

SUMMARY
Angiogenesis, the emergence of vessels from an existing vascular network, is pathologically associated with tumor progression and is of great interest for therapeutic intervention. PTEN is a frequently mutated tumor suppressor and has been linked to the progression of many types of tumors, including hemangiosarcomas in zebrafish. Here, we report that mutant zebrafish embryos lacking functional Pten exhibit enhanced angiogenesis, accompanied by elevated levels of phosphorylated Akt (pAkt). Inhibition of phosphoinositide 3-kinase (PI3K) by LY294002 treatment and application of sunitinib, a widely used anti-angiogenic compound, suppressed enhanced angiogenesis in Pten mutants. Vegfaa has a crucial role in angiogenesis and vegfaa expression was upregulated in embryos lacking functional Pten. Interestingly, vegfaa expression was also upregulated in hemangiosarcomas from haploinsufficient adult zebrafish Pten mutants. Elevated vegfaa expression in mutant embryos lacking functional Pten was suppressed by LY294002. Surprisingly, sunitinib treatment dramatically enhanced vegfaa expression in Pten mutant embryos, which might account for tumor relapse in human patients who are treated with sunitinib. Combined treatment with suboptimal concentrations of sunitinib and LY294002 rescued enhanced angiogenesis in pten mutant embryos without the dramatic increase in vegfaa expression, suggesting a new approach for therapeutic intervention in VEGFR-signaling-dependent tumors.
INTRODUCTION
PTEN is one of the most frequently mutated tumor suppressor genes found in cancer (Stokoe, 2001). Somatic deletion of PTEN leads to tissue-specific tumor formation and germline deletion of PTEN is associated with syndromes such as Cowden’s disease, Bannayan-Zonana and Lhermitte-Duclos disease (Liaw et al., 1997; Marsh et al., 1997; Zhou et al., 2003). Individuals with those syndromes share pathological features, including the formation of benign tumors and enhanced susceptibility to malignant cancer. PTEN, a lipid and protein phosphatase, antagonizes the phosphoinositide 3-kinase (PI3K)-Akt (also called PKB) pathway by balancing the cellular phosphatidylinositol (3,4,5)-trisphosphate [PtdIns(3,4,5)P3; also known as PIP3] level (Maehama and Dixon, 1998; Myers et al., 1998). Loss of PTEN increases PIP3 levels, resulting in constitutive activation of Akt signaling. Cell survival and proliferation are linked to activated Akt and thus uncontrolled activation of Akt leads to enhanced cell survival and proliferation, the hallmarks of cancer.
The zebrafish genome encodes two pten genes, designated ptena and ptenb (Croushore et al., 2005; Faucherre et al., 2008). Single mutants are viable and fertile, suggesting redundant function during development. Concomitant loss of Ptena and Ptenb results in embryonic lethality (Faucherre et al., 2008), reminiscent of loss-of-function of PTEN in mice (Di Cristofano et al., 1998), Caenorhabditis elegans (Mihaylova et al., 1999) and Drosophila (Goberdhan et al., 1999). We recently reported that haploinsufficiency of Pten in zebrafish (ptena+/−ptenb−/− and ptena−/−ptenb+/−) results in hemangiosarcoma formation during adult life (Choorapoikayil et al., 2012). The mechanism underlying uncontrolled endothelial growth resulting in hemangiosarcoma is not understood.
In vitro studies showed that inhibition of endogenous PTEN in cultured endothelial cells enhances vascular endothelial growth factor (VEGF) signaling (Huang and Kontos, 2002). VEGFs, secreted ligands binding to VEGF receptors (VEGFRs), are key players in vasculogenesis and angiogenesis. VEGF signaling promotes proliferation and differentiation of endothelial cells. The human VEGF family consists of five related growth factors, VEGFA, VEGFB, VEGFC, VEGFD and PIGF (placental growth factor). From these five secreted ligands, VEGFA was shown to be the main factor during angiogenesis, functioning as a mitogen, acting specifically on endothelial cells (Koch et al., 2011). It has been demonstrated that VEGFB promotes fatty acid uptake in endothelial cells (Hagberg et al., 2010; Li et al., 2008) and the role of VEGFB during angiogenesis is not fully elucidated yet. VEGFC is, together with VEGFD, crucial for lymphangiogenesis and has a minor role in vasculogenesis and angiogenesis (Koch et al., 2011).
We set out to study the function of Pten in endothelial cells in vivo. To this end, we investigated angiogenesis during embryonic development in ptena−/−ptenb−/− mutants. Here we report that ptena−/−ptenb−/− mutants displayed ectopic vessel growth. Inhibition of PI3K signaling rescued hyperplasia of endothelial cells. Moreover, treatment of ptena−/−ptenb−/− mutants with sunitinib, an angiogenesis inhibitor that selectively inhibits receptor tyrosine kinases (RTKs), also rescued enhanced angiogenesis. We found that elevated overall phosphorylated Akt (pAkt) levels in embryos were suppressed by PI3K inhibitors, and to a lesser extent by sunitinib.
TRANSLATIONAL IMPACT.
Clinical issue
The PTEN gene is the second most frequently mutated tumor suppressor gene in human cancer. The gene encodes PTEN, a lipid and protein phosphatase, loss of which is associated with enhanced cell survival and proliferation, both hallmarks of cancer. Previously, this group reported that zebrafish (which express two pten genes, ptena and ptenb) that retain only a single wild-type allele of ptena or ptenb develop hemangiosarcomas, tumors of endothelial origin. The mechanisms underlying uncontrolled endothelial cell growth in the development of hemangiosarcomas are unknown. However, in vitro studies have suggested that loss of endogenous PTEN augments vascular endothelial growth factor (VEGF) signaling, which is involved in angiogenesis and vasculogenesis. The role of PTEN in endothelial cells in vivo has not yet been examined.
Results
Here, the authors generated a zebrafish model lacking functional Pten to analyze the role of the protein in cancer and development. Zebrafish embryos lacking functional Pten (ptena−/–ptenb−/–) displayed increased angiogenesis. The authors showed that hypervascularization could be rescued by exogenous ptena RNA and also by a phosphoinositide 3-kinase (PI3K) inhibitor, LY294002, and an angiogenesis inhibitor, sunitinib. Sunitinib acts by inhibiting receptor tyrosine kinases, including angiogenesis-promoting receptors in the VEGF signaling pathway. The authors also report that Pten mutants display enhanced expression of vegfaa, a ligand of VEGF receptors (VEGFRs). Interestingly, enhanced vegfaa expression was also observed in hemangiosarcomas from Pten haploinsufficient adult mutants. In the embryos, vegfaa expression was suppressed by LY294002, but, surprisingly, sunitinib treatment dramatically enhanced vegfaa expression. However, combination treatment of Pten mutant zebrafish embryos with low concentrations of LY294002 and sunitinib fully rescued the hypervascularization phenotype without enhancing vegfaa expression.
