

Abstract
Aims: Genotyping is a prerequisite for tyrosine kinase inhibitor therapy in high risk and malignant GIST. About 10% of GISTs are wild-type for KIT but carry PDGFRA mutations. Applying the traditional approach, mutation analysis of these cases is associated with higher costs if all hotspots regions in KIT (exon 9, 11, 13, 17) are performed at first. Our aim was to evaluate the predictive value of a combined histomorphological-immunohistochemical pattern analysis of PDGFRA-mutated GISTs to efficiently direct KIT and PDGFRA mutation analysis. Methods: The histomorphology and PDGFRA immunostaining pattern was studied in a test cohort of 26 PDGFRA mutants. This was then validated on a cohort of 94 surgically resected GISTs with mutations in KIT (n=72), PDGFRA (n=15) or with wild-type status (n=7) on a tissue microarray. The histological subtype (spindled, epithelioid, mixed), PDGFRA staining pattern (paranuclear dot-like/Golgi, cytoplasmic and/or membranous), and extent of staining were determined without knowledge of the genotype. The combination of histomorphology and immunophenotype were used to classify tumors either as PDGFRA- or non-PDGFRA phenotype. Results: PDGFRA-mutated GISTs were significantly more often epithelioid (p<0.001) and had a higher PDGFRA expression, compared to KIT-mutants (p<0.001). Paranuclear PDGFRA immunostaining was almost exclusively observed in PDGFRA mutants (p<0.001). The sensitivity and specificity of this combined histological-immunohistochemical approach to predict the PDGFRA-genotype was 100% and 99%, respectively (p=6x10-16). Conclusion: A combination of histomorphology and PDGFRA immunostaining is a reliable predictor of PDGFRA genotype in GIST. This approach allows direct selection of the “gene/exons of relevance” to be analyzed and may help to reduce costs and work load and shorten processing time of GIST genotyping by mutation analysis.
Keywords: GIST, PDGFRA, immunohistochemistry, genotype, dot-like
Introduction
Although generally uncommon, gastrointestinal stromal tumors (GISTs) are the most frequent mesenchymal GI neoplasms with an estimated annual incidence of 1.4-2 cases per 100.000/year [1]. GISTs occur at any site along the tubular GI tract from the esophagus to the anorectum but they are more common in the stomach (50-60%) and small bowel (20-30%) while a minority of cases originates at other sites including anorectum, colon, appendix and esophagus (together 5-10%) [2]. GISTs display spindle cell morphology more commonly (70%) than epithelioid (20%), mixed (10%) or pleomorphic (2%) phenotypes [2]. The great majority of GISTs (~80%) harbor mutations in KIT exon 11 (70%), exon 9 (8%), exon 13 (1%) and exon 17 (1%) [3]. The remainder show either mutated platelet-derived growth factor receptor alpha PDGFRA (~10%) or they are wild type for both (~10%) [3]. Of the latter, a small subset harbor the V600E BRAF mutation (2-4%) [4].
