

Jane Carlton, Kazuyuki Tanabe and colleagues report the draft genome sequences of three Plasmodium cynomolgi strains isolated from infected monkeys. Their comparative genomic analysis with P. vivax and P. knowlesi offers insights into these simian malaria parasites.
Supplementary information
The online version of this article (doi:10.1038/ng.2375) contains supplementary material, which is available to authorized users.
Subject terms: Genomic analysis, Phylogenetics, Disease genetics
Abstract
P. cynomolgi, a malaria-causing parasite of Asian Old World monkeys, is the sister taxon of P. vivax, the most prevalent malaria-causing species in humans outside of Africa. Because P. cynomolgi shares many phenotypic, biological and genetic characteristics with P. vivax, we generated draft genome sequences for three P. cynomolgi strains and performed genomic analysis comparing them with the P. vivax genome, as well as with the genome of a third previously sequenced simian parasite, Plasmodium knowlesi. Here, we show that genomes of the monkey malaria clade can be characterized by copy-number variants (CNVs) in multigene families involved in evasion of the human immune system and invasion of host erythrocytes. We identify genome-wide SNPs, microsatellites and CNVs in the P. cynomolgi genome, providing a map of genetic variation that can be used to map parasite traits and study parasite populations. The sequencing of the P. cynomolgi genome is a critical step in developing a model system for P. vivax research and in counteracting the neglect of P. vivax.
Supplementary information
The online version of this article (doi:10.1038/ng.2375) contains supplementary material, which is available to authorized users.
Main
Human malaria is transmitted by anopheline mosquitoes and is caused by four species in the genus Plasmodium. Of these, P. vivax is the major malaria agent outside of Africa, annually causing 80–100 million cases1. Although P. vivax infection is often mistakenly regarded as benign and self-limiting, P. vivax treatment and control present challenges distinct from those of the more virulent Plasmodium falciparum. Biological traits, including a dormant (hypnozoite) liver stage responsible for recurrent infections (relapses), early infective sexual stages (gametocytes) and transmission from low parasite densities in the blood2, coupled with emerging antimalarial drug resistance3, render P. vivax resilient to modern control strategies. Recent evidence indicates that P. falciparum derives from parasites of great apes in Africa4, whereas P. vivax is more closely related to parasites of Asian Old World monkeys5,6,7, although not itself infective of these monkeys.
P. vivax cannot be cultured in vitro, and the small New World monkeys capable of hosting it are rare and do not provide an ideal model system. P. knowlesi, an Asian Old World monkey parasite recently recognized as a zoonosis for humans8, has had its genome sequenced9, but the species is distantly related to P. vivax and is phenotypically dissimilar. In contrast, P. cynomolgi, a simian parasite that can infect humans experimentally10, is the closest living relative (a sister taxon) to P. vivax and possesses most of the same genetic, phenotypic and biological characteristics—notably, periodic relapses caused by dormant hypnozoites, early infectious gametocyte formation and invasion of Duffy blood group–positive reticulocytes. P. cynomolgi thus offers a robust model for P. vivax in a readily available laboratory host, the Rhesus monkey, whose genome was recently sequenced11. Here, we report draft genome sequences of three P. cynomolgi strains and comparative genomic analyses of P. cynomolgi, P. vivax12 and P. knowlesi9, three members of the monkey malaria clade.
We sequenced the genome of P. cynomolgi strain B, isolated from a monkey in Malaysia and grown in splenectomized monkeys (Online Methods). A combination of Sanger, Roche 454 and Illumina chemistries was employed to generate a high-quality reference assembly at 161-fold coverage, consisting of 14 supercontigs (corresponding to the 14 parasite chromosomes) and ∼1,649 unassigned contigs, comprising a total length of ∼26.2 Mb (Supplementary Table 1). Comparing genomic features of P. cynomolgi, P. knowlesi and P. vivax reveals many similarities, including GC content (mean GC content of 40.5%), 14 positionally conserved centromeres and the presence of intrachromosomal telomeric sequences (ITSs; GGGTT(T/C)A), which were discovered in the P. knowlesi genome9 but are absent in P. vivax (Fig. 1, Table 1 and Supplementary Table 2).
Figure 1. Architecture of the P. cynomolgi genome and associated genome-wide variation data.

Data are shown for each of the 14 P. cynomolgi chromosomes. The six concentric rings, from outermost to innermost, represent (i) the location of 5,049 P. cynomolgi genes, excluding those on small contigs (cyan lines); (ii) genome features, including 14 centromeres (thick black lines), 43 telomeric sequence repeats (short red lines), 43 tRNA genes (red lines), 10 rRNAs (dark blue lines) and several gene family members, including 53 cyir (dark green lines), 8 rbp (brown lines), 13 sera (serine-rich antigen; pink lines), 25 trag (tryptophan-rich antigen; purple lines), 12 msp3 (merozoite surface protein 3; light gray lines), 13 msp7 (merozoite surface protein 7; gray lines), 25 rad (silver lines), 8 etramp (orange lines), 16 Pf-fam-b (light blue lines) and 7 Pv-fam-d (light green lines); (iii) plot of dS/dN for 4,605 orthologs depicting genome-wide polymorphism within P. cynomolgi strains B and Berok (black line) and divergence between P. cynomolgi strains B and Berok and P. vivax Salvador I (red line); a track above the plot indicates P. cynomolgi genes under positive selection (red) and purifying selection (blue), and a track below the plot indicates P. cynomolgi–P. vivax orthologs under positive selection (red) and purifying selection (blue); (iv) heatmap indicating SNP density of 3 P. cynomolgi strains plotted per 10-kb windows: red, 0–83 SNPs per 10 kb (regions of lowest SNP density); blue, 84–166 SNPs per 10 kb; green, 167–250 SNPs per 10 kb; purple, 251–333 SNPs per 10 kb; orange, 334–416 SNPs per10 kb; yellow, 417–500 SNPs per10 kb (regions of highest SNP density); (v) log2 ratio plot of CNVs identified from a comparison of P. cynomolgi strains B and Berok; and (vi) map of 182 polymorphic intergenic microsatellites (MS, black dots). The figure was generated using Circos software (see URLs).
