

Abstract
Microglia are regarded as macrophages in the central nervous system (CNS) and play an important role in neuroinflammation in the CNS. Microglial activation has been strongly implicated in neurodegeneration in the brain. Increasing evidence also suggests an important role of spinal cord microglia in the genesis of persistent pain, by releasing the proinflammatory cytokines tumor necrosis factor-alpha (TNFα), Interleukine-1beta (IL-1β), and brain derived neurotrophic factor (BDNF). In this review, we discuss the recent findings illustrating the importance of microglial mediators in regulating synaptic plasticity of the excitatory and inhibitory pain circuits in the spinal cord, leading to enhanced pain states. Insights into microglial-neuronal interactions in the spinal cord dorsal horn will not only further our understanding of neural plasticity but may also lead to novel therapeutics for chronic pain management.
1. Microglia-Synapse Interactions in Healthy CNS
Microglia are derived from myeloid precursor cells in the periphery and penetrate the central nervous system (CNS) during embryogenesis [1]. They account for approximately 10–20% of all cells in the CNS, however their distribution varies from one region to another [2, 3]. Microglial density is particularly high in the hippocampus, basal ganglia, substantia nigra, and spinal cord [2, 4]. Microglia are regarded as the resident macrophages in the CNS and, similar to peripheral macrophages, they display different morphology depending upon their physiological states. In the resting physiological state, microglial cells are ramified with slender, radially projecting processes with similar thickness, length, and ramification, whereas in pathological states, microglia can be activated presenting an amoeboid morphology characterized by large soma, short/thick, and radially projecting processes with few ramifications [4–6]. Although most studies have focused on the role of activated microglia and synaptic transmission, both resting and activated microglia dynamically interact with synapses shaping their connectivity and function [7].
Microglial processes constantly and dynamically survey their environment and interact with nearby synapses [8, 9]. In mature CNS, it has been observed that microglial processes interact with axon terminals and dendritic spines in a transient manner, for an average of five minutes and at a rate of approximately one microglial contact per hour [10]. Notably, microglia processes are driven by neuronal activity and can simultaneously interact with both presynaptic and postsynaptic elements. Reducing neural activity by inhibiting sensory inputs or lowering body temperature results in retraction of microglial processes and decreases the frequency of contacts between microglial processes and synapses [10]. It is well known that astrocyte processes envelop synapses and actively modulate physiological synaptic transmission; however, whether and how microglia directly influence physiological synaptic transmission is still unclear.
Several studies demonstrated that microglial processes can engulf synapses and participate to their phagocytic elimination in an experience-dependent manner in the mature healthy brain [11–13]. Interestingly, a progressive accumulation of microglial phagocytic-like structures was observed in both mouse visual and auditory cortices by age-related loss of vision and hearing, respectively [14]. This suggests that microglia shape neuronal circuits not only during postnatal development but aslo along the lifespan. Together, these observations suggest that periodic interactions between microglia and synapses exist in the absence of pathological insult. These interactions may be compromised following nervous system injury or disease. The physiological role of microglia in spinal cord circuitry development and pain transmission remains to be investigated.
2. Nociceptive Pain and Persistent Pain
Our bodies play host to a wide variety of sensory information that is detected every moment by the peripheral nervous system. Primary sensory neurons that are responsible for the detection and transduction of painful stimuli (e.g., cold, heat, mechanical, and chemical), which are somatosensory stimuli that cause potential danger to the organism, are called nociceptors [15]. Nociception is of vital importance for survival; thus, it has become a highly regulated pathway within the nervous system of humans. Nociceptive input elicits pain as well as emotional, neuroendocrine, and autonomic responses. Persistent nociceptive input in this system after intense noxious stimulation or tissue injury results in activity-dependent plasticity or a progressive increase in the response of the system to repeated stimuli [16]. As a result of the neural plasticity, a normally innocuous low-threshold stimulation such as light touch could trigger a painful response (mechanical allodynia). Pathological pain or chronic pain results from neural plasticity both in the peripheral nervous system (i.e., peripheral sensitization) and CNS (central sensitization) [17]. The circuitry in the spinal cord dorsal horn connects incoming primary afferents to outgoing projection neurons that ascend to the brain. The projection neurons in the superficial dorsal horn (lamina I) also receive input from interneurons in the lamina II [18]. Importantly, spinal cord dorsal horn neurons undergo marked plastic changes in the pathological conditions, leading to hyperactivity of the projections neurons. Thus, dorsal horn is not only a critical relay center in nociceptive transmission [19] but also an important player in the development and maintenance of central sensitization [16, 17]. Several animal models are used for the study of neuropathic pain. In chronic constriction injury (CCI), the sciatic nerve is constricted by several loose ligatures. In spared nerve injury (SNI), the tibial and common peroneal divisions of the sciatic nerve are ligated and cut while sparing the sural division. In spinal nerve ligation (SNL), a spinal nerve, usually L5, is ligated where it exits the spinal column. Each of these models produces robust mechanical allodynia for weeks following injury; however, the underlying mechanisms may vary. Here we will focus on the contribution of spinal cord microglia to central sensitization in nerve injury-induced neuropathic pain and tissue injury-induced inflammatory pain states.
3. Spinal Cord Microglial Activation in the Context of Persistent Pain
Under pathological conditions, especially nerve injury conditions, microglia undergo “microgliosis”, a complex set of changes that allow the cell to respond rapidly and perform a broad range of functions such as shielding injury sites, phagocytosing cellular debris, and releasing inflammatory signals to initiate and/or propagate the immune response. Traditionally, microgliosis has been determined by a change in morphology from ramified to amoeboid [20]. Several pain models of persistent neuropathic pain including CCI, SNI, SNL, or spinal cord injury as well as chronic morphine-induced hyperalgesia and tolerance are associated with the development of microgliosis in the dorsal horn of the spinal cord [21–23]. Figure 1 shows microglial reaction in the dorsal horn of the lumbar spinal cord seven days following CCI of the sciatic nerve in CX3CR1-GFP mice. CX3CR1 promoter activity is restricted to microglia [24]. GFP expression revealed evenly distributed resident microglia in the contralateral side of the spinal cord that exhibited a quiescent or resting type morphology (left, Figure 1). However on the side ipsilateral to injury (right, Figure 1), microglia of the dorsal horn and ventral horn showed enlarged and amoeboid morphology, indicating their activation. Nerve injury also induces marked increases in the density and number of microglia, due to proliferation and possible migration (Figure 1) [25]. Of interest, nerve injury-induced microgliosis is very mild in young animals (2-3 weeks old) [26]. It should be noted, however, that there are also a number of persistent pain conditions that are not associated with spinal cord microgliosis, such as adjuvant-induced inflammatory pain and chemotherapy-induced neuropathic pain [27, 28].
