3.46 (ovlp m, 2H), 4.03–4.04 (m, 1H), 4.07 (dd, J = 5.6, 2.9 Hz, 1H), 4.58 (t, J = 5.9 Hz, 1H), 5.67 (d, J = 6.2 Hz, 1H), 7.51 (t, J = 7.9 Hz, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.86 (t, J = 7.9 Hz, 1H), 7.98 (d, J = 8.2 Hz, 1H), 8.12 (s, 1H), 8.19 (s, 1H); 13C NMR (150 MHz, 1:10 D2O–DMSO-d6) δ 46.0, 71.2, 73.0, 84.0, 88.2, 116.7, 119.5, 120.8, 124.8, 127.2, 137.2, 140.6, 148.9, 152.4, 156.0, 157.4, 161.4, 164.4; HRMS (ESI+) calcd for C18H18N7O7S+ [M + H]+ 476.0983, found 476.0996 (error 2.7 ppm).
Fluorescence Polarization Assays
The overexpression, and purification of MbtA,10 BasE,33, 16 EntE,33, 37 and VibE33, 38 was performed as previously described. The FP assays were performed using a modification of our previously described protocol.33 Briefly, FP measurements were performed on a microplate reader with excitation and emission wavelengths of 485 and 530 nm, respectively, using PMT sensitivity set to high and 100 readings per well. Assays were performed in triplicate in flat bottom, black polystyrene 384-well plates (3575 Corning Inc.) in a final volume of 50 µL. To determine the equilibrium dissociation constant KD1 of our fluorescent probe Fl-Sal-AMS, a direct binding experiment in which the probe was titrated with enzyme was performed. Specifically, a three-fold serial dilution of enzyme (10 µL, from ~0.1–1000 nM MbtA and EntE and ~2–2000 nM BasE and VibE final concentrations) was added to a 40 µL solution of Fl-Sal-AMS (20 nM final concentration) in FP buffer (30 mM Tris•HCl [pH 7.5], 1 mM MgCl2, 0.0025% Igepal CA-630, and 1 mM final concentrations). The fluorescence anisotropy was measured after a 30 min incubation at 22 °C. Experimentally measured anisotropies AOBS were fit to Equations 1 and 2 below using Mathematica 8 (Wolfram Research Inc.) to give the KD1. To determine the equilibrium dissociation constant KD2 of each compound, a competitive binding experiment in which each was titrated into Fl-Sal-AMS and enzyme was performed. Specifically, a three-fold serial dilution of each compound (0.5 µL, ~1–100000 nM final concentrations) was added to a 49.5 µL solution of Fl-Sal-AMS (20 nM final concentration), enzyme (50 nM MbtA and 200 nM BasE, EntE, and VibE final concentrations), FP buffer, and water. The fluorescence anisotropy was measured after a 30 min incubation at 22 °C. Displacement curves of measured fluorescent anisotropies versus varied compound concentrations were fit to Equations 2 and 3 below to give the KD2.
| (1) |
| (2) |
| (3) |
[32P]PPi-ATP Exchange Assay
This assay was performed as previously described.10 Briefly, reactions were performed under initial velocity conditions in a total volume of 101 µL. The reaction was set up in a volume of 90 µL and contained 250 µM salicylic acid (SAL), 10 mM ATP, 1 mM PPi, and 7 nM MbtA in assay buffer (75 mM Tris•HCl [pH 7.5], 10 mM MgCl2, 2 mM DTT). The inhibitors (1 µL) in DMSO or DMSO only as a control were added. The reaction components were allowed to equilibrate for 10 min at 22 °C. Reactions were initiated by the addition of 10 µL (0.5 µCi 32PPi, Perkin-Elmer 84.12Ci/mmol) in 50 mM sodium phosphate buffer (pH 7.8) and placed at 37 °C for 20 min. Reactions were quenched by the addition of 200 µL quenching buffer (350 mM HClO4, 100 mM PPi, 1.8% w/v activated charcoal). The charcoal was pelleted by centrifugation and washed once with 500 µL water. The washed pellet was resuspended in 200 µL water, transferred to a scintillation vial, mixed with 15 mL scintillation fluid (RPI), and counted on a scintillation counter. The counts from the bound γ-[32P]-ATP were directly proportional to initial velocity of the reaction and the data were fit to Morrison’s quadratic equation for fitting concentration–response data for tight binding inhibitors as described by Copeland.39
M. tuberculosis H37Rv MIC Assay
This assay was performed as previously described.8c Briefly, MICs were determined in quadruplicate in iron-deficient GAST according to the broth microdilution method6c using drugs from DMSO stock solutions or with control wells treated with an equivalent amount of DMSO. All measurements reported herein used an initial cell density of 104–105 cells/assay, and growth was monitored at 10 and at 14 days, with the untreated and DMSO-treated control cultures reaching an OD620 ~0.2–0.3. Plates were incubated at 37 °C (100 µL/well) and growth was recorded by measurement of optical density at 620 nm.