Table 1.
Comparison of genome features between P. cynomolgi, P. vivax and P. knowlesi, three species of the monkey malaria clade
| Feature | P. cynomolgi | P. vivax 12 | P. knowlesi 9 |
|---|---|---|---|
| Assembly | |||
| Size (Mb) | 26.2 | 26.9 | 23.7 |
| Number of scaffoldsa | 14 (1,649) | 14 (2,547) | 14 (67) |
| Coverage (fold) | 161 | 10 | 8 |
| GC content (%) | 40.4 | 42.3 | 38.8 |
| Genes | |||
| Number of genes | 5,722 | 5,432 | 5,197 |
| Mean gene length (bp) | 2,240 | 2,164 | 2,180 |
| Gene density (bp per gene)b | 4,428.2 | 4,950.5 | 4,416.1 |
| Percentage codingb | 51.0 | 47.1 | 49.0 |
| Structural RNAs | |||
| Number of tRNA genes | 43 | 44 | 41 |
| Number of 5S rRNA genes | 3 | 3 | 0c |
| Number of 5.8S, 18S and 28S rRNA units | 7 | 7 | 5 |
| Nuclear genome | |||
| Number of chromosomes | 14 | 14 | 14 |
| Number of centromeres | 14 | 14 | 14 |
| Isochore structured | + | + | − |
| Mitochondrial genome | |||
| Size (bp)e | 5,986 (AB444123) | 5,990 (AY598140) | 5,958 (AB444108) |
| GC content (%) | 30.3 | 30.5 | 30.5 |
| Apicoplast genome | |||
| Size (bp) | 29,297f | 5,064g | N/A |
| GC content (%) | 13.0 | 17.1 | N/A |
N/A, not available.
aSmall unassigned contigs indicated in parentheses.
bSequence gaps excluded.
cNot present in P. knowlesi assembly version 4.0.
dRegions of the genome that differ in density and are separable by CsCl centrifugation; isochores correspond to domains differing in GC content.
eIdentified in other studies (GenBank accessions given in parentheses).
fPartial sequence (∼86% complete) generated during this project.
gPartial sequence of reference genome only published12; actual size is ∼35 kb.
We annotated the P. cynomolgi strain B genome using a combination of ab initio gene prediction programs trained on high-quality data sets and sequence similarity searches against the annotated P. vivax and P. knowlesi genomes. Not unexpectedly for species from the same monkey malaria clade, gene synteny along the 14 chromosomes is highly conserved, although numerous microsyntenic breaks are present in regions containing multigene families (Fig. 2 and Table 2). This genome-wide view of synteny in six species of Plasmodium also identified two apparent errors in existing public sequence databases: an inversion in chromosome 3 of P. knowlesi and an inversion in chromosome 6 of P. vivax. The P. cynomolgi genome contains 5,722 genes, of which approximately half encode conserved hypothetical proteins of unknown function, as is the case in all the Plasmodium genomes sequenced to date. A maximum-likelihood phylogenetic tree constructed using 192 conserved ribosomal and translation- and transcription-related genes (Supplementary Fig. 1) confirms the close relationship of P. cynomolgi to P. vivax compared to five other Plasmodium species. Approximately 90% of genes (4,613) have reciprocal best-match orthologs in all three species (Fig. 3), enabling refinement of the existing P. vivax and P. knowlesi annotations (Supplementary Table 3). The high degree of gene orthology enabled us to identify specific examples of gene duplication (an important vehicle for genome evolution), including a duplicated homolog of P. vivax Pvs28—which encodes a sexual stage surface antigen that is a transmission-blocking vaccine candidate13—in P. cynomolgi (Supplementary Table 4). Genes common only to P. cynomolgi and P. vivax (n = 214) outnumber those that are restricted to P. cynomolgi and P. knowlesi (n = 100) or P. vivax and P. knowlesi (n = 17). Such figures establish the usefulness of P. cynomolgi as a model species for studying the more intractable P. vivax.
Figure 2. Genome synteny between six species of Plasmodium parasite.
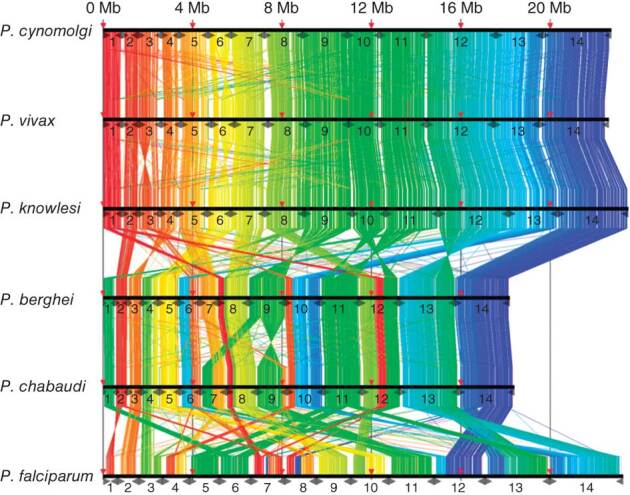
Protein-coding genes of P. cynomolgi are shown aligned with those of five other Plasmodium genomes: two species belonging to the monkey malaria clade, P. vivax and P. knowlesi; two species of rodent malaria, P. berghei and P. chabaudi; and P. falciparum. Highly conserved protein-coding regions between the genomes are colored in order from red (5′ end of chromosome 1) to blue (3′ end of chromosome 14) with respect to genomic position of P. cynomolgi.