Figure 1.
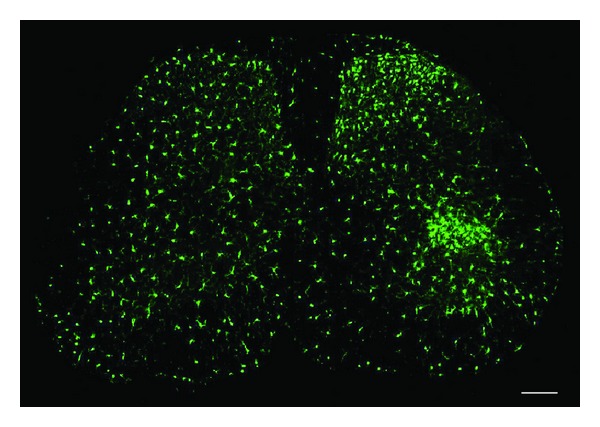
Nerve injury induces marked microglial reaction in the ipsilateral lumbar spinal cord 7 days after chronic constriction injury (CCI). Microglia as demonstrated by CX3CR1 expression in CX3CR1-GFP mice. The contralateral side (left) shows the typical resting microglial morphology, and the ipsilateral side (right) illustrates the enlarged and amoeboid morphological features of activated microglia cell bodies. Also note that on the ipsilateral side the number and density of microglia in the medial superficial dorsal horn and lateral ventral horn, where injured primary afferent axons terminate, are significantly increased. Scale, 100 μm.
Mitogen-activated kinase (MAPK) pathways are important for intracellular signal transduction and play critical roles in neuronal plasticity and inflammatory responses. The MAPK family consists of three separate signaling pathways: extracellular signal-regulated kinases (ERK), p38, and c-Jun N-terminal kinase (JNK). MAPK activation is correlated with most if not all persistent pain conditions [29, 30]. Several neuropathic pain models, including spinal nerve injury and spared nerve injury, exhibit a robust increase in p38 activation (phosphorylation) in microglia of the dorsal horn beginning at twelve hours, peaking at three days, and slowly declining over several weeks [29, 31–33]. Intrathecal administration of p38 inhibitors has been shown to attenuate neuropathic pain [29, 33]. Minocycline, a broad-spectrum antimicrobial tetracycline compound that inhibits microglial activation, also decreases pain behavior following nerve injury [34, 35], possibly by inhibiting p38 activation [33]. However, minocycline is not able to reverse existing pain states [36]. Interestingly, intrathecal administration of minocycline in models of inflammatory pain, where morphological activation of microglia is not evident, also prevents the development of mechanical sensitization, by inhibiting spinal cord microglial p38 activation [37, 38]. Thus the release of inflammatory mediators from microglia may also occur without morphological alteration. The morphological changes associated with microgliosis may be mediated by the activation of ERK/MAPK [30], and nerve injury was shown to induce ERK activation in spinal microglia in the early phase [39]. It seems there may be multiple activation states whereby microglia do change the manner with which they participate in neural plastic changes, but do not reach a morphologically activated phenotype.
The p38 MAPK pathway can be activated by a host of molecules known to increase pain sensitivity, including the proinflammatory cytokines TNFα and IL-1β, CCL2 (also known as monocyte chemoattractant protein 1 (MCP-1)), fractalkine (CX3CL1), inducible nitric oxide synthase (iNOS), and matrix metalloprotease-9 (MMP-9) as well as the ATP receptors P2X4 and P2X7 [40–47]. As shown in Figure 2, some of these microglial activators, such as ATP, CCL2, fractalkine, and MMP-9, could be released from primary afferent neurons [48, 49]. Once activated, the p38 pathway induces the expression of proinflammatory transcription factors, enzymes, and signaling molecules, including NFkappaB, COX2, iNOS, BDNF, TNFα, IL-1β, and IL-6 [38, 41, 50]. p38 activation in microglia also results in increased release of BDNF and TNFα in microglia [22]. Microglial production of proinflammatory cytokines can further recruit microglia, activate surrounding astrocytes, and promote the sensitization of central nervous system nociceptive circuits.
Figure 2.
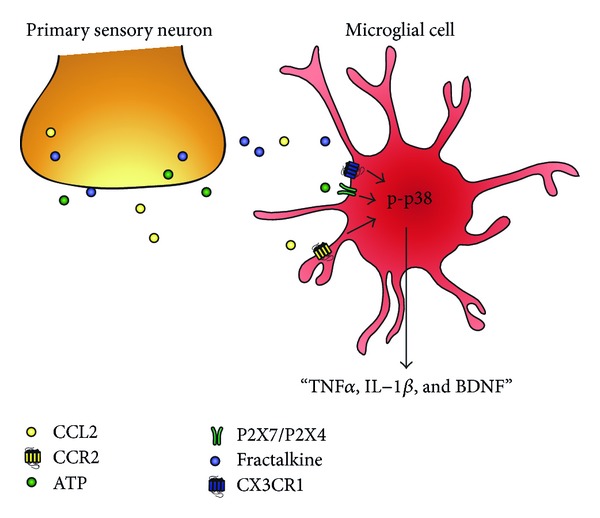
Schematic of primary afferent releasing factors that are responsible for microglial activation. Microglial cells are activated by factors released from hyperexcitable primary afferents such as CCL2 (MCP-1), ATP, and CX3CL1 (fractalkine). Respective activation of CCR2, P2X4/P2X7, and CX3CR1 receptors on microglia causes phosphorylation of p38 in microglia, leading to increased synthesis and release of TNFα, IL-1β, and BDNF.
4. Dorsal Horn Microglial-Synapse Interactions in the Context of Persistent Pain
The development of central sensitization in persistent pain is characterized by increased excitatory synaptic transmission and decreased inhibitory synaptic transmission in the dorsal horn of the spinal cord. In order to modulate pain sensitivity and participate in central sensitization, glia must interact with neural pain circuits via modulation of neurotransmission. Glial mediators can modulate synaptic transmission at very low concentrations. While neurotransmitters such as glutamate, GABA, and glycine produce synaptic effects at μM concentrations, glial cytokines, chemokines, and growth factors can affect synaptic activity at nM concentrations [51–53]. Accumulating evidence indicates a critical role of TNFα, IL-1β, and BDNF, all of which are released from activated microglia, in inducing the hyperactivity of dorsal horn neurons and thus in the development of pain hypersensitivity primarily in the setting of neuropathic pain, through modulation of both excitatory and inhibitory neurotransmission in the dorsal horn [40, 54, 55].
