

Abstract
Ischemic retinopathies, such as diabetic retinopathy (DR), retinopathy of prematurity and retinal vein occlusion are a major cause of blindness in developed nations worldwide. Each of these conditions is associated with early neurovascular dysfunction. However, conventional therapies target clinically significant macula edema or neovascularization, which occur much later. Intraocular injections of anti-VEGF show promise in reducing retinal edema, but the effects are usually transient and the need for repeated injections increases the risk of intraocular infection. Laser photocoagulation can control pathological neovascularization, but may impair vision and in some patients the retinopathy continues to progress. Moreover, neither treatment targets early stage disease or promotes repair. This review examines the potential role of the ureahydrolase enzyme arginase as a therapeutic target for the treatment of ischemic retinopathy. Arginase metabolizes L-arginine to form proline, polyamines and glutamate. Excessive arginase activity reduces the L-arginine supply for nitric oxide synthase (NOS), causing it to become uncoupled and produce superoxide and less NO. Superoxide and NO react and form the toxic oxidant peroxynitrite. The catabolic products of polyamine oxidation and glutamate can induce more oxidative stress and DNA damage, both of which can cause cellular injury. Studies indicate that neurovascular injury during retinopathy is associated with increased arginase expression/activity, decreased NO, polyamine oxidation, formation of superoxide and peroxynitrite and dysfunction and injury of both vascular and neural cells. Furthermore, data indicate that the cytosolic isoform arginase I (AI) is involved in hyperglycemia-induced dysfunction and injury of vascular endothelial cells whereas the mitochondrial isoform arginase II (AII) is involved in neurovascular dysfunction and death following hyperoxia exposure. Thus, we postulate that activation of the arginase pathway causes neurovascular injury by uncoupling NOS and inducing polyamine oxidation and glutamate formation, thereby reducing NO and increasing oxidative stress, all of which contribute to the retinopathic process.
Keywords: arginase, diabetic retinopathy, retinopathy of prematurity, polyamine, oxidative stress, nitric oxide, peroxynitrite, superoxide
1. Introduction
1.1. Need for new therapies for ischemic retinopathy
Ischemic retinopathy develops in a variety of retinal disorders, including diabetic retinopathy, retinopathy of prematurity and retinal vein occlusion and is the most frequent cause of vision loss and blindness in the industrialized world. The retina is the most metabolically active tissue in the body and when retinal blood flow is insufficient to match the metabolic demands, retinopathy will occur. At the cellular level, the pathological process represents a destructive cycle involving neuronal depolarization, calcium influx and oxidative stress initiated by energy failure and increased glutamatergic stimulation. Diabetic retinopathy is the most frequent cause of blindness in working age adults in the US and affects over 40% of diabetic patients (NEI, 2008). By 2030, 366 million people are expected to have diabetes and over 146 million will have diabetic retinopathy. Retinal-vein occlusion is the second most common retinal vascular disease (Wong and Scott, 2010), with a prevalence of 1 to 2% in persons older than 40 years, affecting 16 million persons worldwide (Joussen et al., 2007). Retinopathy of prematurity (ROP) affects ~16,000 US infants yearly; many have lifelong vision impairment (Hartnett, 2010). Each of these conditions is associated with neuronal as well as vascular injury (Barber et al., 2011; Fulton et al., 2009; Osborne et al., 2004). So far laser photocoagulation is the only recommended treatment for advanced retinopathy. Laser treatment is usually effective, but can impair vision and in some patients the retinopathy continues to progress. Clinical trials of anti-VEGF intra-ocular injections show promise in reducing diabetic macular edema (Nicholson and Schachat, 2010). However, the beneficial effects are usually transient and the treatment does not promote tissue repair. Therefore, there is great need for new strategies and therapies to prevent retinal injury and enhance repair. The mechanisms underlying early vascular and neural injury during retinopathy are not understood and no treatments are available yet to prevent the damage. Understanding the mechanisms that underlie the initiation of vascular and neural injury in the ischemic retina is a critical step toward developing new therapies to reduce vision loss.
Our studies in models of retinopathy have revealed that the urea cycle enzyme arginase is critically involved in both vascular and neuronal injury. Arginase metabolizes L-arginine to form proline, polyamines and glutamate. Excessive arginase activity reduces the L-arginine supply for nitric oxide synthase (NOS), causing it to become uncoupled and produce superoxide and less nitric oxide (NO). Superoxide and NO react rapidly and form the toxic oxidant peroxynitrite. Glutamate and the catabolic products of polyamine oxidation can induce more oxidative stress and DNA damage, both of which are damaging to retinal cells. Our studies indicate that neurovascular dysfunction and injury during retinopathy are associated with increased arginase expression/activity, decreased NO, increased polyamine oxidation, oxidative stress and death of vascular and neural cells. Thus, we postulate that activation of the arginase/polyamine pathway plays an important role in neurovascular injury during retinopathy (Fig. 1). The scientific literature and experimental data supporting this concept will be developed in the subsequent sections.
Figure 1.

Working model for suggested mechanisms of arginase-induced retinal neurovascular dysfunction and death.
1.2. Actions of arginase
Arginase is a member of the ureohydrolase family of enzymes. In the liver, arginase catalyzes the final step in the urea cycle, in which the body disposes of toxic ammonia produced by protein catabolism. Specifically arginase converts L-arginine into urea and L-ornithine (Fig. 2). Arginase processes ammonia and ornithine into L-citrulline by activity of carbamoyl phosphate synthase-1 (CPS-1) and ornithine carbamoyl-transferase (OCT). L-citrulline can be recycled back to L-arginine by argininosuccinate synthase (ASS) and argininosuccinate lyase (ASL) to complete the urea cycle (Osowska et al., 2004). However, most non-hepatic tissues lack OCT or CPS-1 and therefore do not have the complete urea cycle.
Figure 2.

Scheme for synthesis of L-arginine from L-glutamine. Also shown are catabolism of L-arginine to L-ornithine/ urea or L-citrulline/NO, production of polyamines and anabolism and catabolism of proline. Abbreviations: ASL, aminosuccinate lyase; ASS, aminosuccinate synthase; Asp, aspartate; NOS, nitric oxide synthase; OAT; ornithine aminotransferase; ODC, ornithine decarboxylase; OCT, orthinine carbomoyltraferase; P5CS, pyrroline-5-carboxylate synthase.
When arginase activity is elevated it can compete with NOS for their common substrate, L-arginine. L-arginine is a semi-essential amino acid and when the supply needed for NOS activity is insufficient, NOS can become uncoupled (Romero et al., 2008; White et al., 2006). Uncoupled NOS will use more molecular oxygen to form superoxide which will react with any available NO to form peroxynitrite, leading to further decreases in NO (Fig. 3). These effects of arginase become pathologically significant when its activity and expression are elevated in diseases such as diabetes, hypertension and aging. In these conditions endothelial-dependent vasorelaxation is impaired and vascular smooth muscle cell proliferation and vascular and perivascular fibrosis can occur (Yang and Ming, 2006). Arginase activity and expression have been shown to be enhanced by inflammatory processes and reactive oxygen species associated with disease states (Morris, 2009; Munder, 2009). Superoxide, peroxynitrite and other reactive oxygen species increase arginase expression and activity (Chandra et al., 2012).
Figure 3.

Impact of the arginase pathway on NOS function and ROS formation
Synthesis of ornithine by arginase leads primarily to the formation of polyamines (putrescine, spermidine, and spermine) by ornithine decarboxylase (ODC) and to the formation of proline by ornithine aminotransferase (OAT). The actions of polyamines are not fully understood. However they are critically involved in cell cycling and proliferation. Polyamines play an important role in neural development, wound healing and tissue regeneration (Lange et al., 2004), but polyamine metabolism has been implicated in cell death under some conditions (Durante et al., 1998; Li et al., 2001). Proline is essential for the synthesis of collagen and for the organization of collagen into fibrous protein (Li et al., 2001). While polyamines and proline are needed for wound repair and structural integrity of fibrous tissue (Witte and Barbul, 2003), excessive activity of these pathways can lead to thickened and stiff blood vessels and airways, hypertrophied and fibrotic hearts and kidneys and growth of cancers. Furthermore, polyamine metabolism has been shown to be involved in the pathogenesis of ischemic brain damage (Ivanova et al., 2002; Takano et al., 2005; Wood et al., 2006). Amino aldehydes, acrolein and hydrogen peroxide, which are generated as byproducts during the oxidation of spermine and spermidine by polyamine oxidases (Fig. 4) have been found to be toxic during ischemic brain injury (Seiler, 2000).
Figure. 4.

