

SUMMARY
Mutations in the human NOTCH3 gene cause CADASIL syndrome (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy). CADASIL is an inherited small vessel disease characterized by diverse clinical manifestations including vasculopathy, neurodegeneration and dementia. Here we report two mutations in the zebrafish notch3 gene, one identified in a previous screen for mutations with reduced expression of myelin basic protein (mbp) and another caused by a retroviral insertion. Reduced mbp expression in notch3 mutant embryos is associated with fewer oligodendrocyte precursor cells (OPCs). Despite an early neurogenic phenotype, mbp expression recovered at later developmental stages and some notch3 homozygous mutants survived to adulthood. These mutants, as well as adult zebrafish carrying both mutant alleles together, displayed a striking stress-associated accumulation of blood in the head and fins. Histological analysis of mutant vessels revealed vasculopathy, including: an enlargement (dilation) of vessels in the telencephalon and fin, disorganization of the normal stereotyped arrangement of vessels in the fin, and an apparent loss of arterial morphological structure. Expression of hey1, a well-known transcriptional target of Notch signaling, was greatly reduced in notch3 mutant fins, suggesting that Notch3 acts via a canonical Notch signaling pathway to promote normal vessel structure. Ultrastructural analysis confirmed the presence of dilated vessels in notch3 mutant fins and revealed that the vessel walls of presumed arteries showed signs of deterioration. Gaps in the arterial wall and the presence of blood cells outside of vessels in mutants indicated that compromised vessel structure led to hemorrhage. In notch3 heterozygotes, we found elevated expression of both notch3 itself and target genes, indicating that specific alterations in gene expression due to partial loss of Notch3 function might contribute to the abnormalities observed in heterozygous larvae and adults. Our analysis of zebrafish notch3 mutants indicates that Notch3 regulates OPC development and mbp gene expression in larvae, and maintains vascular integrity in adults.
INTRODUCTION
The Notch signaling pathway is widely conserved among metazoans. Vertebrate genomes contain several Notch paralogs, which control key developmental decisions at numerous stages and in diverse tissues and organs (Andersson et al., 2011; Gazave et al., 2009). The Notch pathway is ideally suited for signaling between adjacent cells because both the Notch receptor and its ligands are transmembrane proteins (Kopan and Ilagan, 2009). This feature allows Notch signals to assign different fates to adjacent cells with similar developmental potential. According to prevailing models, feedback mechanisms amplify small differences in the expression levels of Notch receptor and ligand in neighboring cells, resulting in active Notch signaling in one cell and suppression of Notch signaling in the other. This lateral inhibition mechanism directs binary fate decisions in many species and developmental contexts, including neurogenesis in Drosophila (Ruohola et al., 1991), development of the inner ear and the intestine in vertebrates (Cotanche and Kaiser, 2010; Crosnier et al., 2005; Haddon et al., 1998a; Heath, 2010), and development of the vertebrate vasculature (Gridley, 2010). In the developing vasculature, Notch signaling inhibits endothelial cells from adopting a tip cell fate during sprouting angiogenesis (Sainson et al., 2005). Accordingly, inhibition of Notch signaling in zebrafish and mouse leads to increased sprouting and branching of vessels due to supernumerary tip cells (Leslie et al., 2007; Siekmann and Lawson, 2007). The Notch pathway also confers arterial identity to endothelial cells and vascular mural cells [e.g. vascular smooth muscle cells (VSMCs) and pericytes] (Domenga et al., 2004; Lawson et al., 2001).
In Drosophila, notch pathway mutants have neurogenic phenotypes characterized by an excess of neuronal cells (Campos-Ortega, 1985). The role of Notch signaling in limiting neurogenesis is conserved in vertebrates (Lewis, 1996). Mutants disrupting vertebrate pathway components also display neurogenic phenotypes (Imayoshi et al., 2010; Yoon et al., 2008). These phenotypes have been attributed to excessive differentiation of neural progenitor cells (NPCs) into neurons, which depletes the progenitor pool in notch pathway mutants. Thus, Notch regulates the balance between neuronal differentiation and maintenance of progenitor cell fates, including of radial glia. Notch also drives differentiation of other glial cell types (e.g. Müller glia of the retina), and Notch activity is thought to broadly control the balance between neurogenesis and gliogenesis (Pierfelice et al., 2011). In addition, Notch activity promotes expansion of oligodendrocyte precursor cells (OPCs), while inhibiting their terminal differentiation (Park and Appel, 2003; Wang et al., 1998). Outside of the nervous system, the Notch pathway contributes to the maintenance of other progenitor and stem cell populations, such as Leydig progenitor cells of the testis and melanocyte stem cells (Moriyama et al., 2006; Tang et al., 2008).
TRANSLATIONAL IMPACT.
Clinical issue
Mutations in the human NOTCH3 gene cause cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). CADASIL is an inherited disease characterized by progressive degeneration of small blood vessels, which leads to a diverse set of clinical manifestations including vasculopathy, neurodegeneration and dementia. None of the existing CADASIL mouse models fully recapitulates the human disease. In light of the variability among patients and the limitations of currently available NOTCH3 and CADASIL animal models, the mechanism by which NOTCH3 mutations cause CADASIL symptoms – including vascular, white matter and other disease phenotypes – remain to be clarified.
Results
In this study, zebrafish notch3 mutants were analyzed to gain insight into the mechanisms underlying CADASIL pathophysiology. The authors report that mutations in notch3 reduce the expression of myelin basic protein (mbp) and the number of oligodendrocyte precursor cells (OPCs). Despite displaying an early neurogenic phenotype, some notch3 homozygous mutants survived to adulthood. These mutants, as well as adult zebrafish carrying both mutant alleles together, displayed a striking stress-associated accumulation of blood in the head and fins. The authors’ histological analysis revealed vasculopathy in the mutants, including enlarged vessels in the brain and peripheral circulatory system and an apparent loss of arterial morphological structure. Moreover, ultrastructural analysis revealed signs of vessel deterioration in notch3 mutants, including gaps in the arterial wall and the presence of blood cells outside of vessels. Subsequent examination of Notch pathway target genes indicated that Notch3 acts via a canonical Notch signaling pathway to promote normal vessel structure. In notch3 heterozygotes, expression of notch3 and its target genes was elevated, indicating that alterations in gene expression occur in response to partial loss of Notch3.
Implications and future directions
These analyses of zebrafish notch3 mutants indicate that Notch3 regulates OPC development and mbp gene expression in larvae, and plays a conserved role in maintaining vascular tone and vessel integrity in adults. Changes in gene expression and cell death in the CNS were observed specifically in notch3 heterozygotes, suggesting that a partial loss of Notch3 function could contribute to aspects of the pathophysiology of CADASIL. These changes were not detected using previously reported animal models that completely lack Notch3 expression. Zebrafish notch3 mutants provide a valuable tool for future screens aiming to identify Notch signaling modifiers that affect the phenotype, and for studies of the activity of CADASIL mutant alleles and the signaling and cellular mechanisms that contribute to Notch3-associated pathologies.
Mutations in the human NOTCH3 gene cause CADASIL syndrome (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy). CADASIL is a small vessel disease that has been diagnosed in more than 500 families worldwide (Hervé and Chabriat, 2010). Clinical symptoms comprise migraine with aura, leukoaraiosis, subcortical infarcts, mood disturbances, apathy and dementia (Hervé and Chabriat, 2010). Many pathological mutations in CADASIL result in an odd number of cysteines in one of the EGF repeats of NOTCH3, which is thought to cause aggregation of the NOTCH3 extracellular domain (NECD) in individuals with CADASIL (Joutel et al., 1997; Opherk et al., 2009). The majority of individuals with CADASIL display leukoaraiosis and ischemic events that are particularly prominent in the white matter of the CNS, a region characterized by low blood flow that consists primarily of glial cells and myelinated axons (Lee et al., 2012). In the CNS, oligodendrocytes form myelin, a multi-lamellar sheath that allows for rapid impulse propagation by saltatory conduction in the vertebrate nervous system. Damage to oligodendrocytes and demyelination of axons can cause or contribute to neurodegenerative pathology, as occurs in the CNS in multiple sclerosis (MS) (Prineas and Parratt, 2012). The demyelination of axons might contribute to the cerebral atrophy seen in individuals with CADASIL (Viswanathan et al., 2006); however, axonal damage and atrophy in the white matter has also been viewed as a secondary defect to vascular disruption.
Histopathological analysis revealed that CADASIL is a vasculopathy of small cerebral and systemic arteries (Baudrimont et al., 1993; Ruchoux et al., 1995). In CADASIL, the VSMCs show signs of degeneration and eventually are lost. Endothelial dysfunction and ultrastructural endothelial changes also have been reported in CADASIL. Although vascular changes are observed all along the arterial tree, and in liver, muscle, spleen and skin, organ failure is only apparent in the brain and retina (Robinson et al., 2001; Ruchoux et al., 1995). It is not clear why widespread arteriopathy does not cause more global organ defects.
In an effort to better understand CADASIL pathologies, Notch3 knockout, knock-in and transgenic mouse models have been developed (Arboleda-Velasquez et al., 2011; Arboleda-Velasquez et al., 2005; Joutel, 2011; Joutel et al., 2004; Krebs et al., 2003; Monet-Leprêtre et al., 2009; Ruchoux et al., 2003). The defective constrictive responses upon experimental challenge by proximal middle cerebral artery occlusion and the enlarged arteries with thinner walls that are observed in Notch3 knockout mice indicate roles for Notch3 in arterial VSMC specification and maturation in tail vessels, and in maintaining vascular tone (Arboleda-Velasquez et al., 2008; Belin de Chantemèle et al., 2008; Domenga et al., 2004; Krebs et al., 2003). Although arteriopathies in the tail and, when challenged, cortical vessels (Arboleda-Velasquez et al., 2008) have been observed in knockout mice, important pathologies and hallmarks of CADASIL – including Notch3 ectodomain deposits, leukoencephalopathy and lacunar infarcts – have not been found. Nevertheless, Notch3 knockout mice show a higher susceptibility for ischemic stroke (Arboleda-Velasquez et al., 2008). None of the existing CADASIL mice fully recapitulates the human disease. In light of this variability among the available Notch3 and CADASIL animal models, the mechanism by which Notch3 mutations cause CADASIL symptoms is not known, and the mechanism and extent of interdependence of the vascular, white matter and other disease phenotypes remain to be clarified.
In a genetic screen to identify genes that are required for the development of myelinated axons in zebrafish, st51 was discovered as a mutation that reduced myelin basic protein (mbp) mRNA expression in the CNS (Pogoda et al., 2006). Here we show that st51 disrupts the zebrafish notch3 gene. Analysis of notch3st51 and an insertional allele of notch3 revealed that notch3 is required for OPC development and myelin gene expression during larval development. At later stages, mbp expression increased in the CNS of notch3 mutants, and some homozygous mutants survived to adulthood. In these adult mutants, we observed bleeding in response to mild stress. Histological and electron microscopy (EM) analyses revealed enlarged vessels in the telencephalon and fins, which indicated that blood accumulation was due to dilation and leaking of the vessels in notch3 mutant adults. Our studies of zebrafish notch3 mutants provide evidence that Notch3 plays a conserved role in maintaining vascular tone and vessel integrity in vertebrate adult vessels. The presence of oligodendrocyte abnormalities before vascular pathology provides evidence that notch3 might function in parallel in myelination and vasculogenesis.
