

Abstract
Ghrelin/GHS-R axis is known as its role in stimulating growth hormone release. Besides, it is also implicated in the regulation of atherosclerosis (AS), a chronic vascular disease that has been recognized as the main cause of coronary heart disease and cerebrovascular disease. It has been reported that both Ghrelin and AMPK play protective roles in AS by inhibiting the inflammatory response as well as cell proliferation. However, it remains unclear whether AMPK pathway is involved in Ghrelin/GHS-R-mediated inhibition of the inflammatory response and cell proliferation in AS. Here, we established the GHS-R gene knockout mice (GHS-R-/-) and found that AMPK activity is notably down-regulated in endothelial cells (ECs) of GHS-R-/- mice and the ECs from GHS-R-/- mice possess higher proliferative capability than the ECs from wild-type mice. Moreover, AMPK is activated in primary ECs upon Ghrelin induction in vitro. Taking together, the present study unravels that Ghrelin/GHS-R could efficiently activate AMPK in ECs, suggesting a possible mechanism that the roles of Ghrelin/GHS-R in the inhibition of inflammatory response and cell proliferation in AS disease may be partially mediated by activating AMPK.
Keywords: GHS-R-/-, AMPK, atherosclerosis, ECs, cell proliferation
Introduction
Atherosclerosis (AS) is a chronic vascular disease closely associating with a variety of risk factors such as high blood pressure, high cholesterol, diabetes and obesity [1,2]. On the basis of the inflammatory response, complex signaling pathways are activated to regulate the proliferation and apoptosis of specific cells, to facilitate the formation and development of atherosclerotic plaques [1,2]. AS is the main cause of coronary heart disease and cerebrovascular disease, and is one of the leading reasons for the death of elderly. Study of AS has been the focus of attention, even though extensive progresses have been achieved in recent years, but the mechanisms remain elusive.
Ghrelin was first found and isolated by Kojima from the acid secreting part of rat stomach tissue in 1999, capable of stimulating human and other mammals’ pituitary growth hormone (GH) release [3]. Ghrelin is identified as a small peptide comprising 28 amino acids and often modified at a post-translational level. Especially, the octanoylation on Ser-3 of the N-terminal of Ghrelin is the mainly and most activated form in mammals [3]. The Ghrelin receptor, also known as the growth hormone secretagogue receptor (GHS-R) mediating the actions of Ghrelin belongs to the G protein-coupled receptor (GPCR) family. So when Ghrelin conjugates to the GHS-R, PKC kinase is activated and the intracytoplasmic concentration of Ca2+ would be elevated via the classical pathway, leading to the activation of relative physiological functions [4]. Ghrelin is mainly secreted by the gastric gland and the concentration of Ghrelin in an adult’s blood is 117.0 ± 37.2 pM/L [3,5]. Ghrelin and GHS-R are widely distributed in many types of human tissues such as stomach, thyroid gland, heart, lung, pancreas, kidney, placenta, immune system, as well as ovaries and testis [6].
Recent studies have found that Ghrelin is massively distributed in the cardiovascular system, and its receptor GHS-R is highly expressed in the myocardium, blood vessel and atrium. Especially in the atherosclerotic plaques, expression levels of GHS-R are significantly up-regulated, and studies reveal that Ghrelin might have the putative therapeutic implications for AS [7]. Animal experiments showed that the treatment of Hexarelin, the agonist of GHS-R, can inhibit the occurrence of AS in the high-fat diet rats, and induction of Ghrelin can significantly reduce the inflammatory response of the vascular wall in the LDLR-/- mice [8,9]. In addition, subcutaneous injection of Ghrelin in mice significantly relieved the interstitial fibrosis of ventricular remodeling after myocardial infarction and non-infarcted area, and mitigated the myocardial ischemia-reperfusion injury, and these effects were growth hormone (GH)-independent [10]. Clinical studies found that in the elderly patients of coronary syndrome, diabetes and high blood pressure, plasma Ghrelin levels are negatively correlated with carotid intima-media thickness, an important symbol of the AS, suggesting that plasma Ghrelin levels can be used as a new predictor for carotid atherosclerosis [11,12]. Moreover, our previous studies reveal that plasma Ghrelin levels are closely associated with stenosis severity and morphology of angiographically-detected coronary atherosclerosis in patients with coronary artery disease and diabetes mellitus [13,14].
AMP-activated protein kinase (AMPK) is a metabolic sensor directly sensing the cellular AMP/ATP ratio in mammalian cells, comprising a catalytic α subunit and the regulatory β and γ subunits. Under the moderate energy stress, ADP or AMP replaces ATP by binding to the γ subunit, promoting the phosphorylation of Thr-172 on the α-catalytic subunit of AMPK by liver kinase B1 (LKB1), leading to an 100-fold increase in AMPK kinase activity [15]. CaMKKβ also can directly trigger the activation of AMPK kinase in response to the increased cellular Ca2+ without necessarily requiring the alternation of ADP or AMP levels. AMPK has been identified as a multi-functional kinase involved in regulating a variety of cell activities such as energy metabolism, cell growth, cell cycle, cell structure, cell polarity and apoptosis [15,16]. In recent years, studies have shown that AMPK signaling pathways are involved in the endothelial injury-mediated inflammatory and proliferative responses in atherosclerotic plaques. Activation of AMPK by the AMP analogue AICAR may play protective roles in AS, including the improvement of endothelial function, inhibition of the inflammatory response as well as proliferation and migration of SMCs [17].