Implications and future directions
These results indicate that angiogenesis and vegfaa expression are enhanced in Pten zebrafish mutants, which could have important implications for humans with tumors that lack functional PTEN. Sunitinib has been used to suppress angiogenesis in cancer patients; however, successful treatment is followed by severe relapse in some cases. An increase in the expression of the human homolog of vegfaa in response to sunitinib treatment might explain this relapse. Moreover, this work provides evidence that combined treatment with a PI3K inhibitor and sunitinib suppresses hypervascularization without enhancing vegfaa expression, suggesting a new approach for therapeutic intervention in VEGFR-signaling-dependent tumors such as hemangiosarcomas.
Vegfaa expression was upregulated in ptena−/−ptenb−/− mutants and inhibition of PI3K abolished upregulation of vegfaa. Surprisingly, vegfaa expression was dramatically upregulated by sunitinib treatment. Combining PI3K inhibitors and sunitinib cooperatively rescued hypervascularization in ptena−/−ptenb−/− zebrafish embryos, revealing a tentative therapeutic approach to combat neovascularization in cancer.
RESULTS
ptena−/−ptenb−/− mutants display enhanced angiogenesis
Haploinsufficiency of Pten leads to uncontrolled proliferation of endothelial cells, resulting in the formation of hemangiosarcomas in zebrafish (Choorapoikayil et al., 2012). To investigate how loss of Pten supports tumor growth and in particular how loss of Pten affects endothelial cells, we visualized the vasculature in zebrafish ptena−/−ptenb−/− mutant embryos using the Tg(kdrl:eGFP) line (Jin et al., 2005). The anatomy of the vasculature in the trunk was monitored between 2 and 4 dpf. We observed excessive sprouting of endothelial cells of the intersegmental vessels, in that these cells developed excessive filopodia from 72 hpf onwards (Fig. 1), resulting in ectopic vessel growth at 4 dpf (Fig. 2A–B′). Time-lapse imaging revealed that endothelial cells lacking Pten display protruding filopodia in a highly dynamic manner, whereas endothelial cells in siblings remain quiescent (supplementary material Movies 1, 2 and Fig. S1). Examination of three intersegmental vessels in 11 ptena−/−ptenb−/− mutant embryos at 4 dpf revealed that, on average, each intersegmental vessel formed two ectopic sprouts. At this stage, no sprouting was observed in the intersegmental vessels of siblings. Enhanced angiogenesis in ptena−/−ptenb−/− embryos was not restricted to the trunk and tail region, and was also observed in the head (supplementary material Fig. S2). Using confocal microscopy, we observed that newly formed vessels are perfused at 3 and 4 dpf (data not shown). Mutants retaining one wild-type allele (ptena+/−ptenb−/− or ptena−/−ptenb+/−) do not display any detectable malformations in vasculogenesis or angiogenesis during embryonic development (supplementary material Fig. S3). Taken together, we found that angiogenesis was enhanced in ptena−/−ptenb−/− mutants, resulting in hypervascularization.
Fig. 1.
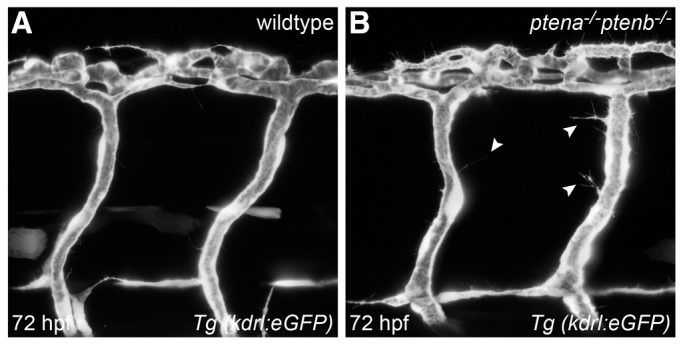
Loss of Ptena and Ptenb leads to excessive filopodia formation in endothelial cells at 72 hpf. Endothelial cells in living wild-type (A) and ptena−/−ptenb−/− mutant (B) embryos were visualized using Tg(kdrl:eGFP) and confocal imaging was performed at 70–72 hpf. Intersegmental vessels along the trunk in ptena−/−ptenb−/− mutants (4/4) show excessive filopodia formation (arrowheads), whereas no filopodia were observed in wild-type (0/4) embryos. Anterior to the left, 40× + 1.5 zoom, 0.5 μm step size.
Fig. 2.
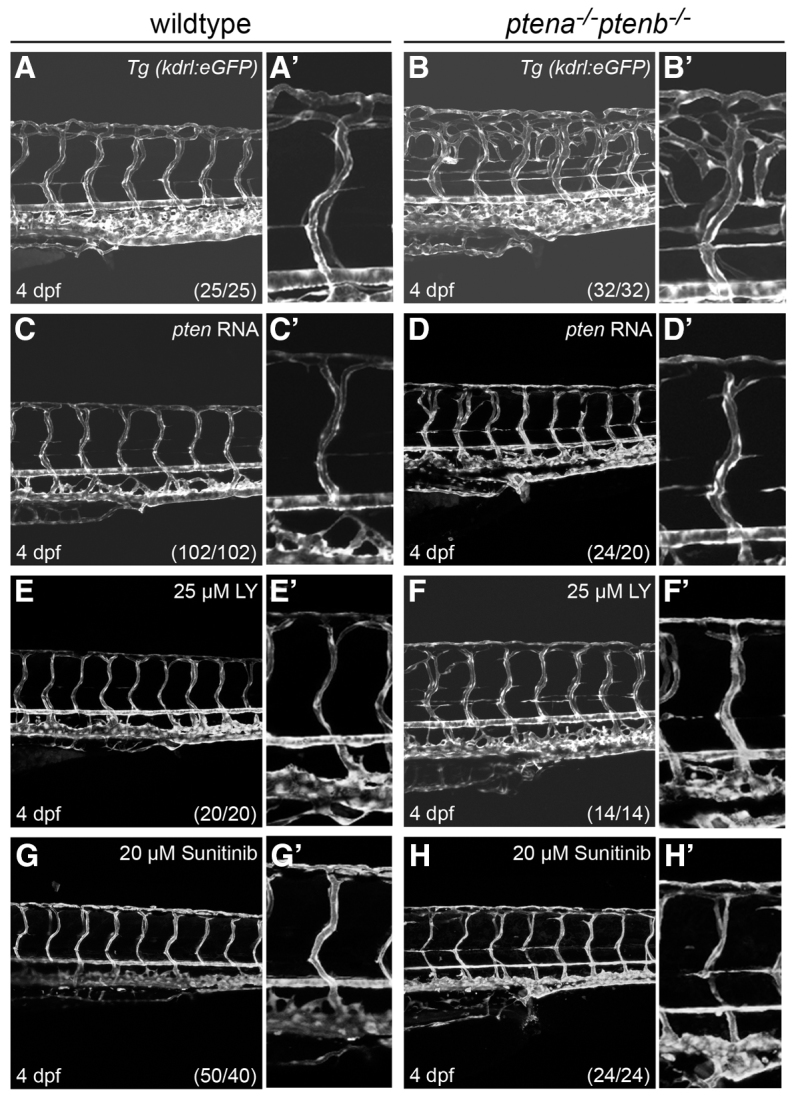
Rescue of enhanced angiogenesis in ptena−/−ptenb−/− mutants by exogenous ptena mRNA, LY294002 or sunitinib. The transgenic line, Tg(kdrl:eGFP), was used to visualize the vasculature at 4 dpf in wild-type (A,C,E,G) and ptena−/−ptenb−/− (B,D,F,H) embryos. Close-ups of the intersegmental vessel above the urogenital opening are shown in adjacent panels. (A–B′) ptena−/−ptenb−/− mutants display ectopic vessel growth compared with wild-type embryos. (C–D′) 10 pg synthetic ptena mRNA was injected at the one-cell stage into wild-type and ptena−/−ptenb−/− embryos. (E–F′) 25 μM LY294002 (LY) was applied from 70–72 hpf onwards. (G–H′) 20 μM sunitinib was applied from 70–72 hpf onwards. Images were taken using a confocal microscope with 20×. The numbers in the bottom right corner represent the total number of embryos treated/the number of embryos showing the phenotype depicted here. Anterior to the left, 20×, 2 μm step size.