RESULTS
Identification of mutations in notch3
In a screen for zebrafish mutants with abnormal myelination, Pogoda et al. identified st51 as a mutation that reduced mbp expression in the CNS at 5 dpf (Pogoda et al., 2006). Previous mapping experiments localized the st51 mutation to linkage group 3 (Pogoda et al., 2006). To identify the gene disrupted by st51, we conducted additional mapping experiments and localized the mutation to an interval bordered by the simple sequence length polymorphism (SSLP) markers Z3725 and Z20058, which contains the notch3 gene (Woods et al., 2005). Sequence analysis of genomic DNA from st51 mutants revealed a G-to-A transition in the mutant allele that disrupts the splice donor site after exon 13 in the notch3 gene (Fig. 1A). We detected no recombination between this lesion and the st51 mutation among 348 meioses (data not shown), indicating that st51 was tightly linked to notch3. Reverse transcriptase PCR (RT-PCR) analysis of RNA isolated from wild-type and mutant larvae at 3 dpf revealed that the notch3 pre-mRNA was aberrantly spliced in st51 homozygotes, such that intron 13was retained in the majority of mutant transcripts (Fig. 1B,C). Retention of this intron in the predominant mutant transcript caused truncation of the predicted notch3 open reading frame in the extracellular region, after the 17th EGF repeat (Fig. 1B). In addition, a minority of the mutant notch3 mRNAs contained aberrantly spliced sequences that result in partial deletions of exon 13. Sequence analysis identified two such deletions, a 15 bp deletion that maintained the reading frame, and a 103 bp deletion that induced a frameshift, truncating the predicted notch3 reading frame after the 16th EGF repeat (Fig. 1B,C).
Fig. 1.

Mutations in notch3 reduce mbp mRNA expression. (A) Schematic depicting the notch3 gene and the locations of the retroviral insertion (zm allele) and of the st51 mutation. Introns are not to scale. (B) Schematic of the wild-type Notch3 protein domains and the truncated protein produced owing to the st51 mutation. (C) RT-PCR of the wild-type (WT) and st51 notch3 transcripts. (D–I) In (D,G) wild-type zebrafish, (E) notch3st51/+ and (H) notch3zm/+ heterozygotes, and (F) notch3st51/st51 mutants, notch3 transcripts were detected in the CNS. In contrast to heterozygotes and notch3st51/st51 mutants, notch3 expression was barely detectable in the CNS of (I) notch3zm/zm mutants. Panels D–F and G–I are age-matched siblings from st51 and zm clutches, respectively. (D–F) Short staining reaction. (G–I) Long staining reaction. (J–Q) Expression of mbp was similar in (J,N) wild-type zebrafish and (K,O) notch3st51/+ or notch3zm/+ heterozygotes, but was reduced in the CNS (compare yellow dashed areas in the hindbrain) of (L) notch3st51/st51 and (M,Q) notch3zm/zm homozygotes and (P) notch3st51/zm transheterozygotes, indicating that the st51 and zm mutations failed to complement. (D–Q) Dorsal views. Anterior (A) and posterior (P) are indicated.
To further investigate the function of notch3, we obtained and examined a commercially available notch3 allele, ZM_197263, produced by insertional mutagenesis. The ZM_197263 mutation (referred to as ‘zm’ hereafter) contains a retroviral insertion in the 5′UTR of the notch3 gene, 158 nucleotides upstream of the open reading frame (Fig. 1A). In wild-type animals, notch3 was expressed throughout the nervous system, including in the hindbrain (Fig. 1D). Compared with wild type (Fig. 1D,G; n=5 each), notch3st51/+ and notch3zm/+ heterozygous siblings (Fig. 1E,H; n=11 each) and notch3st51/st51 mutants (Fig. 1F; n=7) showed enhanced expression of notch3 in the CNS, a finding we return to below. In contrast, little if any notch3 mRNA was detectable in notch3zm/zm mutants (Fig. 1I; n=9), indicating that notch3zm is a strong loss-of-function mutation.
To examine the phenotype of notch3zm/zm mutants and determine whether st51 and zm fail to complement, we examined mbp expression in early larvae (Fig. 1J–Q). Expression of mbp was similar to wild type in notch3st51/+ heterozygotes (Fig. 1K), but was reduced in the CNS of notch3st51/st51 homozygotes (Fig. 1L) and notch3zm/zm homozygotes (Fig. 1M). The reduction of mbp expression in the CNS of notch3zm/zm mutants, notch3st51/st51 homozygotes and notch3st51/zm transheterozygotes at 5 dpf (Fig. 1N–Q and data not shown) indicated that the st51 and zm mutations failed to complement. Moreover, the reduction seemed to be more severe in notch3zm/zm mutants, suggesting that the notch3st51 mutation might have more residual notch3 function than does notch3zm. On the basis of mapping data, the identification of lesions that disrupt the splicing or expression level of notch3 and complementation analysis, we conclude that both the st51 and zm mutations disrupt the function of notch3. Phenotypic and molecular studies suggest that notch3zm is a null or near-null mutation, and that notch3st51 is a hypomorphic allele that probably retains weak notch3 function. The residual Notch3 activity in notch3st51/st51 homozygotes is probably provided by the minor splice variant with the 15 bp deletion that maintains the reading frame in these mutants. These results demonstrate that notch3 is essential for normal expression of mbp in the larval CNS.
Transient reduction in OPCs in notch3 mutant larvae
Mutational analysis shows that notch3 is essential for normal expression of mbp mRNA in the CNS of zebrafish larvae (Pogoda et al., 2006) (and this work). To investigate the possibility that an early deficit in oligodendrocytes underlies reduced mbp expression in notch3 mutants, we examined oligodendrocyte transcription factor 2 (olig2), which is expressed in OPCs and motor neurons (Park et al., 2002; Park et al., 2007; Shin et al., 2003; Takebayashi et al., 2000; Zannino and Appel, 2009). At 24 hpf, expression of olig2 was comparable between wild type, notch3st51/+ heterozygotes and notch3st51/st51 homozygotes (data not shown). However, olig2 expression was reduced in OPCs of notch3st51/st51 mutants at 50 hpf (Fig. 2C,C′; n=5) compared with wild type (Fig. 2A,A′; n=2) and notch3st51/+ heterozygotes (Fig. 2B,B′; n=8). At 52 hpf, expression of sox10, an independent marker of cells of the oligodendrocyte lineage, was also reduced in the midline of the hindbrain in notch3st51/st51 mutants (Fig. 2F, compared with 2D,E). These results suggested that a partial loss of OPCs resulted in the reduced expression of mbp in the CNS of notch3 mutants. This analysis indicates that notch3 is required for OPC development and myelin gene expression in the embryo and early larva. Despite these defects at earlier stages, some notch3 mutants achieve normal levels of mbp expression in the hindbrain by 7 dpf (data not shown).
Fig. 2.
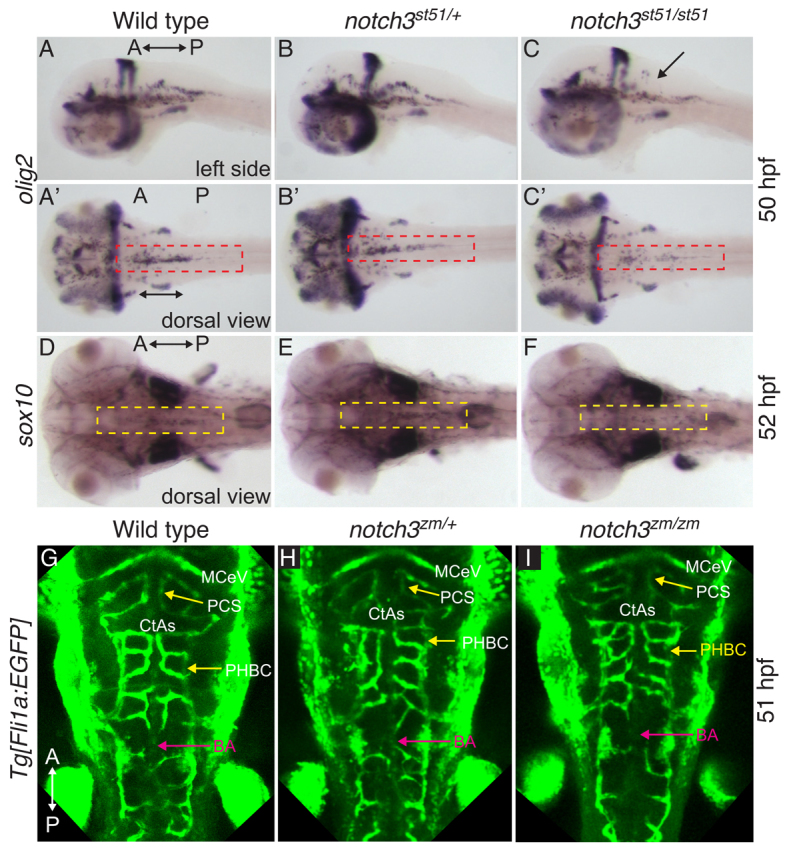
Transient reduction in OPCs in notch3 mutants at stages at which vessel patterning is intact. (A–C) olig2 at 50 hpf was similar in (A,A′) wild type and (B,B′) notch3st51/+ heterozygotes, but was reduced in OPCs (compare red dashed areas) of (C,C′) notch3st51/st51 mutants. Arrow indicates region devoid of olig2-expressing cells. (D–F) sox10 expression at 52 hpf was comparable in (D) wild type and (E) notch3st51/+ heterozygotes, but was reduced in the midline of the hindbrain (compare yellow dashed areas) in (F) notch3st51/st51 mutants. (A–C) Lateral views. (A′–C′,D–I) Dorsal views. (G–I) Vessel pattern at the anatomical level is intact in notch3zm/zm mutants. Max projection images of Tg[fli1a:EGFP] labeled vasculature of (G) wild type (n=9), (H) notch3zm/+ heterozygotes (n=9) and (I) notch3zm/zm mutants (n=5). CtAs, central arteries; PHBC, primordial hindbrain channel; BA, basilar artery; MCeV, mid-cerebral vein; PCS, posterior connecting segment. The orientation of the anterior (A) and posterior (P) axis is indicated.
In individuals with CADASIL, loss of myelin accompanies and has been proposed to be caused by vascular defects (Okeda et al., 2002), and adult notch3 knockout mice have vascular defects. Although swim bladder inflation was delayed at 5 days post-fertilization (dpf), no overt evidence of edema or compromised circulation was detected morphologically at 3–5 dpf (stages when OPC defects were apparent among progeny) in notch3st51/st51 or notch3zm/zm mutants from five independent heterozygous incrosses. To more closely examine vessel patterning, we conducted microangiography and crossed the notch3 mutations into the Tg[fli1a:EGFP] background (Lawson and Weinstein, 2002b). As compared with wild-type genotypes, microangiography (supplementary material Fig. S1A–C) and expression of Tg[fli1a:EGFP] (Fig. 2G–I and supplementary material Fig. S2) indicated that, at the anatomical level, overall sprouting, vessel branching and morphogenesis were intact in notch3 mutants at stages before and after OPC deficits were already apparent. Consistent with Tg[fli1a:EGFP] analysis and microangiography, vascular markers [deltaC (dlC), ephrinB2 (efn2ba/ephB2), fms-related tyrosine kinase 4 (flt4) and disabled homolog 2 (dab2)] were indistinguishable between wild type, notch3zm/+ heterozygotes and notch3zm/zm mutants at 28 hpf (supplementary material Fig. S3A–L), providing additional evidence that, at the anatomical level, arterial venous identity and arterial venous patterning of the vasculature is intact at stages when OPCs and oligodendrocytes are disrupted in notch3 mutants.