4.1. TNFα
TNFα is present both in healthy brain tissue and in disease states. TNFα is known to play a role in synaptic plasticity, which has been studied mainly in hippocampal slices. Glial TNFα has been shown to enhance synaptic efficacy by increasing the surface expression of GluR1-possessing AMPA receptors via TNFR1-mediated PI3 K activation [56, 57]. Glial TNFα also causes endocytosis of GABAA receptors resulting in a decrease in inhibitory synaptic currents [57]. Homeostatic synaptic scaling of excitatory synapses increases their strength in response to network activity reduction or decreases their strength in response to increased network activity. In response to decreases in network activity, glial TNFα was shown to increase AMPA-mediated currents by increasing the number of calcium permeable AMPA receptors at the cell surface [58]. However, its role may be more permissive rather than instructive in this change [59].
The effects of TNFα have also been studied in the dorsal horn of the spinal cord (see Figure 3). Intrathecal injection of TNFα causes the development of thermal and mechanical hyperalgesia [51]. To investigate the synaptic mechanisms of TNFα, many studies have used ex vivo spinal cord slice preparation. Incubation with TNFα increases the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) in lamina II excitatory interneurons [60]. This could be indicative of a change in presynaptic glutamate release. TNFα increases sEPSC frequency via activation of transient receptor potential cation channel subfamily V member 1 (TRPV1) in presynaptic terminals, possibly through activation of adenylyl cyclase, protein kinase (PKA), or extracellular signal-related kinase (ERK) [60]. Activation of TRPV1 results in increased presynaptic calcium influx and, therefore, increased vesicular glutamate release [60].
Figure 3.
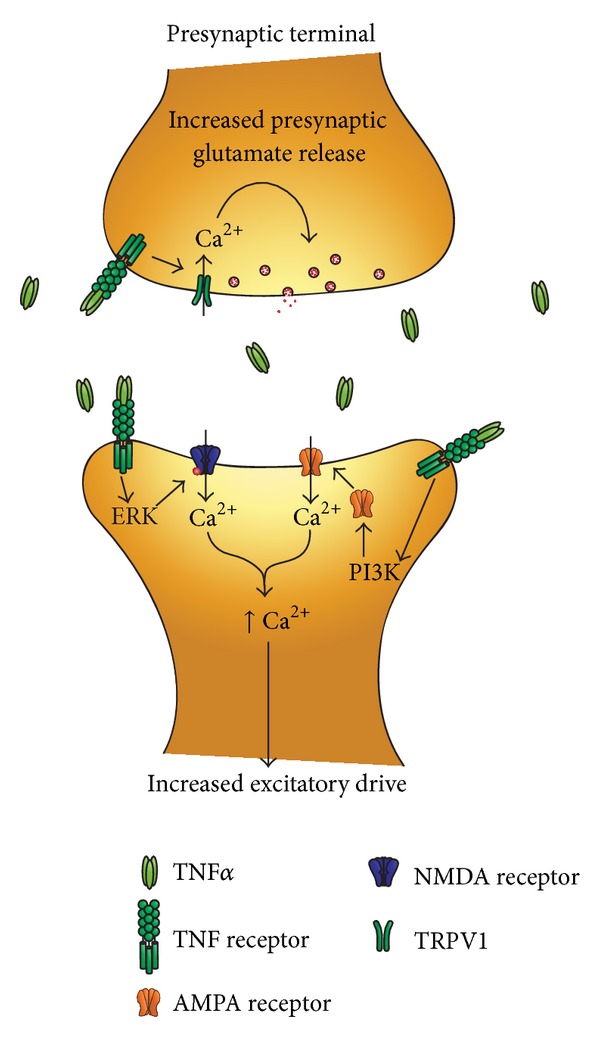
Schematic of TNFα induced potentiation of spinal cord synaptic transmission. Microglial release of TNFα increases excitatory neurotransmission in the dorsal horn via both presynaptic and postsynaptic mechanisms. At presynaptic sites, TNFα increases glutamate release via TRPV1 activation and there will be a subsequent increase in intracellular Ca2+. At postsynaptic sites, TNFα increases the activity of AMPA and NMDA receptors via activation of PI3 K and ERK on glutamatergic neurons to increase excitatory drive.
TNFα also acts on the postsynaptic neurons in the spinal cord. In a carrageenan model of inflammation, TNFα recruited Ca2+ permeable AMPA receptors to dorsal horn neurons resulting in increased sEPSC amplitude [61]. NMDA currents in lamina II neurons are also enhanced by application of TNFα [51], and TNFα increases NMDA receptor (NMDAR) activity through phosphorylation of ERK in dorsal horn, neurons [62]. Thus via pre- and post synaptic mechanisms TNFα increases excitatory neurotransmission in the dorsal horn.
In spinal cord slices, TNFα not only enhances sEPSCs but also suppresses the frequency of spontaneous inhibitory postsynaptic currents (sIPSCs) [63]. This was found to be mediated by a decrease in spontaneous action potentials in GABAergic neurons via activation of TNF receptor 1 (TNFR1) and activation of p38 MAPK [63]. Neurons in the dorsal horn possess both TNFR1 and 2 (TNFR2), however, TNFR1 seems to make a greater contribution to enhancing nociceptive signaling in the dorsal horn [64].
In spinal cord slices from TNFR1 KO mice, TNFα was unable to elicit increases in sEPSCs or increases in NMDA currents [64]. However in TNFR2 KO mice, TNFα was still able to produce a small increase in sEPCS, and it elicited a normal increase in NMDA currents [64]. Both TNFR1 and TNFR2 knockout (KO) animals show decreased pain behavior in response to complete Freund's adjuvant and formalin induced inflammatory pain as well as intrathecal injection of TNFα [64]. Thus, microglial release of TNFα in the dorsal horn both enhances excitatory neuronal/synaptic activity and suppresses inhibitory neuronal/synaptic activity to enhance central sensitization primarily through the activation of TNFR1 on nociceptive dorsal horn neurons.