Hypervascularization in ptena−/−ptenb−/− mutants is rescued by LY294002 and sunitinib
To investigate the signaling underlying hypervascularization in ptena−/−ptenb−/− mutant embryos, we performed rescue experiments. Although vascularization throughout the embryo was affected, we focused on the trunk and tail region. Microinjection of ptena mRNA in ptena−/−ptenb−/− mutants at the one-cell stage suppressed the enhanced angiogenic phenotype at 4 dpf (Fig. 2D,D′). Similar rescues were obtained with microinjection of ptenb mRNA (data not shown). Ectopic expression of moderate amounts of Ptena in wild-type embryos did not affect the vasculature grossly (Fig. 2C,C′). The overall morphology of ptena−/−ptenb−/− mutant embryos is distinct from wild-type embryos in that the mutants are shorter and particularly the trunk and tail region is wider. These defects are largely, but not completely, rescued by injection of ptena mRNA (supplementary material Fig. S4). Morphological analysis revealed that, at 4 dpf, wild-type embryos injected with ptena mRNA displayed mild defects in body length (supplementary material Fig. S4A,C).
To investigate whether enhanced PI3K signaling is associated with enhanced angiogenesis in loss of Pten mutants, we treated embryos with the PI3K inhibitor LY294002 from the earliest time point at which we observed defects (70–72 hpf) onwards. Earlier treatment with LY294002 induces severe defects in the vasculature (Herbert et al., 2009) as well as defects as early as gastrulation (Montero et al., 2003). The overall morphology and vasculature of treated embryos was examined at 4 dpf. Wild-type embryos displayed mild defects in head size, and body length was reduced compared with non-treated embryos (supplementary material Fig. S4A,E). Consistent with our previous report (Faucherre et al., 2008), the morphological phenotype of ptena−/−ptenb−/− mutants was largely rescued by LY294002 treatment (supplementary material Fig. S4F,F′). In addition, the excessive sprouting phenotype in ptena−/−ptenb−/− mutants was largely rescued at 4 dpf after treatment with LY294002 (Fig. 2F,F′). Wild-type embryos treated with LY294002 displayed mild defects in vessel morphology, suggesting that endothelial cells are highly responsive to altered PI3K/Akt levels (Fig. 2E,E′). Thus, antagonizing the PI3K pathway suppressed ectopic vessel growth in ptena−/−ptenb−/− mutants, indicating that PI3K signaling has a central role in angiogenesis.
Next, we investigated whether inhibition of angiogenesis in ptena−/−ptenb−/− mutants suppressed the phenotype. To this end, we used the angiogenesis inhibitor sunitinib, which selectively inhibits RTKs (Roskoski, 2007), including VEGFRs in embryos. Wild-type embryos that were treated from 70–72 hpf onwards with sunitinib displayed no obvious morphological malformation in the vasculature (Fig. 2G,G′). Examination of the vasculature in ptena−/−ptenb−/− mutants at 4 dpf revealed that enhanced angiogenesis was suppressed by sunitinib treatment (Fig. 2G–H′). Our results suggest that signaling by sunitinib-sensitive RTKs has a crucial role in hypervascularization in Pten mutants.
Elevated pAkt level in ptena−/−ptenb−/− mutants is suppressed by LY294002 and to a lesser extent by sunitinib
Pten antagonizes PI3K signaling upstream of the Akt pathway and, consequently, loss of Pten leads to constitutive activation of Akt. We assessed pAkt levels by immunoblotting of individual embryos at 4 dpf. As expected, ptena−/−ptenb−/− mutants displayed dramatically enhanced levels of pAkt compared with wild-type embryos at 4 dpf (Fig. 3; supplementary material Fig. S5). Whereas pAkt levels varied from embryo to embryo, pAkt levels were consistently elevated in ptena−/−ptenb−/− embryos, compared with wild-type embryos. Re-expression of Ptena resulted in downregulation of elevated pAkt in ptena−/−ptenb−/− mutant embryos (Fig. 3). Similarly, we observed suppressed levels of pAkt in ptena−/−ptenb−/− mutant embryos upon treatment with the PI3K inhibitor LY294002 (Fig. 3). Sunitinib treatment reduced elevated pAkt levels in ptena−/−ptenb−/− mutants to a much lesser extent than did Ptena expression or LY294002 treatment (Fig. 3). pAkt levels were also reduced in wild-type embryos by expression of Ptena and by LY294002 or sunitinib treatment (Fig. 3). In summary, elevated pAkt levels in Pten mutants were suppressed by expression of Ptena and by treatment with LY294002 or sunitinib.
Fig. 3.