Approximately 30-50% of GISTs behave malignant during the clinical course of the disease; half of them have already metastasized at initial diagnosis [2]. Currently, tyrosine kinase inhibitor (TKI) therapy with imatinib mesylate (Glivec) represents the gold standard treatment for patients with inoperable or metastatic disease [5]. However, several studies have demonstrated that this targeted molecular therapy is largely dependent on the mutational status (genotyping) of individual tumors [6]. Consequently, genotyping has emerged as a powerful predictive test for patients with planned or anticipated TKI treatment. However, exploring all relevant exons in KIT (9, 11, 13, 17) and PDGFRA (12, 14, 18) based on the variable frequencies of the different mutations in GISTs is both time-consuming and is associated with high cost which represents a real burden on health systems, particularly in countries with limited resources. To date, there have been no general agreements or recommendations as to the rationale order of sequentially analyzing the relevant hotspot exons in KIT and PDGFRA in a given GIST tumor. Accordingly, most institutions start with the most commonly involved exons of KIT and get to PDGFRA only if all 4 exons of KIT showed a wild-type sequence. Still other institutions start the molecular analysis looking for the more common mutations (exon 9 and exon 11 of KIT), followed by rare kinase domain mutations (exon 13 and 17) if the former displayed a wild-type. Accordingly, molecular sequencing of the hotspots in PDGFRA (exon 12, 14, 18) are usually performed retrospectively after demonstration of wild-type KIT. An alternative method adopted by some investigators is to perform parallel analysis of KIT exon 9 and 11 and PDGFRA exon 18 initially and get to other rare exons if these turned out to be of wild-type sequence. Both approaches cannot reduce the financial burden on health insurance companies or the patients themselves. The most certain causes of this practice are the limited familiarity with the phenotype-genotype correlations in GISTs and analysis in laboratories without morphological experience, in which case a DNA might be sent for analysis without the tumor being assessed by an experienced pathologists prior to molecular investigation. Thus, it would be of great value to establish an approach that allows for direct sequencing of the highly expected exons based on a combined histomorphological and immunohistochemical approach.
In a recent study, we demonstrated upregulated expression of KIT in KIT-mutated GISTs, in contrast to upregulated PDGFRA expression in PDGFRA-mutated GISTs, on mRNA (qRT-PCR) and protein (Western Blot) level [7]. However, most routinely processed GISTs are formalin-fixed and paraffin-embedded, so these methods are not applicable in daily pathology routine. Thus, reliable determination of PDGFRA expression by immunohistochemistry might help to identify GISTs with PDGFRA mutation. This could enable sequential mutational analysis of PDGFRA and KIT genes, with reduced costs and shorter processing time. In the current study we analyzed the different patterns of PDGFRA immunostaining in a well characterized cohort of PDGFRA mutated GISTs. Then, a validation cohort spanning the morphological and genotypic spectrum of GISTs was used to predict the PDGFRA genotypic status based on the histomorphology of tumors and their PDGFRA immunostaining pattern.
Material and methods
First, a test cohort of 26 GISTs with known PDGFRA mutations was used to evaluate specific growth patterns as well as characteristics of PDGFRA immunohistochemistry. Second, a validation cohort of 94 surgically resected GISTs with known mutation status of KIT (n=72), PDGFRA (n=15) or with wild-type status (n=7) were used to conduct a tissue microarray. The histomorphological phenotype (spindled, epithelioid and mixed) was determined on H&E sections without knowledge of the genotype. PDGFRA immunostaining was performed on fresh-cut 3 μm sections using a Ventana automated staining system (VENTAGE) and a monoclonal antibody purchased from Cell Signalling, (#3164, dilution: 1:100). The staining intensity (negative, weak, intermediate, strong), extension of staining (0, negative; 1-25%, 1+; 26-50%, 2+ and >50%, 3+) and the predominant staining pattern (paranuclear dot-like/Golgi, cytoplasmic or membranous) of PDGFRA were assessed by two pathologists experienced in GIST (A.A., F.H.) without knowledge of the genotype. The combination of growth pattern and immunophenotype were used to classify tumors either as PDGFRA-phenotype or non-PDGFRA phenotype, and this was correlated to the mutation status in the whole validation cohort. This study and the related translational research activities are covered by ethical vota of the medical faculty of the Friedrich-Alexander University Erlangen-Nuremberg.