Table 2.
Components of multigene families of P. cynomolgi, P. vivax and P. knowlesi differ in copy number
| Family | Multigene family | Localization | Arrangement | P. cynomolgi | P. vivax | P. knowlesi | Putative function and other information |
|---|---|---|---|---|---|---|---|
| 1 | pir (vir-like) | Subtelomeric | Scattered and clustered | 254 | 319a | 4 | Immune evasion |
| 2 | pir (kir-like) | Subtelomeric and central | Scattered and clustered | 11 | 2 | 66a | Immune evasion |
| 3 | SICAvar | Subtelomeric and central | Scattered and clustered | 2 | 1 | 242a | Antigenic variation, immune evasion |
| 4 | msp3 | Central | Clustered | 12 | 12 | 3 | Merozoite surface protein |
| 5 | msp7 | Central | Clustered | 13 | 13 | 5 | Merozoite surface protein |
| 6 | dbl (dbp/ebl) | Subtelomeric | Scattered | 2 | 1 | 3 | Host cell recognition |
| 7 | rbl (rbp/nbp/rh) | Subtelomeric | Scattered | 8a | 10a | 3a | Host cell recognition |
| 8 | Pv-fam-a (trag) | Subtelomeric | Scattered and clustered | 36 | 36 | 26a | Tryptophan-rich |
| 9 | Pv-fam-b | Central | Clustered | 3 | 6 | 1 | Unknown |
| 10 | Pv-fam-c | Subtelomeric | Unknownb | 1 | 7 | 0 | Unknown |
| 11 | Pv-fam-d (hypb) | Subtelomeric | Scattered | 18 | 16 | 2 | Unknown |
| 12 | Pv-fam-e (rad) | Subtelomeric | Clustered | 27 | 44 | 16 | Unknown |
| 13 | Pv-fam-g | Central | Clustered | 3 | 3 | 3 | Unknown |
| 14 | Pv-fam-h (hyp16) | Central | Clustered | 6 | 4 | 2 | Unknown |
| 15 | Pv-fam-i (hyp11) | Subtelomeric | Scattered | 6 | 6 | 5 | Unknown |
| 16 | Pk-fam-a | Central | Scattered | 0 | 0 | 12a | Unknown |
| 17 | Pk-fam-b | Subtelomeric | Scattered | 0 | 0 | 9 | Unknown |
| 18 | Pk-fam-c | Subtelomeric | Scattered | 0 | 0 | 6a | Unknown |
| 19 | Pk-fam-d | Central | Scattered | 0 | 0 | 3a | Unknown |
| 20 | Pk-fam-e | Subtelomeric | Scattered | 0 | 0 | 3a | Unknown |
| 21 | PST-A | Subtelomeric and central | Scattered | 9a | 11a | 7 | αβ hydrolase |
| 22 | ETRAMP | Subtelomeric | Scattered | 9 | 9 | 9 | Parasitophorous vacuole membrane |
| 23 | CLAG (RhopH-1) | Subtelomeric | Scattered | 2 | 3 | 2 | High-molecular-wieght rhoptry antigen complex |
| 24 | PvSTP1 | Subtelomeric | Unknownb | 3 | 10a | 0 | Unknown |
| 25 | PHIST (Pf-fam-b) | Subtelomeric | Scattered and clustered | 21 | 20 | 15 | Unknown |
| 26 | SERA | Central | Clustered | 13a | 13a | 8a | Cysteine protease |
aPseudogenes, truncated genes and gene fragments included.
bGene arrangement could not be determined due to localization on unassigned contigs.
Figure 3. Comparison of the genes of P. cynomolgi, P. vivax and P. knowlesi.
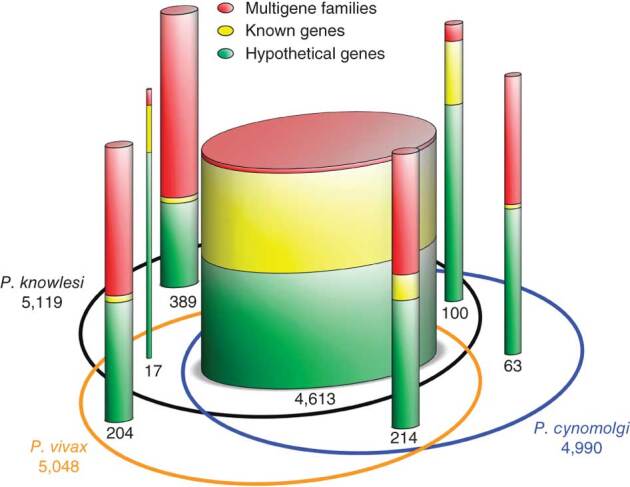
The Venn ellipses represent the three genomes, with the total number of genes assigned to the chromosomes indicated under the species name. Cylinders depict orthologous and non-orthologous genes between the three genomes, with the number of genes in each indicated and represented graphically by cylinder relative width. In each cylinder, genes are divided into three categories whose thickness is represented by colored bands proportional to category percentage.
Notably, most of the genes specific to a particular species belong to multigene families (excluding hypothetical genes; Table 2 and Supplementary Table 5). This suggests repeated lineage-specific gene duplication and/or gene deletion in multigene families within the three monkey malaria clade species. Moreover, copy numbers of the genes composing multigene families were generally greater in the P. cynomolgi–P. vivax lineage than in P. knowlesi, suggesting repeated gene duplication in the ancestral lineage of P. cynomolgi and P. vivax (or repeated gene deletion in the P. knowlesi lineage). Thus, the genomes of P. cynomolgi, P. vivax and P. knowlesi can largely be distinguished by variations in the copy number of multigene family members. Examples of such families include those that encode proteins involved in evasion of the human immune system (vir, kir and SICAvar) and invasion of host red blood cells (dbp and rbp).