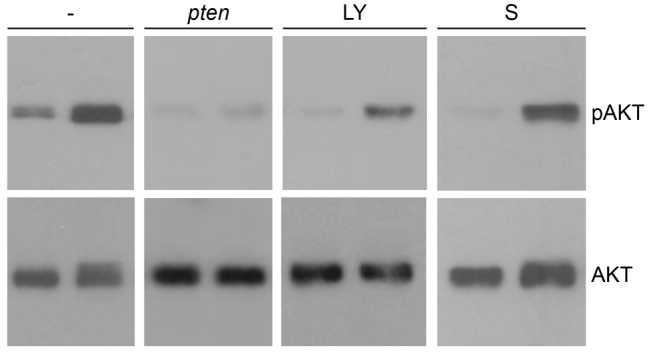
Elevated pAkt level in ptena−/−ptenb−/− mutants is suppressed by LY294002 and to a lesser extent by sunitinib. Wild-type (left lane of each blot) and ptena−/−ptenb−/− mutant (right lane of each blot) embryos were left untreated (–), were injected at the one-cell stage with ptena mRNA (pten), or were treated with 25 μM LY294002 (LY) or 20 μM sunitinib (S) from 72 hpf onwards. Single embryos were lysed at 4 dpf and the protein from individual embryos was isolated. The proteins were run on a denaturing SDS-polyacrylamide gel and transferred to PVDF membranes. After blocking, the blot was probed with phosphospecific anti-pAkt antibody (directed against pSer473), stripped and probed with Akt-specific antibody as a loading control. The number of individual embryos that were analyzed is: wild type, 24; mutant, 23; wild type + pten, 10; mutant + pten, 15; wild type + LY, 2; mutant + LY, 6; wild type + S, 5; mutant + S, 5. Representative blots are depicted here. The intensities of the bands were quantified (supplementary material Fig. S5).
ptena−/−ptenb−/− mutants display enhanced expression of vegfaa
VEGF signaling, in particular that of vegfaa, is indispensable for angiogenesis. To address whether VEGF signaling is involved in enhanced angiogenesis in Pten mutants, we examined vegfaa expression levels at 4 dpf by quantitative PCR. Vegfaa expression was dramatically upregulated (eightfold) in ptena−/−ptenb−/− mutants compared with wild type (Fig. 4A). To assess at which developmental stage vegfaa expression is elevated in ptena−/−ptenb−/− mutants, we performed time course analysis at 1, 2 and 3 dpf. At 1 and 2 dpf of development, no difference in expression was detected between mutants and wild types. We found that vegfaa is significantly upregulated (threefold) from 3 dpf onwards (Fig. 4B), which coincides with the onset of enhanced filopodia formation in ptena−/−ptenb−/− mutant embryos (cf. Fig. 1). In order to verify upregulation of vegfaa expression, we performed whole-mount in situ hybridization. Consistent with the results obtained by quantitative PCR, we found elevated vegfaa expression at 3 and 4 dpf in mutants lacking Pten (Fig. 4C–F). The vegfaa expression pattern was rather diffuse and predominantly in the anterior region of the embryos. Next, we addressed whether the rescued angiogenic phenotype in ptena−/−ptenb−/− mutants after re-expression of Pten is associated with downregulation of vegfaa. We found that restoring Ptena expression in ptena−/−ptenb−/− mutants significantly downregulated the elevated vegfaa level (from eightfold to 2.5-fold) (Fig. 4A). Similarly, we found that vegfaa expression was significantly downregulated in ptena−/−ptenb−/− mutants by LY294002 (from eightfold to twofold; Fig. 4A). Surprisingly, vegfaa expression was dramatically enhanced by sunitinib in ptena−/−ptenb−/− mutants (from eightfold to 40-fold, compared with untreated wild type). In wild-type embryos, sunitinib treatment induced a modest increase in vegfaa expression (fourfold; Fig. 4A). Taken together, loss of Pten led to elevated vegfaa expression, which was rescued by inhibition of PI3K. Inhibition of angiogenesis using sunitinib greatly enhanced vegfaa expression in wild-type and ptena−/−ptenb−/− embryos, suggesting a feedback loop.
Fig. 4.
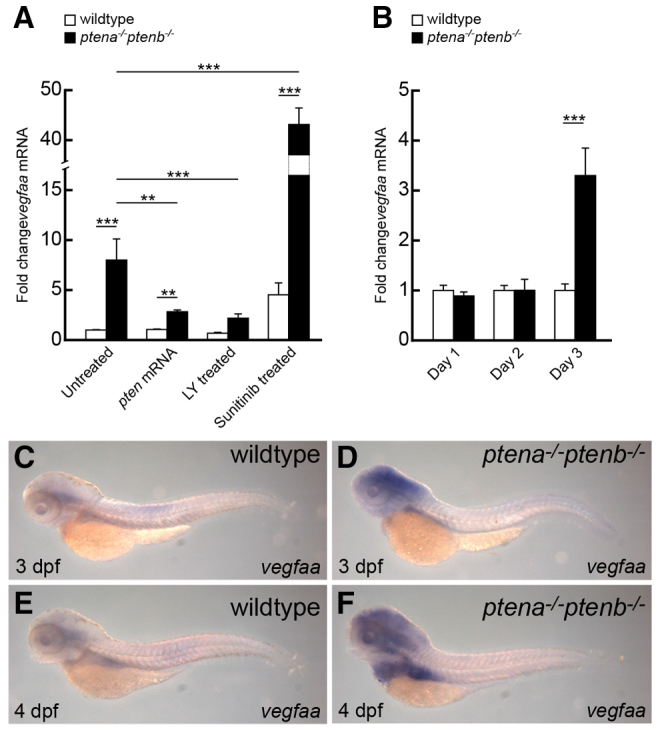
Upregulated vegfaa expression in ptena−/−ptenb−/− mutants is diminished by LY294002 and enhanced by sunitinib. (A,B) Quantitative PCR was performed to determine vegfaa expression levels in ptena−/−ptenb−/− mutants compared with wild type at 4 dpf (A) and at 1, 2 and 3 dpf (B). Rescue experiments were done by microinjection of ptena mRNA at the one-cell stage or by treatment with 25 μM LY294002 (LY) or 20 μM sunitinib from 72 hpf onwards. Wild-type control was set to 1 and all values were determined relative to the wild-type control at 3 dpf. Three embryos were pooled per condition and the data represent the results of at least two independent experiments. Statistical analysis (Kruskal-Wallis with Dunn’s post-hoc test) was performed using Excel and SPSS 20 (IBM); significance is indicated: **P<0.01; ***P<0.001. Note that the y-axis is discontinuous to accommodate the 40-fold increase in vegfaa expression upon sunitinib treatment of ptena−/−ptenb−/− mutants. (C–F) In situ hybridization was performed with a vegfaa-specific probe on 3-dpf or 4-dpf wild-type and ptena−/−ptenb−/− mutant embryos, as indicated. The number of embryos analyzed is: wild type 3 dpf, 6; wild type 4 dpf, 4; ptena−/−ptenb−/− 3 dpf, 4; ptena−/−ptenb−/− 4 dpf, 5. Representative pictures are depicted here. Pictures were taken with a 4.5× objective.