Results
In the test cohort, all 26 PDGFRA-mutated GIST displayed epithelioid (n=20, 77%) or mixed (n=6, 23%) growth pattern (Table 1), while there was no case with pure spindled morphology (Figure 1A, 1B). All 26 cases showed a paranuclear dot-like staining (Golgi pattern) for PDGFRA, commonly accompanied with variable cytoplasmic or membranous staining, in either >50% (n=22, 85%), or 26-50% (n=4, 15%) of the cells (Figure 1C, 1D). In the validation cohort, PDGFRA-mutated GISTs were significantly more often of epithelioid or mixed (n=12, 80%) phenotype compared to KIT-mutated GISTs (n=16, 22%) (p<0.001) (Figure 2A, 2C). Immunostaining with PDGFRA displayed a predominant paranuclear dot-like (Golgi-like) staining in >50% of the cells in all 15 PDGFRA-mutated tumors (100%), usually combined with variable cytoplasmic or membranous staining similar to the pattern seen in the test cohort (Figure 1C, 1D). In contrast, none of the KIT mutants or the wild-type GIST showed a predominant paranuclear dot-like PDGFRA staining like that seen in cases with PDGFRA mutation (p<0.001). Approximately one half of KIT mutants and wild-type GISTs lacked any PDGFRA expression, and in the remaining cases the staining intensity was mostly weak or intermediate, with a membranous and/or cytoplasmic pattern (Figure 2B, 2D). In two of the KIT-mutated cases, a focal paranuclear dot-like staining was seen in 26-50% of the cells, however, in both cases, there was also a strong KIT expression. Interestingly, in both of these cases, a hemizygous status of the KIT mutation was observed in the Sanger chromatogram, most likely corresponding to an amplification of the chromosomal region 4q comprising the mutated KIT allele as well as the PDGFRA gene in close proximity. Of the seven wild-type GISTs, six were correctly identified as non-PDGFRA genotype and one case was estimated to harbor a PDGFRA mutation based on staining pattern. Thus, combining histomorphology with the PDGFRA staining intensity and staining pattern, the sensitivity and specificity of the combined PDGFRA phenotype predictor in identifying PDGFRA mutants was 100% and 99%, respectively (Figure 3 and Table 2).
Table 1.
Distribution of the histological types, genotypes and PDGFRA immunostaining in the test set and the validation cohort
| Test set | Validation set | P-value | |||
|---|---|---|---|---|---|
|
|
|||||
| PDGFRA mutation (n=26) | PDGFRA mutation (n=15) | KIT mutation (n=72) | wild-type (n=7) | ||
| Morphology | <0.001 | ||||
| Epithelioid | 20 (77%) | 12 (80%) | 5 (7%) | 1 (14%) | |
| Mixed | 6 (23%) | 0 (0%) | 11 (15%) | 1 (14%) | |
| Spindled | 0 (0%) | 3 (20%) | 56 (78%) | 5 (72%) | |
| PDGFRA staining intensity | <0.001 | ||||
| Negative | 0 (0%) | 0 (0%) | 34 (47%) | 4 (57%) | |
| Weak | 0 (0%) | 0 (0%) | 8 (11%) | 2 (29%) | |
| Intermediate | 4 (15%) | 0 (0%) | 18 (25%) | 1 (14%) | |
| Strong | 22 (85%) | 15 (100%) | 12 (17%) | 0 (0%) | |
| predominant PDGFRA staining pattern | <0.001 | ||||
| dot-like | 26 (100%) | 15 (100%) | 2 (3%) | 0 (0%) | |
| cytoplasmic/membranous | 0 (0%) | 0 (0%) | 36 (50%) | 3 (43%) | |
| Negative | 0 (0%) | 0 (0%) | 34 (47%) | 4 (57%) | |
Figure 1.