Flow chart of the ornithine pathway showing formation of polyamines and glutamate and polyamine oxidation. Abbreviations: ODC; ornithine de-carboxylase, OAT; ornithine aminotransferase, SMO; spermine oxidase, APAO; N (1)-acetyl polyamine oxidase SSAT; spermine spermidine acetyl transferase.
1.3. Arginase isoforms
There are two isoforms of arginase: the cytosolic isoform, arginase I and the mitochondrial isoform, arginase II. Arginase I is strongly expressed in the liver, where it is the central player in the urea cycle (Morris, 2002). Arginase II is expressed primarily in extra-hepatic tissues, especially the kidney (Miyanaka et al., 1998), but is also found in other tissues including prostate and brain.
The molecular structure of arginase I and II is 58% homologous (Munder, 2009). Human arginase I consists of 322 amino acids (Dizikes et al., 1986) and arginase II has 354 amino acids (Gotoh et al., 1996). The gene for human arginase I is mapped on chromosome 6q23 (Sparkes et al., 1986) and that for arginase II is mapped on chromosome 14q24.1–24.3 (Gotoh et al., 1997). High-resolution crystallography has shown that each isoform consists of 3 identical subunits with an active site at the bottom of a 15 Ǻ deep cleft. Manganese ions, which are essential for enzyme activity, are located at the bottom of the cleft, separated by approximately 3.3 Ǻ and bridged by oxygen derived from 2 aspartic acid residues and a solvent-derived hydroxide. The metal bridging hydroxide is proposed to be the nucleophile that attacks the guanidinium carbon of L-arginine. The overall fold of each subunit belongs to the α/β family and consists of a parallel, 8 stranded β-sheet flanked on both sides by numerous α-helices (Ash, 2004).
1.4. Arginase in health
1.4.1. Ammonia detoxification
In the liver, the urea cycle removes excess ammonia accumulated from the diet or breakdown of endogenous protein. Various urea cycle enzymes convert ammonia to urea which is non-toxic, water-soluble and excreted by the kidney in the form of urine. Hyperammonemia results from deregulation of urea cycle enzymes, including arginase (Deignan et al., 2008) (Fig. 2). Patients with urea cycle disorders are healthy at birth but develop pathological symptoms due to the progressive accumulation of ammonia. Deficiency in arginase I, the last enzyme in urea cycle, causes a severe form of hyperammonemia. If left untreated, ammonia toxicity is manifested by seizures, mental disorders and early morbidity (Deignan et al., 2008). Lack of arginase I in mice has been associated with hyperammonemia, decerebrate posture, encephalopathy, tremors and death within 10–12 days after birth (Iyer et al., 2002). Arginase II knockout mice do not present any pathological abnormalities, but develop mild hypertension at 8 weeks of age due to increased sympathetic tone (Huynh et al., 2009a). Treatment of patients with arginase I deficiency involves reduction of protein intake, dietary supplementation with essential amino acids other than arginine, hemodialysis and in severe cases orthotropic liver transplant and gene replacement therapy (Deignan et al., 2008).
1.4.2. Wound healing
The acute phase of wound repair involves oxidative insult through activation of resident macrophages (M1 phase). These cells express high levels of NOX2 NADPH oxidase and inducible nitric oxide synthase (iNOS) and produce cytotoxic levels of superoxide and NO. Both are important for eradication of pathogens (Satriano, 2004). The acute phase is followed within 3–5 days by a repair phase (M2) wherein arginase is upregulated (Kampfer et al., 2003). Arginase converts L-arginine to L-ornithine and urea. L-ornithine is metabolized by OAT to form proline needed for collagen synthesis and by ODC to form polyamines which enhance cell proliferation. The balance between rate of consumption of L-arginine by iNOS (for NO production) and arginase (for synthesis of collagen and polyamines) determines the course of wound repair.
1.4.3. Neuroprotection
Arginase also has been implicated in neuroprotection and neural regeneration via production of polyamines which are important for neural growth, development, and regeneration (Esch et al., 1998; Filbin, 1996; Lange et al., 2004). Other work suggests that elevated arginase activity can promote axon regeneration. In one study, combination of nerve grafts and acidic fibroblast growth factor treatment led to improved locomotor function after spinal transaction (Kuo et al., 2007). This beneficial effect was associated with upregulation of arginase I expression and increased spermine formation within motor neurons and macrophages of the graft area. However, as is discussed in the sections that follow, activation of the arginase/polyamine pathway can be neurotoxic in some contexts.
1.5. Arginase in Disease
1.5.1. Brain disease and injury
Enhanced arginase activity and polyamine synthesis are not always neuroprotective. Alterations in expression of spermine, spermidine and putrescine have been reported in Alzheimer’s disease (Morrison and Kish, 1995) and Parkinson’s disease (Gomes-Trolin et al., 2002). Studies in a variety of models have implicated polyamines in the pathogenesis of ischemic brain damage (Ivanova et al., 2002; Takano et al., 2005; Wood et al., 2006). Amino aldehydes, acrolein and hydrogen peroxide are generated as byproducts during the oxidation of spermine and spermidine by polyamine oxidases and can be toxic during ischemic brain injury (Seiler, 2000). Studies in vitro and in vivo have also demonstrated key roles for reactive aldehydes in apoptotic and necrotic mechanisms leading to both neuronal and glial cell death (Ivanova et al., 1998; Kruman et al., 1997; McCracken et al., 2000; Ong et al., 2000). Polyamine oxidase and acrolein have been suggested as biomarkers for diagnosis of cerebral stroke (Tomitori et al., 2005; Yoshida et al., 2009) Polyamine catabolism was found to be enhanced in a rat model of traumatic brain injury (Zahedi et al. 2010). Acrolein has been shown to exert direct mitochondrial toxicity (Pocernich and Butterfield, 2003) while aminoaldehydes like 3-amino propanal act at the premitochondrial phase of apoptosis, via promoting lysosomal leakage or lysis (Yu et al., 2003; Yu et al., 2004). Polyamine regulated neurotoxicity is not well studied in retinopathy. However, elevated arginase activity and polyamine production have been linked to retinal ganglion cell death due to excessive activation of the excitotoxic NMDA receptors (Pernet et al., 2007). Recent data from our laboratory has linked altered polyamine metabolism to neuronal death in the mouse model of oxygen-induced retinopathy (Narayanan et al., unpublished) which will be discussed later in this article. Our ongoing studies in retinal models have implicated arginase II in ischemia-induced neurovascular degeneration in the retina (Narayanan et al., 2011). This work will be discussed in the sections that follow.
1.5.2. Diabetic vascular complications
Decreased levels of L-arginine have been reported in plasma of diabetic animals and patients (Hagenfeldt et al., 1989; Pieper and Dondlinger, 1997) and in vascular tissue of diabetic rats (Pieper and Dondlinger, 1997). Increased arginase activity seems to be involved in these conditions. In diabetic patients, arginase activity is also increased in red blood cells (Jiang et al., 2003). Our group has shown that increases in arginase activity and elevated expression of arginase I are involved in diabetes and high glucose-induced dysfunction of aorta, coronary and retinal arteries (Elms et al., 2013; Romero et al., 2012; Romero et al., 2008; Tawfik et al., 2006). Evidence supporting the role of arginase in retinopathy will be discussed below.
1.5.3. Hypertension
Elevated arginase activity has been reported in aorta, heart and lung of spontaneously hypertensive rats (SHR). Furthermore, treatment with the arginase inhibitor Nω-hydroxy-nor-L-arginine was shown to reduce systemic blood pressure, improve vascular function and reduce cardiac fibrosis in SHR (Bagnost et al., 2010). Pulmonary hypertension is also associated with increased arginase activity. Hypoxia-induced pulmonary hypertension is reported to involve increases in arginase II expression and activity (Chen et al., 2009; Jin et al., 2010). This elevation in arginase expression/activity is associated with decreased NO production and proliferation of pulmonary artery endothelial cells (Xu et al., 2004).
1.5.4. Sickle Cell Disease
In sickle cell disease, erythrocytes contain a mutant form of hemoglobin, hemoglobin-S. Cells with hemoglobin-S are rigid, sickle shaped and become entrapped in the microcirculation. This leads to repetitive cycles of ischemia-reperfusion injury and infarction (Bunn, 1997). Primary and secondary inflammation, oxidant stress, endothelial activation and expression of adhesion molecules contribute to this process. Sickle cell disease is characterized by low tissue levels of NO, slowing and clumping of red blood cells, hemolysis, vaso-occlusive disorders and pulmonary hypertension. Arginase activity and expression are substantially elevated in the plasma, red blood cells and platelets from these patients (Morris et al., 2008). Hemolysis releases large amounts of both free hemoglobin and arginase. Hemoglobin consumes endothelial NO, while arginase limits L-arginine availability to NOS, further reducing NO. Treatment of sickle cell patients with supplemental L-arginine or L-citrulline has been shown to reduce red blood cell sludging, decrease vaso-occlusion and prevent pulmonary hypertension (Fasipe et al., 2004; Morris et al., 2005; Waugh et al., 2001).
1.5.5. Erectile Dysfunction
Normal erectile function is mediated by NO-induced relaxation of smooth muscle cells in the corpus cavernosum (Masuda et al., 2004). Both neuronal NOS (nNOS) and endothelial NOS (eNOS) serve as sources of the NO. Arginase I and II are also expressed in corpus cavernosum. Inhibition of arginase enhances relaxation of cavernosal smooth muscle cells (Bivalacqua et al., 2001; Cox et al., 1999). Whereas NO production from the endothelium and nitrergic nerves regulates intrinsic tone, arginase activity in these tissues modulates tone by inhibiting NO production, presumably by competition with NOS for L-arginine. Both increased arginase activity and reduced NO bioavailability have been strongly associated with erectile dysfunction in many conditions including diabetes (Bivalacqua et al., 2001). Further, diabetic mice lacking the gene for arginase II do not exhibit the impaired nitrergic nerve and endothelial dependent relaxation of the corpus cavernosum observed in wild type diabetic mice (Toque et al., 2010).
1.5.6. Asthma
Allergic asthma is a chronic inflammatory disorder of the airways characterized by allergen-induced bronchoconstriction and inflammation. Decreased availability of L-arginine for NOS and reduced NO production appear to be centrally involved (Maarsingh et al., 2008b). Supplemental L-arginine or treatment with the arginase inhibitor N-ω-hydroxy-nor-l-arginine (nor-NOHA) reduces bronchoconstriction in response to allergens or other stimulants (de Boer et al., 2001) and enhances nitrergic nerve-mediated airway smooth muscle relaxation (Maarsingh et al., 2006). Both arginase I and II are constitutively expressed in airways tissues (Lindemann and Racke, 2003; Maarsingh et al., 2008a). Arginase activity and expression are elevated in asthmatic humans and animal models (Kenyon et al., 2008; Zimmermann et al., 2003). During asthma, chronic inflammation also leads to airway remodeling including thickening of basement membranes, fibrosis and enhanced smooth muscle mass. These changes can reduce lung function and are thought to involve elevated production of arginase products, polyamines and proline (Meurs et al., 2008).
1.5.7. Ischemia/reperfusion injury
Following ischemic insult, reperfusion is necessary to minimize tissue damage. However, ischemia/reperfusion (I/R) can cause further injury due to induction of reactive oxygen species and pro-inflammatory factors. Microvascular dysfunction can result due to decreases in NO. As was explained above, arginase competes with NOS for L-arginine, thereby uncoupling NOS which reduces NO production and increases oxidative stress. Arginase has been shown to be up-regulated during I/R injury in cardiac tissue and kidney as well as in liver (Hein et al., 2003; Jeyabalan et al., 2008; Jung et al., 2010; Reid et al., 2007). Treatment with the arginase inhibitor nor-NOHA has been reported to protect myocardium from I/R injury (Jung et al., 2010).
1.5.8. Atherosclerosis
The pathogenesis of atherosclerosis involves inflammation, vasoconstriction, and thrombus formation. Diabetes and hyperglycemia contribute significantly to atherosclerosis by increasing pro-inflammatory and pro-thrombotic factors that lead to plaque formation and increase the risk of thromboembolism (Retnakaran and Zinman, 2008). The central mechanism for endothelial dysfunction in atherosclerosis is thought to involve uncoupling of NOS, leading to decreased NO levels and increased production of reactive oxygen species (Ozaki et al., 2002; Yang and Ming, 2006). Vascular arginase activity has been shown to be increased with high cholesterol diet. This has been attributed to an increase in the expression of arginase II, as deletion of this gene prevents vascular dysfunction and oxidative stress (Ming et al., 2004; Ryoo et al., 2008). Arginase II has been reported to be increased in endothelial cells of atherosclerotic mice (Zhang et al., 2001) and in human aortic endothelial cells following exposure to oxidized LDL (Ryoo et al., 2006). Increasing NO bioavailability by arginase inhibition has been found to reduce atherosclerosis in a model of diabetes (Hayashi and Iguchi, 2010).
1.5.9. Nephropathy
Nitric oxide levels are significantly reduced in chronic and end stage kidney disease (Zharikov et al., 2008). One mechanism involves reduced availability of L-arginine for NOS. Arginase II is the predominant isoform in kidney and if elevated, it can reduce the L-arginine supply for NOS. Inhibition of arginase was found to protect the kidney from structural damage in a model of chronic nephropathy (Sabbatini et al., 2003). In models of nephrotoxic nephritis, both iNOS and arginase are elevated. Up-regulation of proline and polyamine production through the arginase II pathway has been implicated in glomerulonephritis (Waddington et al., 1998). Early stages of diabetes are characterized by hyperfiltration postulated to be due to increased L-arginine uptake and polyamine synthesis in the renal glomerulus (Schwartz et al., 2004). Both arginase I and II were found to be elevated in this model which could affect NO levels as well as increase polyamines through ornithine formation.
1.5.10. Cancer
Arginase plays a major role in tumor survival, growth and metastasis by several mechanisms and the effects can vary depending on tissue type. Arginase activity and arginase II expression have been shown to be increased in breast, colon and prostate cancers (Buga et al., 1998; Mumenthaler et al., 2008; Singh et al., 2000). Arginase increases ornithine levels which can enhance cell replication through polyamine synthesis. A reduction of iNOS and up-regulation of arginase in macrophages causes a phenotypic shift from a tumor-repressive to tumor-supportive microenvironment in which factors are released that promote tumor growth (Mills et al., 1992; Weigert and Brune, 2008). Indeed, elevated expression of arginase I has been reported in macrophages isolated from tumors (Davel et al., 2002; Kusmartsev and Gabrilovich, 2005; Sinha et al., 2005) as well as in tumor cells lines (Lechner et al., 2005). Knockdown of arginase II was found to cause apoptosis of thyroid cancer cells (de Sousa et al., 2010). In many tumor settings, enhanced arginase activity and resultant reduction of L-arginine levels impairs T cell-mediated immune function and promotes tumor growth. Inhibition of arginase can enhance T cell function and suppress tumor growth (Narita et al., 2012). On the other hand, a number of tumors develop an arginine auxotrophy (Delage et al., 2010). That is, they become dependent on extracellular L-arginine for growth, usually because of epigenetic down regulation of the enzyme which recycles L-citrulline to L-arginine – aminosuccinase synthase. Tumors of this type, including melanoma, mesothelioma and prostate and renal cancers, are very susceptible to the starvation-like stress responses – cell cycle arrest, apoptosis and autophagy - which occur with reductions in extracellular L-arginine levels. This understanding has become the basis of novel L-arginine depletion/lowering therapies with recombinant arginase or arginine deiminase (Glazer et al., 2011).
1.5.11. Aging
Age-related vascular changes have been linked to arginase activity in humans and animals. There is increasing evidence that upregulation of arginase contributes to impaired vascular endothelial function in aging (Berkowitz et al., 2003). Studies indicate that there is higher arginase activity, lower NO and higher superoxide production in old rats compared with young rats (Kim et al., 2009). Furthermore, acute inhibition of NOS and arginase can reduce superoxide production in old rats and prevent uncoupling of the eNOS dimer. Aging is also associated with accelerated senescence of vascular endothelial cells which can contribute to atherosclerosis as well as endothelial dysfunction. Indeed, knocking down arginase I (White et al., 2006) or increasing NO bioavailability by citrulline therapy (Hayashi and Iguchi, 2010) prevents aging-related endothelial senescence and improves vascular function. Increased arginase activity can also cause vessel stiffness with age due to excessive collagen deposition and fibrosis associated with activation of the proline and polyamine pathways.
1.6. Regulation of arginase expression and activity
Reactive oxygen species have been implicated in increasing arginase activity and expression. Studies have shown that hydrogen peroxide causes arginase activation in endothelial cells leading to decreased NO production which was restored by antioxidant treatment (Thengchaisri et al., 2006). In a model of wound healing, arginase effects in promoting excessive scar formation were mitigated by peroxynitrite inhibitors (Kapoor et al., 2004) Furthermore, peroxynitrite treatment increased arginase activity and mRNA and protein expression in coronary and aortic endothelial cells (Chandra et al., 2012). Reactive oxygen species can activate the janus kinases (JAKs) and signal transducers and activators of transcription (STATs) (Marrero et al., 1997; Schieffer et al., 2000). Activation of the JAK/STAT pathway has been shown to promote vascular smooth muscle proliferation, collagen deposition and fibrosis and to be involved in up-regulation of arginase (Wei et al., 2000). Studies in our lab have shown that treatment of endothelial cells with angiotensin II increases both arginase activity and arginase I protein expression (Shatanawi et al., 2011).
Enhanced arginase function can result from increases in specific activity/enzyme efficiency or protein expression levels. So far, little is known about the mechanisms underlying alterations in enzyme kinetics. It has been reported that NO produced by iNOS causes S-nitrosylation of arginase I which increases its affinity for L-arginine, thereby reducing the Km (Santhanam et al., 2008). Uric acid also has been reported to increase arginase activity by increasing its affinity for L-arginine. This effect is apparently operative for both isoforms (Zharikov et al., 2008).