Neurogenic phenotype of notch3 mutants and apoptosis in notch3 heterozygotes
To investigate whether the reduction of OPCs in notch3 mutants was due to loss of progenitors via cell death, we used Acridine Orange (AO) to label cells undergoing apoptosis. At 52 hpf, AO staining was similar in the CNS of wild-type siblings (Fig. 3A; n=12) and notch3st51/st51 mutants (Fig. 3C; n=8), indicating that the reduction of OPCs in notch3 mutants was not due to cell death. Because apoptosis did not account for reduced OPCs in mutants, we examined the expression of progenitor and differentiation markers. During neuronal development, deltaD expression in precursors prevents neighboring notch-expressing cells from adopting neuronal fates (Haddon et al., 1998b; Hans and Campos-Ortega, 2002; Mahler et al., 2010; Takke et al., 1999). We observed abundant deltaD mRNA (Fig. 3D–I) in lateral regions of notch3st51/st51 (Fig. 3F; n=11) and notch3zm/zm (Fig. 3I; n=2 at 48 hpf and 4 at 54 hpf) mutants, with holes in deltaD expression in medial regions similar to the gaps in notch3 and notch1a expression that were present in notch3st51/st51 mutants (Fig. 1F; n=11 and supplementary material Fig. S4). To further investigate the development of neural progenitors, we examined gfap expression at 52 hpf, which marks radial glial progenitors at this stage (Fig. 3J–L). As we observed for notch3 and deltaD, there were medial gaps in gfap expression in notch3 mutants (Fig. 3L; st51: n=8; zm: n=9), indicating that OPCs might be reduced owing to aberrant differentiation of medial progenitors.
Fig. 3.
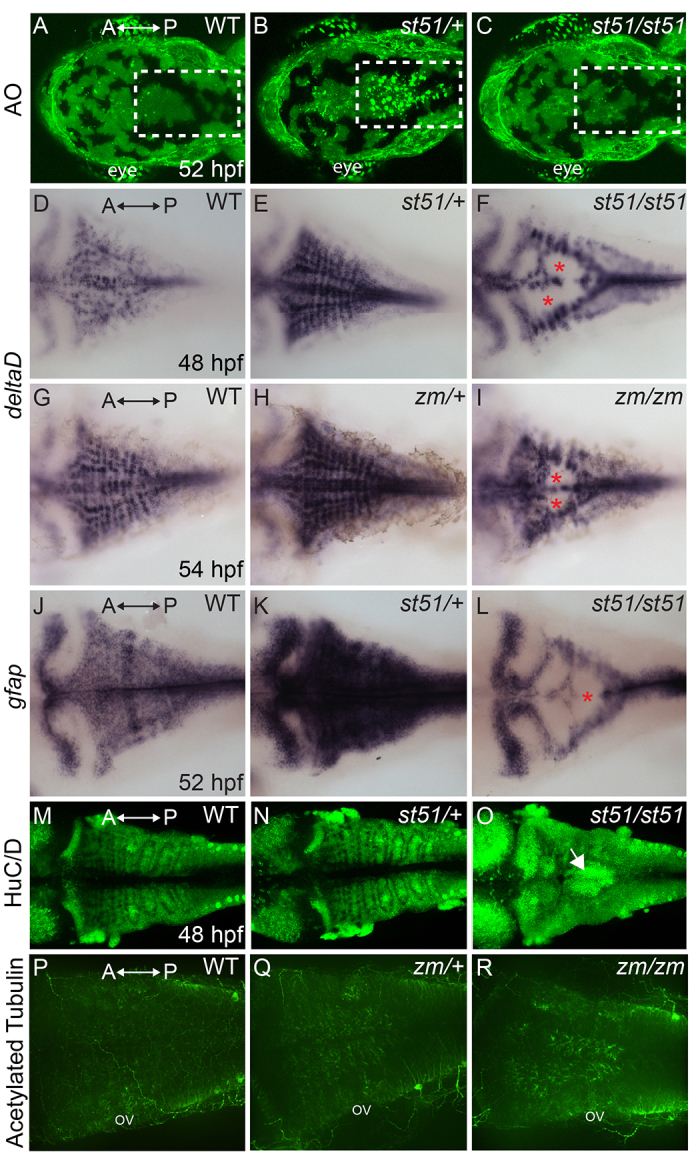
Apoptosis in notch3 heterozygotes and neurogenic phenotype of notch3 mutants. (A–C) Acridine orange (AO)-labeled embryos. The hindbrains (white dashed areas) of (A) wild type (WT) and (C) notchst51/st51 mutants were largely devoid of AO-positive cells at 52 hpf. In contrast, AO-positive cells were abundant in the hindbrain region of (B) notch3 heterozygotes. (D–I) Dorsal views of deltaD expression. (D,G) In wild type, deltaD mRNA was abundant in the CNS. deltaD expression was more pronounced in (E) notch3st51/+ and (H) notch3zm/+ heterozygotes. deltaD expression was robust in lateral regions and showed gaps in medial regions (asterisks) of (F) notch3st51/st51 and (I) notch3zm/zm mutants. (J,K) Expression of another progenitor marker, gfap, which marks neural progenitor cells, was more robust in (K) notch3st51/+ heterozygotes compared with in (J) wild type. (L) In notch3st51/st51 mutants, gfap was present in lateral regions and showed medial gaps (asterisk). (M–O) HuC/D, a marker of post-mitotic neurons, expression was comparable in (M) wild type and (N) notch3st51/+ heterozygotes, but was more robust in the medial hindbrain (arrow) of notch3st51/st51 mutants. (P-R) Acetylated-tubulin-labeled (P) wild-type (n=5), (Q) notch3zm/+ heterozygotes (n=11) and (R) notch3zm/zm mutant (n=9) larvae. (P-R) Max projections of confocal images. ov, otic vesicle. All panels are dorsal views, anterior (A) is to the left and posterior (P) is to the right.
To investigate the possibility that the reduction of OPCs results from a neurogenic phenotype in notch3 mutants, we examined HuC/D, which marks post-mitotic neurons. In contrast to wild type (Fig. 3M) and notch3 heterozygotes (Fig. 3N and data not shown), notch3 mutants had abundant HuC/D expression (Fig. 3O and data not shown) and acetylated tubulin staining (compare with Fig. 3P–R) in medial regions of R3/R5, consistent with the possibility that OPCs are reduced because progenitors are inappropriately allocated to neuronal fates in notch3 mutants. In other regions of the embryo, major axonal projections were comparable between wild type and notch3 mutants (supplementary material Fig. S5).
A striking feature of the foregoing analysis is that notch3 heterozygotes expressed highly elevated levels of Notch pathway components, including deltaD [Fig. 3E (80% of notch3st51/+ heterozygotes; n=26); Fig. 3H (n=9 notch3zm/+ heterozygotes at 48 hpf and 6 notch3zm/+ heterozygotes at 54 hpf)] and notch3 itself (Fig. 1E), and the progenitor marker gfap (Fig. 3K). In addition, AO staining revealed elevated cell death in the CNS of notch3st51/+ (Fig. 3B; n=12) and notch3zm/+ (n=2) heterozygotes that was not seen in wild type or homozygous mutants (Fig. 3A, n=12; Fig. 3C, n=8). Despite these changes in gene expression and cell death, notch3 heterozygotes were fully viable and fertile. Indeed, even some notch3 homozygous mutants achieved normal levels of mbp expression in the hindbrain by 7 dpf (data not shown) and survived to adulthood.
Compromised vascular tone in notch3 mutant adults
Despite the reduced numbers of OPCs in notch3st51/st51 and notch3zm/zm mutants, a subset of the mutants recovered from these early zygotic phenotypes and survived to adulthood. Homozygous notch3st51/st51 and notch3zm/zm males that survived to adulthood were fertile. However, in response to mild stress (such as moving the tank from the rack, or mating), the homozygous mutant adults displayed an accumulation of blood within the head and fins that was not observed in wild-type or heterozygous siblings (Fig. 4). In notch3zm/+ (Fig. 4A,E) and notch3st51/+ (Fig. 4C,E) heterozygous siblings, blood was evident at the base of the fins, but did not extend into the caudal fin rays. In contrast, blood extended from the base to the tip of the fins in notch3zm/zm (Fig. 4B,E) and notch3st51/st51 (Fig. 4D,E) homozygotes and notch3st51/zm transheterozygotes (data not shown), indicating that vascular tone might be compromised in notch3 mutant adults. As observed for the reduction in mbp expression in the larvae, notch3zm/zm mutants showed more pronounced phenotypes compared with notch3st51/st51 mutants.
Fig. 4.

Vascular phenotype of notch3 mutants. (A–D) Lateral views of (A) notch3zm/+ heterozygotes, (B) notch3zm/zm mutants, (C) notch3st51/+ heterozygotes and (D) notch3st51/st51 mutants. (B) notch3zm/zm mutant adults show craniofacial defects (black arrow) and accumulation of blood (light and dark blue arrows and arrowheads) in the head, pectoral anal and caudal fins. (E) Quantification of the bleeding phenotypes. Degree of shading indicates penetrance of phenotype: white cell in the table indicates less than 50% of animals showed the phenotype, light gray indicates that 50–75% of individuals were affected, and dark gray denotes greater than 75% of animals had the phenotype.
CADASIL is a vasculopathy of small cerebral and systemic arteries caused by mutations in NOTCH3 (Baudrimont et al., 1993; Ruchoux et al., 1995). We conducted histopathological analysis to investigate whether adult zebrafish notch3 mutants showed any evidence of vasculopathy in the brain. Compared with wild-type (Fig. 5A,B,G) and heterozygous (Fig. 5C,D,I) genotypes, vessels were more prominent in the telencephalon of notch3zm/zm (Fig. 5E,H) and notch3st51/st51 mutants (Fig. 5F,J). In contrast, the tectum of wild-type zebrafish had highly branched large- and small-diameter vessels (Fig. 5A), whereas the tectum of notch3 mutants was covered with small-diameter vessels and capillaries (Fig. 5E,F). Histological analysis of hematoxylin and eosin (H&E)-stained sections within the medial telencephalon confirmed the presence of small vessels (probably capillaries) traversing the tissue of notch3zm/+ and notch3st51/+ heterozygotes (Fig. 5K,M). In contrast, abnormally large vessels were present in notch3zm/zm and notch3st51/st51 mutant telencephalon (Fig. 5L,N), indicative of vasculopathy in notch3 mutant brain tissue.
Fig. 5.
