

Abstract
A lethal and extensively characterized familial form of hypertrophic cardiomyopathy (HC) is due to a point mutation (Arg403Gln) in the cardiac β-myosin heavy-chain (MHC) gene. Although this is associated with abnormal energy metabolism and progression to heart failure in an animal model, in vivo cardiac energetic shave not been characterized in patients with this mutation. Noninvasive phosphorus saturation transfer magnetic resonance spectroscopy was used to measure the adenosine triphosphate (ATP)supplied by the creatine kinase (CK) reaction and phosphocreatine (PCr), the heart’s primary energy reserve, in 9 of 10 patients from a single kindred with HC caused by Arg403GIn mutation, and 17 age-matched healthy controls. Systolic and diastolic function was assessed by echocardiography in all 10 HC patients. HC patients had impairment of diastolic function as well as mild systolic dysfunction, when assessed using global systolic longitudinal strain. Myocardial [PCr] was significantly decreased by 24% in patients (7.1±2.3μmol/g) compared to controls (9.4±1.2μmol/g; p=0.003). The pseudo-first-order CK rate-constant was 26% lower (0.28±0.15 vs. 0.38±0.07s−1, p=0.035) and the forward CK flux was 44% lower (2.0±1.4 vs3.6±0.9 μmol/g/s, p=0.001) than controls. The contractile abnormalities did not correlate with metabolic indices. In conclusion, myocardial PCr and CK ATP delivery are significantly reduced in patients with HC due to Arg403Gln mutation, akin to prior results from mice with the same mutation. Lack of a relation between energetic and contractile abnormalities suggests the former are due to the sarcomeric mutation and not a late consequence of mechanical dysfunction.
Keywords: familial hypertrophic cardiomyopathy, energy metabolism, creatine kinase, ATP, myosin mutation
INTRODUCTION
The myosin heavy chain Arg403Gln mutation has a clinically important phenotype of ventricular hypertrophy, heart failure, arrhythmias and sudden death 1. Because the murine Arg403Gln model manifests an increased energetic cost of contraction2,3, we report here the first noninvasive energetic 31P magnetic resonance spectroscopy study of cardiac creatine kinase (CK) metabolite concentrations and ATP (adenosine tri-phosphate) kinetics in patients from a single family whose affected members carry this mutation4. We used 31P magnetic resonance spectroscopy with saturation transfer5–7 in a single exam to test the hypothesis that patients with the Arg403Gln mutation and hypertrophy who are at risk of developing heart failure exhibit energetic changes consistent with an increased energetic cost of contraction (reduced cardiac creatine phosphate, preserved adenosine triphosphate (ATP) and decreased free energy of ATP hydrolysis) as observed in the myosin heavy chain Arg403Gln murine model3. We also test whether the ATP energy supply through CK is reduced in these patients, as observed in other cardiomyopathies6,7.
METHODS
All studies were approved by the Johns Hopkins Institutional Review Board on human investigation and all subjects provided informed written consent. We examined 10 patients with a clinical history of hypertrophic cardiomyopathy from a single family (4 men, 6women; mean age 40±13years) confirmed as carrying a missense Arg-403 to glutamine mutation in the beta myosin heavy chain gene4. All 10 HC patients underwent echocardiography, but only 9 patients underwent magnetic resonance spectroscopy; one patient was claustrophobic and could not undergo magnetic resonance spectroscopy. Patients had no history of hypertension, chest pain, coronary disease or diabetes and were classified according to New York Heart Association functional class at the time of imaging. Seventeen healthy subjects (12 men, 5 women; mean age 40±8 yrs) with no history of heart disease, hypertension or diabetes served as controls for magnetic resonance studies.
We used the same magnetic resonance protocol as described previously6,7. Nine HC patients and 17 controls underwent magnetic resonance spectroscopy. Image-guided 31P magnetic resonance spectroscopy was performed on a 1.5-T General Electric(Milwaukee, WI) Signamagnetic resonance scanner. Briefly, the patient cardiac protocol consisted of the following steps:
Conventional scout proton 1H magnetic resonance imaging acquired with the body coil of the scanner to position the anterior myocardium over the coil and for shimming;
Acquisition of 4 31P four-angle saturation transfer data sets localized by 1-dimensional chemical shift imaging to measure CK flux;5,7
Acquisition of a fifth 31P 1-dimensional chemical shift imaging set with saturation turned off for phosphate metabolite quantification and “spillover” correction5,7,8;
Acquisition of a sixth proton (1H)1-dimensional chemical shift imaging data set using the 31P coil to provide a water concentration reference for metabolite quantification9;
Repetition of the 1-dimensional chemical shift imaging acquisitions of Steps 3 and 4 under fully-relaxed conditions from a 0.15 mol/L inorganic phosphate (Pi) reference phantom to determine the metabolite concentrations7,9,10.