There is extensive evidence indicating a role for protein expression levels in regulating enzyme activity. Levels of arginase I are increased in smooth muscle cells in response to interleukins and this effect is apparently mediated through cyclic AMP/protein kinase A and JAK/STAT6 pathways (Wei et al., 2000). In macrophages, cAMP has been shown to activate arginase I via activation of protein kinase A type I and histone deacetylase (Haffner et al., 2008). We and others have shown that RhoA and Rho kinase trigger upregulation of arginase activity/expression in atherosclerosis, inflammatory bowel disease and diabetes (Horowitz et al., 2007; Ming et al., 2004; Romero et al., 2008). RhoA-GDP is activated by Rho GEF (guanosine nucleotide exchange factor), which promotes exchange of GDP for GTP. Active RhoA-GTP stimulates ROCK to trigger downstream signaling (Loirand et al., 2008). Reactive oxygen species have been implicated in the enhancement of arginase activity (Boor et al., 2007; Horowitz et al., 2007; Ming et al., 2004; Romero et al., 2008; Zhang et al., 2009a). We have shown that oxidative radicals can increase arginase activity/expression in endothelial cells through a pathway involving protein kinase C (PKC) activated p115-Rho GEF and subsequent activation of RhoA/Rho kinase (Chandra et al., 2012).
Active RhoA has also been indicated as an upstream activator of mitogen activated protein kinase (MAPK) family members, including p38 MAPK (Guo et al., 2009; Lovett et al., 2006). Activation of p38 MAPK has been implicated in increasing arginase I activity and expression in macrophages (Chang et al., 2000; Stempin et al., 2004). Studies also indicate a role of p38 MAPK in increasing arginase activity/expression in endothelial cells upon exposure to angiotensin II and reactive oxygen species by a mechanism involving activation of RhoA/ROCK (Chandra et al., 2012; Shatanawi et al., 2011). These signal transduction intermediates may be considered targets for the therapeutic limitation of arginase activity.
1.7. Arginase inhibitors
Several inhibitors of arginase are commercially available. The first class of inhibitors to be developed had non-specific actions and many side effects owing to the high concentrations required (Morris, 2009). For example, norvaline is a substrate for amidotransferases (Davoodi et al., 1998). Similarly, while Nω-hydroxy-L-arginine (NOHA) is a potent inhibitor for arginase, it is also a precursor for NO synthesis from NOS. Nor-NOHA is also a potent arginase inhibitor and it neither inhibits NOS nor is it an intermediate in NO synthesis. It has a much longer half-life than NOHA. α-Difluoromethylornithine (DFMO) is a non-specific weak inhibitor of arginase, but is a potent inhibitor for ornithine decarboxylase (Morris, 2009). Thus, the effects of DFMO in increasing NO production are likely due to increased accumulation of ornithine which is known to inhibit arginase. Besides producing urea, arginase is also involved in synthesis of polyamines and amino acids such as ornithine, proline and glutamate. In fact, ornithine, leucine, valine, lysine, isoleucine and nor-valine inhibit arginase with ornithine being the most potent (Huynh et al., 2009b). Also, L-citrulline is an allosteric inhibitor of arginase (Shearer et al., 1997), but it also increases NO formation. Recently, competitive inhibitors of arginase have been developed which have greater specificity for the enzyme. Boronic acid analogs of L-arginine [S-(2-boronoethyl)-L-cysteine (BEC) and 2(S)-amino-6-boronohexanoic acid (ABH)] are highly selective arginase inhibitors. Both compounds contain a N-hydroxyguanidinium or boronic acid head which binds the manganese cluster in arginase (Berkowitz et al., 2003).
Although several specific arginase inhibitors have been developed and investigated, none are isoform specific. RNA interference has been used to inhibit expression of specific isoform. For example, administration of short hairpin RNA (shRNA) against arginase I greatly reduced IL-13 induced airway hyper-responsiveness with a concomitant decrease in arginase I mRNA and protein levels in the lungs of mice (Yang et al., 2006). Direct delivery of anti-sense arginase I using adeno-virus vector in corpus cavernosum improved erectile function in aged mice (Bivalacqua et al., 2007). Anti-sense arginase I also increased NO production in endothelial cells exposed to high glucose (Romero et al., 2008).
2. Arginase and retinopathy
Surprisingly, in spite of the extensive literature demonstrating involvement of the arginase pathway in disease, relatively little is known about its role in retinopathy. A PubMed search cross referencing arginase with retina, retinal disease or retinopathy revealed only 19 papers published since 1974. Many of these are descriptive reports on the distribution of the enzymes of arginine metabolism in ocular structures. However, one paper describes the coinduction of iNOS and arginine metabolic enzymes in rats with endotoxin-induced uveitis (Koga et al., 2002). Another reports on a role for L-ornithine metabolites (polyamines) in excitotoxic cell death (Pernet et al., 2007). In both reports, arginase I was increased and localized mainly to Müller cells. Polyamines have been shown to be toxic for microglial cells, but not for astrocytes or Müller cells (Pernet et al., 2007; Takano et al., 2005a; Takano et al., 2005b).
In contrast with the relatively limited literature on the role of arginase in retinopathy, numerous works have addressed the role of L-ornithine and its metabolites in retinal disease and injury conditions. As has been explained above, L-ornithine is metabolized by ornithine decarboxylase (ODC) to the polyamine putrescine, which is further metabolized to spermine and spermidine (Fig. 2). Polyamines regulate cell cycle progression and growth and have been shown to promote the growth of a variety of cell types, including endothelial cells, vascular smooth muscle cells, glial cells and tumor cells (Bachetti et al., 2004; Durante et al., 2007; Jeglinski et al., 1996; Shantz and Levin, 2007). Analyses of vitreous samples from patients with proliferative retinopathy have revealed dramatic increases of the level of spermidine and spermine suggesting their involvement in retinopathy (Nicoletti et al., 2003). In support of this concept, studies have shown that polyamine antagonists block both retinal and choroidal angiogenesis (Lima e Silva et al., 2006; Lima e Silva et al., 2005).
In addition to promoting abnormal cell growth, alterations in the L-ornithine pathway have been demonstrated to play a role in gyrate atrophy of the retina and choroid. Gyrate atrophy is a rare, autosomal recessive disease that occurs due to mutations in the ornithine aminotransferase (OAT) gene. OAT is localized within mitochondria, where it converts L-ornithine into pyrroline-5-carboxylate (P5C). P5C is further metabolized to L-proline. The OAT gene mutation results in a decrease in OAT activity, leading to excessive accumulation of L-ornithine which is toxic to RPE cells [for review, please see (Hayasaka et al., 2011)]. Fundus examination of patients with gyrate atrophy shows retinal lesions similar to that seen in age-related macular degeneration with chorioretinal atrophy that becomes larger with age (Takki, 1974). The primary site of the degeneration is thought to be at the level of the retinal pigment epithelium (RPE) and choriocapillaris.
Polyamine metabolism has also been found to have a role in retinal microvascular function. Puro and colleagues have shown that ATP-sensitive potassium (KATP) channels in retinal capillaries are critically important for the responses of the retinal microvasculature to hypoxia in that activation of these channels generates the hyperpolarization which produces a vasodilator response to adenosine (Ishizaki et al., 2009; Li and Puro, 2001). Adenosine is released by metabolically compromised retinal cells and is believed to provide protective effects during ischemia (Roth et al., 1997). The Puro group’s study of isolated retinal capillary/arteriolar segments showed that KATP currents are generated primarily in the capillary portion of the segment (Ishizaki et al., 2009). Further, they showed that these capillary channels are redox-sensitive and their function depends on cellular production of oxidants during the catabolism of polyamines. Thus, the vasodilator response to adenosine in the retinal capillaries depends on polyamine-driven oxidation within the endothelial cells. On the other hand, in the retinal arterioles spermine inhibits potassium efflux via inwardly rectifying potassium channels (Matsushita and Puro, 2006). This process limits vasodilator responses and is enhanced in microvessels isolated from the diabetic retina. Consistent with the suggested role of spermine in diabetes-induced vascular dysfunction, its concentration has been shown to be elevated in the diabetic eye (Nicoletti et al., 2003).
Thus, there is some evidence supporting a role for the arginase/ornithine pathway in retinal inflammation, neurotoxicity, pathological angiogenesis, gyrate atrophy and retinal vascular dysfunction. The following sections will present additional evidence for the specific involvement of arginase and the arginase/ornithine pathway in models of diabetic retinopathy and retinopathy of prematurity.
3. Arginase and diabetic retinopathy
3.1. Clinical features of diabetic retinopathy
The clinical progression of diabetic retinopathy follows a pattern characteristic of ischemic retinopathy [for review, see (Aiello et al., 1998; Klein and Klein, 1997)]. The disease begins with biochemical and cellular alterations that are not clinically evident. Vascular alterations during this period include leukocyte attachment to the vessel wall, alterations in blood flow, and death of retinal pericytes. With time, the vascular endothelial cells are lost, leading to capillary closure and formation of acellular, non-perfused vessels. Loosening of the endothelial cell-cell junctions results in subtle increases in vascular permeability. Blockage of the retinal capillaries causes focal hypoxia, leading to increased tissue production of angiogenic factors. The permeability increase and growth factor release set the stage for pathological neovascularization. As the disease progresses, obvious alterations in vascular structure, including microaneurysms, dot/blot hemorrhage, cotton-wool spots, venous beading and vascular loops, become clinically evident. At later stages the vascular leakage is prominent, allowing blood and fluid to accumulate within the retinal tissue, forming exudate deposits. The retinal edema causes reduced vision when the macula is affected, but is otherwise asymptomatic. The condition of macular edema is also referred to as non-proliferative retinopathy. In many patients, the retinopathy progresses to proliferative retinopathy, characterized by neovascular growth at the vitreo-retinal interface. The walls of the new blood vessels are easily broken and blood may leak out. This can cloud the vitreous and compromise vision. In advanced stages of proliferative retinopathy, newly formed fibro-vascular tissue grows from the retinal surface into the vitreous cavity. This can cause retinal detachment and blindness, if untreated. Proliferative retinopathy is more commonly seen in Type I diabetes, whereas non-proliferative retinopathy with macular edema is more common in Type II diabetes.
Along with the vascular pathology, neuronal dysfunction and injury are evident early in the course of diabetes. Dysfunction of the blue light sensitive S-cone photoreceptors has been observed prior to the development of clinical signs of retinopathy (Yamamoto et al., 1996). In humans with diabetic retinopathy, the S-cones selectively die, which may contribute to the color vision deficit (Cho et al., 2000). Other signs of photoreceptor dysfunction, including defects in dark adaptation and alterations in the multifocal electroretinogram (ERG) have been reported in patients with no clinical signs of retinopathy (Harrison et al., 2011; Jackson et al., 2012). Death of retinal ganglion cells has been reported in both humans and animal models of diabetes (Barber et al., 1998; Hammes et al., 1995). Other signs of ganglion cell dysfunction include decreases in contrast sensitivity (Gualtieri et al., 2011), decreased amplitude of the pattern ERG (Prager et al., 1990), decreased amplitude of the scotopic threshold of the full field ERG (Aylward, 1989) and thinning of the nerve fiber and ganglion cell layers as shown by optical coherence tomography (Park et al., 2011).
3.2 Pathophysiology of diabetic retinopathy
Types I and II diabetes differ in the clinical manifestation of their retinal complications. However, both types are characterized by early signs of neurovascular dysfunction and injury. Moreover, both have the same pathophysiological basis in that the occurrence and progression of complications is closely correlated with the degree of hyperglycemia. Clinical trials have shown that intensive control of blood glucose delays the onset and reduces the severity of the retinal complications in both type I and type II diabetes (DCCT, 1993; UKPDS, 1998). Furthermore, the beneficial effects of intensive blood glucose control achieved by early intervention can persist for several years, whereas the damaging effects of poor control persist beyond the point when glycemic control had been achieved. This phenomenon has also been termed “metabolic memory” [for review, please see (Ceriello, 2012)].
The molecular mechanisms by which high glucose triggers retinal neurovascular injury and dysfunction are not fully understood. However, increased reactive oxygen species formation is clearly involved. During diabetes, oxidative stress occurs in response to formation of advanced glycation end products, glucose auto-oxidation (Vlassara and Palace, 2002), activation of protein kinase C (Curtis and Scholfield, 2004; Inoguchi et al., 2003) and decreased activity of antioxidant enzymes (Bhatia et al., 2003; Merzouk et al., 2003). Activation of each of these pathways is thought to be initiated by overproduction of superoxide by the mitochondrial electron-transport chain (Nishikawa et al., 2000). Studies have shown that the mitochondrial antioxidant defense systems are also compromised during diabetes. Activities of MnSOD, glutathione peroxidase and catalase are all decreased and the levels of glutathione are below normal (Santos et al., 2011). In addition to these mitochondrial alterations, the superoxide generating enzyme NADPH oxidase is an important source of reactive oxygen species in diabetic patients and experimental models (Ellis et al., 2000; Griendling et al., 2000; Inoguchi et al., 2003; Sonta et al., 2004). Furthermore, NADPH oxidase-dependent formation of peroxynitrite has been shown to contribute to endothelial dysfunction by causing mitochondrial injury and activation of mitochondrial oxidase (Doughan et al., 2008; Guzik and Harrison, 2006). Thus, mitochondria can be a target as well as a source of reactive oxygen species.
Our studies in models of diabetic retinopathy, acute retinal inflammation and ischemic-reperfusion injury have shown that activation of the NOX2 NADPH oxidase is critically involved in the initiation of retinopathy. Moreover NOX2 deletion or inhibition reduces signs of retinal inflammation and vascular and neuronal injury, implying a key role of NOX2-derived superoxide in the pathology (Al-Shabrawey et al., 2008a; Al-Shabrawey et al., 2005; Al-Shabrawey et al., 2008b; Yokota et al., 2011; Zhang et al., 2009a; Zhang et al., 2009b).
Under normal conditions superoxide levels are controlled by superoxide dismutase (SOD). However superoxide reacts with NO to produce peroxynitrite at a rate three times faster than it can react with SOD. Under conditions of oxidative stress, this will lead to a net decrease in NO with an increase in peroxynitrite (Beckman et al., 1990; Darley-Usmar et al., 1995). Peroxynitrite is a major contributor to cellular injury either through direct oxidative reactions with lipids, DNA and proteins or indirect, radical-mediated mechanisms (Pacher et al., 2007). Protein tyrosine nitration is now recognized as a biomarker for peroxynitrite formation and has been linked to vascular injury and dysfunction in multiple diseases, including diabetic retinopathy (El-Remessy, 2003; Pacher et al., 2007). Inhibiting nitric oxide synthase (NOS) has been shown to prevent diabetes-induced vascular dysfunction (Du et al., 2002; El-Remessy, 2003; Kowluru et al., 2000), implying that reactive nitrogen species play a key role in the pathology.
3.3. Involvement of arginase in diabetic retinopathy
3.3.1. Retinal inflammation
As has been noted above, diabetic retinopathy is characterized by increases in oxidative stress and inflammatory reactions, together with blood flow alterations, breakdown of the blood-retinal barrier and death of vascular cells. Diabetes-induced decreases in retinal blood flow have been observed in both humans and animal models (Clermont and Bursell, 2007; Curtis et al., 2009; Nagaoka et al., 2010). This is proposed to be mediated by numerous mechanisms, but decreased NO formation due to dysfunction of the vascular endothelium is very likely involved. The requirement of adequate levels of NO for maintaining blood flow is well established (De Caterina et al., 1995; Furchgott and Zawadzki, 1980; Gruetter et al., 1979). NO-induced activation of guanylyl cyclase within the vascular smooth muscle cells dilates the vessels, increasing blood flow. Adequate production of NO by eNOS is necessary for the maintenance of healthy vessels, proper blood flow, prevention of leukocyte adhesion and platelet aggregation and control of smooth muscle cell growth (Cooke et al., 1991; Furchgott and Zawadzki, 1980; Gruetter et al., 1979). The role of endothelial cell dysfunction and decreased NO in diabetes-induced peripheral vascular dysfunction is well established (Cohen and Tong, 2010; Guzik et al., 2002; Lopez-Lopez et al., 2008). As explained above, NOS requires an adequate supply of L-arginine to produce NO. When L-arginine is limited, the NOS homodimer becomes uncoupled, causing it to produce superoxide (Xia and Zweier, 1997). Superoxide will react with NO to form peroxynitrite (ONOO−), further reducing NO and increasing oxidative stress (Pacher et al., 2007).
Our studies have demonstrated increases in arginase activity in retinas of mice and rats with streptozotocin-induced diabetes, retinal endothelial cells treated with high glucose, retinas of mice with endotoxin-induced retinal inflammation, and retinal glia and microglia treated with endotoxin (Elms et al., 2013; Patel et al., In Press; Zhang et al., 2009a). These studies have shown that arginase I is expressed in retinal neurons, vascular cells, glia and microglia (Fig. 5). Our studies in mice with endotoxin-induced acute retinal inflammation have shown that the increases in arginase activity and arginase I expression are associated with increased formation of superoxide, increased expression of inflammatory genes and attachment of leukocytes to the retinal vessels (Fig. 6). These alterations were prevented in double knockout mice lacking one copy of arginase I (AI) and both copies of arginase II (AI+/−AII−/−). Furthermore the endotoxin-induced increase in arginase I expression was blunted by blockade of NADPH oxidase or deletion of NOX2. Thus endotoxin induced retinal inflammation is mediated by NADPH oxidase-dependent increases in arginase. Similar results have been observed in streptozotocin-induced diabetic mice and high glucose-treated retinal endothelial cells (Patel et al., In Press). Moreover, the diabetes-induced increases in oxidative stress and retinal inflammation were found to be associated with a significant decrease in levels of bioavailable NO.
Figure 5.