In malaria-causing parasites, invasion of host erythrocytes, mediated by specific interactions between parasite ligands and erythrocyte receptors, is a crucial component of the parasite lifecycle. Of great interest are the ebl and rbl gene families, which encode parasite ligands required for the recognition of host erythrocytes. The ebl genes encode erythrocyte binding–like (EBL) ligands such as the Duffy-binding proteins (DBPs) that bind to Duffy antigen receptor for chemokines (DARC) on human and monkey erythrocytes. The rbl genes encode the reticulocyte binding–like (RBL) protein family, including reticulocyte-binding proteins (RBPs) in P. cynomolgi and P. vivax, and normocyte-binding proteins (NBPs) in P. knowlesi, which bind to unknown erythrocyte receptors14. We confirmed the presence of two dbp genes in P. cynomolgi15 (Supplementary Table 6), in contrast to the one dbp and three dbp genes identified in P. vivax and P. knowlesi, respectively. This raises an intriguing hypothesis that P. vivax lost one dbp gene, and thus its infectivity of Old World monkey erythrocytes, after divergence from a common P. vivax–P. cynomolgi ancestor. This hypothesis is also supported by our identification of single-copy dbp genes in two other closely related Old World monkey malaria-causing parasites, Plasmodium fieldi and Plasmodium simiovale, which are incapable of infecting humans16. These two Old World monkey species lost one or more dbp genes during divergence that confer infectivity to humans, whereas P. cynomolgi and P. knowlesi retained dbp genes that allow invasion of human erythrocytes (Supplementary Fig. 2).
We found multiple rbp genes, some truncated or present as pseudogenes, in the P. cynomolgi genome (Fig. 1 and Table 2). Phylogenetic analysis showed that rbl genes from P. cynomolgi, P. vivax and P. knowlesi can be classified into three distinct groups, RBP/NBP-1, RBP/NBP-2 and RBP/NBP-3 (Supplementary Fig. 3), and suggests that these groups existed before the three species diverged. All three groups of RBP/NBP are represented in P. cynomolgi, whereas P. vivax and P. knowlesi lack functional genes from the RBP/NBP-3 and RBP/NBP-1 groups, respectively. Thus, rbl gene family expansion seems to have occurred after speciation, indicating that the three species have multiple species-specific erythrocyte invasion mechanisms. Notably, we found an ortholog of P. vivax rbp1b in some strains of P. cynomolgi but not in others (Supplementary Table 6). To our knowledge, this is the first example of a CNV for a rbp gene between strains of a single Plasmodium species, highlighting how repeated creation and destruction of rbl genes, a signature of adaptive evolution, may have enabled species of the monkey malaria clade to expand or switch between monkey and human hosts.
The largest gene family in P. cynomolgi, consisting of 256 cyir (cynomolgi-interspersed repeat) genes, is part of the pir (plasmodium-interspersed repeat) superfamily that includes P. vivax vir genes (n = 319) and P. knowlesi kir genes (n = 70) (Table 2). Pir-encoded proteins reside on the surface of infected erythrocytes and have an important role in immune evasion17. Most cyir genes have sequence similarity to P. vivax vir genes (n = 254; Supplementary Table 7) and are found in subtelomeric regions (Fig. 1), but, notably, 11 cyir genes have sequence similarity to P. knowlesi kir genes (Supplementary Table 7) and occur more internally in the chromosomes, as do the kir genes in P. knowlesi. As with 'molecular mimicry' in P. knowlesi (mimicry of host sequences by pathogen sequences)9, one CYIR protein (encoded by PCYB_032250) has a region of 56 amino acids that is highly similar to the extracellular domain of primate CD99 (Supplementary Fig. 4), a molecule involved in the regulation of T-cell function. A new finding is that P. cynomolgi has two genes whose sequences are similar to P. knowlesi SICAvar genes (Supplementary Table 7) that are expressed on the surfaces of schizont-infected macaque erythrocytes and are involved in antigenic variation18.
The ability to form a dormant hypnozoite stage is common to both P. cynomolgi and P. vivax and was first shown in laboratory infections of monkeys by mosquito-transmitted P. cynomolgi19. In a search for candidate genes involved in the hypnozoite stage, we identified nine coding for 'dormancy-related' proteins that had the upstream ApiAP2 motifs20 necessary for stage-specific transcriptional regulation at the sporozoite (pre-hypnozoite) stage (Supplementary Table 8). The candidates include kinases that are involved in cell cycle transition; hypnozoite formation may be regulated by phosphorylation of proteins specifically expressed at the pre-hypnozoite stage. Our list of P. cynomolgi candidate genes represents an informed starting point for experimental studies of this elusive stage.
We sequenced P. cynomolgi strains Berok (from Malaysia) and Cambodian (from Cambodia) to 26× and 17× coverage, respectively, to characterize P. cynomolgi genome-wide diversity through analysis of SNPs, CNVs and microsatellites. A comparison of the three P. cynomolgi strains identified 178,732 SNPs (Supplementary Table 9) at a frequency of 1 SNP per 151 bp, a polymorphism level somewhat similar to that found when P. falciparum genomes are compared21,22. We calculated the pairwise nucleotide diversity (π) as 5.41 × 10−3 across the genome, which varies little between the chromosomes. We assessed genome-wide CNVs between the P. cynomolgi B and Berok strains, using a robust statistical model in the CNV-seq program23, by which we identified 1,570 CNVs (1 per 17 kb), including 1 containing the rbp1b gene on chromosome 7 (Supplementary Fig. 5). Finally, mining of the P. cynomolgi B and Berok strains identified 182 polymorphic intergenic microsatellites (Supplementary Table 10), the first set of genetic markers developed for this species. These provide a toolkit for studies of genetic diversity and population structure of laboratory stocks or natural infections of P. cynomolgi, many of which have recently been isolated from screening hundreds of wild monkeys for the zoonosis P. knowlesi24.