Supplementary Material
Acknowledgments
This research was supported by a grant from the National Institutes of Health (AI070219). We thank João Neres for providing FP probe, Fl-Sal-AMS. We thank Daniel Wilson for performing the [32P]PPi-ATP exchange assay and Dr. Helena Boshoff for whole-cell screening against M. tuberculosis H37Rv.
Footnotes
Supporting Information
1H, 13C, and 19F NMR spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.World Health Organization, Tuberculosis Fact Sheet No. 104. Oct; http://www.who.int/mediacentre/factsheets/fs104/en/index.html. [Google Scholar]
- 2.Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. Nature. 2011;469:483. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 3.Ratledge C, Dover LG. Annu. Rev. Microbiol. 2000;54:881. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- 4.(a) Miethke M, Marahiel MA. Microbiol. Mol. Biol. Rev. 2007;71:413. doi: 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crosa JH, Walsh CT. Microbiol. Mol. Biol. Rev. 2002;66:223. doi: 10.1128/MMBR.66.2.223-249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sandy M, Butler A. Chem. Rev. 2009;109:4580. doi: 10.1021/cr9002787. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hider RC, Kong X. Nat. Prod. Rep. 2010;27:637. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 5.Snow GA. Bacteriol. Rev. 1970;34:99. doi: 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vergne AF, Walz AJ, Miller MJ. Nat. Prod. Rep. 2000;17:99. doi: 10.1039/a809397k. [DOI] [PubMed] [Google Scholar]; (c) De Voss JJ, Rutter K, Schroeder BG, Barry CE. 3rdJ. Bacteriol. 1999;181:4443. doi: 10.1128/jb.181.15.4443-4451.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a Tullius MV, Harmston CA, Owens CP, Chim N, Morse RP, McMath LM, Iniguez A, Kimmey JM, Sawaya MR, Whitelegge JP, Horwitz MA, Goulding CW. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5051. doi: 10.1073/pnas.1009516108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jones CM, Niederweis M. J. Bacteriol. 2011;193:1767. doi: 10.1128/JB.01312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE. 3rdProc. Natl. Acad. Sci. U. S. A. 2000;97:1252. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. Chem. Biol. 1998;5:631. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]; (b) Chavadi SS, Stirrett KL, Edupuganti UR, Vergnolle O, Sadhanandan G, Marchiano E, Martin C, Qiu WG, Soll CE, Quadri LE. J. Bacteriol. 2011;193:5905. doi: 10.1128/JB.05811-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McMahon MD, Rush JS, Thomas MG. J. Bacteriol. 2012;194:2809. doi: 10.1128/JB.00088-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Nat. Chem. Biol. 2005;1:29. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]; (b) Miethke M, Bisseret P, Beckering CL, Vignard D, Eustache J, Marahiel MA. FEBS J. 2006;273:409. doi: 10.1111/j.1742-4658.2005.05077.x. [DOI] [PubMed] [Google Scholar]; (c) Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, 3rd, Aldrich CC. J. Med. Chem. 2006;49:31. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- 9.Isono K, Uramoto M, Kusakabe H, Miyata N, Koyama T, Ubukata M, Sethi SK, McCloskey JA. J. Antibiot. 1984;37:670. doi: 10.7164/antibiotics.37.670. [DOI] [PubMed] [Google Scholar]