Immunoreactivity for arginase I (A) and arginase activity (B) are increased in the diabetic retina. Arginase I is also increased in high glucose treated retinal endothelial cells (C).
Figure 6.

Increases in arginase I are associated with increased GFAP expression in Müller glia during endotoxin-induced retinal inflammation (A). DHE imaging shows marked increase in superoxide reaction in retinas of the endotoxin-treated wild type mice as compared with wild type control mice and the endotoxin-treaed AI+/−AII−/− mice (B). Lectin labeling shows numerous attached leukocytes in the endotoxin-treated wild type mouse retina. This effect is prevented in AI+/− AII−/− retinas or by treatment with the arginase inhibitor BEC (C) (Zhang et al., 2009a).
3.3.2. Retinal vascular dysfunction
In order to assess the impact of overactive arginase on retinal blood flow, we developed a protocol for using the Micron III Fundus Microscope Imaging System to measure vasodilation in responses to the endothelial-dependent vasodilator acetylcholine (ACh) and the endothelial-independent vasodilator sodium nitroprusside (SNP). Because our work comparing arginase I hemizygous knockout mice wild type for arginase II (AI+/−) with mice deficient in arginase II and wild type for arginase I (AII−/−) has shown that diabetes- and high glucose-induced dysfunction of aortic endothelial cells is mediated by arginase I and not arginase II (Romero et al., 2012), we conducted studies to assess on the role of arginase I in diabetes-induced retinal vascular dysfunction (Elms et al., 2013). These experiments used diabetic and control wild type mice and knockout mice lacking one copy of arginase I (AI+/−). We used heterozygous arginase I knockout mice for these studies because homozygous deletion of arginase I is lethal around 10 days postnatal due to hyperammonemia. Retinal arteries were selected based on morphology and branching patterns and diameter was measured at a location two optic disc diameters from the nerve head (Fig. 7). Measurement of vessel diameters prior to the infusion showed that neither diabetes nor mouse genotype had any effect on vessel size. Infusion of vehicle alone had no effect on the retinal vessels. Retinal vascular dilation in response to ACh was significantly reduced in the wild type diabetic mice (Fig. 8A) as compared to the normoglycemic controls. The diabetes-induced impairment of the vasodilatory response to ACh was significantly blunted in the AI+/− diabetic mice, implying the involvement of arginase I in the diabetes-induced vascular dysfunction. Vasodilation to the endothelial-independent vasodilator sodium nitroprusside was similar in all groups indicating that NO-mediated relaxation of the smooth muscle cells was not affected by diabetes. Control studies measuring blood pressure and heart rate showed no differences among the groups, indicating that the effects of diabetes on retinal vasodilation are not due to changes in blood pressure or heart rate.
Figure 7.