Combined LY294002 and sunitinib treatment abolished enhanced vegfaa expression and reduced hypervascularization
Sunitinib is a widely used anti-angiogenic compound that prevents neovascularization (Roskoski, 2007). Our results demonstrate that sunitinib treatment led to dramatic upregulation of vegfaa expression, particularly in ptena−/−ptenb−/− mutant embryos (Fig. 4). LY294002 treatment rescued elevated vegfaa expression to some extent. We hypothesized that LY294002 treatment might suppress sunitinib-induced vegfaa expression and the two inhibitors might cooperate to suppress enhanced angiogenesis. To test this, we combined LY294002 and sunitinib at suboptimal doses. A suboptimal concentration of LY294002 (5 μM) did not fully repress enhanced angiogenesis (Fig. 5B–E), but did suppress enhanced vegfaa expression in ptena−/−ptenb−/− mutant embryos (Fig. 5A), suggesting that vegfaa expression is tightly regulated by PI3K signaling. A suboptimal concentration of sunitinib (5 μM) did not fully repress enhanced angiogenesis in ptena−/−ptenb−/− mutant embryos (Fig. 5G) and still led to an eightfold increase in vegfaa expression (Fig. 5A), indicating that a slight modification of VEGFR signaling still has a dramatic effect on vegfaa expression. Concomitant application of suboptimal concentrations of LY294002 and sunitinib significantly suppressed vegfaa expression and fully inhibited hypervascularization in ptena−/−ptenb−/− mutant embryos (Fig. 5A,I). Analysis of pAkt levels following treatment with suboptimal concentrations of LY294002 or sunitinib indicated that these treatments did not fully suppress enhanced pAkt levels in ptena−/−ptenb−/− mutants. Combined treatment led to further downregulation of pAkt, but still did not completely suppress pAkt levels (supplementary material Fig. S6). Our data indicate that simultaneous partial inhibition of PI3K and VEGFR signaling cooperatively suppressed enhanced angiogenesis in ptena−/−ptenb−/− mutant embryos.
Fig. 5.
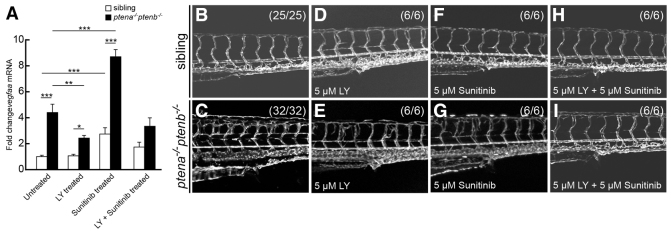
Combined treatment with LY294002 and sunitinib rescued hypervascularization. (A) Quantitative PCR was performed to determine vegfaa expression at 4 dpf in ptena−/−ptenb−/− mutants compared with siblings following treatment with suboptimal concentrations of LY294002 (LY, 5 μM) or sunitinib (5 μM), or both, from 72 hpf onwards. Three embryos per condition were pooled and used for quantitative PCR; at least two independent experiments were performed. Relative amounts were determined with wild type untreated set to 1.0. Statistical analysis (Kruskal-Wallis with Dunn’s post-hoc test) was performed using Excel and SPSS 20 (IBM); significance is indicated: **P<0.01; ***P<0.001. (B–I) Vasculature of wild-type and ptena−/−ptenb−/− embryos at 4 dpf was imaged in the Tg(kdrl:eGFP) line by confocal microscope with 20×. The embryos were treated with suboptimal concentrations of LY294002 (LY), sunitinib or both as indicated. Representative embryos are depicted; anterior to the left.
Hemangiosarcoma formation in Pten haploinsufficient fish is accompanied by elevated vegfaa expression
Ptena+/−ptenb−/− and ptena−/−ptenb+/− mutant adult fish are prone to develop hemangiosarcomas during their lifetime (Choorapoikayil et al., 2012). We have established that these hemangiosarcomas are preferentially formed in the rete mirabile, a highly vascularized tissue that is connected to the eye bulb. In general, hemangiosarcomas are associated with the vasculature and consist of perfused endothelial lumens. We investigated whether vegfaa expression was enhanced in hemangiosarcomas of pten mutant adult fish by quantitative PCR. We isolated RNA from the tumors and from contralateral tissue of the same animals and, as a control, we isolated RNA from roughly the same tissue in wild-type zebrafish. Vegfaa expression was threefold higher in the hemangiosarcoma than in wild-type tissue. Vegfaa expression in the contralateral tissue from the tumor-bearing fish was not significantly different from vegfaa expression in wild type (Fig. 6). Taken together, we show that vegfaa expression is enhanced in hemangiosarcomas, which might enhance tumor growth.
Fig. 6.
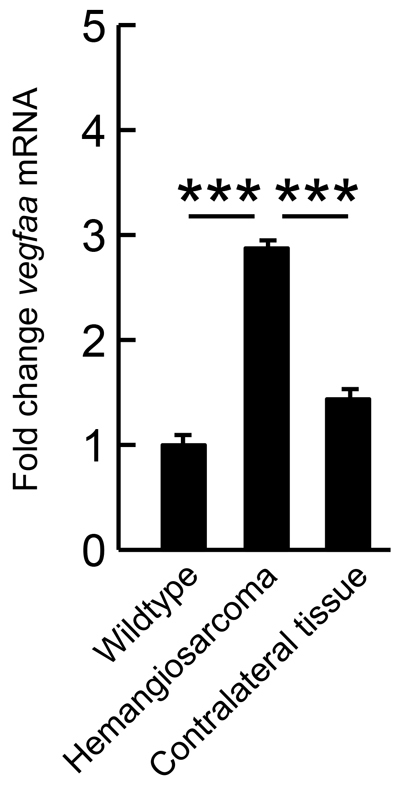
Elevated expression of vegfaa in hemangiosarcoma. Hemangiosarcoma tumor material of ptena+/−ptenb−/− mutants (n=3) was isolated. Contralateral tissue from the tumor-bearing fish and tissue from the same area in wild-type adult fish were isolated as control. RNA was isolated and quantitative PCR was performed to establish vegfaa expression. Statistical analysis was performed using Excel and fold-change values were determined with wild type set to 1.0; significance is indicated: ***P<0.001.