As the reports above show, both Ghrelin and AMPK play protective roles in AS by inhibiting the inflammatory response as well as cell proliferation, and it has been reported that Ghrelin exerts its roles in anti-inflammatory, anti-oxidant and metabolic regulation via AMPK-mediated pathway [18,19]. However, it remains unclear whether AMPK pathway is involved in Ghrelin/GHS-R-mediated inhibition of the inflammatory response and cell proliferation in AS. In this regard, here we examined the AMPK activity in primary endothelial cells of GHS-R-/- mice, trying to elucidate the roles of AMPK in Ghrelin/GHS-R pathway.
Materials and methods
Generation of GHS-R knockout mice (GHS-R-/-) mice
GHS-R knockout mice (GHS-R-/-) were generated in Shanghai Research Center for Model Organisms according to the approaches as described previously [20]. In brief, PGK-neo cassette on X-pPNT vector was replaced by TK-neo cassette to form the X-pPNT-TK-neo vector, and then 3.2 kb of 5’ homologous chromosome arm and 2.5 kb of 3’ homologous chromosome arm of GHS-R gene were inserted into the X-pPNT-TK-neo vector to constitute the GHS-R gene targeting vector. Thereafter, the GHS-R gene targeting vector was transfected into ES cells of mice to perform the homologous recombination with the endogenous GHS-R gene, and the exon 1, exon 2 and a part of exon 3 of GHS-R gene were replaced by TK-neo cassette on X-pPNT vector via homologous recombination. Subsequently, the positive clones of recombinant mice ES cells were subjected to transplantation into pseudopregnant mice to produce the heterozygote mice. After the hybridization between the heterozygote mice, the homozygote of GHS-R-/- mice were selected and identified. Mice were kept under specific pathogen-free conditions and all animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (the “NIH Guide”). The protocols for the use of animals were approved by Shanghai Jiao Tong University.
Primary endothelial cells (ECs) culture
8-week-old mice were killed and then dissected in biological safety cabinet. The lung tissues were removed and rinsed thoroughly with PBS containing penicillin and streptomycin antibiotics, and then cut with scissors into small pieces of 1 × 1 × 1 mm3. These small pieces of lung tissues were placed into 24-well plate pre-treated with gelatin. 2 h after the consolidation of gelatin, DMEM (Invitrogen) culture medium containing 20% FBS (Gibco) and 90 U/ml of heparin (Sigma) was added and cultured in a humidified incubator with 5% CO2 at 37°C. The culture medium was changed with fresh medium everyday and 2-3 days later, many adherent cells spread around the tissues can be observed under a microscopy. Until the cells covered the well, tissues were then removed out and cells were washed 3 times with PBS containing penicillin and streptomycin antibiotics and then passaged for further experiments.
PCR and RT-PCR
Genomic DNA was extracted from the tails of hybridized mice and used for GHS-R gene analysis by PCR using the following primers: P1 (5’-GTGCGCACTGTCTCCTCTGATTTG-3’), P2 (5’-GTGCTTTGGGGTGCGTGTGATGGA-3’) and P3 (5’-CACGCCCACCAGCACGAAGA-3’). PCR reactions were performed as following: one cycle, 95°C for 5 min; 35 cycles: 94°C for 50 s, 61°C for 50 s and then 72°C for 3 min; the final elongation: 72°C for 10 min. For RT-PCR, total RNA was extracted from hypothalamus tissue of the hybridized mice with TRIzol Reagent (Invitrogen, USA) according to the manufacturer’s instructions. The cDNAs were synthesized by reverse transcriptase and subjected to PCR using specific primers for mouse GHS-R gene: RT1 (CGACCTGCTCTGCAAACTC), RT2 (CACGCCCACCAGCACGAAGA) and primers for mouse β-actin gene: Actin1 (TACCCAGGCATTGCTGACAGG), Actin2 (ACTTGCGGTGCA CGATGGA). RT-PCR reactions were performed as following: one cycle, 95°C for 3 min; 40 cycles: 94°C for 30 s, 55°C for 30 s and then 72°C for 2 min; the final extension: 72°C for 10 min. The PCR products were analyzed with 2% agarose gel electrophoresis. These experiments were performed more than three times.
Western blot
The hypothalamus tissue and endothelial cells (ECs) were lysed with lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EGTA, 1% Triton X-100, 1 mM PMSF, 10 nM phosphatase inhibitor microcystin, 1 μg/ml aprotinin and 1 μg/ml leupeptin). Equal amounts of protein were separated on 10% SDS-PAGE and then transferred onto nitrocellulose (NC) membranes (Amersham). After blocking with 1% BSA in TBST (0.05% Tween 20 in Tris-buffered saline) for 1 hour at room temperature, the membranes were separately incubated with anti-phospho-AMPK (Cell Signaling), anti-phospho-ERK1/2 (Cell Signaling), anti-β-actin (Cell Signaling), anti-GHS-R (Santa Cruz) and anti-GAPDH (Cell Signaling) antibodies at a 1:1,000 dilution in TBST for 1 hour at room temperature. Then membranes were incubated with HRP-conjugated secondary antibodies (Jackson ImmunoResearch, PA) at a 1:10,000 dilution for 1 h at room temperature before visualizing with ECL detection reagents. These assays were performed three times and similar results were acquired.
MTT assay
For MTT assay, primary ECs isolated from wild-type and GHS-R-/- mice were placed into 96-well plate at a density of 5,000 cells per well and cultured in a humidified incubator with 5% CO2 at 37°C. 24, 48 and 72 h later, 20 μl of 5 g/L MTT (Sigma) dissolved in PBS were added to each well and incubated for 4 h, followed by the addition of 100 μl dimethyl sulfoxide (DMSO) (Sigma) and left for 10 min in the incubator for color development. Optical density was determined using 570 nm filter on a microplate reader (Tecan, Austria). Each group was set in triplicate and the experiment was repeated for three times.