The pseudo-first-order CK reaction rate constant kf was determined from the ratio of the peak heights of phosphocreatine (PCr) acquired in Step 2, from the spillover-corrected Eq. [11] of Ref.8 and as described in the Online Methods of Ref11. The myocardial phosphocreatine and ATP concentrations, [PCr] and [ATP] were determined from data generated as previously described6,7,9,12. The forward CK flux or unidirectional CK ATP synthesis rate, is given by the product, kf.[PCr], in μmol/g wet wt/s7. The free-energy of ATP hydrolysis (−ΔG~ATP, kJ/mol) was determined per reference13 with conservative assumptions of an inorganic phosphate concentration of~1μmol/g wet weight6,7 in control subjects and HC patients. Cytosolicadenosine diphosphate (ADP) concentration was calculated as described earlier11.
All 10 HC patients underwent conventional transthoracic and tissue Doppler echocardiography. Standard para-sternal long axis view, 4-,2-, and 3- chamber apical views, and a mid-ventricular short-axis view were obtained at end-expiration using a 3.5 MHz transducer (Vivid 7, General Electric Vingmed Ultrasound, Milwaukee, Wisconsin) at a frame rate of 40–70 fps for B-mode images. Mitral inflow was evaluated in apical 4-chamber views by sampling 1–2 mm volumes at the tips of the mitral leaflets with pulsed-Doppler to assess left ventricular diastolic function. The left ventricular outflow tract was also evaluated with pulsed-Doppler, from apical chamber views with sample volumes placed just proximal to the aortic valve. Tissue Doppler images were obtained in apical projections, including narrow-sector images with a minimum frame rate of 200 fps.
Off-line analysis was performed using Echopac Software (V. 7.1.1 GE Medical Systems, Milwaukee, Wisconsin) to determine left ventricular dimensions and volumes. Left ventricular mass was calculated using the Penn formula14 and ejection fraction was calculated using the biplane modified Simpsons method15. Conventional indices of diastolic function such as mitral early (E) and late (A) diastolic flow velocities and ratio (E/A), and deceleration time were measured from mitral inflow pulsed-Doppler recordings. Deceleration time was measured by extending the deceleration slope from peak early wave velocity to the zero velocity baseline.
Myocardial longitudinal strain was derived from the tissue velocity data. Prior to analysis, left ventricular ejection timing was measured from the left ventricular outflow track pulsed-Doppler recordings. Using narrow-sector tissue Doppler images, color-coded velocity analysis was performed with 12×6 mm sample volumes at the septal annulus to measure mitral annular early diastolic tissue velocity. Sample volumes placed at the basal and mid-segments of the anterior and inferior septum, lateral, posterior, anterior, and inferior left ventricular walls were used to assess regional tissue velocities, longitudinal strain rate, and longitudinal strain. Peak negative global longitudinal strain was taken as the average of the 12-segment peak negative regional longitudinal strain within the entire cardiac cycle. Measurements were obtained from 3 cardiac cycles, excluding segments corrupted by reverberations, drop-outs and/or poor alignment with Doppler signals (≥20° angle of insonation).
Continuous variables are presented as mean±SD, and categorical variables are expressed as N (%). Study groups were compared using unpaired t-test and Fisher’s exact test for continuous and categorical variables, respectively. Univariate and multivariate linear regression analysis was performed to test the association of New York Heart Association class, E/Ea, left ventricular mass, peak global longitudinal strain, brain natriuretic peptide with 31P metabolic measures of [PCr], kf, and CK flux. A p-value of <0.05 was considered significant.