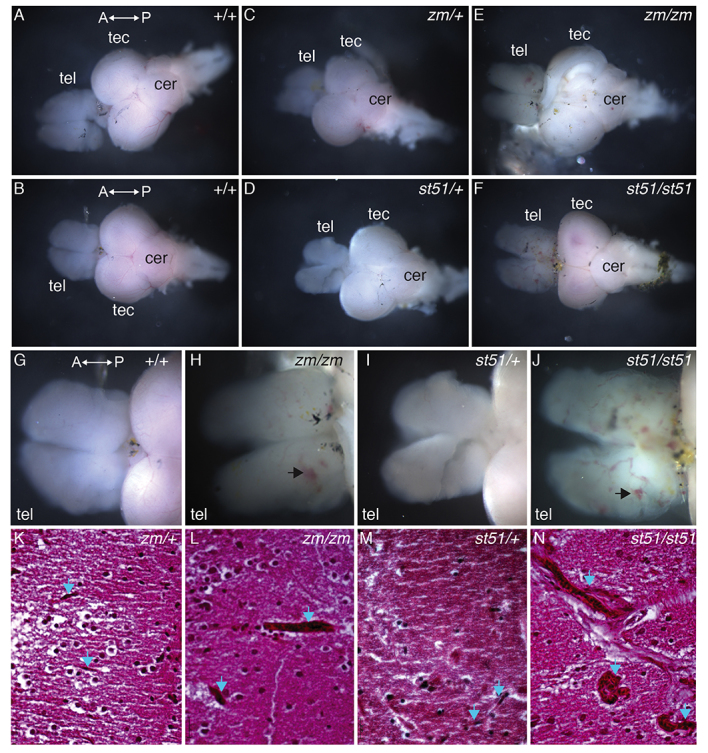
Vessels are more prominent in the telencephalon of notch3 mutants compared with wild type. (A–F) Overview images of dissected adult brains. (G–J) High-magnification images of the telencephalon. Compared with (A,B,G) wild-type and (C,D,I) heterozygous genotypes, vessels are more prominent in the telencephalon of (E,H) notch3zm/zm and (F,J) notch3st51/st51 mutants. (G–J) Large vessels and accumulation of blood (arrows) are apparent in mutant telencephalons. (K–N) H&E-stained sections of the medial telencephalon of (K) notch3zm/+, (L) notch3zm/zm, (M) notch3st51/+ and (N) notch3st51/st51 mutants. Small vessels traverse the tissue in heterozygotes, whereas large dilated vessels are present in homozygous mutant telencephalon. Blue arrows denote vessels. tel, telencephalon; tec, tectum; cer, cerebellum. The orientation of the anterior (A) and posterior (P) axis is indicated. (K–N) Scale bars: 20 μm.
Defective organization and vasodilation of vessels in notch3 mutants
To further investigate the basis for the accumulation of blood in notch3 mutants, we conducted histological analysis of caudal fin tissue isolated from notch3 mutants. Wild-type fins had the expected highly ordered reiterative structure of dermal, vascular and nervous tissues, with veins occupying the joints between and arteries within bony regions (Fig. 6A,B; n=2). This ordered pattern was largely intact in notch3zm/+ and notch3st51/+ heterozygotes (Fig. 6C,E; n=2 each). By contrast, in notch3st51/st51 mutants (Fig. 6D; n=2) veins were not limited to the joints and extended into the bony regions, which were co-occupied by multiple distended vessels. Similar but more severe phenotypes were observed in notch3zm/zm mutants (Fig. 6F; n=2). Even with higher magnification, it was difficult to unambiguously identify arteries in the notch3zm/zm mutant fins (Fig. 6F).
Fig. 6.
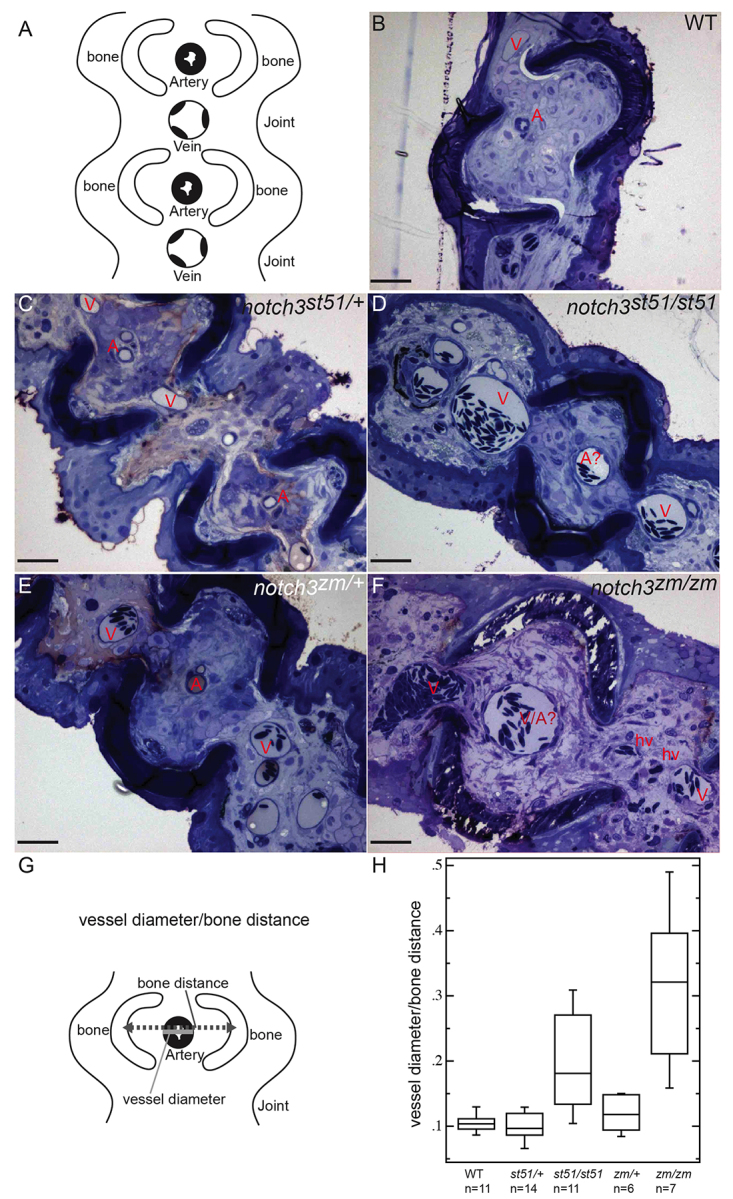
Distension of vessels in notch3 mutants. (A) Schematic depicting cross-section and the ordered arrangement of arteries within the bony regions and larger caliber veins within the joints of the fin rays observed in (B) wild type, (C) notch3st51/+ and (E) notch3zm/+ heterozygotes. This pattern is moderately disrupted in (D) notch3st51/st51 mutants and severely disrupted in (F) notch3zm/zm mutants. (B–F) Toluidine-blue-stained sections. Scale bars: 50 μm. A, artery; A?, probable artery based on position; V, vein; hv, hemorrhaged vessel. (G) Schematic depicting the strategy for quantification of vessel defects (detailed in the Materials and Methods). (H) Box plot of results reveals significantly larger vessels within the bony segments of the fin of notch3 mutants compared with wild-type adults. ANOVA results: mean squares between=6.7821×10−2; mean squares error=3.4565×10−3; P<0.0001.
In order to quantitatively assess the vessel defects given the difficulty in distinguishing arteries from veins in notch mutants, we determined the ratio of the vessel diameter within the bony segments of the fin ray, where arteries are typically located, to the distance between the bones (Fig. 6G). This ratio was not significantly different between wild-type and notch3st51/+ or notch3zm/+ heterozygotes (Fig. 6H). In contrast, notchst51/st51 and notch3zm/zm mutants showed statistically significant distension of the vessels residing within bony regions of the fin ray, such that vessels in notch3zm/zm mutants showed a threefold increase in relative diameter.
Consistent with the vascular phenotypes of notch3 mutants, notch3 expression was prominent in arteries of adult fin (Fig. 7A). In notch3 mutants, the vessels within bony regions, where arteries normally reside, showed morphological features that were more reminiscent of veins. To investigate whether the notch3 mutant vessels within bony segments showed aberrant morphology because they had lost their arterial identity, we examined the expression of deltaC, a marker of arterial identity. As in notch3zm/+ heterozygotes (Fig. 7B), notch3zm/zm mutants (Fig. 7C) expressed deltaC in the enlarged vessels within bony segments. Importantly, deltaC was not expressed in the vessels that run adjacent to the bony segments, indicating that those vessels kept their venous identity. We also examined flt4, which was expressed in arteries and veins within the fin of notch3zm/+ heterozygotes (Fig. 7D) and notch3zm/zm mutants (Fig. 7E). These analyses indicate that, although the mutant arteries displayed morphological features of veins, their arterial identity was intact in notch3zm/zm mutants.
Fig. 7.

Expression of notch3, Notch targets, and arterial and venous markers in adult fin, and analysis of compromised vessel integrity and hemorrhage of vessels in notch3 mutants. (A) Consistent with the vascular phenotypes of notch3 mutants, we detected notch3 expression in association within arteries of adult wild-type (WT) fin. (B) deltaC is expressed in the arteries within the bony segments, but not in the veins, which are located in the joints of notch3zm/+ heterozygotes fins as expected. (C) In notch3zm/zm mutants, deltaC is also expressed in the enlarged vessels (note the enlarged lumen) within bony segments but not in the veins; thus, arterial and venous identity does not require notch3. (D,E) No difference in the pattern of flt4 expression between (D) notch3zm/+ heterozygotes and (E) notch3zm/zm mutant fins was detected, although the mutant vessels were distended. (F,G) qRT-PCR of Notch target expression in caudal fin. (F) qRT-PCR hey1. (G) qRT-PCR her9. y-axis indicates % of expression normalized to WT. (H–O) TEM analysis. (H) Wild-type artery. Pink arrows indicate dense plaques. (I) Wild-type vein. (J) Intact dense plaques surrounded arteries of notch3zm/+ heterozygotes and (K) veins show normal morphology, but appear slightly enlarged. (L) Breaks in the walls and reduced dense plaques (pink arrows) of notch3zm/zm mutant vessels with features reminiscent of arteries. Blood cells (red asterisk) are located outside of the damaged vessel. (M) notch3zm/zm mutant veins are comparable to those of notch3zm/+ heterozygotes. (N) Wild-type vein. The purple arrows in N and O indicate the vessel wall. (O) Hemorrhage and inflammation of notch3zm/zm vessel. Note the red blood cell (red asterisk in O) outside of the compromised vessel wall indicated by purple arrows. Scale bars: 1 μm. ec, endothelial cell; mc, vascular mural cells; RBC, red blood cell. *n=2; **P=0.06; ***P=0.02.
Previous studies of mouse Notch3 knockout mice suggest distinct mechanisms by which Notch3 functions. One proposed mechanism involves the regulation of canonical Notch targets such as Hey and Hes genes (Arboleda-Velasquez et al., 2008), whereas the second points to a newly identified pathway whereby Notch3 activates unknown targets (Domenga et al., 2004). Moreover, human CADASIL mutations show a differing ability to bind Jagged1 ligand and to activate canonical Notch targets (e.g. RBP-Jκ) (Joutel et al., 2004). To investigate the pathway underlying compromised vascular tone in zebrafish notch3 mutants, we surveyed the expression of canonical Notch targets in mutant fins (Fig. 7F,G). We found that the canonical notch target hey1 was significantly reduced in notch3zm/zm and notch3st51/st51 mutant fins (Fig. 7F), and that levels of her9 were not significantly changed (Fig. 7G). These results indicate that Notch3 acts via a mechanism that involves the activation of canonical Notch targets to promote vascular tone and integrity.