Measurement of endothelial vasodilation of secondary retinal arterioles using Micron III fundus microscope. B. Vessel diameter at baseline is equal in all groups (Elms et al., 2013).
Figure 8.
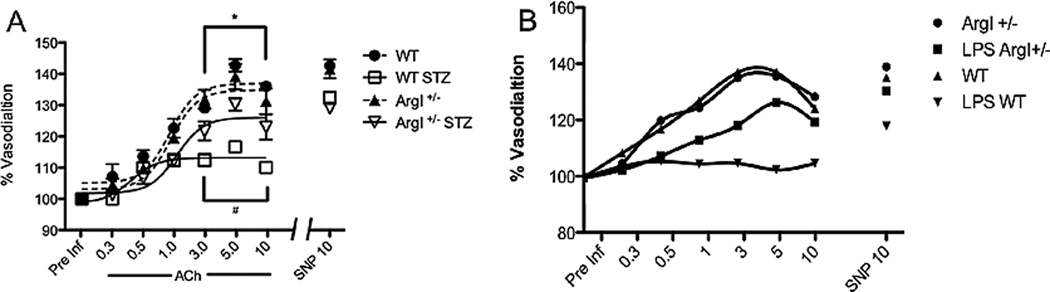
Vasodilation to ACh is markedly impaired in mice with streptozotocin (STZ)-induced diabetes (A, Elms et al., 2013) or endotoxin-induced retinal inflammation (B). The impairment is attenuated in mice lacking one copy of arginase I. Vasorelaxation to SNP is not altered in either model.
Parallel studies using the specific arginase inhibitor ABH confirmed the involvement of arginase in the diabetes-induced vascular dysfunction (Elms et al., 2013). The ABH treatment reduced retinal arginase activity to a level comparable with wild type and AI+/− non-diabetic control. The ABH-induced reduction in arginase activity was accompanied by a significant improvement in ACh-mediated endothelium dependent vasodilation in wild type diabetic mice as compared to the normal control mice. These beneficial effects of arginase blockade on retinal vascular reactivity were confirmed by ex vivo studies using pressure myography on central retinal arteries isolated from streptozotocin diabetic rats. Vessels from diabetic rats showed significant impairment of endothelial-dependent vasodilation. The vasorelaxation responses were partially restored by pre-treatment of the isolated vessels with the arginase inhibitor BEC. Parallel studies in mice with endotoxin-induced retinal inflammation showed a prominent impairment of endothelial-dependent vasodilation that was largely prevented by hemizygous deletion of arginase I (Fig. 8B). These experiments indicate that upregulation of arginase I is necessary for diabetes or inflammation-induced retinal vascular dysfunction. To directly assess whether increases in arginase I expression are sufficient to induce endothelial cell-dependent vascular dysfunction, we transduced retinal endothelial cells or explants with adenovirus carrying arginase I, empty vector containing red fluorescent protein (RFP) or an inactive mutant of arginase I (D128G). The data showed that overexpression of arginase I causes a prominent increase in arginase activity that is accompanied by decreased NO release and significant impairment of endothelial cell-dependent vasodilation (Fig. 9).
Figure 9.