RESULTS
Baseline demographics are presented in Table 1. Despite being from the same family and carrying the same mutation, New York Heart Association class varied among HC patients: 8 patients were class I, 1 patient was class II and 1 patient was class III. Four patients had severe septal hypertrophy (maximum septal wall thickness >2.0 cm) and 3 patients had moderate septal hypertrophy (maximum septal wall thickness >1.2–1.5 cm); 3patients had normal septal thickness. Left ventricular dimensions and ejection fraction were within the normal range in all patients except in 1 patient who had a moderately reduced ejection fraction (Table 1).
Table 1.
Patient demographics and echocardiography measurements
| Age (Years) | Sex | NYHA Class | VS (cm) | PWT (cm) | LVIDd (cm) | LVIDs (cm) | EF % | NYHA Class at F/U |
|---|---|---|---|---|---|---|---|---|
| 22 | Male | I | 1.5 | 1.0 | 5.4 | 3.6 | 62 | I |
| 29 | Male | I | 1.2 | 1.2 | 5.2 | 3.3 | 69 | I |
| 33 | Female | I | 1.3 | 0.9 | 5.4 | 3.2 | 54 | I |
| 34 | Female | I | 1.1 | 0.8 | 4.1 | 2.8 | 66 | I |
| 43 | Male | I | 1.2 | 1.0 | 5.6 | 3.8 | 65 | I |
| 50 | Male | I | 1.4 | 0.8 | 5.3 | 3.9 | 56 | I |
| 52 | Female | II | 2.3 | 1.1 | 4.2 | 2.9 | 58 | III |
| 52 | Male | I | 2.1 | 1.0 | 3.7 | 2.4 | 65 | I |
| 57 | Female | I | 2.2 | 1.2 | 4.1 | 2.6 | 61 | II |
| 62 | Female | III | 2.0 | 1.2 | 5.4 | 3.9 | 37 | Transplant |
VS = ventricular septum; PWT= posterior wall thickness; LVIDs= Left ventricular internal dimension at end systole; LVIDd= Left ventricular internal dimension at end diastole; EF= left ventricular ejection fraction; NYHA= New York Heart Association; F/U= follow-up
HC patients had increased diastolic septal wall thickness and left ventricular mass in the setting of normal diastolic posterior wall thickness, indicating asymmetric septal hypertrophy (Table 1). Left ventricular systolic and diastolic volumes as well as ejection fraction were normal16. However, peak global longitudinal strain was mildly reduced (p<0.001) when compared to historic controls17, indicating systolic dysfunction. There was evidence of global diastolic dysfunction as indicated by reduced mitral E/A ratio and mitral annular tissue velocity (Table 1). E/Ea ratio was increased (15 ±6) suggesting elevated left ventricular filling pressure18. Fig. 1 shows representative strain curves from one HC subject.
Fig. 1.
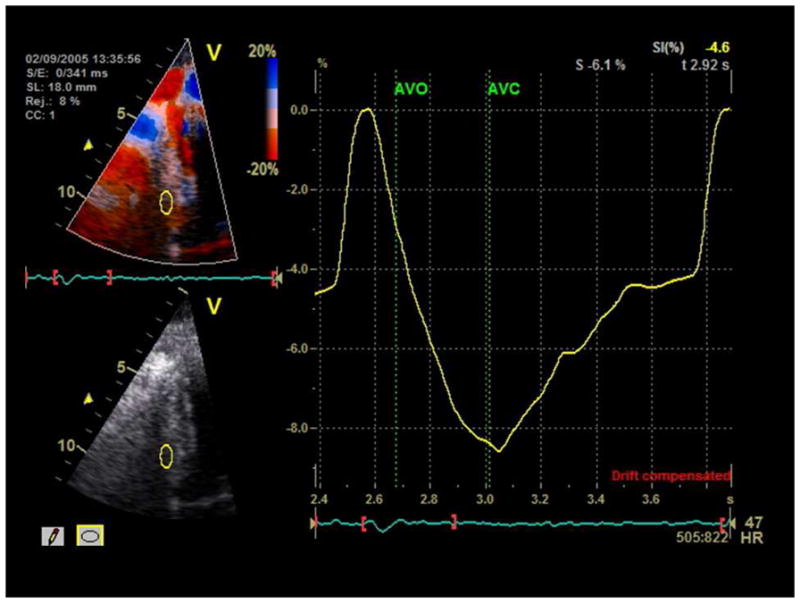
Representative strain curves from an HC subject with strain value of 8.15% with sample volumes placed at the basal septum. AVO-aortic valve opening. AVC-aortic valve closure.