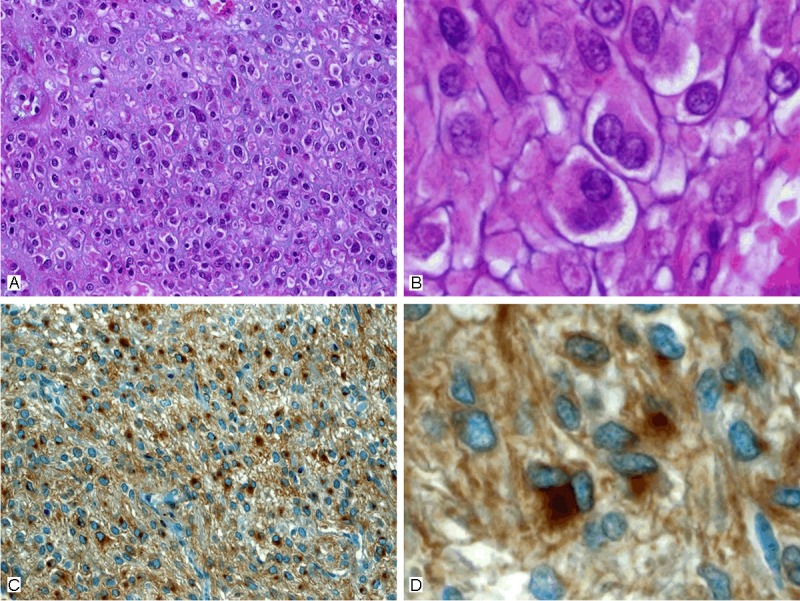
Example of the typical features of a PDGFRA-mutant GIST. A: Large epithelioid cells with peripheral cytoplasmic clearance and distinct cell borders. B: Higher magnification showed typical binucleated cells. C: Golgi-type immunoreactivity for PDGFRA was seen in almost all tumor cells. D: Higher magnification, binucleated cells usually display the strongest reactivity for PDGFRA.
Figure 2.
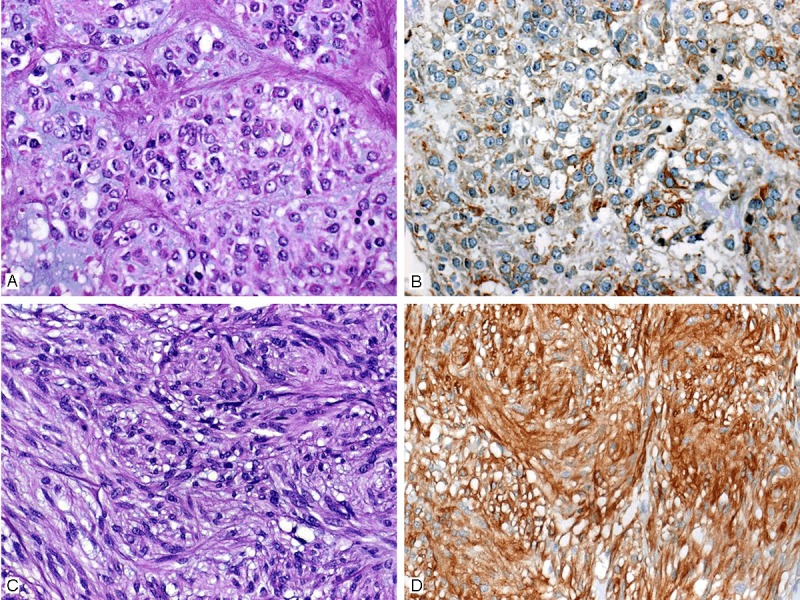
Examples of KIT mutant GISTs with epithelioid (A) and spindle (C) morphology. Expression of PDGFRA varied from generally weak and focal (B) to wide-spread and strong (D) cytoplasmic staining. Note absence of unequivocal Golgi pattern.
Figure 3.
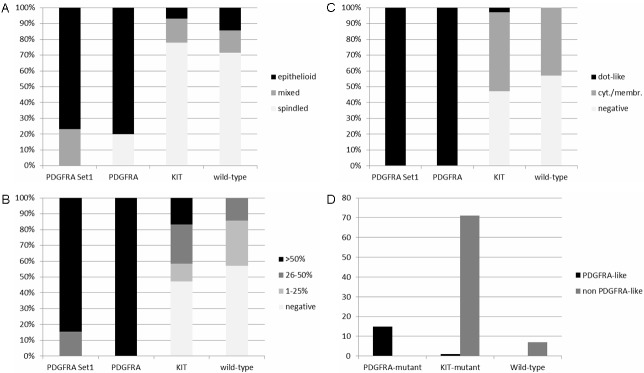
Sensitivity and specificity of histomorphology and PDGFRA immunohistochemistry in predicting the PDGFRA genotype. A: Epithelioid/mixed morphology vs. spindled morphology. B: PDGFRA staining intensity. C: PDGFRA staining pattern. D: Combined predictor according to morphology, immunostaining intensity and staining pattern.