Statistical analysis
All data were statistically analyzed by GraphPad Prism 5 software (GraphPad, San Diego, USA). Comparisons between two groups were performed by Student’s t test, while comparisons among multiple groups were performed by one-way analysis of variables (ANOVA). Statistical significance was defined as P < 0.05.
Results
Establishment of GHS-R-/- mice model
The gene targeting vector map is shown in Figure 1A as the previous PGK-neo cassette on X-pPNT vector has been replaced by TK-neo cassette. The 3.2 kb of 5’ homologous chromosome arm was inserted into X-pPNT-TK-neo vector by BamHI-EcoRI digestion while the 2.5 kb of 3’ homologous chromosome arm was inserted into X-pPNT-TK-neo vector by NotI-SalI digestion (Figure 1A). Then the DNA of X-pPNT-TK-neo vector containing 5’ and 3’ homologous chromosome arms was transfected into mice ES cells to perform the homologous recombination with the endogenous GHS-R gene as described in Figure 1B. The exon 1, exon 2 and a part of exon 3 of GHS-R gene were replaced by TK-neo cassette on X-pPNT vector via homologous recombination (Figure 1B). Then the ES cell clones survived in medium containing G418 and GANC were collected for further PCR identification by the strategy shown in Figure 1B. For PCR identification of the 3’ homologous chromosome arm in recombinant ES cells, The 6.4 kbp of PCR products were observed in lane 1, 2, 3 and 4 representing the wild-type GHS-R gene, while another expected 4.2 kbp was observed only in lane 4 indicating the fourth sample (clone No. 120) was the positive clone with GHS-R gene deletion (Figure 1C). For PCR identification of the 5’ homologous chromosome arm, the expected 3.8 kbp of PCR products were observed in lane 1 (No. 120), lane 2 (No. 200) and lane 3 (No. 237) presenting the positively recombinant clones and the 3.8 kbp fragments were confirmed by sequencing (Figure 1D). Then those positively recombinant clones of mice ES cells that were subjected to transplantation into pseudopregnant mice for further process to generate the GHS-R gene knockout mice.
Figure 1.
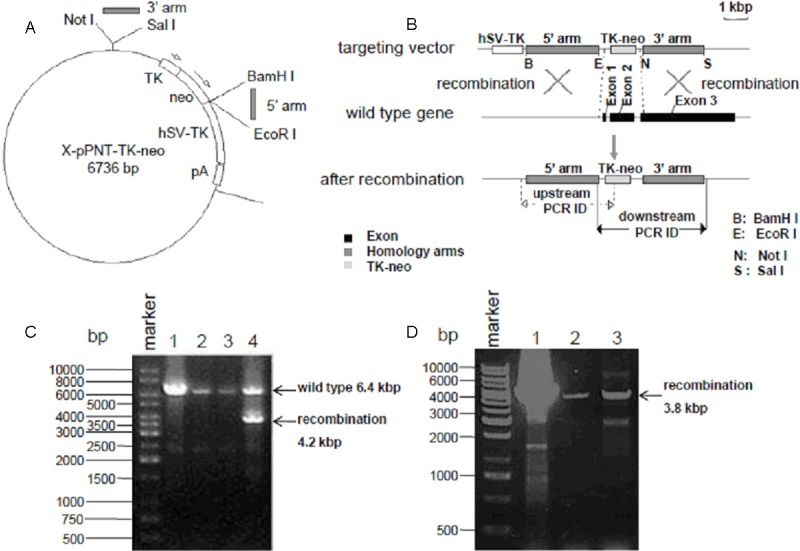
Generation of GHS-R-/- mice. A: Vector map for gene targeting. PGK-neo on X-pPNT vector was replaced by TK-neo. The 5’ homologous chromosome arm was inserted into BamHI-EcoRI site while 3’ homologous chromosome arm was inserted into NotI-SalI site. B: The schematic diagram of GHS-R gene deletion. The exon 1, exon 2 and a part of exon 3 of GHS-R gene were replaced by TK-neo cassette on X-pPNT vector via homologous recombination. C: PCR identification of the 3’ homologous chromosome arm in recombinant ES cells after GHS-R gene knockout. The 6.4 kbp of PCR products were observed in lane 1, 2, 3 and 4 while another expected 4.2 kbp was observed only in lane 4 indicating the fourth sample (clone No. 120) was the positive clone. D: PCR identification of the 5’ homologous chromosome arm in recombinant ES cells after GHS-R gene knockout. The expected 3.8 kbp of PCR products were observed in all lanes indicating those samples were the positive clones.
Identification of GHS-R gene knockout mice
To investigate whether GHS-R gene was successfully knocked out, the genomic DNA acquired from the tails of different genotype mice were used for PCR analysis using 3 primers (P1, P2 and P3) targeting to the relative sites as described in Figure 2A. As the PCR results shown, the 1.2 kbp of PCR product bands yielded by P1 and P3 primers were observed in lane 4, 7 and 9 representing GHS-R+/+, while the 1.9 kbp of PCR product bands yielded by P1 and P2 primers were observed in lane 8, 10 and 11 representing GHS-R-/-, and both sizes of bands were observed in lane 2, 3, 5 and 6 representing GHS-R+/- (Figure 2B). To further examine whether GHS-R gene was successfully deleted, RT-PCR assays were performed to analyzed GHS-R gene expression in hypothalamus of these hybridized mice. The β-actin mRNA expression represented by 260 bp of PCR products were observed in all the three lanes and the levels were equal. The GHS-R mRNA expression represented by 217 bp of PCR products were observed in wild-type (GHS-R+/+) and heterozygote (GHS-R+/-), while not detected in homozygote (GHS-R-/-). Moreover, the levels of GHS-R mRNA in heterozygote were obviously lower compared with those in wild-type (Figure 2C). To make sure the GHS-R gene was thoroughly knocked out in GHS-R-/- mice, the hypothalamus tissues acquired from these hybridized mice were lysed for western blot analysis using GHS-R antibody to examine the GHS-R expression. Two main bands were observed in samples from wild-type mice, presenting the two isoforms of GHS-R, GHS-R-1a and GHS-R-1b. Obviously, the expression levels of GHS-R in samples from heterozygote mice (GHS-R+/-) were lower than those in wild-type mice (GHS-R+/+). No detectable signals were observed in samples from homozygote mice (GHS-R-/-), indicating that GHS-R gene was successfully knocked out in GHS-R-/- mice (Figure 2D).