DISCUSSION
PTEN is one of the most frequently mutated tumor suppressor genes in cancer. Concomitant loss of both pten genes in zebrafish leads to hyperplasia and dysplasia, resulting in embryonic lethality by 5 dpf (Faucherre et al., 2008). Mutants that retain one wild-type pten allele (ptena+/−ptenb−/− or ptena−/−ptenb+/−) are prone to develop endothelial-derived hemangiosarcomas later in life. Here, we investigated angiogenesis in the absence of functional Pten during zebrafish embryogenesis and found a dramatic hypervascularization in the vasculature throughout the embryo. Single pten mutants and mutants retaining one active pten allele (ptena+/−ptenb−/− or ptena−/−ptenb+/−) do not display any malformation in the vasculature. Hence, we conclude that Ptena and Ptenb have redundant functions in angiogenesis/vasculogenesis. In ptena−/−ptenb−/− mutants, we observed enhanced sprouting from 3 dpf onwards, resulting in the formation of ectopic blood vessels at 4 dpf. Normally, once the vasculature has been established, endothelial cells are quiescent and rarely form new branches. The absence of defects in vasculature at earlier time points in ptena−/−ptenb−/− embryos might be due to maternally provided Pten. However, this is unlikely because immunoblotting demonstrated that maternally contributed Pten was not detectable anymore from 60 hpf onwards (data not shown), well before the stage at which we observed enhanced angiogenesis. Perhaps Pten is not essential for vasculogenesis, i.e. de novo formation of blood vessels, and it only has a role in angiogenesis. Interestingly, it has been reported that PI3K signaling is essential for angiogenesis in mouse and fish development. Mouse mutant embryos with a homozygous mutation in the PI3K catalytic subunit (p110αD933A/D933A) show regular heartbeat and blood flow in central vessels until E10.5, indicating that vasculogenesis is normal. However, at E12.5, phosphorylation of Akt in p110αD933A/D933A mutants is absent and embryos are lethal, exhibiting primary angiogenic remodeling defects (Graupera et al., 2008). We conclude that the loss of Pten induced defects in angiogenesis, not vasculogenesis.
Hypervascularization was not limited to the trunk area. We also observed massive increases in blood vessels in other areas of the embryo, including the head, by imaging ptena−/−ptenb−/− and wild-type embryos in the Tg(kdrl:eGFP) line (supplementary material Fig. S2). However, we focused on hypervascularization in the trunk and tail and investigated the molecular basis for upregulated endothelial proliferation in Pten mutant embryos by treatment with the inhibitors LY294002 and sunitinib. Treatment of ptena−/−ptenb−/− embryos with the PI3K inhibitor LY294002 from 72 hpf onwards rescued the hypervascularization phenotype at 4 dpf, indicating that these defects were caused by enhanced PI3KAkt signaling. Consistent with this notion is that elevated pAkt levels in Pten mutant embryos were suppressed by LY294002 treatment. The morphological defects in ptena−/−ptenb−/− mutants were also largely rescued by LY294002 treatment, which is consistent with our earlier report in which we treated embryos from 2 dpf onwards (Faucherre et al., 2008). Inhibition of PI3K at very early stages induced severe gastrulation defects (Montero et al., 2003), which precludes a full rescue of the loss of Pten phenotype by early treatment with LY294002.
Sunitinib treatment led to a full rescue of hypervascularization at 4 dpf. Yet, sunitinib did not fully suppress enhanced pAkt levels in ptena−/−ptenb−/− mutants. Sunitinib selectively inhibits a subset of RTKs, including the angiogenic VEGFR1, VEGFR2 and PDGFRβ (Roskoski, 2007). PI3K-Akt signaling downstream of other RTKs is not affected by sunitinib. Therefore, it is not surprising that sunitinib treatment did not fully suppress pAkt levels in ptena−/−ptenb−/− mutants. Apparently, inhibition of the angiogenic RTKs by sunitinib fully rescued hypervascularization in ptena−/−ptenb−/− mutants.
It seems that endothelial cells are particularly sensitive to loss of Pten. Previously, we reported that pten haploinsufficient zebrafish predominantly developed hemangiosarcomas, tumors of endothelial origin (Choorapoikayil et al., 2012). Moreover, recent work demonstrated that mouse endothelial cells lacking Pten are hypersensitive to vascular growth factor stimulation (Hamada et al., 2005). Enhanced sensitivity of endothelial cells to loss of Pten might be intrinsic to these cells. However, the finding that vegfaa expression is enhanced in ptena−/−ptenb−/− embryos suggests that this might contribute to enhanced sensitivity of endothelial cells to loss of Pten, because these cells express VEGFRs, providing positive feedback. Upregulation of VEGF expression in response to deletion of Pten is not unprecedented. siRNA-mediated knockdown of PTEN in a panel of pancreatic cell lines led to upregulation of VEGF expression (Ma et al., 2009). Moreover, ectopic expression of PTEN in the chronic myelogenous leukemia cell line, K562, led to reduced expression of VEGF (Zhiyong et al., 2012), which is consistent with our data in zebrafish. Elevated vegfaa expression in ptena−/−ptenb−/− zebrafish embryos is suppressed by treatment with LY294002, indicating that upregulation of vegfaa expression in ptena−/−ptenb−/− embryos is dependent on PI3K signaling. Sunitinib treatment led to a dramatic increase in vegfaa expression, particularly in ptena−/−ptenb−/− mutant embryos, suggesting a feedback mechanism. Inhibition of VEGFR1 and VEGFR2 and a subset of other RTKs enhanced expression of the VEGFR ligand Vegfaa. The mechanism underlying transcriptional regulation of vegfaa in Pten mutants and in response to inhibitors remains to be determined.
VEGF signaling is crucial for vascular development during embryogenesis. Elevated levels of vegfaa mRNA expression were detected from 72 hpf onwards, which is concomitant with the onset of enhanced angiogenesis, suggesting a causal relation. To address directly whether elevated vegfaa expression induced enhanced angiogenesis, we used morpholinos to knockdown Vegfaa expression. Unfortunately, Vegfaa knockdown induced massive defects in vasculature in wild-type embryos, consistent with previous reports (Nasevicius et al., 2000; Weijts et al., 2012), precluding assessment of the effect of Vegfaa knockdown on angiogenesis in ptena−/−ptenb−/− embryos. Elevated expression of vegfaa was not limited to ptena−/−ptenb−/− embryos. Significant upregulation of vegfaa expression was also observed in hemangiosarcomas that were isolated from adult zebrafish mutants that retained one wild-type allele of pten. Hemangiosarcomas are tumors that consist of endothelial cells and exhibit constitutive expression of Vegfr2 (kdrl) (Jinnin et al., 2008). Elevated vegfaa expression will result in a positive feedback loop, which might account for the hyperproliferation of endothelial cells in the hemangiosarcoma and hence contribute to tumor growth.