Compromised vessel integrity leads to hemorrhage of vessels in notch3 mutants
To investigate whether the accumulation of blood in the fins of notch3 mutants was due to compromised vascular tone or reflected a more severe defect (hemorrhaging of the vessels), we used EM to examine the vascular ultrastructure. In wild type (Fig. 7H,I) and notch3zm/+ heterozygotes (Fig. 7J,K), arteries and veins were readily distinguishable, albeit that veins seemed to be larger in notch3zm/+ heterozygotes, suggesting a mild defect in vascular tone. In notch3zm/zm mutants the vessels were severely dilated and arteries were difficult to distinguish from veins (Fig. 7L,M and data not shown). Mural cells associated with vessels were easily identifiable in wild-type and heterozygous genotypes, but could not be unambiguously identified in notch3 mutants. Moreover, gaps in the vessel walls and the presence of blood cells outside of the vessels indicated that the pooling of blood in the fins of notch3zm/zm mutants was probably due to both loss of vascular tone and hemorrhage of the notch3 mutant vessels (compare Fig. 7N and 7O).
DISCUSSION
The Notch pathway is highly conserved and is widely used to mediate cell fate decisions in embryonic and adult tissues (reviewed in Kopan and Ilagan, 2009). Mutations disrupting human NOTCH3 cause CADASIL, an inherited small vessel disease associated with neurodegeneration and cognitive decline with advanced patient age (Hervé and Chabriat, 2010). Prevailing models indicate that cognitive decline might be secondary to vascular pathology, but this remains an open question because the existing mouse knockins of common patient NOTCH3 alleles recapitulate some but not all of the CADASIL pathologies. The two reported Notch3 knockout mouse models are susceptible to stroke when challenged, but no other neural deficits have been reported. Studies of these knockout mouse mutants point to opposing models of Notch3 function in establishing vascular structure. Based on their analysis of one knockout mutation, Domenga et al. presented evidence for a vascular defect and argue for a non-canonical mechanism (Domenga et al., 2004), whereas Arboleda-Velasquez et al. did not report vascular structure defects in a different knockout strain and argued that a canonical pathway was important for the regulation of gene expression in vascular smooth muscle cells (VSMCs) (Arboleda-Velasquez et al., 2011; Arboleda-Velasquez et al., 2008). Although the mouse analyses present different views of the role of notch3 in establishing vascular structure, our analysis of zebrafish notch3 mutants provides strong evidence that notch3 has a conserved function in sustaining mature vessel structure and maintaining vascular integrity, probably via a canonical pathway, as indicated by reduced hey1 in notch3 mutants. In addition, our studies of zebrafish notch3 mutants provide evidence that notch3 regulates OPC development and mbp gene expression in larvae and promotes mural cell and artery maturation in adults. Our analysis of neural development reveal that notch3 mutant larvae have a neurogenic phenotype and that notch3 heterozygous larvae have abnormalities in development of the CNS that are not seen in homozygous mutants.
We demonstrate that notch3 is essential for normal differentiation of neural progenitors during embryogenesis. In the developing hindbrain of notch3 mutants, there were prominent gaps in the expression of neural progenitor markers, and excess neurons developed at the expense of OPCs. When Notch signaling is disrupted, excess neurons often differentiate at early stages at the expense of later-born cell types, including OPCs (Bingham et al., 2003; Haddon et al., 1998a; Itoh et al., 2003; Park et al., 2000; Shih and Keller, 1992). In the zebrafish embryo, Qiu et al. reported that Notch3 and Notch1a have redundant functions in controlling hindbrain patterning and neurogenesis (Qiu et al., 2009). They found that excess neurons developed in notch1a mutants that also had reduced notch3 function (via morpholino injection), and rhombomere boundaries were not maintained. In combination, our analysis and the results of Qiu et al. suggest that notch3 mutants have reduced OPCs in the early larval stage because of misallocation of the fates of some early neural progenitors (Qiu et al., 2009). Furthermore, the relatively mild and transient notch3 mutant phenotype might reflect redundancy with notch1a. It is possible that, in the mouse, Notch3 has a similar role in OPC development that is masked by overlapping functions of other notch genes.
The elevated expression of notch3, deltaD and gfap and increased apoptosis in the CNS of notch3 heterozygotes was a striking finding of our analysis. The elevation of progenitor markers suggests that the differentiation of some progenitors might be delayed by the partial reduction of Notch signaling in notch3 heterozygotes. The elevated cell death in the heterozygotes suggests that some of these delayed progenitors undergo apoptosis, but the majority must differentiate properly because the pattern of neurons and OPCs is apparently normal and the fish are fully viable. The finding of changes in gene expression and cell death in the CNS specific to notch3 heterozygotes suggests that a partial loss of Notch3 function could contribute to aspects of the pathophysiology of CADASIL that have not been observed in animals that completely lack notch3 function.
Based largely on the temporal manifestation and detection of phenotypes, leukoaraiosis, subcortical ischemic events, mood disturbances, apathy and dementia have been thought to be secondary to vascular defects and reduced capillary density (reviewed in Hervé and Chabriat, 2010; Kalimo et al., 2002; Pantoni, 2010). Analysis of zebrafish cloche mutants, which lack endothelium but have normal hindbrain neuron clusters and axon tracts, provides evidence that endothelium is not required for proper patterning and organization of neurons in the early hindbrain (Ulrich et al., 2011). In zebrafish notch3 mutants, OPCs are already reduced at stages when gross vascular patterning is intact, as assessed by the Tg[fli1a:EGFP] transgene, microangiography and endothelial markers. In addition, the OPC deficits are evident at stages when the other cell type of the vessel, the mural cell, has not been specified or undergone maturation (Miano et al., 2006; Santoro et al., 2009). Taken together, this indicates that notch3 is likely to have parallel roles in regulating OPC development and mbp gene expression in larvae, and in promoting vascular integrity. Future investigation is required to identify the notch3 targets involved in the abnormalities in OPC development and vasculopathy in brain and peripheral tissues, and to determine whether the brain phenotypes involve communication between neural and vascular cells, which has been proposed to account for the non-autonomous neuropathology in CADASIL phenotypes. Alternatively, in mouse and zebrafish, notch3 is expressed widely in neural progenitors (Irvin et al., 2001; Lindsell et al., 1996; Qiu et al., 2009); therefore, the effects might instead be later manifestations of a notch3 function in neural development rather than a secondary consequence of vascular defects.
Some zebrafish notch3 mutants survive to adulthood. This is probably due to compensation by other notch family members such as notch1a, because adult mutants that are homozygous even for the presumed null notch3zm allele were recovered. Those notch3 mutants that do survive show enlarged vessels in the telencephalon and accumulation of blood in peripheral vessels along the rostral caudal axis (in particular within the pectoral, anal and caudal fins), indicating loss of vascular tone within peripheral vessels. Cardiovascular development in zebrafish, as in mammals, involves specification of endothelial cell fates during the earliest stages of vessel development. Arterial differentiation is regulated by Shh, vascular endothelial growth factor (Vegf) and Notch signaling in zebrafish and in other vertebrates (Lawson et al., 2001; Lawson et al., 2002; Lawson and Weinstein, 2002a; Torres-Vázquez et al., 2003; Weinstein and Lawson, 2002). Complete loss of Notch signaling, as occurs in mutants for mindbomb (mib), an E3 ubiquitin ligase involved in Notch activation, results in deficits of arterial-specific and ectopic vein marker expression within the dorsal aorta (Lawson et al., 2002). Arterial and venous fates are intact in notch3 mutant embryos and in adults, indicating that Notch3 is not involved or that other notch genes compensate for its loss.
In addition to endothelial cell specification, cardiovascular development in mammals and zebrafish involves recruitment of vascular mural cells (vMCs) during a process known as vascular myogenesis (Miano et al., 2006; Owens, 1995; Owens, 1998; Owens et al., 2004; Owens and Wise, 1997; Santoro et al., 2009). vMCs, VSMCs and pericytes initially act as support cells, but subsequently become highly specialized cells that control vasodilation and vessel caliber. During larval development in zebrafish, immature VSMCs are detected adjacent to the dorsal aorta by ultrastructural criteria and marker expression (Miano et al., 2006; Santoro et al., 2009). However, differentiated VSMCs are only apparent after 1 month of development, and definitive VSMCs, defined by dense plaques (hallmarks of a mature actin cytoskeleton), myofilament bundles and a basement membrane, are only apparent at 3 months (Miano et al., 2006; Santoro et al., 2009). Histological and ultrastructural analyses revealed vasodilation of both peripheral arteries and veins of notch3 mutants, indicating that notch3, which is expressed in the walls of both vessel types, has an essential role in limiting vasodilation. Vascular smooth muscle cell contraction and relaxation regulate vessel caliber and local blood pressure in response to demand for oxygenated blood. Differentiated VSMCs display contractile properties, whereas undifferentiated VSMCs are proliferative (Owens, 1995). Pooling of blood and vasodilation in notch3 mutants are consistent with a role for notch3 in promoting the mature, contractile VSMC state to regulate vascular tone. notch3 seems to play an additional role in regulating arterial architecture, because vessels positioned where arteries are typically located had thinner vessel walls, a morphology resembling that of veins rather than the typical thick cell walls of arteries, despite normal arterial venous patterning in vessels of embryos and adults. The absence of obvious patterning defects in embryos is consistent with adult-onset vasculopathy due to defective VSMC maturation or function in notch3 mutants. Mural cells and VSMCs both contribute to the normal vascular response to Vegf in newly forming vessels and during regeneration (Carmeliet and Collen, 1999; Carmeliet and Storkebaum, 2002). We were unable to detect definitive pericytes associated with vessels in bony regions of mutant fin rays (where arteries reside in wild type); thus, impaired pericyte development or recruitment might contribute to impaired vascular integrity. Similarly, VSMC destruction associated with reduced pericytes has been reported in individuals with CADASIL (Kalimo et al., 2002; Ruchoux et al., 2003). Thus, zebrafish notch3 mutant phenotypes show aspects of CADASIL pathology, specifically compromised VSMC and vascular integrity despite the different nature of the mutations in notch3.
Two Notch3 knockout mice have been reported. In contrast to the sub-viable zebrafish mutants, both mouse knockout models are viable and fertile (Arboleda-Velasquez et al., 2008; Domenga et al., 2004; Kitamoto et al., 2005; Krebs et al., 2003). One Notch3 knockout shows deficits in differentiation and maturation of VSMCs of small systemic vessels (Domenga et al., 2004). Similar to zebrafish notch3 mutants, the tail arteries of this Notch3 knockout are enlarged with a thin media reminiscent of veins. Domenga et al. suggested that Notch3 might act through a non-canonical pathway in VSMCs, because expression of classical Notch targets such as Hes genes were normal in Notch3 mutants (Domenga et al., 2004). Instead, they hypothesized that Notch3 acts as a luminal pressure gauge or sensor that modulates the myogenic contractile response via regulation of the actin cytoskeleton (Domenga et al., 2004). In the second mouse knockout, ultrastructural vessel defects were not reported, but transcriptional profiling of purified populations of cerebral VSMCs did show deficits in regulators of muscle contraction, including regulators of cell motility and architecture and modest reductions of some canonical Notch targets, such as Hes1 and Hey1 in isolated VSMCs (Arboleda-Velasquez et al., 2008). Notably, the resting cerebral blood flow (CBF) in both Notch3 mice was normal, but middle cerebral artery occlusion led to an exacerbated stroke phenotype. Specifically, CBF defects and more frequent ischemic lesions without vasoconstriction were observed, thus supporting a role for Notch3 in regulating contractile tone in cerebral vessels (Arboleda-Velasquez et al., 2011; Arboleda-Velasquez et al., 2008; Ayata, 2010; Domenga et al., 2004; Krebs et al., 2003). Similarly, evidence of compromised vascular tone and accumulation of blood in vessels in zebrafish notch3 mutants was prominent in response to mild stress, but difficult to detect otherwise. Thus, notch3 seems to have a conserved function in vascular myogenesis and vessel maturation via a mechanism involving canonical Notch pathway targets. Notably, in both zebrafish and mammals, VSMC differentiation is a Notch3-dependent process that occurs during post-larval (zebrafish) and postnatal (mammals) development. In contrast to mouse knockouts, some of the zebrafish notch3 mutants do not survive to adulthood, perhaps owing to incomplete recovery from early CNS defects.