Table 2.
Sensitivity and specificity of histological type, staining pattern and staining intensity of PDGFRA to predict the PDGFRA genotype in GIST
| PDGFRA-mutant | KIT-mutant | Sensitivity | Specificity | P-value* | |
|---|---|---|---|---|---|
| Histomorphology | |||||
| Epithelioid/mixed | 12 | 16 | 80% | 78% | 4x10-5 |
| Spindled | 3 | 56 | |||
| PDGFRA staining intensity | |||||
| >25% | 15 | 30 | 100% | 58% | 1.7x10-5 |
| <25% | 0 | 42 | |||
| PDGFRA staining pattern | |||||
| dot-like | 15 | 2† | 100% | 97% | 5.2x10-15 |
| non dot-like | 0 | 70 | |||
| PDGFRA genotype predictor | |||||
| PDGFRA-phenotype | 15 | 1 | 100% | 99% | 6x10-16 |
| non PDGFRA-phenotype | 0 | 71 | |||
Fisher’s exact test.
Both tumors showed an amplification of the mutated KIT allele, as demonstrated by hemizygous status in the Sanger sequencing reaction.
One of these two tumors corresponds to the only misclassified case by our PDGFRA genotype predictor.
Discussion
PDGFRA immunostaining in GIST has been studied by several groups [8-11]. However, all these studies have generally focused on the value of this staining in confirming diagnosis of PDGFRA mutants KIT-negative cases and their distinction from intraabdominal lesions including desmoid fibromatosis and other neoplasms that may closely mimic GISTs. In the United States, PDGFRA immunostaining did not receive sufficient attention and is not recommended for use in routine surgical pathology practice. This is probably because of disappointing observations made in initial studies using antibodies that were not sufficiently purified at that time [12]. Thus, almost all of published studies on PDGFRA immunohistochemistry in GIST were from European study groups [8-11]. Although some studies have pointed out the characteristic PDGFRA pattern in GISTs and its usefulness as an adjunct in recognizing KIT-negative cases, to our knowledge, no study has evaluated the usefulness of this antibody to predict the PDGFRA genotype in GIST in the way we did in the present study.
In this study, we evaluated the pattern of PDGFRA immunostaining in a cohort of well characterized PDGFRA mutant GISTs. The majority of these tumors (84%) showed predominantly epithelioid morphology and the remainder were of mixed type consistent with previous studies [13]. The epithelioid pattern of these tumors was consistent with the 4 subtypes defined by Miettinen et al for gastric epithelioid GISTs [2]. Overall, these tumors were characterized by copious cytoplasm of variable quality (clear, rhabdoid, plasmacytoid retracted spider-like or vacuolated) with either distinct cell borders or syncytial growth pattern [14]. A common feature of these tumors was the presence of bi- and multinucleated tumor cells (giant cells) with paranuclear cytoplasmic condensations [13,14].
In our study, immunostaining with PDGFRA displayed a strong/moderate paranuclear (Golgi-like) staining in 100% of these cases, usually combined with variable weak cytoplasmic or membranous staining. Applying these morphological and immunohistochemical criteria predicted the PDGFRA mutant genotype in 100% of the mutated cases in our study. However, a small subset of KIT mutants showed moderate to strong diffuse cytoplasmic PDGFRA expression that is most likely the consequence of gene amplification. Of note, almost all wild-type GISTs and approximately one half of KIT mutants lacked any PDGFRA expression.