Ex vivo transduction of rat retinal arteries with AV vector carrying arginase I increases arginase I protein levels and arginase activity (A) and decreases EC-dependent vasorelaxation (B) compared with controls carrying RFP alone or inactive A1 (D128G) *= p<0.01, n=3. Transduction of retinal endothelial cells with arginase I also reduces NO release in response to treatment with the calcium ionophore thapsigargan (C). * = p<0.05 vs RFR and D1286
4. Arginase and retinopathy of prematurity
4.1. Clinical features of retinopathy of prematurity
Retinopathy of prematurity (ROP) is a devastating disease in premature infants and a major cause of childhood vision impairment. Approximately 68% of children born weighing less than 1.250 Kg are at risk of developing ROP (Good and Gendron, 2005) and around 50% of these children have long term visual problems (O'Connor et al., 2002). Despite the improvements in neonatal care, the incidence of ROP does not decline, probably due to increased survival of low birth weight, premature infants. From the clinical perspective, ROP is considered to be a vascular disease and current treatments target abnormal retinal angiogenesis. However, despite effective treatment of the vascular injury, many children have impaired vision suggesting disruption of neuronal development as well as vascular injury. The current treatments for ROP are cryotherapy and laser photocoagulation, which are intended to normalize the pathological blood vessels by destroying the ischemic tissue and reducing metabolic demand. However, these approaches only partially reduce the incidence of blindness in infants with severe late stage disease effects. Since the development of laser surgery there has been little progress in the treatment of ROP. Anti-VEGF therapy is being tested for patients with severe ROP (Mintz-Hittner et al., 2011). However, long term effects of inhibiting VEGF in the developing retina are a significant concern. Given that VEGF has potent neurotrophic activity, blocking its function may further impair retinal neuronal function.
4.2. Pathophysiology of retinopathy of prematurity
ROP is a complex condition of the developing retina, in which various factors participate at different stages of the disease leading to neurovascular degeneration followed by neovascularization and glial injury. In humans, the retinal vessels begin to form around 16 weeks of gestation in a central to peripheral wave at a rate of about 0.1 mm/day (Ashton et al., 1970). Since the retinas of premature infants are not completely vascularized, the severity of ROP is directly proportional to the degree of prematurity (Rivera et al., 2011). ROP develops in two phases. When a premature infant is exposed to conditions of relative hyperoxia in the extrauterine environment and when supplemental oxygen must be administered to support the immature respiratory system, maturation of the developing vessels is interrupted, resulting in obliteration of developing retinal capillaries. This leaves the peripheral retina avascular. The neural retina continues to develop during this period. Thus, when the newborn experiences relative normoxia at a later stage of development, retinal hypoxia occurs, causing pathological angiogenesis in the avascular retina (Fletcher et al., 2010). During this hypoxic phase, an overproduction of growth factors, particularly VEGF, and growth hormones occurs, resulting in growth, proliferation and migration of endothelial cells (Pollan, 2009).
Considerable evidence suggests that ROP is not only a vascular disorder, but is associated with functional visual deficits as well. Visual acuity is reduced in patients with ROP, even in regressed or mild forms of ROP (O'Connor et al., 2002). It has been shown that the age of onset of ROP coincides with the time of photoreceptor development. Studies have reported that rod photoreceptors are affected in ROP patients (Barnaby et al., 2007; Fulton and Hansen, 1996; Fulton et al., 2001). Deficits in rod and rod-bipolar cell sensitivity have been observed several years after ROP has resolved (Fulton et al., 2001; Hansen and Fulton, 2000).
4.3. Animal models of retinopathy of prematurity
A widely used animal model of ROP is the oxygen induced retinopathy (OIR) model, generated by exposing neonates to hyperoxia for certain periods and then returning them to normoxia conditions. Since ROP has been considered as a vascular disease, pathological angiogenesis in the retina was considered as the integral response in the development of animal models of ROP. This model was developed first in the kitten (Ashton et al., 1954) and later was extended to other species such as puppy (McLeod et al., 1998), rat (Penn et al., 1993), mouse (Smith et al., 1994) and zebrafish (Cao et al., 2008). These different animal models follow the same basic paradigm in which newborns are exposed to hyperoxia during early retinal development, causing an arrest/delay in vascular development. Later, when the animals are returned to normoxia, their retinas undergo a relative hypoxic situation. This ischemic situation results in uncontrolled neovascularization. The neovascularization occurs at the junction between the vascularized and nonvascularized retina and may be followed by normalization of the vascular pattern with regression of the abnormal vessels (Smith et al., 1994). Retinal detachment which can be seen in late stage of ROP in the puppy OIR model, but not in the mouse, rat or kitten models (Ashton et al., 1954; Madan and Penn, 2003; McLeod et al., 1998; Smith et al., 1994).
The most widely used OIR models are the mouse model developed by Smith and colleagues (Smith et al., 1994) and the rat model developed by Penn and colleagues (Penn et al., 1993). The major advantages of the mouse OIR model is the ease of genetic manipulation, the reproducibility of the phenotype, relative cost, small size of the animals and reliable methods for quantifying retinal neovascularization (Aguilar et al., 2008; Smith et al., 1994). The main disadvantage of this OIR model is the pattern of vaso-obliteration in the retina, where the central retinal vessels, rather than the peripheral vessels, are obliterated during hyperoxia treatment. This is different from what happens in ROP patients where the peripheral retinal vessels fail to develop. Whereas the mouse model utilizes exposure to the constant hyperoxia for 5 days before returning to the room air, the rat OIR model uses alternating hyperoxia-hypoxia cycles. The main advantage of the rat OIR model is its similarity to the human ROP, where the peripheral blood vessels become obliterated upon hyperoxia exposure (Madan and Penn, 2003). Removal from oxygen and exposure to normoxia results in pathological neovascularization between vascular and avascular areas in the mid-peripheral retina.
The mouse OIR model has been extensively used by various laboratories. The use of this model has been extended to the general study of ischemic retinopathies and related antiangiogenic treatments. Apart from neovascularization, retinal degeneration (Downie et al., 2007; Sennlaub et al., 2001; Sennlaub et al., 2002) and glial activation (Downie et al., 2007; Gu et al., 2002) have also been demonstrated in mouse OIR model. Figure 10 summarizes the experimental protocol developed by Smith et al (1994) and the parameters we have investigated using this model. Briefly, litters of mice with the nursing dam are placed in 70% oxygen on P7, returned to normal atmosphere on P12 and maintained for various periods until euthanasia and tissue harvesting. We have modified the protocol by reducing the oxygen concentration to 70% because our adult mutant mice are relatively sensitive to the hyperoxia treatment.
Figure 10.