Long-term potentiation (LTP) in the spinal cord is implicated in pathological pain [65]. LTP in the spinal cord can be triggered by stimulation of the primary afferent fibers with the typical high-frequency titanic stimulation [66] and also by low-frequency stimulation (a more physiological firing pattern of nociceptors) [67] as well as by formalin or capsaicin administration to the paw or by nerve injury [16, 17, 67]. TNFα is also important for the induction of spinal LTP [68], and both TNFR1 and TNFR2 are required for titanic stimulation-induced LTP [60, 69]. While the prevailing view is that TNFα release from glia activates TNF receptors on neurons to promote LTP, new evidence has recently been found that TNF receptor expression on glial cells is also necessary for the generation of spinal LTP [69]. In the presence of fluorocitrate, a pharmacological blocker of glial activation, TNFα failed to potentiate AMPAR-mediated synaptic currents [69]. This suggests that there is an intermediate step which may facilitate TNFα-induced sensitization of dorsal horn neurons. TNFα could act on glial cells to elicit the release of additional glial mediators to facilitate its pronociceptive effects. Furthermore, pretreatment of slices with the microglial selective inhibitor minocycline inhibits LTP produced by high-frequency stimulation, however, application of TNFα reversed the effect [70], again suggesting that microglial TNFα release is important for long-term synaptic plasticity.
It is important to note that the effects of TNFα vary among regions of the CNS particularly at high pathological concentrations. While constitutive TNFα release may be permissive for plastic changes in neurotransmission, the activation of microglia can result in 10-fold higher TNFα concentrations and, at these high concentrations (greater than 0.3 nM), TNFα may change its mode of action [71]. When cultured in the presence of microglia, astrocytic glutamate released is dramatically amplified [72]. In hippocampal slices, high concentrations of TNFα may cause prostaglandin E2 mediated glutamate release from astrocytes [73] to the degree that it causes excitotoxic damage in neurons [72]. It has been repeatedly demonstrated that at high concentrations (10–100 ng/mL), TNFα inhibited hippocampal LTP [74–76]. However, in spinal dorsal, horn both low and high concentrations of TNFα increased C-fiber induced LTP in nerve-injured rats [68].
4.2. IL-1β
IL-1β is induced in astrocytes, neurons, and microglia in inflammatory and neuropathic pain [77–80]. Intrathecal administration of IL-1β induces both thermal and mechanical hyperalgesia [81, 82]. Inhibition of IL-1β signaling through administration of IL-1 receptor antagonists decreased allodynia in neuropathic pain models [77, 83–85]. IL-1β enhances nociceptive activity in the dorsal horn through some of the same mechanisms as TNFα. Application of IL-1β to spinal cord slices increases sEPSC frequency, which indicates enhanced excitatory neurotransmission through increased release of neurotransmitter [51]. Importantly, IL-1β, but not TNFα, increased the amplitude of sEPSCs, suggesting additional postsynaptic regulation [51]. Also similar to TNFα, IL-1β enhances NMDA receptor currents in dorsal horn neurons [86], albeit by a different mechanism. IL-1β induces phosphorylation of the NR1 subunit of the NMDA receptor [87, 88]. NR1 is an essential subunit of the NMDA receptor, and phosphorylation of NR1 increases NMDA-mediated inward currents. In both inflammatory and neuropathic pain models, phosphorylation of NR1 increases NMDA receptor activity, facilitating excitatory neurotransmission in the dorsal horn [89–91].
IL-1β also suppresses inhibitory neurotransmission in the dorsal horn. Application of IL-1β to spinal cord slices inhibits both the frequency and the amplitude of spontaneous postsynaptic currents (sIPSCs) [51]. Therefore it functions via both pre- and postsynaptic mechanisms. Furthermore, application of IL-1β is capable of reducing both GABA and glycine mediated currents in dorsal horn neurons [51]. Overall, IL-1β enhances pronociceptive neurotransmission both through enhancing excitatory neurotransmission and suppressing inhibitory neurotransmission.
4.3. BDNF
Brain derived neurotrophic factor (BDNF) is a secreted protein and part of the family of neurotrophins which act on neurons to promote survival, growth, and differentiation of new neurons and synapses [92]. In the brain, BDNF is important for synaptic plasticity and long-term memory [93, 94]. However, in the spinal cord following nerve injury, it plays a deleterious role.
Following nerve injury, microglia upregulate their expression of the ionotropic ATP receptor P2X4 concurrently with the development of allodynia [95]. P2X4 receptor stimulation results in the activation of the p38 MAPK pathway and increases the synthesis and release of BDNF from microglial cells [96]. Application of BDNF to the spinal cord is capable of producing mechanical allodynia in otherwise naïve animals [97]. Furthermore, intrathecal administration of microglia activated by ATP into naïve animals produces mechanical allodynia, which can be alleviated by blockade of the BDNF receptor TrkB [95]. Thus microglial produce BDNF following nerve injury and BDNF is sufficient to produce pain behavior.
Microglial derived BDNF contributes to pain hypersensitivity through the disinhibition of nociceptive processing in the dorsal horn. BDNF acts on lamina I pain transmission neurons; neurons that carry the output message of the dorsal horn to higher brain centers where pain is perceived. BDNF, through the activation of its receptor TrkB, decreases expression of the potassium-chloride cotransporter 2 (KCC2) resulting in a rise in intracellular chloride [53, 98]. This shift in the neuronal anion gradient is sufficient to suppress inhibition in the majority of lamina I projection neurons, and in some neurons the shift is large enough that their response to application with GABA becomes excitatory rather than inhibitory [53, 99, 100]. The outcome is suppression of the intrinsic inhibitory circuit in the dorsal horn. Protein levels of KCC2 are down-regulated in the spinal cord following both spinal cord injury and nerve-injury models in parallel with the development of thermal and mechanical hypersensitivity [101, 102].
In naïve animals, lamina I projection neurons respond to painful but not innocuous stimuli. The suppression of inhibitory drive and thus exaggerated responses of lamina I neurons to normally noxious stimuli explains the development of hyperalgesia, an exaggerated response to normally noxious stimuli, but it does not explain the development of allodynia, the painful response to normally innocuous stimuli. Persistent pain sufferers report three cardinal features of their pain: hyperalgesia, allodynia, and spontaneous pain. In vivo recordings from lamina I projection neurons show that following nerve-injury, these neurons begin to respond to nonnoxious stimuli, increase their response to noxious stimuli, and discharge spontaneously [103]. Though the mechanism is not fully elucidated, these results can be recapitulated by disrupting the chloride homeostasis of lamina I projection neurons as well as by the addition of ATP stimulated microglia, which presumably secrete BDNF [103]. Thus microglial derived BDNF may participate further in the development of persistent pain conditions, yet the entire mechanism remains to be elucidated.
5. Concluding Remarks
Hyperactivity of peripheral nociceptive fibers due to inflammation or injury causes the release of factors such as CCL2, ATP, and fractalkine into the dorsal horn of the spinal cord [48, 49]. The binding of these factors to their respective receptors CCR2, P2X4, P2X7, and CX3CR1 on microglia causes activation of proinflammatory cascades and often gliosis [40–47]. p38 MAPK acts as a signal integrator of microglia stimulation. Activation of p38 results in the production and release of cytokines, chemokines, and proinflammatory mediators [22] that amplify pronociceptive signals in the dorsal horn.