Representative image-guided, spatially-localized, cardiac 31P four-angle saturation transfer spectra from a 52 yr old male patient with HC, who was New York Heart Association Class I, are shown in Fig. 2. Spectra from the chest wall exhibit a high PCr/ATP ratio due to the higher concentration of PCr in skeletal muscle (~25μmol/g) compared to that of heart (≤10 μmol/g)12. Peak areas in (b) are proportional to metabolite concentrations which are determined after calibration. Application of saturation at control and γ-ATP locations (c,d) result in a reduction in the phosphocreatine (PCr) signal in direct proportion to the forward CK rate constant, kf5,7. In this subject, kf is lower in heart at 0.13/s than in the chest muscle (0.23/s).
Fig. 2.

(a)Transaxial scout magnetic resonance image of 52 yr old male HC patient, annotated to show location of 5 adjacent tissue volumes sampled by 31P magnetic resonance spectroscopy (b–d) in the chest wall (blue arrows), and anterior myocardium (red arrows). Spectra in (b) were acquired with no saturation applied for the purpose of measuring absolute concentrations. Spectra in (c) and (d) were acquired with control saturation (green arrow) and with γ-ATP saturated (amber arrow). The change in the height of the PCr peak (blue and red lines) is due to the forward flux through CK, and is proportional to the pseudo first-order rate constant. Each magnetic resonance spectroscopy data set was acquired in about 6min.
The 31P magnetic resonance spectroscopy results from all subjects are summarized in Table 2. Of the myocardial metabolite pool sizes, [PCr] was significantly decreased by approximately 24% in HC patients as compared to that in control subjects (p=0.003, Table 2). However, owing to a downward trend in [ATP] in HC (p=0.10), a corresponding 16% reduction in PCr/ATP ratio was not statistically significant (p=0.054). The mean pseudo-first-order rate-constant for the CK reaction, kf, was reduced 26% in HC compared to that of control subjects (p=0.035). This, in combination with the [PCr] reduction, resulted in a highly significant 44% decrease in the forward flux of myocardial ATP generated by the CK reaction in HC patients compared to controls (p=0.001). There were no significant correlations between 31P metabolic measures and New York Heart Association class, the ratio of early diastolic mitral inflow velocity to that of the mitral annulus, left ventricular mass, peak global longitudinal strain, or brain naturetic peptide. Fig. 3 plots the individual [PCr], kf, and CK flux measurements for all subjects.
Table 2.
Spectroscopy results
| Parameters | HC (n=9) | Controls (n=17) | p-value | Change |
|---|---|---|---|---|
| Creatine Phosphate (μmol/g) | 7.1 ± 2.3 | 9.4 ± 1.2 | 0.003 | −24% |
| ATP (μmol/g) | 5.0± 0.8 | 5.8 ± 1.2 | 0.10 | −13% |
| PCr/ATP | 1.4 ± 0.4 | 1.7 ± 0.3 | 0.054 | −15% |
| CK Rate constant, Kf (s−1) | 0.28 ± 0.15 | 0.38 ± 0.07 | 0.035 | −26% |
| CK flux (μmol/g/s) | 2.0 ± 1.4 | 3.6 ± 0.9 | 0.001 | −44% |
Fig. 3.

[PCr], kf and CK flux data in HC patients and healthy controls.
Mean cytosolicadenosine diphosphate[ADP] concentration was similar in control subjects (110±20μM) and in HC patients (110±50μM, p=0.9). With inorganic phosphate [Pi] conservatively estimated at 1μmol/g wet wt6,7 in both control and HC patients, the free-energy release with ATP hydrolysis is similar in both groups (ΔG~ATP. −58.5±0.4 vs −58.3±1.3 kJ/mol). Allowing for a possible 35% increase in inorganic phosphate in HC19 results in a mean free energy release trending lower in HC(−58.5±0.4 in controls vs −57.5 ±1.3 kJ/mol in HC, p=0.05, whereby a more negative ΔG~ATP indicates greater energy release).