Figure 2.
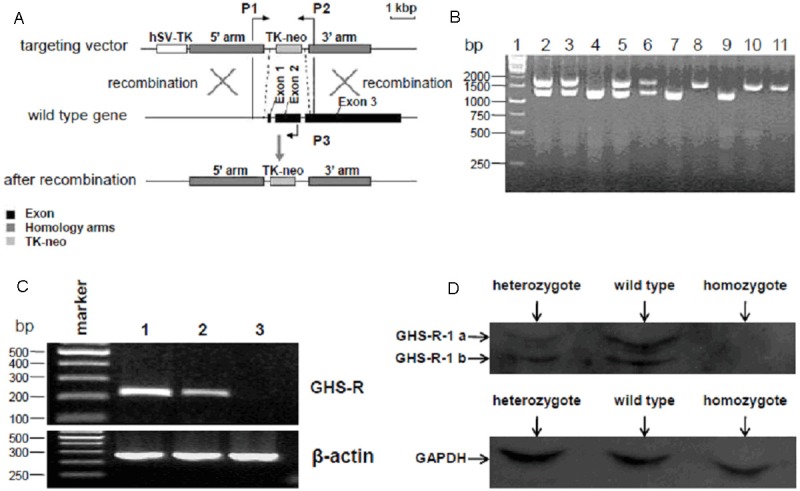
Identification of GHS-R-/- mice. A: Strategy for the PCR identification of GHS-R gene knockout mice. PCR primers (P1, P2 and P3) were designed targeted to the relative sites as shown in the schematic diagram. B: PCR identification of GHS-R gene knockout mice. Genomic DNAs extracted from tails of the hybridized mice were analyzed by PCR identification as described in Materials and Methods. The 1.2 kbp of PCR products observed in lane 4, 7 and 9 represented the GHS-R+/+, and 1.9 kbp of PCR products observed in lane 8, 10 and 11 represented the GHS-R-/-, while both the 1.2 kbp and 1.9 kbp of PCR products observed in lane 2, 3, 5 and 6 represented the GHS-R+/-. C: RT-PCR identification of GHS-R gene knockout mice. Total RNA extracted from the hypothalamus of the hybridized mice was used for RT-PCR identification with specific primers targeting to GHS-R gene and β-actin. The levels of β-actin mRNA represented by 260 bp of PCR products were nearly equal in all the three lanes. The GHS-R mRNA expression represented by 217 bp of PCR products was observed in wild-type and heterozygote, while not detected in homozygote (lane 3). Moreover, the levels of GHS-R mRNA in heterozygote (lane 2) were lower compared with those in wild-type (lane 1). D: Analysis of GHS-R expression in hybridized mice by western blot. Total protein extracted from the hypothalamus of the hybridized mice was quantified and analyzed by western blot using specific GHS-R and GAPDH antibodies. GAPDH expression was detected with the nearly same quantity in all three lanes. GHS-R expression detected in wild-type and heterozygote were presenting in two bands, GHS-R-1a (upper) and GHS-R-1b (lower). Moreover, the levels of GHS-R expression in heterozygote were lower compared with those in wild-type. However, GHS-R expression in homozygote was under detectable.
Endothelial cells (ECs) from GHS-R-/- mice proliferate faster
The endothelial cells isolated from wild-type and GHS-R-/- mice were cultured in vitro and observed under the inverted phase contrast microscopy. Much more round and slender morphology of ECs were observed in wild-type group and these cells seemed not to well adhere to the dish. However, ECs from GHS-R-/- grew very well and the cytoplasmic membrane was largely spread on the dish (Figure 3A). Meanwhile, the cell density of GHS-R-/- group seemed higher than that of wild-type group. To confirm this, MTT assays were conducted to analyze the proliferative rate of ECs from wild-type and GHS-R-/- mice. As the results shown, the proliferative rate of ECs from GHS-R-/- mice was kept at the same levels of ECs from wild-type mice at day 1 and day 2, however, the proliferative rate of ECs from GHS-R-/- mice significantly rose up at day 3 (Figure 3B) (**P < 0.01).
Figure 3.