Sunitinib is commonly used as an anti-angiogenic drug to prevent (tumor) angiogenesis. Clinical reports describe cases in which, after administration of sunitinib, tumor relapse occurred with severe growth and increased metastatic behavior (Kikuchi et al., 2012; Tielen et al., 2012; Tonini et al., 2010). Here we discovered that treatment with sunitinib led to upregulation of vegfaa in wild-type embryos and to a further upregulation of vegfaa expression in mutant embryos lacking Pten. Transcriptional upregulation of VEGFA expression in response to sunitinib in patients will result in long-term enhanced VEGFA expression. By the time sunitinib has lost its potency, VEGFA expression is still elevated, leading to hyperactivation of VEGFRs, resulting in hyperproliferation of endothelial cells, hence explaining the tumor relapse after sunitinib treatment. Treatment with suboptimal concentrations of LY294002 and sunitinib did not lead to dramatic increases in vegfaa expression in zebrafish embryos, yet it fully rescued the hypervascularization phenotype. These results suggest that combined treatment might represent a novel approach for therapeutic intervention.
MATERIALS AND METHODS
Zebrafish husbandry
ptena−/−ptenb−/− and Tg(kdrl:eGFP) (Jin et al., 2005) were maintained, crossed, raised and staged as described (Kimmel et al., 1995; Westerfield, 2000). All procedures involving experimental animals were approved by the local animal experiments committee and performed in compliance with local animal welfare laws, guidelines and policies, according to national and European law.
Immunoblotting
Single embryo lysates were obtained from wild type and ptena−/−ptenb−/− at 4 dpf using lysis buffer (50 mM HEPES, pH 7.4, 15 mM NaCl, 1 mM MgCl2, 10% glycerol, 1% Triton X-100, 1% sodium orthovanadate and protease inhibitors, including 5 mM beta glycerophosphate, 1 μg/ml aprotinin, 5 mM NaF, 1 mM Na3VO4 and 1 μg/ml leupeptin). Samples were mixed with 2× Laemmli sample buffer, boiled for 5 minutes and proteins were run on SDS-polyacrylamide gels. Immunoblotting was performed according to standard procedures, using p-Ser473-Akt (1:2000, Cell Signaling) and Akt (1:1000, Cell Signaling) antibodies.
Confocal and brightfield microscopy
Fluorescence images of transgenic embryos were acquired using TCS-SPE and processed with ImageJ. Embryos were anesthetized with Tricaine and mounted on a glass cover dish with 0.7% low melting agarose and covered with standard E3 medium. Whole-mount brightfield images were taken with a Leica DC 300F stereomicroscope or a Zeiss Axioplan microscope connected to a Leica DFC480 camera.
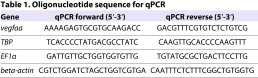
RNA isolation, cDNA synthesis and quantitative PCR
Total RNA was extracted using the RNeasy Mini Kit according to the manufacturer’s instructions (Qiagen). cDNA was synthesized with random hexamer primers according to the manufacturer’s instructions (Fermentas). Quantitative PCR was performed on a MyiQ cycler (Bio-Rad) using SYBRgreen chemistry (Bio-Rad). Three reference genes were used: tata box binding protein (TBP), elongation factor 1α (EF1α) and β-actin. Sequences of oligonucleotide are listed in Table 1. MIQE standards were applied to our protocols (Bustin et al., 2009). RNA extraction has been performed from three pooled embryos for each condition. For statistical analysis of two groups, unpaired t-test, or in case of unequal variances, Mann-Whitney U-test were used. For statistical analysis of multiple groups, 1-way ANOVA, or in case of unequal variances, Kruskal-Wallis test was used. Dunn’s post-hoc test was used to compare between selected groups. P-values <0.05 were considered significant. Statistical analysis was performed using SPSS 20 (IBM).
Table 1.
Oligonucleotide sequence for qPCR Gene
LY294002 and sunitinib treatment, and pten RNA injection
Embryos were treated from 70–72 hpf onwards with 25 μM LY294002 (Calbiochem) or 20 μM sunitinib malate (Sigma), unless stated otherwise. Control embryos were mock treated with DMSO and the presence of ptena−/−ptenb−/− mutations was confirmed by genotyping as described. Embryos were kept in the dark during treatment. ptena and ptenb cDNA was cloned in pCS2+. 5′ capped sense RNA was synthesized using the mMessage mMachine kit from Ambion according to the manufacturer’s instructions and 10 pg/nl injected at the one-cell stage.
In situ hybridization
Whole-mount in situ hybridization on 3-dpf and 4-dpf embryos was performed as described (Thisse and Thisse, 2008) using a vegfaa-specific probe (Liang et al., 2001).
Supplementary Material
Acknowledgments
The authors thank Mark Reijnen for excellent animal care and Stefan Schulte-Merker for providing the vegfaa probe. Microscopy was performed at the Hubrecht Imaging Centre.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
S.C. and J.d.H. conceived the experiments; S.C., B.W., A.d.B. and J.d.H. designed the experiments; S.C., R.K. and B.W. performed the experiments; S.C., B.W., A.d.B. and J.d.H. analyzed the data; S.C. and J.d.H. wrote the manuscript.
FUNDING
This work was supported in part by an EU (FP7) grant, ZF-CANCER (HEALTH-F2-2008-201439).