DISCUSSION
This is the first study to characterize the in vivo CK and cardiac energetic consequences of the Arg403Gln point mutation in the myosin heavy chain in HC patients, and to relate the 31P magnetic resonance spectroscopy-derived energetic findings to cardiac mechanics. This mutation is important because it was the first HC mutation identified, is associated with high morbidity and has been extensively investigated in animal models2,4. There are 2 main findings. First, there is a reduction in myocardial [PCr] with preserved [ATP] and calculated [ADP] which would suggest an increased cost of contraction as previously reported in the mouse model carrying this mutation2,3. Second, there is reduced ATP delivery by creatine kinase (CK), the primary cardiac energy reserve, which would further limit energy availability at the myofibril.
In heterozygous mice bearing the Arg403Gln mutation there is a decrease in myocardial [PCr] and increase in inorganic phosphate [Pi] with unchanged [ATP] under conditions associated with diastolic dysfunction3. Because hypertrophy is not present in the mouse model, these are not secondary consequences of hypertrophy but must instead be primary consequences of the myosin mutation3. Such an increased energetic cost of contraction with the Arg403Gln mutation in the intact mouse heart3 is thought to be consistent with several molecular studies suggesting a decrease in actomyosin motility20,21 that effectively puts an energetic “drag” on the wild-type myosin during shortening3,19,22. We demonstrate here that human HC hearts with the Arg403Gln mutation exhibit similar energetic changes of a decreased PCr and a preserved ATP associated with diastolic dysfunction, albeit in the presence of hypertrophy.
The CK reaction serves as the prime energy reserve of the heart, rapidly and reversibly transferring high-energy phosphate between ATP and creatine 23,24. In addition to acting as a temporal ATP buffer during work-related or cyclic contractile-related increases in energy demand, CK is also thought to act as a spatial buffer in transferring high-energy phosphates between the mitochondrial sites of ATP generation and the cytosolic sites of ATP utilization 22,25,26 and to be a major part of an integrated metabolic network that supports intracellular ATP transfer and buffering25,27. Because CK is the most abundant and important ATP buffering phospho-transfer reaction, the 43% decrease in forward CK flux at rest reported here for the first time in HC patients, would be expected to reduce cardiac metabolic energy reserve. Indeed, the magnitude of the reduction is comparable, albeit less than, the 50–65% reductions observed previously in other heart failure patients6,7.
Diastolic dysfunction is a consistent finding in Arg403Gln HC2,4 and was manifest in this cohort (Table 1). In theory, several energetic abnormalities could contribute to the diastolic dysfunction in human Arg403Gln HC and these include ATP depletion, increased cytosolic [ADP], and altered free energy of ATP hydrolysis. However, none of these were observed in these patients. In a prior study, step-wise inhibition of CK by iodoacetamide in isolated normal rat hearts resulted in progressive diastolic dysfunction with preserved systolic function at baseline28. In the current study, HC patients have reduced mean CK flux and diastolic dysfunction. It will be important in future work, to investigate whether a causal relationship exists between diastolic dysfunction, CK flux, and more precise measures of the free energy release during ATP hydrolysis in these patients.
Previous human 31P magnetic resonance spectroscopy studies of HC have reported either unchanged cardiac PCr/ATP ratios29, or mild reductions 30 in HC patients. Although in our study mean myocardial PCr/ATP ratio trended lower by about 16% in HC patients as compared to controls, the differences were not strictly statistically significant (P=0.054). We did observe a significant 24% reduction in myocardial [PCr], with [ATP] trending 13% lower in HC which are consistent with, albeit lower than, a HC study reporting no change in PCr/ATP but 37% reductions in [PCr] and [ATP]29.
The same quantitative 31P magnetic resonance spectroscopy protocols for measuring human myocardial metabolite concentrations and CK kinetics used here, previously revealed significant reductions in myocardial CK flux inpatients with non-ischemic dilated cardiomyopathy7, and left ventricular hypertrophy secondary to hypertension6. The present work extends the observation of reduced kf and CK flux in patients who have both heart failure and hypertrophy produced by pressure overload6, to those with hypertrophy of genetic origin–specifically those with an Arg403Gln point mutation in MH. However, it is important to point out that the pathophysiology of the conditions is quite different. In contrast to pressure-overload left ventricular hypertrophy, HC is a genetic disorder caused by mutations typically in sarcomeric proteins, some of which increase the cost of force production resulting in increased ATP turnover3,22. That we were unable to detect any correlation between bioenergetic indices and contractile abnormalities suggests that the former are due to the sarcomeric mutation and not a late consequence of mechanical dysfunction.