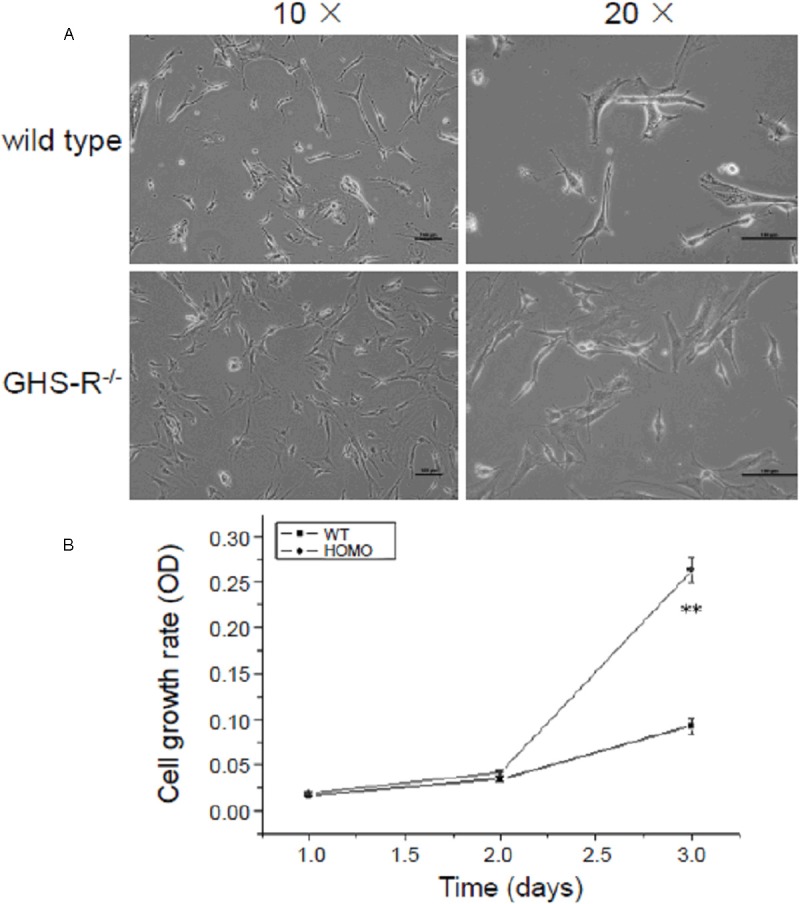
Morphology observation and cell growth analysis of endothelial cells (ECs) from GHS-R-/- mice. A: Morphology observation of in vitro cultivated ECs from GHS-R+/+ and GHS-R-/- mice. Primary ECs were acquired from GHS-R+/+ and GHS-R-/- mice and cultivated in vitro as described in Materials and Methods, and then observed with inverted phase contrast microscopy. ECs from GHS-R+/+ mice were not well adherent to the dish and presented as the round and slender morphology, whereas ECs from GHS-R-/- grew very well and the cytoplasmic membrane was largely spread on the dish. Bar = 100 μm. B: MTT analysis of proliferative rate of ECs from GHS-R-/- mice. The proliferative rate of ECs from GHS-R-/- mice significantly rose up at day 3 compared with that of ECs from wild-type mice. **P < 0.01.
AMPK is activated in ECs upon Ghrelin induction
It has been reported that Ghrelin exerts its roles in inhibition of vascular smooth muscle cells (VSMCs) proliferation and atherosclerotic plaques formation via classical Akt/ERK pathway [21,22], however, it remains unclear whether AMPK activation is involved in Ghrelin/GHS-R pathway to exhibit the relative roles in AS disease. Here, we first try to explore whether AMPK is activated in ECs exposed to Ghrelin. Primary ECs isolated from wild-type mice were cultivated in vitro and incubated with 100 nM Ghrelin for 10, 20, 30, 45 and 60 min. Then cells were harvested for western blot analysis using phospho-ERK1/2 and phospho-AMPK antibodies. As shown in Figure 4A, the variations of activated AMPK protein expression were similar to the activated ERK, while no obvious changes were observed in β-actin expression (Figure 4A). The expression levels of both phospho-ERK and phospho-AMPK increased immediately in 10 min upon Ghrelin induction and achieved the highest levels in 30 min. Thereafter, the expression levels decreased drastically and arrived at the normal levels 45 min after induction and declined to the lowest levels 60 min after induction (Figure 4B) (**P < 0.01, ***P < 0.001). The results above revealed that AMPK is also activated upon Ghrelin induction in ECs, indicating that AMPK may be involved in Ghrelin/GHS-R-mediated regulation in AS disease.
Figure 4.
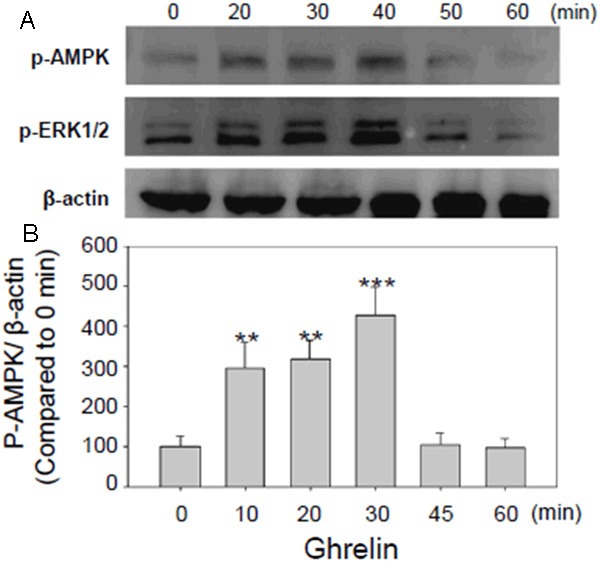
Ghrelin induction activated AMPK in primary endothelial cells (ECs) cultivated in vitro. A: Western blot analysis of AMPK activity induced by Ghrelin in primary ECs. ECs isolated from wild-type mice were cultivated in vitro and treated with 100 nM Ghrelin for 10, 20, 30, 45 and 60 min. Then cells were harvested for western blot analysis using β-actin, phospho-ERK1/2 and phospho-AMPK antibodies. AMPK and ERK1/2 activity increased significantly in 10 min after Ghrelin induction, and achieved the highest levels in 30 min. B: Statistical analysis of the AMPK activity induced by Ghrelin in primary ECs. The p-AMPK/β-actin values of each time point were calculated and analyzed statistically compared to value of 0 min. **P < 0.01, ***P < 0.001.