We examined the mechanisms of three key signaling molecules released by microglial cells in the context of pain: TNFα, IL-1β, and BDNF. TNFα and IL-1β increase presynaptic release of excitatory neurotransmitter as well as increasing excitatory postsynaptic currents through the recruitment of AMPA receptors and enhancing NMDA currents [51, 60–62, 86]. Both also suppress inhibitory neurotransmission through distinct mechanisms. TNFα decreases spontaneous activity in GABAergic neurons while IL-1β suppresses GABA and glycine mediated inhibitory currents [51, 63]. BDNF in the dorsal horn functions a wholly different mechanism to suppress inhibitory neurotransmission. Microglial release of BDNF decreases expression of KCC2 [53, 98]. This shifts the chloride gradient of the cell sufficiently to make the normally inhibitory currents resulting from GABA application negligible or even excitatory. Therefore, these proinflammatory mediators promote central sensitization through multiple mechanisms to increase excitatory drive and decrease inhibitory drive in dorsal horn nociceptive circuitry.
TNFα, IL-1β, and BDNF also affect long-term neuronal plasticity in the dorsal horn. Activation of their respective receptors, TNFR1, IL-1, and trkB, on neurons leads to the phosphorylation of ERK which can enter the nucleus and produce phosphorylation of cAMP response element binding protein (CREB) [51, 104–106]. Activation of the transcription factor CREB occurs following nerve injury [107]. Its activation leads to the transcription of genes such as COX-2, NK-1, and trkB which increase neuronal excitability and further promote sensitization [106, 108, 109]. These proinflammatory mediators encourage both short-term and long-term sensory plasticity in the dorsal horn resulting in an increase in ascending nociceptive neurotransmission to the brain. This central sensitization is a key component in the development of hyperalgesia and allodynia associated with persistent pain disorders, and microglia are emerging as a key contributor to central sensitization.
6. Future Directions
Recently, studies have found that microglia, like peripheral macrophages, are capable of distinct functional states referred to as classical or alternative activation. Classical activation is initiated by IFNγ, IL-1β, and LPS which increase production and release of proinflammatory molecules such as TNFα and IL-1β [110, 111]. Alternative activation is induced by IL-4, IL-13, IL-10, and TGFβ which activate anti-inflammatory cascades or tissue repair mechanisms in microglia [110, 112]. The anti-inflammatory actions of IL-4 and IL-13 are mainly a result of their ability to antagonize changes induced by proinflammatory cytokines and suppress the production of IL-1β and TNFα from microglia [113–117]. Additionally, IL-4 treatment increases the mRNA expression of repair genes, which are also markers of alternative activation in macrophages, such as arginase 1and mannose receptor 1, found in inflammatory zone 1 and Ym1 [118, 119]. Administration of IL-10 and TGFβ or uptake of apoptotic cells induces an alternative activation state sometimes called acquired deactivation, that as well as being anti-inflammatory is also an immunosuppressive state [110]. However, the majority of studies on alternative activation have been performed using peripheral macrophages, and anti-inflammatory signaling cascades in microglia have not been widely investigated and remain poorly understood. Furthermore, it also remains to be seen if these anti-inflammatory cytokines are capable of regulating synaptic plasticity in the dorsal horn.
A second class of anti-inflammatory, proresolution mediators are the lipid derivatives resolvins and neuroprotectins. Administration of resolvin E1 (RvE1) or neuroprotectin/protectin D1 (NPD1/PD1) can block TNFα induced synaptic plasticity as well as long-term potentiation in the dorsal horn [120–122]. Additionally, RvE1 is capable of blocking TNFα induced increases of postsynaptic NMDA currents via regulation of ERK [121, 122]. RvE1 and NPD1 can further block LPS-induced TNFα release in microglia [123, 124]. While blocking proinflammatory cytokine production remains a major avenue of clinical pain management, anti-inflammatory and proresolving lipid mediators may provide new avenues for controlling clinical pain.
Conflict of Interests
All the authors have no competing financial interests in this study.
Acknowledgments
This work was supported in part by the National Institutes of Health Grants R01DE17794 and R01DE22743, and NS67686 to R.R.J.
References
- 1.Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annual Review of Immunology. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 2.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 3.Perry VH, Gordon S. Macrophages and the nervous system. International Review of Cytology. 1991;125:203–244. doi: 10.1016/s0074-7696(08)61220-6. [DOI] [PubMed] [Google Scholar]
- 4.Zhang F, Vadakkan KI, Kim SS, Wu L-J, Shang Y, Zhuo M. Selective activation of microglia in spinal cord but not higher cortical regions following nerve injury in adult mouse. Molecular Pain. 2008;4, article 15 doi: 10.1186/1744-8069-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayoub AE, Salm AK. Increased morphological diversity of microglia in the activated hypothalamic supraoptic nucleus. Journal of Neuroscience. 2003;23(21):7759–7766. doi: 10.1523/JNEUROSCI.23-21-07759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrison HW, Filosa JA. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. Journal of Neuroinflammation. 2013;10, article 4 doi: 10.1186/1742-2094-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tremblay M-È, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. Journal of Neuroscience. 2011;31(45):16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 9.Nimmerjahn A, Kirchhoff F, Helmchen F. Neuroscience: resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 10.Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. Journal of Neuroscience. 2009;29(13):3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 12.Schafer DP, Lehrman EK, Stevens B. The, “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. GLIA. 2013;61(1):24–36. doi: 10.1002/glia.22389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tremblay M-È, Zettel ML, Ison JR, Allen PD, Majewska AK. Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. GLIA. 2012;60(4):541–558. doi: 10.1002/glia.22287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherrington CS. Observations on the scratch-reflex in the spinal dog. Journal of Physiology. 1906;34(1-2):1–50. doi: 10.1113/jphysiol.1906.sp001139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288(5472):1765–1768. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 17.Ji R-R, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends in Neurosciences. 2003;26(12):696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 18.Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nature Reviews Neuroscience. 2010;11(12):823–836. doi: 10.1038/nrn2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willis W, Coggeshall RE. Sensory Mechanisms of the Spinal Cord. 3rd edition. Vol. 1. New York, NY, USA: Kluwer Academic, Plenum Publishers; 2004. [Google Scholar]