Diagram showing scheme for oxygen treatment of the mouse model for OIR.
4.4. Involvement of arginase in retinopathy of prematurity
So far there is no direct clinical evidence to implicate excessive arginase activity in ROP. However, premature infants undergoing intensive care are frequently deprived of both arginine and glutamine (Neu, 2003; Wu et al., 2004). Both arginine and glutamine are semi-essential in that they can be produced by the body under normal conditions. However, premature infants are highly vulnerable to glutamine and arginine deprivation because they are not able to maintain endogenous synthesis of these amino acids. A randomized trial of arginine supplementation in low-birth-weight infants demonstrated a decreased incidence of necrotizing enterocolitis (Amin et al., 2002). The authors speculated that this effect was a result of improved blood flow to the intestinal microvasculature due to increased NO production through the L-arginine–NOS pathway. While the effects of L-arginine on ROP are as yet unknown, studies in the mouse OIR model have shown a beneficial effect of supplemental treatment with the dipeptide arginyl- glutamine in reducing retinal neovascularization (Neu et al., 2006). Given that both L-glutamine and L-arginine are metabolized in the arginase pathway and in view of the well established action of arginase in reducing L-arginine availability to NOS in other tissues, it is likely that arginase also plays a role in the development and progression of ROP. We are examining this hypothesis by studies in the mouse model for OIR.
4.5. Involvement of arginase in oxygen-induced retinopathy
4.5.1. Arginase and neurodegeneration
NOS signaling has been shown to be involved in apoptosis of retinal cells during retinal ischemia related disorders (Neufeld et al., 2002; Sennlaub et al., 2002). However, not much literature is available on the role of arginase in ROP. Work from our laboratory is the first report to show a role for arginase II in neuroglial injury and retinal neuronal dysfunction in ROP (Narayanan et al., 2011). Previous studies had suggested that arginase II might have a beneficial role in limiting retinal injury in the mouse OIR model (Stevenson et al., 2010). In this study, the authors used the Smith model for OIR in which mice were placed in hyperoxia on P7 and then returned to room air on P12. The retinas were collected for analysis at various times during the period of relative hypoxia. The results demonstrated improved vascular recovery and increased astrocyte survival in TNFα-deficient knockout mice. The authors found that the TNFα-deficient OIR retinas had lower activity of iNOS, increased arginase II mRNA levels and higher arginase activity relative to the wild type OIR retinas. Based on these observations and the known action of arginase in limiting NO formation at the end of the M1 phase of the inflammatory reaction, they speculated that the vascular protection could be mediated through an anti-inflammatory action of arginase in limiting by iNOS activity and reducing NO, superoxide and peroxynitrite formation.
Vascular abnormality characterized by vaso-obliteration and pathological angiogenesis is the most recognized characteristic of ROP. However the vascular pathology represents only one facet of the disorder. ROP causes retinal neuronal degeneration and impaired retinal function as well. Sennlaub and colleagues (Sennlaub et al., 2002) provided the first evidence for retinal neuronal apoptosis and degeneration in the mouse model of OIR. At P17 in that study, a significant thinning of the retina along with reduced thickness in the inner nuclear (INL) and inner plexiform layers (IPL) was observed in the OIR retina compared to room air control. Apoptosis of retinal neurons was studied using TUNEL assay and significantly increased numbers of apoptotic cells were observed in the central retina. At P14 the dying cells were mainly localized in the INL. Other studies including ours, have confirmed neuronal apoptosis and/or retinal degeneration in the OIR retina (Brafman et al., 2004; Downie et al., 2007; Fletcher et al., 2010; Narayanan et al., 2011; Stevenson et al., 2010). While the cellular mechanisms causing this neurodegeneration are not yet understood, iNOS has been shown to play a crucial role in the neuronal death and degeneration (Sennlaub et al., 2002). Significant neuroprotection was observed in the OIR retinas of iNOS deficient mice as compared to controls, suggesting that NO released by iNOS can induce neurotoxicity and apoptosis. As has been explained above, studies using TNFα-deficient mice also indicate that neuronal death in the OIR retina is mediated by iNOS (Stevenson et al. 2010). Retinas from TNFα-deficient OIR mice had significantly reduced neuronal death and decreased levels of reactive oxygen species, reactive nitrogen species, and NO along with increased arginase activity. Further, there was enhanced survival of astrocytes and increased numbers of microglial cells actively participating in phagocytosis.
In contrast with the previously suggested neuroprotective role of arginase during OIR, recent studies from our laboratory using arginase II deficient mice have demonstrated the involvement of arginase signaling in retinal neurodegeneration (Narayanan et al., 2011). We found that deletion of arginase II significantly reduced neuronal degeneration and thinning of the retina during OIR (Fig. 11). Electroretinographic recordings showed that the protective effects of arginase II deletion were accompanied by significant preservation of the b-wave component of the electroretinogram. Partial deletion of arginase I along with arginase II did not confer any additional preservation of the retinal neurons, suggesting that arginase II is the more important arginase isoform involved in the neurodegeneration.
Figure 11.