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.012377/-/DC1
REFERENCES
- Bustin S. A., Benes V., Garson J. A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M. W., Shipley G. L., et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 [DOI] [PubMed] [Google Scholar]
- Choorapoikayil S., Kuiper R. V., de Bruin A., den Hertog J. (2012). Haploinsufficiency of the genes encoding the tumor suppressor Pten predisposes zebrafish to hemangiosarcoma. Dis. Model. Mech. 5, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croushore J. A., Blasiole B., Riddle R. C., Thisse C., Thisse B., Canfield V. A., Robertson G. P., Cheng K. C., Levenson R. (2005). Ptena and ptenb genes play distinct roles in zebrafish embryogenesis. Dev. Dyn. 234, 911–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A., Pesce B., Cordon-Cardo C., Pandolfi P. P. (1998). Pten is essential for embryonic development and tumour suppression. Nat. Genet. 19, 348–355 [DOI] [PubMed] [Google Scholar]
- Faucherre A., Taylor G. S., Overvoorde J., Dixon J. E., Hertog J. (2008). Zebrafish pten genes have overlapping and non-redundant functions in tumorigenesis and embryonic development. Oncogene 27, 1079–1086 [DOI] [PubMed] [Google Scholar]
- Goberdhan D. C., Paricio N., Goodman E. C., Mlodzik M., Wilson C. (1999). Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev. 13, 3244–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupera M., Guillermet-Guibert J., Foukas L. C., Phng L. K., Cain R. J., Salpekar A., Pearce W., Meek S., Millan J., Cutillas P. R., et al. (2008). Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 453, 662–666 [DOI] [PubMed] [Google Scholar]
- Hagberg C. E., Falkevall A., Wang X., Larsson E., Huusko J., Nilsson I., van Meeteren L. A., Samen E., Lu L., Vanwildemeersch M., et al. (2010). Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 464, 917–921 [DOI] [PubMed] [Google Scholar]
- Hamada K., Sasaki T., Koni P. A., Natsui M., Kishimoto H., Sasaki J., Yajima N., Horie Y., Hasegawa G., Naito M., et al. (2005). The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 19, 2054–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert S. P., Huisken J., Kim T. N., Feldman M. E., Houseman B. T., Wang R. A., Shokat K. M., Stainier D. Y. (2009). Arterial-venous segregation by selective cell sprouting: an alternative mode of blood vessel formation. Science 326, 294–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Kontos C. D. (2002). PTEN modulates vascular endothelial growth factor-mediated signaling and angiogenic effects. J. Biol. Chem. 277, 10760–10766 [DOI] [PubMed] [Google Scholar]
- Jin S. W., Beis D., Mitchell T., Chen J. N., Stainier D. Y. (2005). Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development 132, 5199–5209 [DOI] [PubMed] [Google Scholar]
- Jinnin M., Medici D., Park L., Limaye N., Liu Y., Boscolo E., Bischoff J., Vikkula M., Boye E., Olsen B. R. (2008). Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat. Med. 14, 1236–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi H., Miyazaki S., Setoguchi T., Hiramatsu Y., Ohta M., Kamiya K., Sakaguchi T., Konno H. (2012). Rapid relapse after resection of a sunitinib-resistant gastrointestinal stromal tumor harboring a secondary mutation in exon 13 of the c-KIT gene. Anticancer Res. 32, 4105–4109 [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 [DOI] [PubMed] [Google Scholar]
- Koch S., Tugues S., Li X., Gualandi L., Claesson-Welsh L. (2011). Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 437, 169–183 [DOI] [PubMed] [Google Scholar]
- Li X., Tjwa M., Van Hove I., Enholm B., Neven E., Paavonen K., Jeltsch M., Juan T. D., Sievers R. E., Chorianopoulos E., et al. (2008). Reevaluation of the role of VEGF-B suggests a restricted role in the revascularization of the ischemic myocardium. Arterioscler. Thromb. Vasc. Biol. 28, 1614–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D., Chang J. R., Chin A. J., Smith A., Kelly C., Weinberg E. S., Ge R. (2001). The role of vascular endothelial growth factor (VEGF) in vasculogenesis, angiogenesis, and hematopoiesis in zebrafish development. Mech. Dev. 108, 29–43 [DOI] [PubMed] [Google Scholar]
- Liaw D., Marsh D. J., Li J., Dahia P. L., Wang S. I., Zheng Z., Bose S., Call K. M., Tsou H. C., Peacocke M., et al. (1997). Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 16, 64–67 [DOI] [PubMed] [Google Scholar]
- Ma J., Sawai H., Ochi N., Matsuo Y., Xu D., Yasuda A., Takahashi H., Wakasugi T., Takeyama H. (2009). PTEN regulates angiogenesis through PI3K/Akt/VEGF signaling pathway in human pancreatic cancer cells. Mol. Cell. Biochem. 331, 161–171 [DOI] [PubMed] [Google Scholar]
- Maehama T., Dixon J. E. (1998). The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378 [DOI] [PubMed] [Google Scholar]
- Marsh D. J., Dahia P. L., Zheng Z., Liaw D., Parsons R., Gorlin R. J., Eng C. (1997). Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat. Genet. 16, 333–334 [DOI] [PubMed] [Google Scholar]
- Mihaylova V. T., Borland C. Z., Manjarrez L., Stern M. J., Sun H. (1999). The PTEN tumor suppressor homolog in Caenorhabditis elegans regulates longevity and dauer formation in an insulin receptor-like signaling pathway. Proc. Natl. Acad. Sci. USA 96, 7427–7432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero J. A., Kilian B., Chan J., Bayliss P. E., Heisenberg C. P. (2003). Phosphoinositide 3-kinase is required for process outgrowth and cell polarization of gastrulating mesendodermal cells. Curr. Biol. 13, 1279–1289 [DOI] [PubMed] [Google Scholar]
- Myers M. P., Pass I., Batty I. H., Van der Kaay J., Stolarov J. P., Hemmings B. A., Wigler M. H., Downes C. P., Tonks N. K. (1998). The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc. Natl. Acad. Sci. USA 95, 13513–13518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasevicius A., Larson J., Ekker S. C. (2000). Distinct requirements for zebrafish angiogenesis revealed by a VEGF-A morphant. Yeast 17, 294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R., Jr(2007). Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem. Biophys. Res. Commun. 356, 323–328 [DOI] [PubMed] [Google Scholar]
- Stokoe D. (2001). Pten. Curr. Biol. 11, R502. [DOI] [PubMed] [Google Scholar]
- Thisse C., Thisse B. (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59–69 [DOI] [PubMed] [Google Scholar]
- Tielen R., Verhoef C., van Coevorden F., Gelderblom H., Sleijfer S., Hartgrink H. H., Bonenkamp J. J., van der Graaf W. T., de Wilt J. H. (2012). Surgery after treatment with imatinib and/or sunitinib in patients with metastasized gastrointestinal stromal tumors: is it worthwhile? World J. Surg. Oncol. 10, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonini G., Intagliata S., Cagli B., Segreto F., Perrone G., Onetti Muda A., Santini D., Persichetti P. (2010). Recurrent scrotal hemangiomas during treatment with sunitinib. J. Clin. Oncol. 28, e737–e738 [DOI] [PubMed] [Google Scholar]
- Weijts B. G., Bakker W. J., Cornelissen P. W., Liang K. H., Schaftenaar F. H., Westendorp B., de Wolf C. A., Paciejewska M., Scheele C. L., Kent L., et al. (2012). E2F7 and E2F8 promote angiogenesis through transcriptional activation of VEGFA in cooperation with HIF1. EMBO J. 31, 3871–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. (2000). The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio Rerio). Eugene, OR: Institute of Neuroscience, University of Oregon [Google Scholar]
- Zhiyong C., Wentong L., Xiaoyang Y., Ling P. (2012). PTEN’s regulation of VEGF and VEGFR1 expression and its clinical significance in myeloid leukemia. Med. Oncol. 29, 1084–1092 [DOI] [PubMed] [Google Scholar]
- Zhou X. P., Marsh D. J., Morrison C. D., Chaudhury A. R., Maxwell M., Reifenberger G., Eng C. (2003). Germline inactivation of PTEN and dysregulation of the phosphoinositol-3-kinase/Akt pathway cause human Lhermitte-Duclos disease in adults. Am. J. Hum. Genet. 73, 1191–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.