These results are consistent with previous studies that PDGFRA mutated GISTs show predominantly epithelioid or mixed phenotype [2,13,14]. However, the term “epithelioid” has been used inconsistently in GISTs for designation of two different phenomena. First, tumors with the epithelioid features as defined in the current study are almost exclusively of the PDGFRA mutated type and they display consistent paranuclear dot-like PDGFRA immunoreactivity. Second, there exist GISTs with higher cellularity and bland relatively small rounded cells with higher nuclear-cytoplasmic ratio. Although this feature may occasionally be observed in the hypercellular epithelioid subtype described by Miettinen et al for gastric GISTs and thus usually correlate with a PDGFRA phenotype [2], a majority of tumors with this “hypercellular round cell morphology” represent a morphological shift from the spindle cell to round cell morphology as a consequence of tumor progression in KIT mutants [2,15]. Accordingly, the presence of rounded (epithelioid) cells in a mixed KIT mutant neoplasm has been linked to adverse outcome [15]. In our experience, tumors with round cell morphology and KIT mutations lack the characteristic PDGFRA staining pattern of PDGFRA mutants. The predictive power of the combined morphological-immunohistochemical analysis is sufficiently high to allow for selective molecular analysis. This approach allows for direct selection of highly expected mutations (starting with exon 18 and then followed by exons 12 and 14 respectively if wild-type is detected). The advantages of using this method are: 1) significant reduction in the workflow for the molecular pathology laboratories, 2) a significant reduction of waiting time before initiation of appropriate therapy, particularly for patients with advance disease, and 3) significant reduction in the cost burden.
Our observations underlines the importance of reviewing the tumor slides by a pathologist experienced in GIST pathology before initiating any molecular analysis. In our experience, this initial review of the slides by an experienced pathologist to confirm diagnosis are essential gastrofor exclusion of GIST mimics (leiomyosarcoma, desmoids tumors, dedifferentiated liposarcoma and other neoplasms with weak cytoplasmic non-specific KIT expression) that would otherwise be erroneously classified as wild-type GIST with the sequelae of inappropriate and costly TKI treatment if molecular analysis is performed blindly.
In summary, we concluded that the combination of histomorphology (epithelioid phenoptype) and unequivocal paranuclear immunostaining for PDGFRA (Golgi pattern) is a reliable highly sensitive and specific predictor of PDGFRA genotype in GIST. The use of this PDGFRA genotype predictor may help to reduce costs, shorten processing time of GIST genotyping before treatment and also reduce the workload by phenotype-oriented genotyping. This might be even more important in less developed countries with restricted health budgets.
Disclosure of conflict of interest
None.
References
- 1.Nilsson B, Bümming P, Meis-Kindblom JM, Odén A, Dortok A, Gustavsson B, Sablinska K, Kindblom LG. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era--a population-based study in western Sweden. Cancer. 2005;103:821–9. doi: 10.1002/cncr.20862. [DOI] [PubMed] [Google Scholar]
- 2.Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23:70–83. doi: 10.1053/j.semdp.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Lasota J, Miettinen M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs) Semin Diagn Pathol. 2006;23:91–102. doi: 10.1053/j.semdp.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Daniels M, Lurkin I, Pauli R, Erbstößer E, Hildebrandt U, Hellwig K, Zschille U, Lüders P, Krüger G, Knolle J, Stengel B, Prall F, Hertel K, Lobeck H, Popp B, Theissig F, Wünsch P, Zwarthoff E, Agaimy A, Schneider-Stock R. Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST) Cancer Lett. 2011;312:43–54. doi: 10.1016/j.canlet.2011.07.029. [DOI] [PubMed] [Google Scholar]