Although this kindred of 10 individuals with the same MHC Arg403Gln mutation is relatively large for a single family, this sample size may limit our ability to detect small differences in cardiac PCr/ATP and/or weak correlations between energetic and functional parameters. Nevertheless, there are advantages to studying a single kindred (e.g. similar common genetic background and geographic exposure) and, moreover, our cardiac metabolic results agree with invasive measurements of metabolites in the Arg403Gln mouse model. Lastly, the estimates of the free energy of ATP hydrolysis and cytosolic free AD Pare dependent on prior measurements of myocardial inorganic phosphate and creatine from other subjects.
Acknowledgments
This work was supported by NIH grants HL61912, HL63030 and HL56332 and the Clarence Doodeman Endowment.
We thank Dr. Shenghan Lai for reviewing the statistics and Dr. Junaid M. Afzal for help with statistics.
Footnotes
DISCLOSURES: none declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 2.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 3.Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG, Ingwall JS. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest. 1998;101:1775–1783. doi: 10.1172/JCI1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenzweig A, Watkins H, Hwang DS, Miri M, McKenna W, Traill TA, Seidman JG, Seidman CE. Preclinical diagnosis of familial hypertrophic cardiomyopathy by genetic analysis of blood lymphocytes. N Engl J Med. 1991;325:1753–1760. doi: 10.1056/NEJM199112193252501. [DOI] [PubMed] [Google Scholar]
- 5.Bottomley PA, Ouwerkerk R, Lee RF, Weiss RG. Four-angle saturation transfer (FAST) method for measuring creatine kinase reaction rates in vivo. Magn Reson Med. 2002;47:850–863. doi: 10.1002/mrm.10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith CS, Bottomley PA, Schulman SP, Gerstenblith G, Weiss RG. Altered creatine kinase adenosine triphosphate kinetics in failing hypertrophied human myocardium. Circulation. 2006;114:1151–1158. doi: 10.1161/CIRCULATIONAHA.106.613646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci U S A. 2005;102:808–813. doi: 10.1073/pnas.0408962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabr RE, Weiss RG, Bottomley PA. Correcting reaction rates measured by saturation-transfer magnetic resonance spectroscopy. J Magn Reson. 2008;191:248–258. doi: 10.1016/j.jmr.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bottomley PA, Atalar E, Weiss RG. Human cardiac high-energy phosphate metabolite concentrations by 1D-resolved NMR spectroscopy. Magn Reson Med. 1996;35:664–670. doi: 10.1002/mrm.1910350507. [DOI] [PubMed] [Google Scholar]
- 10.Bottomley PA, Hardy CJ, Roemer PB. Phosphate metabolite imaging and concentration measurements in human heart by nuclear magnetic resonance. Magn Reson Med. 1990;14:425–434. doi: 10.1002/mrm.1910140302. [DOI] [PubMed] [Google Scholar]
- 11.Hirsch GA, Bottomley PA, Gerstenblith G, Weiss RG. Allopurinol acutely increases adenosine triphospate energy delivery in failing human hearts. J Am Coll Cardiol. 2012;59:802–808. doi: 10.1016/j.jacc.2011.10.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bottomley PA. John Wiley, editor. In: NMR spectroscopy of the human heart. Harris RKREW, editor. Encyclopedia of Magnetic Resonance; Chichester: 2009. [Google Scholar]
- 13.Gibbs C. The cytoplasmic phosphorylation potential. Its possible role in the control of myocardial respiration and cardiac contractility. J Mol Cell Cardiol. 1985;17:727–731. doi: 10.1016/s0022-2828(85)80034-1. [DOI] [PubMed] [Google Scholar]
- 14.Devereux RB, Alonso DR, Lutas EM, Gottlieb GJ, Campo E, Sachs I, Reichek N. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. 1986;57:450–458. doi: 10.1016/0002-9149(86)90771-x. [DOI] [PubMed] [Google Scholar]