We estimated the difference between the number of synonymous changes per synonymous site (dS) and the number of nonsynonymous changes per nonsynonymous site (dN) over 4,563 pairs of orthologs within P. cynomolgi strains B and Berok and 4,601 pairs of orthologs between these two P. cynomolgi strains and P. vivax Salvador I, using a simple Nei-Gojobori model25. We found 63 genes with dN > dS within the two P. cynomolgi strains and 3,265 genes with dS > dN (Supplementary Table 11). Genes with relatively high dN/dS ratios include those encoding transmembrane proteins, such as antigens and transporters, among which is a transmission-blocking target antigen, Pcyn230 (encoded by PCYB_042090). Notably, the P. vivax ortholog (PVX_003905) does not show evidence for positive selection26, suggesting species-specific positive selection. We explored the degree to which evolution of orthologs has been constrained between P. cynomolgi and P. vivax and found 83 genes under possible accelerated evolution but 3,739 genes under possible purifying selection (Supplementary Table 12). This conservative estimate indicates that at least 81% of loci have diverged under strong constraint, compared with 1.8% of loci under less constraint or positive selection (Fig. 1), indicating that, overall, the genome of P. cynomolgi is highly conserved in single-locus genes compared to P. vivax and emphasizing the value of P. cynomolgi as a biomedical and evolutionary model for studying P. vivax.
Our generation of the first P. cynomolgi genome sequences is a critical step in the development of a robust model system for the intractable and neglected P. vivax species27. Comparative genome analysis of P. vivax and the Old World monkey malaria-causing parasites P. cynomolgi and P. knowlesi presented here provides the foundation for further insights into traits such as host specificity that will enhance prospects for the eventual elimination of vivax-caused malaria and global malaria eradication.
URLs.
PlasmoDB, http://plasmodb.org/; Circos, http://circos.ca/; MIcroSAtelite Identification tool (MISA), http://pgrc.ipk-gatersleben.de/misa/; dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_viewBatch.cgi?sbid=1056645.
Methods
Parasite material.
Details of the origin of the P. cynomolgi B, Berok and Cambodian strains, their growth in macaques and isolation of parasite material are given in the Supplementary Note.
Genome sequencing and assembly.
P. cynomolgi B strain was sequenced using the Roche 454 GS FLX (Titanium) and Illumina/Solexa Genome Analyzer IIx platforms to 161× coverage. In addition, 2,784 clones (6.8 Mb) of a ∼40-kb insert fosmid library in pCC1FOS (EpiCentre Biotechnologies) was sequenced by the Sanger method. A draft assembly of strain B was constructed using a combination of automated assembly and manual gap closure. We first generated de novo contigs by assembling Roche 454 reads using GS De novo Assembler version 2.0 with default parameters. Contigs of >500 bp were mapped to the P. vivax Salvador I reference assembly12 (PlasmoDB; see URLs). P. cynomolgi contigs were iteratively arrayed through alignment to P. vivax–assembled sequences with manual corrections. A total of 1,264 aligned contigs were validated by mapping paired-end reads from fosmid clones using blastn (e <1 × 10−15; identity > 90%; coverage > 200 bp) implemented in GenomeMatcher software version 1.65 (ref. 28). Additional linkages (699 regions) were made using PCR across the intervening sequence gaps with primers designed from neighboring contigs. The length of sequence gaps was estimated from insert lengths of the fosmid paired-end reads, the size of PCR products and homologous sequences of the P. vivax genome. Supercontigs were then manually constructed from the aligned contigs. Eventually, we obtained 14 supercontigs corresponding to the 14 chromosomes of the parasite, with a total length of ∼22.73 Mb, encompassing ∼80% of the predicted P. cynomolgi genome. A total of 1,651 contigs (>1 kb) with a total length of 3.45 Mb was identified as unassigned subtelomeric sequences by searching against the P. vivax genome using blastn. Additionally, to improve sequence accuracy, we constructed a mapping assembly of Illumina paired-end reads and the 14 supercontigs and unassigned contigs as reference sequences using CLC Genomics Workbench version 3.0 with default settings (CLC Bio). Comparison of the draft P. cynomolgi B sequence with 23 P. cynomolgi protein-coding genes (64 kb) obtained by Sanger sequencing showed 99.8% sequence identity (Supplementary Table 13). The P. cynomolgi Berok and Cambodian strains were sequenced to 26× and 17× coverage, respectively, using the Roche 454 GS FLX platform, with single-end and 3-kb paired-end libraries made for the former and a single-end library only made for the latter. For phylogenetic analyses of specific genes, sequences were independently verified by Sanger sequencing (Supplementary Table 14 and Supplementary Note).
Prediction and annotation of genes.
Gene prediction for the 14 supercontigs and 1,651 unassigned contigs was performed using the MAKER genome annotation pipeline29 with ab initio gene prediction programs trained on proteins and ESTs from PlasmoDB Build 7.1. For gene annotation, blastn (e <1 × 10−15; identity > 70%; coverage > 100 bp) searches of P. vivax (PvivaxAnnotatedTranscripts_PlasmoDB-7.1.fasta) and P. knowlesi (PknowlesiAnnotatedTranscripts_PlasmoDB-7.1.fasta) predicted proteomes were run, and the best hits were identified. All predicted genes were manually inspected at least twice for gene structure and functional annotation, and orthologous relationships between P. cynomolgi, P. vivax and P. knowlesi were determined on synteny. A unique identifier, PCYB_######, was assigned to P. cynomolgi genes, where the first two of the six numbers indicate chromosome number. Paralogs of genes that seemed to be specific to either P. cynomolgi, P. vivax or P. knowlesi were searched using blastp with default parameters, using a cutoff e value of 1 × 10−16.