In summary, we have identified mutations disrupting notch3 that cause oligodendrocyte deficits in embryos and vascular pathology in brain and peripheral tissues of adults, revealing roles for Notch3 in glia and vascular cells. It is thus possible that early glial deficits also contribute to white matter defects in individuals with CADASIL. Although arterial and venous fates are properly specified in embryos and adults, the fin vessels of adults show morphologies that are typical of veins. Moreover, adult vessels are swollen and show evidence of compromised vascular tone, including vasodilation and hemorrhage. Zebrafish notch3 mutants will be valuable for future screens to identify Notch signaling modifiers or risk factors affecting the phenotype, and for studies of the activity of CADASIL mutant alleles and the signaling and cellular mechanisms contributing to vascular and other Notch3-associated pathologies.
MATERIALS AND METHODS
Ethics statement
The animal work performed in this study was approved by the Institutional Review Boards of Albert Einstein College of Medicine and Stanford University School of Medicine.
Fish strains, mapping and positional cloning
Fish strains
Zebrafish embryos were raised at 28.5°C and staged according to Kimmel et al. (Kimmel et al., 1995). Identification of the st51 mutation has been described (Pogoda et al., 2006). The notch3zm allele was purchased from Znomics.
Genetic mapping
Wild-type and notch3st51 mutant larvae were sorted based on their mbp expression at 5 dpf. The map position of notch3st51 was refined by scoring SSLP markers from linkage group 3 (LG3) in notchst51 mapping crosses as described (Talbot and Schier, 1999). These mapping studies localized the notch3st51 mutation to a region defined by the markers Z3725 and Z20058, which also contained the notch3 gene (Woods et al., 2005). We sequenced the notch3 gene to find the lesion.
Genotyping
To genotype the notch3 alleles, individual larvae and adults were anesthetized in Tricaine as described and genomic DNA was prepared from adult fin samples or whole embryos and larvae (Westerfield, 1995).
The notch3st51 allele was scored using the following primers to amplify fragments from genomic DNA: forward: 5′-GAGTGCCAGTCGAATCCCTGTC-3′; reverse: 5′-ACACTTGCAGGTGAAACCATTGAGC-3′. The PCR product was digested with RsaI, which cuts the wild-type product but not the mutant.
Two different assays were used to genotype the notch3zm allele. In early experiments, we scored notch3zm by performing two separate PCR assays on each genomic DNA sample. One set of primers (5′-ACTCCGATCCGAAGTGAAGTAG-3′ and 5′-CAGGCAATCACATTAGGATGAA-3′) amplified a 184 bp product from the wild-type allele but no product from the mutant allele. The second set (5′-CTGAAGCCTATAGAGTACGAGCC-3′ and 5′-GTCCGAGCTGCAGAAGGAAATACTG-3′) amplified a product of about 700 bp from the mutant allele but no product from the wild-type allele. In more recent experiments, the following three primers were combined in single PCR reaction to genotype the notch3zm mutation: Common F 5′-GTCCGAGCTGCAGAAGGAAATACTG-3′, ZM R 5′-CTGTGCCTTATTTGAACTAACC-3′ and WT R 5′-GTGGAAACTCGATCCTACTCC-3′. A 283 nt product was amplified from the mutant allele, whereas the wild-type product was 190 nt.
RT-PCR
To examine the notch3 mRNA in notch3st51 mutants, RNA was prepared from the body of individual 3-day-old embryos using the Qiagen Rneasy Micro Kit. cDNA was synthesized using the SuperScript II First-Strand System (Invitrogen) with random hexamer primers following the manufacturer’s protocol. The tail of each embryo was used for genotyping as described above. PCR for notch3 was performed on cDNAs with primers flanking the lesion site: forward 5′-GCACTTCAGGTACCAACTGTGA-3′ (across exons 11 and 12) and reverse 5′-AGAGCAAGGTGTGAGCAGTTCT-3′ (from exon 15). The different PCR products from wild-type, heterozygote and mutant cDNAs were run on an agarose gel and then cloned and sequenced. As a control, ef1a was analyzed as described (Monk et al., 2009).
For examination of Notch target expression, total RNA was isolated from wild-type and mutant fins and larvae at the specified stages using TRIZOL according to standard protocols. cDNA was reverse transcribed using SuperScript II reverse transcriptase (Invitrogen) and oligodT primers according to the manufacturer’s instructions. To control for genomic DNA contamination, reverse transcriptase was omitted using the same RNA samples. The following primers were used for qRT-PCR: hey1f 5′-CGATTTCAGCTCGTCGGACA-3′, hey1r 5′-ATGCAGAACCCCATGCCAAG-3′, her9 (Yang et al., 2009), her9f 5′-CCAGCGTTTGCTTCTGCTACAAC-3′, her9r 5′-GCTCATTGCTTTCTGCTCCG-3′, eef1a1l1(ef1α)f 5′-AGCCTGGTATGGTTGTGACCTTCG-3′, eef1a1l1(ef1α)r 5′-CCAAGTTGTTTTCCTTTCCTGCG-3′.
qRT-PCR was performed using an Eppendorf Realplex3 machine according to standard protocols and cycling conditions. For zm genotypes: n=2 biological replicates and 3 technical replicates; for st51 genotypes: n=5 biological and 3 technical replicates. Percent of wild-type expression was calculated using the ΔΔCT method using ef1a as the standard and wild type as the reference. T-test was calculated using Excel (Microsoft).
In situ hybridization
Whole-mount in situ hybridization was performed as described previously (Thisse and Thisse, 1998), except that for analysis of vascular markers and fins BM purple AP (Roche) was used as the substrate for alkaline phosphatase. To generate the notch3 probe, a region of the gene corresponding to bp 14 to 935 of the notch3 cDNA (GenBank accession number AF152001) was cloned into the pCRII-TOPO vector; the plasmid was digested with EcoRV and transcribed with SP6 RNA polymerase. mbp (Lyons et al., 2005), olig2 (Park et al., 2002), sox10 (Dutton et al., 2001), deltaD (Haddon et al., 1998b), flt4 (Lawson et al., 2002), efnb2a (Durbin et al., 1998), deltaC (Smithers et al., 2000), dab2 (Song et al., 2004) and notch1a (Bierkamp and Campos-Ortega, 1993) probes were prepared as previously described. For gfap, an 899 bp fragment of the gene was amplified from 72 hpf larval cDNA using the following primers: gfap1: 5′-GAGATGATGGGGCTAAACGA-3′; gfap2: 5′-TCCAGCAGCTTCCTGTAGGT-3′. The fragment was inserted into the TopoII (Invitrogen) vector; antisense probe was made by cleaving with XbaI and transcribing with SP6 RNA polymerase. For fin vessel analysis using notch3 and deltaC probes, linear templates were amplified from cDNA using the following primers: T7-notch3F 5′-TAATACGACTCACTATAGGGGGTATTTCGAGACGCACGGC-3′, T3-notch3R 5′-CATTAACCCTCACTAAAGGGAATGTGTTGACACCATCGACGC-3′, T3-deltaCF 5′-CATTAACCCTCACTAAAGGGAAGGAGCACCTCAAACACCAGT-3′, T7-deltaCR 5′-TAATACGACTCACTATAGGGCACAACAGCATCCATCATCC-3′.
Immunohistochemistry and cell death assays
Acetylated tubulin [1:1000 mouse α-acetylated alpha tubulin (Sigma, T6793) and 1:100 FITC-conjugated donkey α-mouse antibody (Jackson, 715-095-151)] and HuC/D (Molecular Probes) immunostainings were carried out as described previously (Lyons et al., 2008). Images were acquired on a Zeiss LSM confocal, Zeiss Observer equipped with Apotome, Perkin-Elmer spinning disk confocal or Leica SP2 confocal microscope. After imaging, larvae were genotyped for the notch3 mutation.
Apoptosis was assessed via AO staining as described previously (Lyons et al., 2005).
Tg[fli-EGFP] transgenic larvae at the specified stages were visualized and imaged live or were fixed in 4% PFA and GFP signals were detected using an anti-GFP antibody. Briefly, 51–52 hpf embryos were fixed overnight (O/N) in 4% PFA and then dehydrated in MeOH via 25%/50%/75% MeOH in PBS. Larvae were rehydrated, treated with 10 ng/μl Proteinase K in PBSTri (PBS, 0.5% for Triton X-100) for 30 minutes and postfixed for 20 minutes in 4% PFA. Following washes with PBSTri, the embryos were preincubated for 3 hours in blocking buffer [10% fetal bovine serum (FBS) and 1% DMSO in PBSTri] before incubation with a 1:1000 dilution of chicken anti-GFP antibody (Invitrogen, A10262) in blocking buffer O/N at 4°C. The next day the embryos were washed with PBSTri and preincubated for 3 hours in blocking buffer. Following incubation O/N at 4°C with a 1:300 dilution of Alexa-Fluor-488-conjugated goat anti-chicken antibody (Jackson, 103-545-155) in blocking buffer, immunostained embryos were washed and then mounted in 1% low melting point agarose (Sigma, A9414) in a glass-bottom dish. Dorsal view images were obtained with a Leica SP2 confocal. After imaging, larvae were genotyped for the notch3 mutation.
Microangiography
Anesthetized 2-dpf progeny from clutches of notch3zm/+ intercrosses were embedded in 1% low melting point agarose and 3–4 nl of 1% Fluorescein dextran (Invitrogen, D-1822) was injected into the sinus venosus, essentially as described previously (Weinstein et al., 1995). After injection, the larvae were cut out of the agarose and remounted under anesthesia (Tricaine) in 1% low melting point agarose on a glass bottom dish. The labeled vasculature was imaged in 60–63 hpf larvae using a Leica SP2 confocal microscope. After imaging, larvae were genotyped for the notch3 mutation.
Histology and transmission electron microscopy
Adults were anesthetized and the brains were dissected and fixed in 4% paraformaldehyde and embedded in paraffin. 10-μm sagittal sections were cut on a microtome, deparaffinized, and stained with H&E.
Fins from wild-type and notch3 mutants were clipped according to standard protocols and fixed with 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 M sodium cacodylate buffer, postfixed with 1% osmium tetroxide followed by 2% uranyl acetate, dehydrated through a graded series of ethanol and embedded in LX112 resin (LADD Research Industries, Burlington, VT). Ultrathin sections (80 nM) were cut on a Reichert Ultracut UCT, stained with uranyl acetate followed by lead citrate and viewed on a JEOL 1200EX transmission electron microscope at 80 kv.
Toluidine-blue-stained sections (1 μM) were coated with permount solution, coverslipped, and images were acquired with an Axioskop2 microscope and Axiocam CCD camera (Zeiss). Images were processed in ImageJ, Adobe Photoshop and Adobe Illustrator.
Quantification of vessel defects
The circumference of vessels within the bony regions of the fin rays (i.e. where arteries are positioned in wild type) was traced and the diameter was determined using ImageJ software. The distance between bony segments was measured using ImageJ (as depicted in Fig. 5A). The ratio of the artery/vessel diameter to length between the bone segments was calculated and plotted using Excel (Microsoft). ANOVA analysis was performed using the calculator at http://www.physics.csbsju.edu/stats/anova_pnp_NGROUP_form.html.