AMPK activity declines in ECs from GHS-R-/- mice
Though previous studies have documented that AMPK could be activated under the Ghrelin/GHS-R pathway, the status of AMPK activity in ECs from GHS-R-/- mice have not been reported. Here, the ECs isolated for 2 wild-type mice and 2 GHS-R-/- mice were cultivated in vitro and then lysed for western blot analysis using specific phospho-AMPK (p-AMPK) antibody to investigate the AMPK activity. Results revealed that AMPK activities were kept at the same levels in wild-type mice (WT1 and WT2), however, the AMPK activities were notably attenuated in GHS-R gene knocked out mice (KO1 and KO2) (Figure 5A). The β-actin expression was set as a control and the statistical analysis showed that compared with p-AMPK/β-actin value in WT1 group, the AMPK activity decreased significantly in GHS-R-/- mice (KO1 and KO2 group) (Figure 5B) (*P < 0.05).
Figure 5.
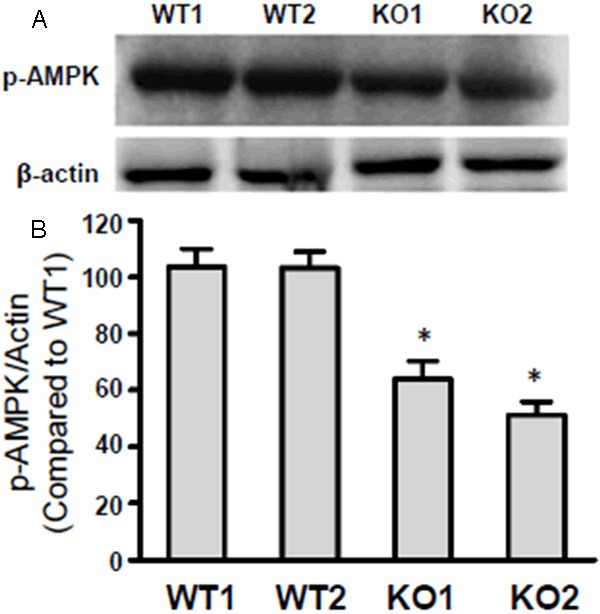
AMPK activity was attenuated in endothelial cells (ECs) from GHS-R-/- mice. A: Western blot analysis of AMPK activity in endothelial cells (ECs) from GHS-R-/- mice. ECs isolated from two wild-type and two GHS-R-/- mice were subjected to western blot using anti-β-actin, and anti-phospho-AMPK antibodies. B: Statistical analysis of AMPK activity as described in A. Compared to p-AMPK/β-actin ratio in wild-type mice 1 (WT1), the AMPK activity was notably down-regulated in ECs from GHS-R-/- mice 1 (KO1) and GHS-R-/- mice 2 (KO2), *P < 0.05.
Discussion
Ghrelin associates with its receptor GHS-R to exert a multiplicity of physiological functions, including inhibiting cell proliferation and inflammatory response in atherosclerosis (AS) disease [23]. Although extensive studies have been focused on the roles of Ghrelin/GHS-R in regulating the AS, the mechanisms remain elusive. Recent studies revealed that AMPK has the similar effects of Ghrelin/GHS-R that plays protective roles in AS by inhibiting the inflammatory response as well as cell proliferation [17], however, no reported studies verify whether AMPK is involved in the Ghrelin/GHS-R pathway to regulate AS progression. It has been reported that Ghrelin/GHS-R could activate AMPK to exhibit the anti-inflammatory, anti-oxidant and apoptosis-promoting effects in rat fatty liver models [19], but it is still unclear whether Ghrelin/GHS-R activates AMPK in endothelial cells (ECs). In the present study, primary ECs isolated from mice were cultivated in vitro and exposed to Ghrelin for relative time. Then cells were subjected to western blot analysis and results reveal that AMPK as well as ERK1/2 is highly activated upon Ghrelin induction immediately, indicating that AMPK functions in the downstream of Ghrelin/GHS-R pathway in ECs. Meanwhile, we also observed that AMPK activity is notably attenuated in ECs of Ghrelin gene knockout (GHS-R-/-) mice, further demonstrating that AMPK is involved in Ghrelin/GHS-R pathway in ECs. LKB1 and CaMKKβ are main upstream kinases responsible for AMPK activation, and previous studies have documented that Ghrelin/GHS-R could increase the intracellular Ca2+ level via phospholipase C (PLC)-PKC pathway, subsequently activating CaMKKβ [24], so we hypothesize that Ghrelin/GHS-R could activate AMPK in ECs via CaMKKβ pathway.
Recent studies revealed that Ghrelin inhibits the proliferation of VSMCs, possibly through the cAMP/PKA pathway and the inhibitory effects of Ghrelin in VSMCs proliferation were also documented in our previous studies [25,26]. So in this study we examined the proliferative status of ECs from GHS-R-/- mice and results show that proliferative rate of ECs from GHS-R-/- is greatly enhanced, indicating that Ghrelin/GHS-R pathway also prevents the ECs growth. This study has unraveled that Ghrelin/GHS-R could activate AMPK in ECs, so it is interesting to explore whether the inhibitory effects of Ghrelin/GHS-R are mediated by AMPK as mounting of evidence show that AMPK is able to strongly inhibit the proliferation of tumor cells and non-tumor cells [27]. Studies have revealed that AMPK can inhibit the proliferation of VSMCs induced by angiotensin II and in the rat femoral artery injury model, sustained activation of AMPK by AICAR can significantly inhibit the neointimal formation at the site of vascular injury as well as the proliferation of ECs and VSMCs [28]. The roles of AMPK in inhibiting cell proliferation may be mediated by promoting the expression of cyclin-dependent kinase inhibitor CDKI as well as p21, and the activation of p53 to arrest cells in G1/S phase of the cell cycle [29]. In this regard, we hypothesize that the roles of Ghrelin/GHS-R in inhibiting ECs proliferation may be partially mediated by AMPK.