Multiple genome sequence alignment.
Predicted proteins of P. cynomolgi B strain were concatenated and aligned with those from the 14 chromosomes of 5 other Plasmodium genomes: P. vivax, P. knowlesi, P. falciparum, P. berghei and P. chabaudi, using Murasaki software version 1.68.6 (ref. 30).
Search for sequence showing high similarity to host proteins.
Eleven P. cynomolgi CYIR proteins (with sequence similarity to P. knowlesi KIR) were subjected to blastp search for regions having high similarity to host Macacca mulatta CD99 protein, with cutoff e value of 1 × 10−12 and compositional adjustment (no adjustment) against the nonredundant protein sequence data set of the M. mulatta proteome in NCBI.
Phylogenetic analyses.
Genes were aligned using ClustalW version 2.0.10 (ref. 31) with manual corrections, and unambiguously aligned sites were selected for phylogenetic analyses. Maximum-likelihood phylogenetic trees were constructed using PROML programs in PHYLIP version 3.69 (ref. 32) under the Jones-Taylor-Thornton (JTT) amino-acid substitution model. To take the evolutionary rate heterogeneity across sites into consideration, the R (hidden Markov model rates) option was set for discrete γ distribution, with eight categories for approximating the site-rate distribution. CODEML programs in PAML 4.4 (ref. 33) were used for estimating the γ shape parameter, α values. For bootstrap analyses, SEQBOOT and CONSENSE programs in PHYLIP were applied.
Candidate genes for hypnozoite formation.
We undertook two approaches. First, genes unique to P. vivax and P. cynomolgi (hypnozoite-forming parasites) and not found in other non-hypnozoite–forming Plasmodium species were identified. We used the 147 unique genes identified in the P. vivax genome12 to search the P. cynomolgi B sequence. For the orthologs identified in both species, ∼1 kb of sequence 5′ to the coding sequence was searched for four specific ApiAP2 motifs20—PF14 0633, GCATGC; PF13_0235_D1, GCCCCG; PFF0670w_D1, TAAGCC; and PFD0985w_D2, TGTTAC—which are involved in sporozoite stage–specific regulation and expression (corresponding to the pre-hypnozoite stage). Second, dormancy-related proteins were retrieved from GenBank and used to search for P. vivax homologs. Candidate genes (n = 128) and orthologs of P. cynomolgi and five other parasite species were searched in the region ∼1 kb upstream of the coding sequence for the presence of the four ApiAP2 motifs. Data for P. vivax, P. knowlesi, P. falciparum, P. berghei, Plasmodium chabaudi and Plasmodium yoelii were retrieved from PlasmoDB Build 7.1.
Genome-wide screen for polymorphisms.
For SNP identification, alignment of Roche 454 data from strains B, Berok and Cambodian was performed using SSAHA2 (ref. 34), with 0.1 mismatch rate and only unique matches reported. Potential duplicate reads generated during PCR amplification were removed, so that when multiple reads mapped at identical coordinates, only the reads with the highest mapping quality were retained. We used a statistical method35 implemented in SAMtools version 0.1.18 to call SNPs simultaneously in the case of duplicate runs of the same strain. SNPs with high read depth (>100) were filtered out, as were SNPs in poor alignment regions at the ends of chromosomes (Supplementary Note).
Nucleotide diversity (π) was calculated as follows. For each site being compared, we calculated allele frequency by counting the two alleles and measured the proportion of nucleotide differences. Letting π be the genetic distance between allele i and allele j, then the nucleotide diversity within the population is
where Pi and Pj are the overall allele frequencies of i and j, respectively. Mean π was calculated by averaging over sites, weighting each by 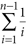 , where n is the number of aligned sites. Average dN/dS ratios were estimated using the modified Nei-Gojobori/Jukes-Cantor method in MEGA 4 (ref. 36).
, where n is the number of aligned sites. Average dN/dS ratios were estimated using the modified Nei-Gojobori/Jukes-Cantor method in MEGA 4 (ref. 36).
CNV-seq23 was used to identify potential CNVs in P. cynomolgi. Briefly, this method is based on a statistical model that allows confidence assessment of observed copy-number ratios from next-generation sequencing data. Roche 454 sequences from P. cynomolgi strain B assembly were used as the reference genome, and the P. cynomolgi Berok strain was used as a test genome; the sequence coverage of the Cambodian strain was considered too low for analysis. The test reads were mapped to the reference genome, and CNVs were detected by computing the number of reads for each test strain in a sliding window. The validity of the observed ratios was assessed by the computation of a probability of a random occurrence, given no copy-number variation.
Polymorphic microsatellites (defined as repeat units of 1–6 nucleotides) between P. cynomolgi strains B and Berok were identified by aligning contigs from a de novo assembly of Berok (generated using Roche GS Assembler version 2.6, with 40-bp minimum overlap, 90% identity) to the B strain using the Burrows-Wheeler Aligner (BWA)37 and allowing for gaps. Using the Phred-scaled probability of the base being misaligned by SAMtools35, indel candidates were called from the alignment. In-house Python scripts were used to then cross-reference with the microsatellites found in the reference strain B assembly identified by MISA (see URLs). All homopolymer microsatellites were discarded to account for potential sequence errors introduced by 454 sequencing.