Supplementary Material
Acknowledgments
The authors thank members of the Marlow and Talbot labs for helpful discussions, Xin Li and Drs Lei Fang and Andreas Jenny for critical evaluation of the manuscript, Spartak Kalinin, Clinton Depaolo and Tuky Reyes for fish care, Alya Raphael for providing the gfap clone, William Joo for assistance with early analysis of the notch3zm mutation, and the Einstein Analytical Imaging Facility of Albert Einstein College of Medicine for histology and TEM services and confocal support.
Footnotes
FUNDING
This work was supported by grants to F.L.M. from the National Institutes of Health (NIH; R01GM089979), and to W.S.T. from NIH (R01 NS050223) and the NMSS (RG-4756-A-3). S.M. was supported by a fellowship from Telethon Italy (GFP03011). N.S. was supported by a predoctoral fellowship from the Howard Hughes Medical Institute (HHMI).
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
Conceived and designed the experiments: A.Z., S.M., N.S., W.S.T. and F.L.M. Performed the experiments: A.Z., S.M. and N.S. Analyzed the data: A.Z., S.M., N.S., W.S.T. and F.L.M. All authors discussed the data and the manuscript. A.Z., W.S.T. and F.L.M. wrote the manuscript.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.012005/-/DC1
REFERENCES
- Andersson E. R., Sandberg R., Lendahl U. (2011). Notch signaling: simplicity in design, versatility in function. Development 138, 3593–3612 [DOI] [PubMed] [Google Scholar]
- Arboleda-Velasquez J. F., Rampal R., Fung E., Darland D. C., Liu M., Martinez M. C., Donahue C. P., Navarro-Gonzalez M. F., Libby P., D’Amore P. A., et al. (2005). CADASIL mutations impair Notch3 glycosylation by Fringe. Hum. Mol. Genet. 14, 1631–1639 [DOI] [PubMed] [Google Scholar]
- Arboleda-Velasquez J. F., Zhou Z., Shin H. K., Louvi A., Kim H.-H., Savitz S. I., Liao J. K., Salomone S., Ayata C., Moskowitz M. A., et al. (2008). Linking Notch signaling to ischemic stroke. Proc. Natl. Acad. Sci. USA 105, 4856–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arboleda-Velasquez J. F., Manent J., Lee J. H., Tikka S., Ospina C., Vanderburg C. R., Frosch M. P., Rodríguez-Falcón M., Villen J., Gygi S., et al. (2011). Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small-vessel disease. Proc. Natl. Acad. Sci. USA 108, E128–E135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C. (2010). CADASIL: experimental insights from animal models. Stroke 41 Suppl, S129–S134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudrimont M., Dubas F., Joutel A., Tournier-Lasserve E., Bousser M. G. (1993). Autosomal dominant leukoencephalopathy and subcortical ischemic stroke. A clinicopathological study. Stroke 24, 122–125 [DOI] [PubMed] [Google Scholar]
- Belin de Chantemèle E. J., Retailleau K., Pinaud F., Vessières E., Bocquet A., Guihot A. L., Lemaire B., Domenga V., Baufreton C., Loufrani L., et al. (2008). Notch3 is a major regulator of vascular tone in cerebral and tail resistance arteries. Arterioscler. Thromb. Vasc. Biol. 28, 2216–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierkamp C., Campos-Ortega J. A. (1993). A zebrafish homologue of the Drosophila neurogenic gene Notch and its pattern of transcription during early embryogenesis. Mech. Dev. 43, 87–100 [DOI] [PubMed] [Google Scholar]
- Bingham S., Chaudhari S., Vanderlaan G., Itoh M., Chitnis A., Chandrasekhar A. (2003). Neurogenic phenotype of mind bomb mutants leads to severe patterning defects in the zebrafish hindbrain. Dev. Dyn. 228, 451–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos-Ortega J. A. (1985). Genetics of early neurogenesis in Drosophila melanogaster. Trends Neurosci. 8, 245–250 [DOI] [PubMed] [Google Scholar]
- Carmeliet P., Collen D. (1999). Role of vascular endothelial growth factor and vascular endothelial growth factor receptors in vascular development. Curr. Top. Microbiol. Immunol. 237, 133–158 [DOI] [PubMed] [Google Scholar]
- Carmeliet P., Storkebaum E. (2002). Vascular and neuronal effects of VEGF in the nervous system: implications for neurological disorders. Semin. Cell Dev. Biol. 13, 39–53 [DOI] [PubMed] [Google Scholar]
- Cotanche D. A., Kaiser C. L. (2010). Hair cell fate decisions in cochlear development and regeneration. Hear. Res. 266, 18–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosnier C., Vargesson N., Gschmeissner S., Ariza-McNaughton L., Morrison A., Lewis J. (2005). Delta-Notch signalling controls commitment to a secretory fate in the zebrafish intestine. Development 132, 1093–1104 [DOI] [PubMed] [Google Scholar]
- Domenga V., Fardoux P., Lacombe P., Monet M., Maciazek J., Krebs L. T., Klonjkowski B., Berrou E., Mericskay M., Li Z., et al. (2004). Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 18, 2730–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin L., Brennan C., Shiomi K., Cooke J., Barrios A., Shanmugalingam S., Guthrie B., Lindberg R., Holder N. (1998). Eph signaling is required for segmentation and differentiation of the somites. Genes Dev. 12, 3096–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton K. A., Pauliny A., Lopes S. S., Elworthy S., Carney T. J., Rauch J., Geisler R., Haffter P., Kelsh R. N. (2001). Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development 128, 4113–4125 [DOI] [PubMed] [Google Scholar]
- Gazave E., Lapébie P., Richards G. S., Brunet F., Ereskovsky A. V., Degnan B. M., Borchiellini C., Vervoort M., Renard E. (2009). Origin and evolution of the Notch signalling pathway: an overview from eukaryotic genomes. BMC Evol. Biol. 9, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gridley T. (2010). Notch signaling in the vasculature. Curr. Top. Dev. Biol. 92, 277–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddon C., Jiang Y. J., Smithers L., Lewis J. (1998a). Delta-Notch signalling and the patterning of sensory cell differentiation in the zebrafish ear: evidence from the mind bomb mutant. Development 125, 4637–4644 [DOI] [PubMed] [Google Scholar]
- Haddon C., Smithers L., Schneider-Maunoury S., Coche T., Henrique D., Lewis J. (1998b). Multiple delta genes and lateral inhibition in zebrafish primary neurogenesis. Development 125, 359–370 [DOI] [PubMed] [Google Scholar]
- Hans S., Campos-Ortega J. A. (2002). On the organisation of the regulatory region of the zebrafish deltaD gene. Development 129, 4773–4784 [DOI] [PubMed] [Google Scholar]
- Heath J. K. (2010). Transcriptional networks and signaling pathways that govern vertebrate intestinal development. Curr. Top. Dev. Biol. 90, 159–192 [DOI] [PubMed] [Google Scholar]
- Hervé D., Chabriat H. (2010). Cadasil. J. Geriatr. Psychiatry Neurol. 23, 269–276 [DOI] [PubMed] [Google Scholar]
- Imayoshi I., Sakamoto M., Yamaguchi M., Mori K., Kageyama R. (2010). Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J. Neurosci. 30, 3489–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvin D. K., Zurcher S. D., Nguyen T., Weinmaster G., Kornblum H. I. (2001). Expression patterns of Notch1, Notch2, and Notch3 suggest multiple functional roles for the Notch-DSL signaling system during brain development. J. Comp. Neurol. 436, 167–181 [PubMed] [Google Scholar]
- Itoh M., Kim C. H., Palardy G., Oda T., Jiang Y. J., Maust D., Yeo S. Y., Lorick K., Wright G. J., Ariza-McNaughton L., et al. (2003). Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev. Cell 4, 67–82 [DOI] [PubMed] [Google Scholar]
- Joutel A. (2011). Pathogenesis of CADASIL: transgenic and knock-out mice to probe function and dysfunction of the mutated gene, Notch3, in the cerebrovasculature. Bioessays 33, 73–80 [DOI] [PubMed] [Google Scholar]
- Joutel A., Vahedi K., Corpechot C., Troesch A., Chabriat H., Vayssière C., Cruaud C., Maciazek J., Weissenbach J., Bousser M. G., et al. (1997). Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 350, 1511–1515 [DOI] [PubMed] [Google Scholar]
- Joutel A., Monet M., Domenga V., Riant F., Tournier-Lasserve E. (2004). Pathogenic mutations associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy differently affect Jagged1 binding and Notch3 activity via the RBP/JK signaling Pathway. Am. J. Hum. Genet. 74, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalimo H., Ruchoux M. M., Viitanen M., Kalaria R. N. (2002). CADASIL: a common form of hereditary arteriopathy causing brain infarcts and dementia. Brain Pathol. 12, 371–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 [DOI] [PubMed] [Google Scholar]
- Kitamoto T., Takahashi K., Takimoto H., Tomizuka K., Hayasaka M., Tabira T., Hanaoka K. (2005). Functional redundancy of the Notch gene family during mouse embryogenesis: analysis of Notch gene expression in Notch3-deficient mice. Biochem. Biophys. Res. Commun. 331, 1154–1162 [DOI] [PubMed] [Google Scholar]
- Kopan R., Ilagan M. X. (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs L. T., Xue Y., Norton C. R., Sundberg J. P., Beatus P., Lendahl U., Joutel A., Gridley T. (2003). Characterization of Notch3-deficient mice: normal embryonic development and absence of genetic interactions with a Notch1 mutation. Genesis 37, 139–143 [DOI] [PubMed] [Google Scholar]
- Lawson N. D., Weinstein B. M. (2002a). Arteries and veins: making a difference with zebrafish. Nat. Rev. Genet. 3, 674–682 [DOI] [PubMed] [Google Scholar]
- Lawson N. D., Weinstein B. M. (2002b). In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307–318 [DOI] [PubMed] [Google Scholar]
- Lawson N. D., Scheer N., Pham V. N., Kim C. H., Chitnis A. B., Campos-Ortega J. A., Weinstein B. M. (2001). Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128, 3675–3683 [DOI] [PubMed] [Google Scholar]
- Lawson N. D., Vogel A. M., Weinstein B. M. (2002). Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev. Cell 3, 127–136 [DOI] [PubMed] [Google Scholar]
- Lee J. H., Bacskai B. J., Ayata C. (2012). Genetic animal models of cerebral vasculopathies. Prog. Mol. Biol. Transl. Sci. 105, 25–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie J. D., Ariza-McNaughton L., Bermange A. L., McAdow R., Johnson S. L., Lewis J. (2007). Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development 134, 839–844 [DOI] [PubMed] [Google Scholar]
- Lewis J. (1996). Neurogenic genes and vertebrate neurogenesis. Curr. Opin. Neurobiol. 6, 3–10 [DOI] [PubMed] [Google Scholar]
- Lindsell C. E., Boulter J., diSibio G., Gossler A., Weinmaster G. (1996). Expression patterns of Jagged, Delta1, Notch1, Notch2, and Notch3 genes identify ligand-receptor pairs that may function in neural development. Mol. Cell. Neurosci. 8, 14–27 [DOI] [PubMed] [Google Scholar]