- 20.Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Progress in Neurobiology. 1999;57(6):563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 21.Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends in Neurosciences. 2005;28(2):101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Wen Y-R, Tan P-H, Cheng J-K, Liu Y-C, Ji R-R. Microglia: a promising target for treating neuropathic and postoperative pain, and morphine tolerance. Journal of the Formosan Medical Association. 2011;110(8):487–494. doi: 10.1016/S0929-6646(11)60074-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou D, Chen M-L, Zhang Y-Q, Zhao Z-Q. Involvement of spinal microglial P2X7 receptor in generation of tolerance to morphine analgesia in rats. Journal of Neuroscience. 2010;30(23):8042–8047. doi: 10.1523/JNEUROSCI.5377-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jung S, Aliberti J, Graemmel P, et al. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Molecular and Cellular Biology. 2000;20(11):4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beggs S, Salter MW. Stereological and somatotopic analysis of the spinal microglial response to peripheral nerve injury. Brain, Behavior, and Immunity. 2007;21(5):624–633. doi: 10.1016/j.bbi.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moss A, Beggs S, Vega-Avelaira D, et al. Spinal microglia and neuropathic pain in young rats. Pain. 2007;128(3):215–224. doi: 10.1016/j.pain.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 27.Lin T, Li K, Zhang F-Y, Zhang Z-K, Light AR, Fu K-Y. Dissociation of spinal microglia morphological activation and peripheral inflammation in inflammatory pain models. Journal of Neuroimmunology. 2007;192(1-2):40–48. doi: 10.1016/j.jneuroim.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng FY, Xiao W-H, Bennett GJ. The response of spinal microglia to chemotherapy-evoked painful peripheral neuropathies is distinct from that evoked by traumatic nerve injuries. Neuroscience. 2011;176:447–454. doi: 10.1016/j.neuroscience.2010.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin S-X, Zhuang Z-Y, Woolf CJ, Ji R-R. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. Journal of Neuroscience. 2003;23(10):4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calvo M, Zhu N, Grist J, Ma Z, Loeb JA, Bennett DLH. Following nerve injury neuregulin-1 drives microglial proliferation and neuropathic pain via the MEK/ERK pathway. GLIA. 2011;59(4):554–568. doi: 10.1002/glia.21124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wen Y-R, Suter MR, Kawasaki Y, et al. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology. 2007;107(2):312–321. doi: 10.1097/01.anes.0000270759.11086.e7. [DOI] [PubMed] [Google Scholar]
- 32.Katsura H, Obata K, Mizushima T, et al. Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. Journal of Neuroscience. 2006;26(34):8680–8690. doi: 10.1523/JNEUROSCI.1771-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. GLIA. 2004;45(1):89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- 34.Ledeboer A, Sloane EM, Milligan ED, et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115(1-2):71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Raghavendra V, Tanga F, Deleo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. Journal of Pharmacology and Experimental Therapeutics. 2003;306(2):624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- 36.Svensson CI, Hua X-Y, Protter AA, Powell HC, Yaksh TL. Spinal p38 MAP kinase is necessary for NMDA-induced spinal PGE2 release and thermal hyperalgesia. NeuroReport. 2003;14(8):1153–1157. doi: 10.1097/00001756-200306110-00010. [DOI] [PubMed] [Google Scholar]
- 37.Hua X-Y, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. European Journal of Neuroscience. 2005;22(10):2431–2440. doi: 10.1111/j.1460-9568.2005.04451.x. [DOI] [PubMed] [Google Scholar]
- 38.Svensson CI, Marsala M, Westerlund A, et al. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. Journal of Neurochemistry. 2003;86(6):1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- 39.Zhuang Z-Y, Gerner P, Woolf CJ, Ji R-R. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114(1-2):149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 40.Svensson CI, Schäfers M, Jones TL, Powell H, Sorkin LS. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neuroscience Letters. 2005;379(3):209–213. doi: 10.1016/j.neulet.2004.12.064. [DOI] [PubMed] [Google Scholar]
- 41.Sung C-S, Wen Z-H, Chang W-K, et al. Inhibition of p38 mitogen-activated protein kinase attenuates interleukin-1β-induced thermal hyperalgesia and inducible nitric oxide synthase expression in the spinal cord. Journal of Neurochemistry. 2005;94(3):742–752. doi: 10.1111/j.1471-4159.2005.03226.x. [DOI] [PubMed] [Google Scholar]
- 42.Zhuang Z-Y, Kawasaki Y, Tan P-H, Wen Y-R, Huang J, Ji R-R. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain, Behavior, and Immunity. 2007;21(5):642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abbadie C, Lindia JA, Cumiskey AM, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(13):7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ikeda H, Tsuda M, Inoue K, Murase K. Long-term potentiation of neuronal excitation by neuron-glia interactions in the rat spinal dorsal horn. European Journal of Neuroscience. 2007;25(5):1297–1306. doi: 10.1111/j.1460-9568.2007.05386.x. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. Journal of Neuroscience. 2008;28(11):2892–2902. doi: 10.1523/JNEUROSCI.5589-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang Q, Svensson CI, Fitzsimmons B, Webb M, Yaksh TL, Hua X-Y. Inhibition of spinal constitutive NOS-2 by 1400W attenuates tissue injury and inflammation-induced hyperalgesia and spinal p38 activation. European Journal of Neuroscience. 2007;25(10):2964–2972. doi: 10.1111/j.1460-9568.2007.05576.x. [DOI] [PubMed] [Google Scholar]
- 47.Kawasaki Y, Xu Z-Z, Wang X, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nature Medicine. 2008;14(3):331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clark AK, Malcangio M. Microglial signalling mechanisms: cathepsin S and Fractalkine. Experimental Neurology. 2012;234(2):283–292. doi: 10.1016/j.expneurol.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 49.Ji R-R, Xu Z-Z, Wang X, Lo EH. Matrix metalloprotease regulation of neuropathic pain. Trends in Pharmacological Sciences. 2009;30(7):336–340. doi: 10.1016/j.tips.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ji R-R, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Molecular Pain. 2007;3, article 33 doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawasaki Y, Zhang L, Cheng J-K, Ji R-R. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1β, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. Journal of Neuroscience. 2008;28(20):5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao Y-J, Zhang L, Samad OA, et al. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. Journal of Neuroscience. 2009;29(13):4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coull JAM, Beggs S, Boudreau D, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438(7070):1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 54.DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90(1-2):1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 55.Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends in Neurosciences. 2001;24(8):450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- 56.Beattie EC, Stellwagen D, Morishita W, et al. Control of synaptic strength by glial TNFα . Science. 