- 5.Casali PG, Blay JY ESMO/CONTICANET/EUROBONET Consensus Panel of Experts. Gastrointestinal stromal tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v98–102. doi: 10.1093/annonc/mdq208. [DOI] [PubMed] [Google Scholar]
- 6.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CD, Silberman S, Dimitrijevic S, Fletcher JA. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 2003;21:4342–9. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 7.Haller F, Happel N, Schulten HJ, von Heydebreck A, Schwager S, Armbrust T, Langer C, Gunawan B, Doenecke D, Füzesi L. Site-dependent differential KIT and PDGFRA expression in gastric and intestinal gastrointestinal stromal tumors. Mod Pathol. 2007;20:1103–11. doi: 10.1038/modpathol.3800947. [DOI] [PubMed] [Google Scholar]
- 8.Pauls K, Merkelbach-Bruse S, Thal D, Büttner R, Wardelmann E. PDGFRalpha- and c-kit-mutated gastrointestinal stromal tumours (GISTs) are characterized by distinctive histological and immunohistochemical features. Histopathology. 2005;46:166–75. doi: 10.1111/j.1365-2559.2005.02061.x. [DOI] [PubMed] [Google Scholar]
- 9.Rossi G, Valli R, Bertolini F, Marchioni A, Cavazza A, Mucciarini C, Migaldi M, Federico M, Trentini GP, Sgambato A. PDGFR expression in differential diagnosis between KIT-negative gastrointestinal stromal tumours and other primary soft-tissue tumours of the gastrointestinal tract. Histopathology. 2005;46:522–31. doi: 10.1111/j.1365-2559.2005.02128.x. [DOI] [PubMed] [Google Scholar]
- 10.Peterson MR, Piao Z, Weidner N, Yi ES. Strong PDGFRA positivity is seen in GISTs but not in other intra-abdominal mesenchymal tumors: Immunohistochemical and mutational analyses. Appl Immunohistochem Mol Morphol. 2006;14:390–6. doi: 10.1097/01.pai.0000203038.33414.a3. [DOI] [PubMed] [Google Scholar]
- 11.Miselli F, Millefanti C, Conca E, Negri T, Piacenza C, Pierotti MA, Tamborini E, Pilotti S. PDGFRA immunostaining can help in the diagnosis of gastrointestinal stromal tumors. Am J Surg Pathol. 2008;32:738–43. doi: 10.1097/PAS.0b013e31815c47e8. [DOI] [PubMed] [Google Scholar]
- 12.Medeiros F, Corless CL, Duensing A, Hornick JL, Oliveira AM, Heinrich MC, Fletcher JA, Fletcher CD. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol. 2004;28:889–94. doi: 10.1097/00000478-200407000-00007. [DOI] [PubMed] [Google Scholar]
- 13.Wardelmann E, Hrychyk A, Merkelbach-Bruse S, Pauls K, Goldstein J, Hohenberger P, Losen I, Manegold C, Büttner R, Pietsch T. Association of platelet-derived growth factor receptor alpha mutations with gastric primary site and epithelioid or mixed cell morphology in gastrointestinal stromal tumors. J Mol Diagn. 2004;6:197–204. doi: 10.1016/s1525-1578(10)60510-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daum O, Grossmann P, Vanecek T, Sima R, Mukensnabl P, Michal M. Diagnostic morphological features of PDGFRA-mutated gastrointestinal stromal tumors: molecular genetic and histologic analysis of 60 cases of gastric gastrointestinal stromal tumors. Ann Diagn Pathol. 2007 Feb;11:27–33. doi: 10.1016/j.anndiagpath.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Haller F, Cortis J, Helfrich J, Cameron S, Schüler P, Schwager S, Gunawan B, Füzesi L, Agaimy A. Epithelioid/mixed phenotype in gastrointestinal stromal tumors with KIT mutation from the stomach is associated with accelerated passage of late phases of the cell cycle and shorter disease-free survival. Mod Pathol. 2011;24:248–55. doi: 10.1038/modpathol.2010.188. [DOI] [PubMed] [Google Scholar]