Recent studies reveal that both Ghrelin/GHS-R and AMPK signaling pathways are involved in regulating endothelial injury mediated inflammation and proliferative responses in the AS plaques. Many studies have demonstrated that Ghrelin improves endothelial function by increasing vascular endothelial nitric oxide synthase (NOS) expression and Ghrelin also inhibits the inflammatory response of the vessel walls after endothelial injury, which is achieved by inhibiting NF-kappa B (NF-κB) activation [30,31]. In addition, NF-κB in endothelial cells (ECs) can be suppressed in vitro exposed to exogenous Ghrelin [31]. Activation of AMPK by the AMP analogue AICAR can improve endothelial function and prevent the inflammatory response as well as SMCs proliferation [17]. It has been reported that AMPK can phosphorylate and activate eNOS in vascular endothelial cells and cardiac myocytes to promote the production and release of NO, thereby improving endothelial function [32,33]. Moreover, the anti-inflammatory effects of AMPK in ECs are achieved by inhibiting NF-κB activation independent of GH pathway [34,35]. Taking together those reports above with the evidence in this study that Ghrelin/GHS-R prevents the ECs cell proliferation and promotes AMPK activation, we hypothesize that Ghrelin/GHS-R plays the key roles in regulating endothelial injury mediated inflammation and proliferative responses in the AS plaques via activating AMPK pathway.
In summary, this study unravels that Ghrelin/GHS-R activates AMPK in ECs and AMPK activity is notably attenuated in GHS-R-/- mice, providing the theoretical and experimental basis for elucidating the mechanisms of Ghrelin/GHS-R-AMPK axis in exhibiting protective roles in the AS disease.
Acknowledgements
This work was financially supported by the Natural Science Foundation of China (No. 81000087).
Disclosure of conflict of interest
None.
References
- 1.Riccioni G, Sblendorio V. Atherosclerosis: from biology to pharmacological treatment. J Geriatr Cardiol. 2012;9:305–317. doi: 10.3724/SP.J.1263.2012.02132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang G, Yin X, Qi Y, Pendyala L, Chen J, Hou D, Tang C. Ghrelin and cardiovascular diseases. Curr Cardiol Rev. 2010;6:62–70. doi: 10.2174/157340310790231662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 4.Kojima M, Hosoda H, Matsuo H, Kangawa K. Ghrelin: discovery of the natural endogenous ligand for the growth hormone secretagogue receptor. Trends Endocrinol Metab. 2001;12:118–122. doi: 10.1016/s1043-2760(00)00362-3. [DOI] [PubMed] [Google Scholar]
- 5.Date Y, Kojima M, Hosoda H, Sawaguchi A, Mondal MS, Suganuma T, Matsukura S, Kangawa K, Nakazato M. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology. 2000;141:4255–4261. doi: 10.1210/endo.141.11.7757. [DOI] [PubMed] [Google Scholar]
- 6.Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P, Bhattacharya S, Carpenter R, Grossman AB, Korbonits M. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J Clin Endocrinol Metab. 2002;87:2988. doi: 10.1210/jcem.87.6.8739. [DOI] [PubMed] [Google Scholar]
- 7.Katugampola SD, Pallikaros Z, Davenport AP. [125I-His(9)] -ghrelin, a novel radioligand for localizing GHS orphan receptors in human and rat tissue: up-regulation of receptors with athersclerosis. Br J Pharmacol. 2001;134:143–149. doi: 10.1038/sj.bjp.0704228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kellokoski E, Kummu O, Serpi R, Lehenkari P, Ukkola O, Kesaniemi YA, Horkko S. Ghrelin vaccination decreases plasma MCP-1 level in LDLR(-/-)-mice. Peptides. 2009;30:2292–2300. doi: 10.1016/j.peptides.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Pang J, Xu Q, Xu X, Yin H, Xu R, Guo S, Hao W, Wang L, Chen C, Cao JM. Hexarelin suppresses high lipid diet and vitamin D3-induced atherosclerosis in the rat. Peptides. 2010;31:630–638. doi: 10.1016/j.peptides.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Kishimoto I, Tokudome T, Hosoda H, Miyazato M, Kangawa K. Ghrelin and cardiovascular diseases. J Cardiol. 2012;59:8–13. doi: 10.1016/j.jjcc.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Kadoglou NP, Sailer N, Moumtzouoglou A, Kapelouzou A, Tsanikidis H, Vitta I, Karkos C, Karayannacos PE, Gerasimidis T, Liapis CD. Visfatin (nampt) and ghrelin as novel markers of carotid atherosclerosis in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2010;118:75–80. doi: 10.1055/s-0029-1237360. [DOI] [PubMed] [Google Scholar]