2002;295(5563):2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 57.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α . Journal of Neuroscience. 2005;25(12):3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-α . Nature. 2006;440(7087):1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 59.Steinmetz CC, Turrigiano GG. Tumor necrosis factor-α signaling maintains the ability of cortical synapses to express synaptic scaling. Journal of Neuroscience. 2010;30(44):14685–14690. doi: 10.1523/JNEUROSCI.2210-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park C-K, Lü N, Xu Z-Z, Liu T, Serhan CN, Ji R-R. Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. Journal of Neuroscience. 2011;31(42):15072–15085. doi: 10.1523/JNEUROSCI.2443-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi JI, Svensson CI, Koehrn FJ, Bhuskute A, Sorkin LS. Peripheral inflammation induces tumor necrosis factor dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. Pain. 2010;149(2):243–253. doi: 10.1016/j.pain.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu Z-Z, Zhang L, Liu T, et al. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nature Medicine. 2010;16(5):592–597. doi: 10.1038/nm.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang H, Nei H, Dougherty PM. A p38 mitogen-activated protein kinase-dependent mechanism of disinhibition in spinal synaptic transmission induced by tumor necrosis factor-alpha. Journal of Neuroscience. 2010;30(38):12844–12855. doi: 10.1523/JNEUROSCI.2437-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang L, Berta T, Xu Z-Z, Liu T, Park JY, Ji R-R. TNF-alpha contributes to spinal cord synaptic plasticity and inflammatory pain: distinct role of TNF receptor subtypes 1 and 2. Pain. 2011;152(2):419–427. doi: 10.1016/j.pain.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sandkühler J, Gruber-Schoffnegger D. Hyperalgesia by synaptic long-term potentiation (LTP): an update. Current Opinion in Pharmacology. 2012;12(1):18–27. doi: 10.1016/j.coph.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu X-G, Sandkuhler J. Long-term potentiation of C-fiber-evoked potentials in the rat spinal dorsal horn is prevented by spinal N-methyl-D-aspartic acid receptor blockage. Neuroscience Letters. 1995;191(1-2):43–46. doi: 10.1016/0304-3940(95)11553-0. [DOI] [PubMed] [Google Scholar]
- 67.Ikeda H, Stark J, Fischer H, et al. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312(5780):1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- 68.Liu Y-L, Zhou L-J, Hu N-W, et al. Tumor necrosis factor-α induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK. Neuropharmacology. 2007;52(3):708–715. doi: 10.1016/j.neuropharm.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 69.Gruber-Schoffnegger D, Drdla-Schutting R, Hönigsperger C, Wunderbaldinger G, Gassner M, Sandkühler J. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-α and IL-1β is mediated by glial cells. Journal of Neuroscience. 2013;33(15):6540–6551. doi: 10.1523/JNEUROSCI.5087-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhong Y, Zhou L-J, Ren W-J, et al. The direction of synaptic plasticity mediated by C-fibers in spinal dorsal horn is decided by Src-family kinases in microglia: the role of tumor necrosis factor-α . Brain, Behavior, and Immunity. 2010;24(6):874–880. doi: 10.1016/j.bbi.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 71.Santello M, Volterra A. TNFalpha in synaptic function: switching gears. Trends in Neurosciences. 2012;35(10):638–647. doi: 10.1016/j.tins.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 72.Bezzi P, Domercq M, Brambilla L, et al. CXCR4-activated astrocyte glutamate release via TNFa: amplification by microglia triggers neurotoxicity. Nature Neuroscience. 2001;4(7):702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- 73.Santello M, Bezzi P, Volterra A. TNFalpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron. 2011;69(5):988–1001. doi: 10.1016/j.neuron.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 74.Tancredi V, D’Arcangelo G, Grassi F, et al. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neuroscience Letters. 1992;146(2):176–178. doi: 10.1016/0304-3940(92)90071-e. [DOI] [PubMed] [Google Scholar]
- 75.Butler MP, O’Connor JJ, Moynagh PN. Dissection of tumor-necrosis factor-α inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early—but not late—Phase LTP. Neuroscience. 2004;124(2):319–326. doi: 10.1016/j.neuroscience.2003.11.040. [DOI] [PubMed] [Google Scholar]
- 76.Cunningham AJ, Murray CA, O’Neill LAJ, Lynch MA, O’Connor JJ. Interleukin-1β (IL-1β) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neuroscience Letters. 1996;203(1):17–20. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- 77.Guo W, Wang H, Watanabe M, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. Journal of Neuroscience. 2007;27(22):6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Clark AK, Staniland AA, Marchand F, Kaan TKY, McMahon SB, Malcangio M. P2X7-dependent release of interleukin-1β and nociception in the spinal cord following lipopolysaccharide. Journal of Neuroscience. 2010;30(2):573–582. doi: 10.1523/JNEUROSCI.3295-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deleo JA, Colburn RW, Rickman AJ. Cytokine and growth factor immunohistochemical spinal profiles in two animal models of mononeuropathy. Brain Research. 1997;759(1):50–57. doi: 10.1016/s0006-8993(97)00209-6. [DOI] [PubMed] [Google Scholar]
- 80.Hanisch U-K. Microglia as a source and target of cytokines. GLIA. 2002;40(2):140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 81.Reeve AJ, Patel S, Fox A, Walker K, Urban L. Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. European Journal of Pain. 2000;4(3):247–257. doi: 10.1053/eujp.2000.0177. [DOI] [PubMed] [Google Scholar]
- 82.Sung C-S, Wen Z-H, Chang W-K, et al. Intrathecal interleukin-1β administration induces thermal hyperalgesia by activating inducible nitric oxide synthase expression in the rat spinal cord. Brain Research. 2004;1015(1-2):145–153. doi: 10.1016/j.brainres.2004.04.068. [DOI] [PubMed] [Google Scholar]
- 83.Milligan ED, O’Connor KA, Nguyen KT, et al. Intrathecal HIV-1 envelope glycoprotein gp120 induces enhanced pain states mediated by spinal cord proinflammatory cytokines. Journal of Neuroscience. 2001;21(8):2808–2819. doi: 10.1523/JNEUROSCI.21-08-02808.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sweitzer S, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience. 2001;103(2):529–539. doi: 10.1016/s0306-4522(00)00574-1. [DOI] [PubMed] [Google Scholar]