- Lyons D. A., Pogoda H. M., Voas M. G., Woods I. G., Diamond B., Nix R., Arana N., Jacobs J., Talbot W. S. (2005). erbb3 and erbb2 are essential for schwann cell migration and myelination in zebrafish. Curr. Biol. 15, 513–524 [DOI] [PubMed] [Google Scholar]
- Lyons D. A., Naylor S. G., Mercurio S., Dominguez C., Talbot W. S. (2008). KBP is essential for axonal structure, outgrowth and maintenance in zebrafish, providing insight into the cellular basis of Goldberg-Shprintzen syndrome. Development 135, 599–608 [DOI] [PubMed] [Google Scholar]
- Mahler J., Filippi A., Driever W. (2010). DeltaA/DeltaD regulate multiple and temporally distinct phases of notch signaling during dopaminergic neurogenesis in zebrafish. J. Neurosci. 30, 16621–16635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano J. M., Georger M. A., Rich A., De Mesy Bentley K. L.(2006). Ultrastructure of zebrafish dorsal aortic cells. Zebrafish 3, 455–463 [DOI] [PubMed] [Google Scholar]
- Monet-Leprêtre M., Bardot B., Lemaire B., Domenga V., Godin O., Dichgans M., Tournier-Lasserve E., Cohen-Tannoudji M., Chabriat H., Joutel A. (2009). Distinct phenotypic and functional features of CADASIL mutations in the Notch3 ligand binding domain. Brain 132, 1601–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk K. R., Naylor S. G., Glenn T. D., Mercurio S., Perlin J. R., Dominguez C., Moens C. B., Talbot W. S. (2009). A G protein-coupled receptor is essential for Schwann cells to initiate myelination. Science 325, 1402–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama M., Osawa M., Mak S. S., Ohtsuka T., Yamamoto N., Han H., Delmas V., Kageyama R., Beermann F., Larue L., et al. (2006). Notch signaling via Hes1 transcription factor maintains survival of melanoblasts and melanocyte stem cells. J. Cell Biol. 173, 333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeda R., Arima K., Kawai M. (2002). Arterial changes in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) in relation to pathogenesis of diffuse myelin loss of cerebral white matter: examination of cerebral medullary arteries by reconstruction of serial sections of an autopsy case. Stroke 33, 2565–2569 [DOI] [PubMed] [Google Scholar]
- Opherk C., Duering M., Peters N., Karpinska A., Rosner S., Schneider E., Bader B., Giese A., Dichgans M. (2009). CADASIL mutations enhance spontaneous multimerization of NOTCH3. Hum. Mol. Genet. 18, 2761–2767 [DOI] [PubMed] [Google Scholar]
- Owens G. K. (1995). Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 75, 487–517 [DOI] [PubMed] [Google Scholar]
- Owens G. K. (1998). Molecular control of vascular smooth muscle cell differentiation. Acta Physiol. Scand. 164, 623–635 [DOI] [PubMed] [Google Scholar]
- Owens G. K., Wise G. (1997). Regulation of differentiation/maturation in vascular smooth muscle cells by hormones and growth factors. Agents Actions Suppl. 48, 3–24 [DOI] [PubMed] [Google Scholar]
- Owens G. K., Kumar M. S., Wamhoff B. R. (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801 [DOI] [PubMed] [Google Scholar]
- Pantoni L. (2010). Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 9, 689–701 [DOI] [PubMed] [Google Scholar]
- Park H. C., Appel B. (2003). Delta-Notch signaling regulates oligodendrocyte specification. Development 130, 3747–3755 [DOI] [PubMed] [Google Scholar]
- Park H. C., Kim C. H., Bae Y. K., Yeo S. Y., Kim S. H., Hong S. K., Shin J., Yoo K. W., Hibi M., Hirano T., et al. (2000). Analysis of upstream elements in the HuC promoter leads to the establishment of transgenic zebrafish with fluorescent neurons. Dev. Biol. 227, 279–293 [DOI] [PubMed] [Google Scholar]
- Park H. C., Mehta A., Richardson J. S., Appel B. (2002). olig2 is required for zebrafish primary motor neuron and oligodendrocyte development. Dev. Biol. 248, 356–368 [DOI] [PubMed] [Google Scholar]
- Park H. C., Shin J., Roberts R. K., Appel B. (2007). An olig2 reporter gene marks oligodendrocyte precursors in the postembryonic spinal cord of zebrafish. Dev. Dyn. 236, 3402–3407 [DOI] [PubMed] [Google Scholar]
- Pierfelice T., Alberi L., Gaiano N. (2011). Notch in the vertebrate nervous system: an old dog with new tricks. Neuron 69, 840–855 [DOI] [PubMed] [Google Scholar]
- Pogoda H. M., Sternheim N., Lyons D. A., Diamond B., Hawkins T. A., Woods I. G., Bhatt D. H., Franzini-Armstrong C., Dominguez C., Arana N., et al. (2006). A genetic screen identifies genes essential for development of myelinated axons in zebrafish. Dev. Biol. 298, 118–131 [DOI] [PubMed] [Google Scholar]
- Prineas J. W., Parratt J. D. (2012). Oligodendrocytes and the early multiple sclerosis lesion. Ann. Neurol. 72, 18–31 [DOI] [PubMed] [Google Scholar]
- Qiu X., Lim C. H., Ho S. H., Lee K. H., Jiang Y. J. (2009). Temporal Notch activation through Notch1a and Notch3 is required for maintaining zebrafish rhombomere boundaries. Dev. Genes Evol. 219, 339–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson W., Galetta S. L., McCluskey L., Forman M. S., Balcer L. J. (2001). Retinal findings in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (cadasil). Surv. Ophthalmol. 45, 445–448 [DOI] [PubMed] [Google Scholar]
- Ruchoux M. M., Guerouaou D., Vandenhaute B., Pruvo J. P., Vermersch P., Leys D. (1995). Systemic vascular smooth muscle cell impairment in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol. 89, 500–512 [DOI] [PubMed] [Google Scholar]
- Ruchoux M. M., Domenga V., Brulin P., Maciazek J., Limol S., Tournier-Lasserve E., Joutel A. (2003). Transgenic mice expressing mutant Notch3 develop vascular alterations characteristic of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Am. J. Pathol. 162, 329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruohola H., Bremer K. A., Baker D., Swedlow J. R., Jan L. Y., Jan Y. N. (1991). Role of neurogenic genes in establishment of follicle cell fate and oocyte polarity during oogenesis in Drosophila. Cell 66, 433–449 [DOI] [PubMed] [Google Scholar]
- Sainson R. C., Aoto J., Nakatsu M. N., Holderfield M., Conn E., Koller E., Hughes C. C. (2005). Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J. 19, 1027–1029 [DOI] [PubMed] [Google Scholar]
- Santoro M. M., Pesce G., Stainier D. Y. (2009). Characterization of vascular mural cells during zebrafish development. Mech. Dev. 126, 638–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih J., Keller R. (1992). Patterns of cell motility in the organizer and dorsal mesoderm of Xenopus laevis. Development 116, 915–930 [DOI] [PubMed] [Google Scholar]
- Shin J., Park H. C., Topczewska J. M., Mawdsley D. J., Appel B. (2003). Neural cell fate analysis in zebrafish using olig2 BAC transgenics. Methods Cell Sci. 25, 7–14 [DOI] [PubMed] [Google Scholar]
- Siekmann A. F., Lawson N. D. (2007). Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 445, 781–784 [DOI] [PubMed] [Google Scholar]
- Smithers L., Haddon C., Jiang Y. J., Lewis J. (2000). Sequence and embryonic expression of deltaC in the zebrafish. Mech. Dev. 90, 119–123 [DOI] [PubMed] [Google Scholar]
- Song H. D., Sun X. J., Deng M., Zhang G. W., Zhou Y., Wu X. Y., Sheng Y., Chen Y., Ruan Z., Jiang C. L., et al. (2004). Hematopoietic gene expression profile in zebrafish kidney marrow. Proc. Natl. Acad. Sci. USA 101, 16240–16245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi H., Yoshida S., Sugimori M., Kosako H., Kominami R., Nakafuku M., Nabeshima Y. (2000). Dynamic expression of basic helix-loop-helix Olig family members: implication of Olig2 in neuron and oligodendrocyte differentiation and identification of a new member, Olig3. Mech. Dev. 99, 143–148 [DOI] [PubMed] [Google Scholar]
- Takke C., Dornseifer P., v Weizsäcker E., Campos-Ortega J. A. (1999). her4, a zebrafish homologue of the Drosophila neurogenic gene E(spl), is a target of NOTCH signalling. Development 126, 1811–1821 [DOI] [PubMed] [Google Scholar]
- Talbot W. S., Schier A. F. (1999). Positional cloning of mutated zebrafish genes. Methods Cell Biol. 60, 259–286 [DOI] [PubMed] [Google Scholar]
- Tang H., Brennan J., Karl J., Hamada Y., Raetzman L., Capel B. (2008). Notch signaling maintains Leydig progenitor cells in the mouse testis. Development 135, 3745–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C., Thisse B. (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59–69 [DOI] [PubMed] [Google Scholar]
- Torres-Vázquez J., Kamei M., Weinstein B. M. (2003). Molecular distinction between arteries and veins. Cell Tissue Res. 314, 43–59 [DOI] [PubMed] [Google Scholar]
- Ulrich F., Ma L. H., Baker R. G., Torres-Vázquez J. (2011). Neurovascular development in the embryonic zebrafish hindbrain. Dev. Biol. 357, 134–151 [DOI] [PubMed] [Google Scholar]
- Viswanathan A., Gray F., Bousser M. G., Baudrimont M., Chabriat H. (2006). Cortical neuronal apoptosis in CADASIL. Stroke 37, 2690–2695 [DOI] [PubMed] [Google Scholar]
- Wang S., Sdrulla A. D., diSibio G., Bush G., Nofziger D., Hicks C., Weinmaster G., Barres B. A. (1998). Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 21, 63–75 [DOI] [PubMed] [Google Scholar]
- Weinstein B. M., Lawson N. D. (2002). Arteries, veins, Notch, and VEGF. Cold Spring Harb. Symp. Quant. Biol. 67, 155–162 [DOI] [PubMed] [Google Scholar]
- Weinstein B. M., Stemple D. L., Driever W., Fishman M. C. (1995). Gridlock, a localized heritable vascular patterning defect in the zebrafish. Nat. Med. 1, 1143–1147 [DOI] [PubMed] [Google Scholar]
- Westerfield M. (1995). The Zebrafish Book. Eugene, OR: University Oregon Press [Google Scholar]
- Woods I. G., Wilson C., Friedlander B., Chang P., Reyes D. K., Nix R., Kelly P. D., Chu F., Postlethwait J. H., Talbot W. S. (2005). The zebrafish gene map defines ancestral vertebrate chromosomes. Genome Res. 15, 1307–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Chan C. Y., Jiang B., Yu X., Zhu G. Z., Chen Y., Barnard J., Mei W. (2009). hnRNP I inhibits Notch signaling and regulates intestinal epithelial homeostasis in the zebrafish. PLoS Genet. 5, e1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K. J., Koo B. K., Im S. K., Jeong H. W., Ghim J., Kwon M. C., Moon J. S., Miyata T., Kong Y. Y. (2008). Mind bomb 1-expressing intermediate progenitors generate notch signaling to maintain radial glial cells. Neuron 58, 519–531 [DOI] [PubMed] [Google Scholar]
- Zannino D. A., Appel B. (2009). Olig2+ precursors produce abducens motor neurons and oligodendrocytes in the zebrafish hindbrain. J. Neurosci. 29, 2322–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.