- 10.Somu RV, Wilson DJ, Bennett EM, Boshoff HI, Celia L, Beck BJ, Barry CE, 3rd, Aldrich CC. J. Med. Chem. 2006;49:7623. doi: 10.1021/jm061068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiao C, Gupte A, Boshoff HI, Wilson DJ, Bennett EM, Somu RV, Barry CE, 3rd, Aldrich CC. J. Med. Chem. 2007;50:6080. doi: 10.1021/jm070905o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vannada J, Bennett EM, Wilson DJ, Boshoff HI, Barry CE, 3rd, Aldrich CC. Org. Lett. 2006;8:4707. doi: 10.1021/ol0617289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, 3rd, Bennett EM, Aldrich CC. J. Med. Chem. 2008;51:5349. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gupte A, Boshoff HI, Wilson DJ, Neres J, Labello NP, Somu RV, Xing C, Barry CE, 3rd, Aldrich CC. J. Med. Chem. 2008;51:7495. doi: 10.1021/jm8008037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Labello NP, Bennett EM, Ferguson DM, Aldrich CC. J. Med. Chem. 2008;51:7154. doi: 10.1021/jm800668u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.May JJ, Kessler N, Marahiel MA, Stubbs MT. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12120. doi: 10.1073/pnas.182156699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drake EJ, Duckworth BP, Neres J, Aldrich CC, Gulick AM. Biochemistry. 2010;49:9292. doi: 10.1021/bi101226n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kathryn Nelson, Courtney Aldrich, unpublished data. [Google Scholar]
- 18.Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. J. Med. Chem. 2002;45:2615. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 19.Föhlisch B. Chem. Ber. 1971;104:348. [Google Scholar]
- 20.Löwe W, Matzanke N. J. Heterocycl. Chem. 1996;33:763. [Google Scholar]
- 21.Heacock D, Forsyth CJ, Shiba K, Musier-Forsyth K. Bioorg. Chem. 1996;24:273. [Google Scholar]
- 22.Winum J-Y, Toupet L, Barragan V, Dewynter G, Montero J-L. Org. Lett. 2001;3:2241. doi: 10.1021/ol0161312. [DOI] [PubMed] [Google Scholar]
- 23.Neustadt BR. Tetrahedron Lett. 1994;35:379. [Google Scholar]
- 24.Chu DT, Fernandes PB, Claiborne AK, Pihuleac E, Nordeen CW, Maleczka RE, Pernet AG. J. Med. Chem. 1985;28:1558. doi: 10.1021/jm00149a003. [DOI] [PubMed] [Google Scholar]
- 25.(a) Abdulla RF, Brinkmeyer RS. Tetrahedron. 1979;35:1675. [Google Scholar]; (b) Abu-Shanab FA, Sherif SM, Mousa SAS. J. Heterocycl. Chem. 2009;46:801. [Google Scholar]; (c) Reiter LA. J. Org. Chem. 1984;49:3494. [Google Scholar]
- 26.Ikeuchi M, Meyer ME, Ding Y, Hiratake J, Richards NG. Bioorg. Med. Chem. 2009;17:6641. doi: 10.1016/j.bmc.2009.07.071. [DOI] [PubMed] [Google Scholar]
- 27.Mitsunobu O. Synthesis. 1981:1. [Google Scholar]
- 28.Mitscher LA. Chem. Rev. 2005;105:559. doi: 10.1021/cr030101q. [DOI] [PubMed] [Google Scholar]
- 29.Touster O. Org. React. 1953;7:327. [Google Scholar]
- 30.Ferris JP, Sanchez RA, Mancuso RW. Org. Synth. 1968;48:1. [Google Scholar]
- 31.2002 Pharmacia & Upjohn Co. WO200200444. [Google Scholar]
- 32.1,2-Benzoxazin-4-ones are rare heterocycles with a mere 20 examples found in SciFinder, searched April 8. 2013 [Google Scholar]
- 33.Neres J, Wilson DJ, Celia L, Beck BJ, Aldrich CC. Biochemistry. 2008;47:11735. doi: 10.1021/bi801625b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.pKa estimated using Marvin 5.12.3. ChemAxon. 2013 ( http://www.chemaxon.com) [Google Scholar]
- 35.Roehrl MHA, Wang JY, Wagner G. Biochemistry. 2004;43:16056. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- 36.Döbele M, Vanderheiden S, Jung N, Bräse S. Angew. Chem. Int. Ed. 2010;49:5986. doi: 10.1002/anie.201001507. [DOI] [PubMed] [Google Scholar]
- 37.Drake EJ, Nicolai DA, Gulick AM. Chem. Biol. 2006;13:409. doi: 10.1016/j.chembiol.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Keating TA, Marshall CG, Walsh CT. Biochemistry. 2000;39:15522. doi: 10.1021/bi0016523. [DOI] [PubMed] [Google Scholar]
- 39.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery. Hoboken, New Jersey: Wiley; 2005. [PubMed] [Google Scholar]
- 40.Sharma SK, Miller MJ, Payne SM. J. Med. Chem. 1989;32:357. doi: 10.1021/jm00122a013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.