- 85.Milligan ED, Twining C, Chacur M, et al. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. Journal of Neuroscience. 2003;23(3):1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Viviani B, Bartesaghi S, Gardoni F, et al. Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. Journal of Neuroscience. 2003;23(25):8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang R-X, Liu B, Li A, et al. Interleukin 1β facilitates bone cancer pain in rats by enhancing NMDA receptor NR-1 subunit phosphorylation. Neuroscience. 2008;154(4):1533–1538. doi: 10.1016/j.neuroscience.2008.04.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang R-X, Li A, Liu B, et al. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain. 2008;135(3):232–239. doi: 10.1016/j.pain.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zou X, Lin Q, Willis WD. Enhanced phosphorylation of NMDA receptor 1 subunits in spinal cord dorsal horn and spinothalamic tract neurons after intradermal injection of capsaicin in rats. Journal of Neuroscience. 2000;20(18):6989–6997. doi: 10.1523/JNEUROSCI.20-18-06989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gao X, Hee KK, Jin MC, Chung K. Enhancement of NMDA receptor phosphorylation of the spinal dorsal horn and nucleus gracilis neurons in neuropathic rats. Pain. 2005;116(1-2):62–72. doi: 10.1016/j.pain.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 91.Zhang X, Wu J, Lei Y, Fang L, Willis WD. Protein phosphatase modulates the phosphorylation of spinal cord NMDA receptors in rats following intradermal injection of capsaicin. Molecular Brain Research. 2005;138(2):264–272. doi: 10.1016/j.molbrainres.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 92.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annual Review of Neuroscience. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamada K, Nabeshima T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. Journal Pharmacological Sciences. 2003;91(4):267–270. doi: 10.1254/jphs.91.267. [DOI] [PubMed] [Google Scholar]
- 94.Bekinschtein P, Cammarota M, Katche C, et al. BDNF is essential to promote persistence of long-term memory storage. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(7):2711–2716. doi: 10.1073/pnas.0711863105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tsuda M, Shigemoto-Mogami Y, Koizumi S, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424(6950):778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- 96.Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. Journal of Neuroscience. 2009;29(11):3518–3528. doi: 10.1523/JNEUROSCI.5714-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thompson SWN, Bennett DLH, Kerr BJ, Bradbury EJ, Mcmahon SB. Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(14):7714–7718. doi: 10.1073/pnas.96.14.7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rivera C, Li H, Thomas-Crusells J, et al. BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. Journal of Cell Biology. 2002;159(5):747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coull JAM, Boudreau D, Bachand K, et al. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424(6951):938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- 100.Prescott SA, Sejnowski TJ, De Koninck Y. Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: towards a biophysical basis for neuropathic pain. Molecular Pain. 2006;2, article 32 doi: 10.1186/1744-8069-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Janssen SP, Truin M, Van Kleef M, Joosten EA. Differential GABAergic disinhibition during the development of painful peripheral neuropathy. Neuroscience. 2011;184:183–194. doi: 10.1016/j.neuroscience.2011.03.060. [DOI] [PubMed] [Google Scholar]
- 102.Cramer SW, Baggott C, Cain J, et al. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Molecular Pain. 2008;4, article 36 doi: 10.1186/1744-8069-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Keller AF, Beggs S, Salter MW, De Koninck Y. Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Molecular Pain. 2007;3, article 27 doi: 10.1186/1744-8069-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ji R-R, Rupp F. Phosphorylation of transcription factor CREB in rat spinal cord after formalin-induced hyperalgesia: relationship to c-fos induction. Journal of Neuroscience. 1997;17(5):1776–1785. doi: 10.1523/JNEUROSCI.17-05-01776.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. Journal of Neuroscience. 1999;19(11):4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Samad TA, Moore KA, Sapirstein A, et al. Interleukin-1 β-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410(6827):471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 107.Ma W, Quirion R. Increased phosphorylation of cyclic AMP response element-binding protein (CREB) in the superficial dorsal horn neurons following partial sciatic nerve ligation. Pain. 2001;93(3):295–301. doi: 10.1016/S0304-3959(01)00335-9. [DOI] [PubMed] [Google Scholar]
- 108.Ji R-R, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. Journal of Neuroscience. 2002;22(2):478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mannion RJ, Costigan M, Decosterd I, et al. Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(16):9385–9390. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. Journal of Neuroimmune Pharmacology. 2009;4(4):399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Block ML, Zecca L, Hong J-S. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature Reviews Neuroscience. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 112.Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. Journal of Neuroinflammation. 2005;2, article 24 doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KHG, Lynch MA. CD200 ligand-receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. Journal of Neuroscience. 2007;27(31):8309–8313. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ledeboer A. Interleukin-10, interleukin-4, and transforming growth factor-beta differentially regulate lipopolysaccharide-induced production of pro-inflammatory cytokines and nitric oxide in co-cultures of rat astrol and microglial cells. GLIA. 2000;30(2):134–142. doi: 10.1002/(sici)1098-1136(200004)30:2<134::aid-glia3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 115.Lee YB, Nagai A, Kim SU. Cytokines, chemokines, and cytokine receptors in human microglia. Journal of Neuroscience Research. 2002;69(1):94–103. doi: 10.1002/jnr.10253. [DOI] [PubMed] [Google Scholar]
- 116.Kitamura Y, Taniguchi T, Kimura H, Nomura Y, Gebicke-Haerter PJ. Interleukin-4-inhibited mRNA expression in mixed rat glial and in isolated microglial cultures. Journal of Neuroimmunology. 2000;106(1-2):95–104. doi: 10.1016/s0165-5728(00)00239-3. [DOI] [PubMed] [Google Scholar]
- 117.Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. Journal of Neuroscience. 2007;27(40):10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. Journal of Neuroinflammation. 2006;3, article 27 doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lyons A, Griffin RJ, Costelloe CE, Clarke RM, Lynch MA. IL-4 attenuates the neuroinflammation induced by amyloid-β in vivo and in vitro. Journal of Neurochemistry. 2007;101(3):771–781. doi: 10.1111/j.1471-4159.2006.04370.x. [DOI] [PubMed] [Google Scholar]
- 120.Park C-K, Lü N, Xu Z-Z, Liu T, Serhan CN, Ji R-R. Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. Journal of Neuroscience. 2011;31(42):15072–15085. doi: 10.1523/JNEUROSCI.2443-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Xu Z-Z, Zhang L, Liu T, et al. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nature Medicine. 2010;16(5):592–597. doi: 10.1038/nm.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ji RR, Berta T, Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain. 2013 doi: 10.1016/j.pain.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xu ZZ, Berta T, Ji RR. Resolvin e1 inhibits neuropathic pain and spinal cord microglial activation following peripheral nerve injury. Journal of NeuroImmune Pharmacology. 2013;8(1):37–41. doi: 10.1007/s11481-012-9394-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xu ZZ, Liu XJ, Berta T, et al. Neuroprotectin/Protectin D1 protects neuropathic pain in mice after nerve trauma. Annals of Neurology. 2013 doi: 10.1002/ana.23928. [DOI] [PMC free article] [PubMed] [Google Scholar]