- 15.Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, Gutgesell H, Reichek N, Sahn D, Schnittger I, et al. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr. 1989;2:358–367. doi: 10.1016/s0894-7317(89)80014-8. [DOI] [PubMed] [Google Scholar]
- 16.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18:1440–1463. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Dalen H, Thorstensen A, Aase SA, Ingul CB, Torp H, Vatten LJ, Stoylen A. Segmental and global longitudinal strain and strain rate based on echocardiography of 1266 healthy individuals: the HUNT study in Norway. Eur J Echocardiogr. 2010;11:176–183. doi: 10.1093/ejechocard/jep194. [DOI] [PubMed] [Google Scholar]
- 18.Nagueh SF, Appleton CP, Gillebert TC, Marino PN, Oh JK, Smiseth OA, Waggoner AD, Flachskampf FA, Pellikka PA, Evangelisa A. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. Eur J Echocardiogr. 2009;10:165–193. doi: 10.1093/ejechocard/jep007. [DOI] [PubMed] [Google Scholar]
- 19.Jung WI, Sieverding L, Breuer J, Hoess T, Widmaier S, Schmidt O, Bunse M, van Erckelens F, Apitz J, Lutz O, Dietze GJ. 31P NMR spectroscopy detects metabolic abnormalities in asymptomatic patients with hypertrophic cardiomyopathy. Circulation. 1998;97:2536–2542. doi: 10.1161/01.cir.97.25.2536. [DOI] [PubMed] [Google Scholar]
- 20.Cuda G, Fananapazir L, Epstein ND, Sellers JR. The in vitro motility activity of beta-cardiac myosin depends on the nature of the beta-myosin heavy chain gene mutation in hypertrophic cardiomyopathy. J Muscle Res Cell Motil. 1997;18:275–283. doi: 10.1023/a:1018613907574. [DOI] [PubMed] [Google Scholar]
- 21.Sweeney HL, Straceski AJ, Leinwand LA, Tikunov BA, Faust L. Heterologous expression of a cardiomyopathic myosin that is defective in its actin interaction. J Biol Chem. 1994;269:1603–1605. [PubMed] [Google Scholar]
- 22.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 23.Ingwall JS, Kramer MF, Fifer MA, Lorell BH, Shemin R, Grossman W, Allen PD. The creatine kinase system in normal and diseased human myocardium. N Engl J Med. 1985;313:1050–1054. doi: 10.1056/NEJM198510243131704. [DOI] [PubMed] [Google Scholar]
- 24.Wallimann T, Dolder M, Schlattner U, Eder M, Hornemann T, O’Gorman E, Ruck A, Brdiczka D. Some new aspects of creatine kinase (CK): compartmentation, structure, function and regulation for cellular and mitochondrial bioenergetics and physiology. Bio factors. 1998;8:229–234. doi: 10.1002/biof.5520080310. [DOI] [PubMed] [Google Scholar]
- 25.Abraham MR, Selivanov VA, Hodgson DM, Pucar D, Zingman LV, Wieringa B, Dzeja PP, Alekseev AE, Terzic A. Coupling of cell energetics with membrane metabolic sensing. Integrative signaling through creatine kinase phosphotransfer disrupted by M-CK gene knockout. J Biol Chem. 2002;277:24427–24434. doi: 10.1074/jbc.M201777200. [DOI] [PubMed] [Google Scholar]
- 26.Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J. 1992;281 (Pt 1):21–40. doi: 10.1042/bj2810021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009;81:412–419. doi: 10.1093/cvr/cvn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian R, Christe ME, Spindler M, Hopkins JC, Halow JM, Camacho SA, Ingwall JS. Role of MgADP in the development of diastolic dysfunction in the intact beating rat heart. J Clin Invest. 1997;99:745–751. doi: 10.1172/JCI119220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okada M, Mitsunami K, Inubushi T, Kinoshita M. Influence of aging or left ventricular hypertrophy on the human heart: contents of phosphorus metabolites measured by 31P MRS. Magn Reson Med. 1998;39:772–782. doi: 10.1002/mrm.1910390515. [DOI] [PubMed] [Google Scholar]
- 30.Jung WI, Hoess T, Bunse M, Widmaier S, Sieverding L, Breuer J, Apitz J, Schmidt O, van Erckelens F, Dietze GJ, Lutz O. Differences in cardiac energetics between patients with familial and non familial hypertrophic cardiomyopathy. Circulation. 2000;101:E121. doi: 10.1161/01.cir.101.12.e121. [DOI] [PubMed] [Google Scholar]