- 12.Yano Y, Toshinai K, Inokuchi T, Kangawa K, Shimada K, Kario K, Nakazato M. Plasma des-acyl ghrelin, but not plasma HMW adiponectin, is a useful cardiometabolic marker for predicting atherosclerosis in elderly hypertensive patients. Atherosclerosis. 2009;204:590–594. doi: 10.1016/j.atherosclerosis.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 13.Zhang M, Fang W, Yuan F, Qu X, Liu H, Chen H, Yu Y, Zheng Z, Shen Y. Plasma ghrelin levels are closely associated with stenosis severity and morphology of angiographically-detected coronary atherosclerosis in patients with coronary artery disease. Int J Cardiol. 2011;151:122–123. doi: 10.1016/j.ijcard.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 14.Zhang M, Fang WY, Yuan F, Qu XK, Liu H, Xu YJ, Chen H, Yu YF, Shen Y, Zheng ZC. Plasma ghrelin levels are closely associated with severity and morphology of angiographically-detected coronary atherosclerosis in Chineses patients with diabetes mellitus. Acta Pharmacol Sin. 2012;33:452–458. doi: 10.1038/aps.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. 2010;6:457–470. doi: 10.2217/fon.09.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu Y, Zhang C, Dong Y, Wang S, Song P, Viollet B, Zou MH. Activation of the AMP-activated protein kinase by eicosapentaenoic acid (EPA, 20:5 n-3) improves endothelial function in vivo. PLoS One. 2012;7:e35508. doi: 10.1371/journal.pone.0035508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barazzoni R, Semolic A, Cattin MR, Zanetti M, Guarnieri G. Acylated ghrelin limits fat accumulation and improves redox state and inflammation markers in the liver of high-fat-fed rats. Obesity (Silver Spring) 2013 doi: 10.1002/oby.20454. doi: 10.1002/oby.20454. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Hai J, Li L, Chen X, Peng H, Cao M, Zhang Q. Administration of ghrelin improves inflammation, oxidative stress, and apoptosis during and after non-alcoholic fatty liver disease development. Endocrine. 2013;43:376–386. doi: 10.1007/s12020-012-9761-5. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci U S A. 2004;101:4679–4684. doi: 10.1073/pnas.0305930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baldanzi G, Filigheddu N, Cutrupi S, Catapano F, Bonissoni S, Fubini A, Malan D, Baj G, Granata R, Broglio F, Papotti M, Surico N, Bussolino F, Isgaard J, Deghenghi R, Sinigaglia F, Prat M, Muccioli G, Ghigo E, Graziani A. Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J Cell Biol. 2002;159:1029–1037. doi: 10.1083/jcb.200207165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dworakowska D, Wlodek E, Leontiou CA, Igreja S, Cakir M, Teng M, Prodromou N, Goth MI, Grozinsky-Glasberg S, Gueorguiev M, Kola B, Korbonits M, Grossman AB. Activation of RAF/MEK/ERK and PI3K/AKT/mTOR pathways in pituitary adenomas and their effects on downstream effectors. Endocr Relat Cancer. 2009;16:1329–1338. doi: 10.1677/ERC-09-0101. [DOI] [PubMed] [Google Scholar]
- 23.van der Lely AJ, Tschop M, Heiman ML, Ghigo E. Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev. 2004;25:426–457. doi: 10.1210/er.2002-0029. [DOI] [PubMed] [Google Scholar]
- 24.Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, Williams LM, Hawley SA, Hardie DG, Grossman AB, Korbonits M. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J Biol Chem. 2005;280:25196–25201. doi: 10.1074/jbc.C500175200. [DOI] [PubMed] [Google Scholar]
- 25.Rossi F, Castelli A, Bianco MJ, Bertone C, Brama M, Santiemma V. Ghrelin inhibits contraction and proliferation of human aortic smooth muscle cells by cAMP/PKA pathway activation. Atherosclerosis. 2009;203:97–104. doi: 10.1016/j.atherosclerosis.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M, Yuan F, Liu H, Chen H, Qiu X, Fang W. Inhibition of proliferation and apoptosis of vascular smooth muscle cells by ghrelin. Acta Biochim Biophys Sin (Shanghai) 2008;40:769–776. [PubMed] [Google Scholar]
- 27.Dandapani M, Hardie DG. AMPK: opposing the metabolic changes in both tumour cells and inflammatory cells? Biochem Soc Trans. 2013;41:687–693. doi: 10.1042/BST20120351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T, Hirata Y. AMP-activated protein kinase inhibits angiotensin II-stimulated vascular smooth muscle cell proliferation. Circulation. 2004;110:444–451. doi: 10.1161/01.CIR.0000136025.96811.76. [DOI] [PubMed] [Google Scholar]
- 29.Igata M, Motoshima H, Tsuruzoe K, Kojima K, Matsumura T, Kondo T, Taguchi T, Nakamaru K, Yano M, Kukidome D, Matsumoto K, Toyonaga T, Asano T, Nishikawa T, Araki E. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ Res. 2005;97:837–844. doi: 10.1161/01.RES.0000185823.73556.06. [DOI] [PubMed] [Google Scholar]
- 30.Caliskan Y, Gorgulu N, Yelken B, Yazici H, Oflaz H, Elitok A, Turkmen A, Bozfakioglu S, Sever MS. Plasma ghrelin levels are associated with coronary microvascular and endothelial dysfunction in peritoneal dialysis patients. Ren Fail. 2009;31:807–813. doi: 10.3109/08860220903151419. [DOI] [PubMed] [Google Scholar]
- 31.Kellokoski E, Kunnari A, Jokela M, Makela S, Kesaniemi YA, Horkko S. Ghrelin and obestatin modulate early atherogenic processes on cells: enhancement of monocyte adhesion and oxidized low-density lipoprotein binding. Metabolism. 2009;58:1572–1580. doi: 10.1016/j.metabol.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem. 2003;278:31629–31639. doi: 10.1074/jbc.M212831200. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki K, Uchida K, Nakanishi N, Hattori Y. Cilostazol activates AMP-activated protein kinase and restores endothelial function in diabetes. Am J Hypertens. 2008;21:451–457. doi: 10.1038/ajh.2008.6. [DOI] [PubMed] [Google Scholar]
- 34.Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, Choi HC, Zou MH. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. 2010;121:792–803. doi: 10.1161/CIRCULATIONAHA.109.900928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 2011;89:667–676. doi: 10.1007/s00109-011-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]