Selective constraint analysis of 4,563 orthologs between P. cynomolgi strains B and Berok and 4,601 orthologs between these strains and P. vivax Salvador I used MUSCLE38 alignments with stringent removal of gaps and missing data (P. cynomolgi Berok orthologs were identified through a reciprocal best-hit BLAST search against strain B genes). Analyses were conducted using the Nei-Gojobori model25. To detect values that could not be explained by chance, we estimated the standard error by a bootstrap procedure with 200 pseudoreplicates for each gene. The expected value for dS/dN is 0 if a given pair of sequences is diverging without obvious effects on fitness. In the case of the comparison within P. cynomolgi, values with a difference of ± 2 s.e.m. from 0 were considered indicative of an excess of synonymous (dS/dN > 0) or nonsynonymous (dS/dN < 0) changes. In the case of the comparison between P. cynomolgi and P. vivax, we used a more stringent criterion of ± 3 s.e.m. from 0.
Accession codes.
Sequence data for the P. cynomolgi B, Cambodian and Berok strains have been deposited in the DNA Data Bank of Japan (DDBJ), the European Molecular Biology Laboratory (EMBL) and the GenBank databases under the following accessions: B strain sequence reads DRA000196, genome assembly BAEJ01000001 – BAEJ01003341 and annotation DF157093 – DF158755; Cambodian strain sequence reads DRA000197; and Berok strain sequence reads SRA047950. SNP calls have been submitted to dbSNP (NYU_CGSB_BIO; 1056645) and may also be downloaded from the dbSNP website (see URLs). Sequences of the dbp genes from P. cynomolgi (Cambodian strain), P. fieldi (A.b.i. strain) and P. simiovale (AB617788 – AB617791) and the P. cynomolgi Berok strain (JQ422035 – JQ422036) and rbp gene sequences from the P. cynomolgi Berok and Cambodian strains (JQ422037 – JQ422050) have been deposited. A partial apicoplast genome of the P. cynomolgi Berok strain has been deposited (JQ522954). The P. cynomolgi B reference genome is also available through PlasmoDB (see URLs).
Supplementary Information for
Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade
List of orthologs between the genomes of P. cynomolgi, P. vivax and P. knowlesi (XLS 1812 kb)
List of multigene families in P. cynomolgi, P. vivax and P. knowlesi (XLS 114 kb)
List of P. cynomolgi cyir genes, P. vivax vir genes, P. knowlesi kir genes, and P. knowlesi SICAvar genes and their homologs in P. cynomolgi, P. vivax and P. knowlesi (XLS 159 kb)
List of polymorphic microsatellite loci identified between P. cynomolgi strains B and Berok. (XLSX 68 kb)
Ds-Dn within 4,605 orthologs of P. cynomolgi strains B and Berok. (XLSX 346 kb)
Ds-Dn between 4,605 orthologs of P. cynomolgi strains B and Berok, and P. vivax Salvador I. (XLSX 373 kb)
Acknowledgements
We thank H. Sawai for suggestions on genome analysis, D. Fisher for help with genome-wide evolutionary analyses and the NYU Langone Medical Center Genome Technology Core for access to Roche 454 sequencing equipment (funded by grant S10 RR026950 to J.M.C. from the US National Institutes of Health (NIH)). Genome and phylogenetic analyses used the Genome Information Research Center in the Research Institute of Microbial Diseases at Osaka University. This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (18073013, 18GS03140013, 20390120 and 22406012) to K.T., an NIH grant (R01 GM080586) to A.A.E. and a Burroughs Wellcome Fund grant (1007398) and an NIH International Centers of Excellence for Malaria Research grant (U19 AI089676-01) to J.M.C. The content is soley the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author Contributions
K.T., J.M.C., A.A.E. and J.W.B. conceived and conducted the study. S.K., Y.K., Y.Y., S.-I.T. and J.W.B. provided P. cynomolgi material. S.N., N.G., T.Y. and H.R.K. constructed a computing system for data processing, and S.-I.T., H.H., P.L.S., S.A.S. and H.R.K. performed scaffolding of contigs and manual annotation of the predicted genes. S.N. performed sequence correction of supercontigs and gene prediction. S.-I.T., S.N., N.G., N.A., M.Y., O.K., K.T., H.R.K., R.S., S.A.S. and J.M.C. analyzed data. S.-I.T., N.M.Q.P., T.T., T.M., K.K., J.M.C., T.H., A.A.E., J.W.B. and K.T. wrote the manuscript.
Accession codes
Primary accessions
DDBJ/GenBank/EMBL
NCBI Reference Sequence
Sequence Read Archive
Referenced accessions
NCBI Reference Sequence
Competing interests
The authors declare no competing financial interests.
Footnotes
Jane M Carlton and Kazuyuki Tanabe: These authors jointly directed this work.
Contributor Information
Jane M Carlton, Email: jane.carlton@nyu.edu.
Kazuyuki Tanabe, Email: kztanabe@biken.osaka-u.ac.jp.
References
- 1.Mendis K, Sina BJ, Marchesini P, Carter R. The neglected burden of Plasmodium vivax malaria. Am. J. Trop. Med. Hyg. 2001;64:97–106. doi: 10.4269/ajtmh.2001.64.97. [DOI] [PubMed] [Google Scholar]
- 2.Mueller I, et al. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect. Dis. 2009;9:555–566. doi: 10.1016/S1473-3099(09)70177-X. [DOI] [PubMed] [Google Scholar]
- 3.Baird JK. Resistance to chloroquine unhinges vivax malaria therapeutics. Antimicrob. Agents Chemother. 2011;55:1827–1830. doi: 10.1128/AAC.01296-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rayner JC, Liu W, Peeters M, Sharp PM, Hahn BH. A plethora of Plasmodium species in wild apes: a source of human infection? Trends Parasitol. 2011;27:222–229. doi: 10.1016/j.pt.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cornejo OE, Escalante AA. The origin and age of Plasmodium vivax. Trends Parasitol. 2006;22:558–563. doi: 10.1016/j.pt.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Escalante AA, et al. A monkey's tale: the origin of Plasmodium vivax as a human malaria parasite. Proc. Natl. Acad. Sci. USA. 2005;102:1980–1985. doi: 10.1073/pnas.0409652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mu J, et al. Host switch leads to emergence of Plasmodium vivax malaria in humans. Mol. Biol. Evol. 2005;22:1686–1693. doi: 10.1093/molbev/msi160. [DOI] [PubMed] [Google Scholar]
- 8.Singh B, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363:1017–1024. doi: 10.1016/S0140-6736(04)15836-4. [DOI] [PubMed] [Google Scholar]
- 9.Pain A, et al. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature. 2008;455:799–803. doi: 10.1038/nature07306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eyles DE, Coatney GR, Getz ME. Vivax-type malaria parasite of macaques transmissible to man. Science. 1960;131:1812–1813. doi: 10.1126/science.131.3416.1812. [DOI] [PubMed] [Google Scholar]
- 11.Gibbs RA, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- 12.Carlton JM, et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455:757–763. doi: 10.1038/nature07327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saxena AK, Wu Y, Garboczi DN. Plasmodium p25 and p28 surface proteins: potential transmission-blocking vaccines. Eukaryot. Cell. 2007;6:1260–1265. doi: 10.1128/EC.00060-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyer J, Gruner AC, Renia L, Snounou G, Preiser PR. Invasion of host cells by malaria parasites: a tale of two protein families. Mol. Microbiol. 2007;65:231–249. doi: 10.1111/j.1365-2958.2007.05791.x. [DOI] [PubMed] [Google Scholar]
- 15.Okenu DM, Malhotra P, Lalitha PV, Chitnis CE, Chauhan VS. Cloning and sequence analysis of a gene encoding an erythrocyte binding protein from Plasmodium cynomolgi. Mol. Biochem. Parasitol. 1997;89:301–306. doi: 10.1016/S0166-6851(97)00118-7. [DOI] [PubMed] [Google Scholar]
- 16.Coatney, G.R., Collins, W.E., Warren, M. & Contacos, P.G. The Primate Malarias (US Department of Health, Education and Welfare, Washington, DC, 1971).
- 17.Cunningham D, Lawton J, Jarra W, Preiser P, Langhorne J. The pir multigene family of Plasmodium: antigenic variation and beyond. Mol. Biochem. Parasitol. 2010;170:65–73. doi: 10.1016/j.molbiopara.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 18.al-Khedery B, Barnwell JW, Galinski MR. Antigenic variation in malaria: a 3′ genomic alteration associated with the expression of a P. knowlesi variant antigen. Mol. Cell. 1999;3:131–141. doi: 10.1016/S1097-2765(00)80304-4. [DOI] [PubMed] [Google Scholar]
- 19.Krotoski WA. The hypnozoite and malarial relapse. Prog. Clin. Parasitol. 1989;1:1–19. [PubMed] [Google Scholar]
- 20.Campbell TL, De Silva EK, Olszewski KL, Elemento O, Llinas M. Identification and genome-wide prediction of DNA binding specificities for the ApiAP2 family of regulators from the malaria parasite. PLoS Pathog. 2010;6:e1001165. doi: 10.1371/journal.ppat.1001165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mu J, et al. Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat. Genet. 2007;39:126–130. doi: 10.1038/ng1924. [DOI] [PubMed] [Google Scholar]
- 22.Volkman SK, et al. A genome-wide map of diversity in Plasmodium falciparum. Nat. Genet. 2007;39:113–119. doi: 10.1038/ng1930. [DOI] [PubMed] [Google Scholar]
- 23.Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009;10:80. doi: 10.1186/1471-2105-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KS, et al. Plasmodium knowlesi: reservoir hosts and tracking the emergence in humans and macaques. PLoS Pathog. 2011;7:e1002015. doi: 10.1371/journal.ppat.1002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 26.Doi M, et al. Worldwide sequence conservation of transmission-blocking vaccine candidate Pvs230 in Plasmodium vivax. Vaccine. 2011;29:4308–4315. doi: 10.1016/j.vaccine.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carlton JM, Sina BJ, Adams JH. Why is Plasmodium vivax a neglected tropical disease? PLoS Negl. Trop. Dis. 2011;5:e1160. doi: 10.1371/journal.pntd.0001160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohtsubo Y, Ikeda-Ohtsubo W, Nagata Y, Tsuda M. GenomeMatcher: a graphical user interface for DNA sequence comparison. BMC Bioinformatics. 2008;9:376. doi: 10.1186/1471-2105-9-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cantarel BL, et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Popendorf K, Tsuyoshi H, Osana Y, Sakakibara Y. Murasaki: a fast, parallelizable algorithm to find anchors from multiple genomes. PLoS ONE. 2010;5:e12651. doi: 10.1371/journal.pone.0012651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Felsenstein, J. PHYLIP, Phylogeny Inference Package, 3.6a3 edn (University of Washington, Seattle, 2005).
- 33.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 34.Ning Z, Cox AJ, Mullikin JC. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade
List of orthologs between the genomes of P. cynomolgi, P. vivax and P. knowlesi (XLS 1812 kb)
List of multigene families in P. cynomolgi, P. vivax and P. knowlesi (XLS 114 kb)
List of P. cynomolgi cyir genes, P. vivax vir genes, P. knowlesi kir genes, and P. knowlesi SICAvar genes and their homologs in P. cynomolgi, P. vivax and P. knowlesi (XLS 159 kb)
List of polymorphic microsatellite loci identified between P. cynomolgi strains B and Berok. (XLSX 68 kb)
Ds-Dn within 4,605 orthologs of P. cynomolgi strains B and Berok. (XLSX 346 kb)
Ds-Dn between 4,605 orthologs of P. cynomolgi strains B and Berok, and P. vivax Salvador I. (XLSX 373 kb)