Arginase II deletion prevents neuronal cell loss and thinning of the neural retina in the mouse OIR model (A). Electroretinographic recording shows a decrease in the b-wave amplitude at low light intensity which is absent in the arginase II-deficient retina. (Naryananan et al., 2011)
Studies in brain have shown arginase II is expressed in neurons (Braissant et al., 1999). Our immunolocalization analyses demonstrated that arginase II is highly expressed in horizontal cells of the retina and in the photoreceptor inner segments (Fig. 12). Arginase II immunoreactivity is also evident in cellular processes throughout the neural retina, consistent with its mitochondrial localization. Our studies in isolated retinal vessels and cultured retinal endothelial cells have shown that arginase II is also expressed in retinal vascular cells in vivo and retinal endothelial cells in vitro (Fig. 13). Western blot analysis of the OIR retinal showed that arginase II protein level was markedly increased immediately after the end of the hyperoxia exposure on P12 and decreased thereafter. Despite these alterations in arginase II protein levels, total arginase activity remained unchanged in the OIR retina. Similar to the report of Sennlaub and colleagues (Sennlaub et al., 2002), we observed that retinal apoptosis peaked at P14 and the majority of the apoptotic cells were localized in the inner nuclear layer (Fig. 14). These included bipolar, amacrine and horizontal cells along with some Müller glia. Additionally a few cells in the ganglion cell and photoreceptor layers were also TUNEL positive. The decrease in arginase II protein levels at P14 and later is likely due to degeneration of arginase II expressing cells.
Figure 12.

Immunohistochemistry of retinal cryostat sections showing the localization of arginase II in the neural retina (A, B). Note intense immunoreactivity in the photoreceptor inner segments (arrows). Retinal flatmount showing increased level of arginase II in the horizontal cells of OIR retina. (Narayanan et al., 2011)
Fig. 13.

Arginase II expression in retina and retinal vessels, studied by immunostaining and western blot.
Figure 14.

TUNEL assay of retinal sections showing numerous degenerating cells (colocalized with DAPI) in wild type OIR compared to room air (RA) controls and AII−/− OIR at P14 (A). Quantification of TUNEL positive cells showing a significant increase in the wild type OIR retina and a decrease in the AII−/− OIR (B). Characterization of TUNEL positive cells in wild type OIR retina by double immunolabelling using NeuN (ganglion and amacrine cells), CRALBP (Müller glia), PKCα (bipolar cells) and recoverin (photoreceptors) antibodies. Insets show magnified images of TUNEL colocalization with respective cellular markers (C). (Narayanan et al., 2011)
In addition to retinal thinning, activation of glial cells is another hallmark of ROP (Downie et al., 2007; Fletcher et al., 2010). This has been confirmed in our study. In normal conditions, glial fibrillary acidic protein (GFAP) is expressed mainly in retinal astrocytes. However, following retinal injury, GFAP is upregulated in Müller cells and astrocytes and is considered a sign of glial cell activation. Müller cells are radial glia whose processes span the retina extending from the nerve fiber layer to the photoreceptor layer. Figure 15 shows that there was a slight increase in the levels of GFAP in the wild type OIR retina as early as P12 compared to the room air control wild type retina. The enhanced expression of GFAP in Müller cells was more evident at P14 in the wild type OIR retinas and was further increased at P17 as indicated by the strong immunoreactivity extending radially from the inner retina through the outer nuclear layer. However this enhanced level of GFAP expression was not observed in OIR retinas of arginase II-deficient mice at any stage of OIR studied. Absence of Müller glial activation in the arginase II-deficient OIR mice suggests an adverse effect of arginase on glia as well as neurons. Our data also demonstrate that glial activation follows neuronal apoptosis. While the peak of apoptosis was seen at P14, glial activation was greatest at P17. It is possible that soluble factors and/or cellular debris released by the dying cells are causing glial cell activation.
Figure 15.

Upregulation of GFAP in the OIR retina. Immunolabeling of retinal cryostat sections from wild type and AII−/− OIR using anti- GFAP antibody at different stages of OIR demonstrating increased gliosis in WT OIR retina. (Narayanan et al., 2011)
Neurochemical changes observed during OIR have been attributed to Müller cell alterations (Fletcher et al., 2010). Our data further demonstrate that significantly reduced cell death in the arginase II-deficient OIR retinas is associated with decreases in glial cell activation. Müller glia are a major source of retinal iNOS and its expression level is reported to be increased during the hypoxic phase of OIR (He et al., 2007; Kaur et al., 2009). This can lead to increased nitrosative stress, which may trigger protein nitration resulting in the loss of function of several proteins especially mitochondrial antioxidant proteins (Aulak et al., 2001; Yamakura et al., 1998). Our immunohistochemistry studies confirmed increased levels of iNOS in the wild type OIR retina compared to normoxic control. This elevation of iNOS was markedly attenuated in the arginase II-deficient OIR retina (Fig. 16). The finding that arginase II deletion is accompanied by a decrease in iNOS expression during OIR suggests the involvement of arginase II in regulating iNOS-mediated oxidative stress and retinal injury. Further study is necessary to elucidate the relationship between arginase II and iNOS expression.
Figure 16.

Expression of iNOS in the OIR retina. A marked increase in level of iNOS is observed in WT OIR retina which is not evident in AII−/− OIR retina.
Clinical studies have shown loss of neuronal function in ROP patients (Barnaby et al., 2007; Fulton and Hansen, 1996; Fulton et al., 2009; O'Connor et al., 2002). In Electroretinographic (ERG) studies of patients with a history of ROP, significant abnormalities in the a- and b-waves are found to continue into adulthood (Fulton and Hansen, 1995; Fulton and Hansen, 1996). Deficits in rod and rod-bipolar cell sensitivity are still evident several years after ROP has resolved (Fulton et al., 2001; Hansen and Fulton, 2000). Recent studies have shown that patients with a history of ROP are more likely to show abnormal development of the fovea and have a poorer visual prognosis (Wu et al., 2012). Since the development of laser surgery there has been little progress in the treatment of ROP.
Defects in retinal function as shown by ERG have been reported for rat and mice OIR models. Rat OIR studies (Akula et al., 2010; Akula et al., 2008; Dembinska et al., 2001; Liu et al., 2006) have shown decreases in the amplitude and sensitivity of the a-wave, suggesting photoreceptor dysfunction, as well as reductions in b-wave amplitudes, indicating that retinal interneurons are affected as well. Recent reports by others (Vessey et al., 2011) and by us (Narayanan et al., 2011) demonstrate functional losses in the mouse OIR model. While our finding demonstrated a decrease in the b-wave amplitude of the ERG at low light intensity in the OIR retina at 6 weeks, Vessey et al. (Vessey et al., 2011) showed that function of both cone and rod pathways are reduced in OIR retina. According to our data, the b-wave amplitudes in arginase II deficient OIR mice were significantly higher than in wild type OIR at lower light intensities, demonstrating improved retinal function (see Fig. 11B). However at higher intensities the responses were similar in the two OIR groups. Others have also reported loss of retinal function in mouse OIR retina (Nakamura et al., 2012). Amplitudes of the a- and b-wave were significantly decreased at 4 weeks in the OIR model, but both recovered after 8 weeks. There were also deficits in oscillatory potential (an index of dysfunction of the inner plexiform layer) which persisted with time. Thus, there is extensive evidence that neurons are adversely affected in ROP, suggesting that retinal neurons are an important target in developing novel therapies for ROP.
The above observations clearly indicate that arginase II plays a key role in retinal neuronal injury during OIR. While the mechanisms of the arginase-mediated injury are not yet known, polyamine metabolism may be involved. Polyamines have been implicated in exitotoxic neuronal death in retina (Pernet et al., 2007) and the neurotoxic actions of glutamate are well established (Lau and Tymianski, 2010). So far no literature is available on the role of polyamines in ROP. Ongoing studies from our laboratory show that polyamines are crucially involved in the neuronal death during the hyperoxic phase of OIR (Narayanan et al., in submission). Increased expression of polyamine oxidase together with decreases in spermine, increases in spermidine and increases in hydrogen peroxide formation are observed in the OIR retina compared to normoxic controls. These changes are consistent with an increase in backward polyamine catabolism and are reversed in the arginase II-deficient OIR retina. Further, treatment with the polyamine oxidase inhibitor MDL-72527 during hyperoxia significantly reduced neuronal death in the OIR retina during the hyperoxia phase. These results demonstrate that polyamine catabolism is involved in neurodegeneration during OIR.
4.4.2. Vascular degeneration and pathological angiogenesis
Vaso-obliteration and neovascularization are the most studied pathologies in ROP. As stated earlier, the OIR retina undergoes hyperoxia followed by relative hypoxia. In response to hyperoxia, the immature vessels undergo massive vasoconstriction and the endothelial cells degenerate, causing subsequent regression of the vascular bed. Various factors participate in this endothelial cell death leading to vaso-obliteration. Some of these include down-regulation of VEGF (Alon et al., 1995; Pierce et al., 1996) and IGF1 (Smith et al., 1999; Smith et al., 1997), increased generation of reactive oxygen species (Aguilar et al., 2008; Niesman et al., 1997; Oguni et al., 1995; Smith et al., 1997) and lipid peroxidation (Beauchamp et al., 2001). Studies from our laboratory have previously demonstrated the involvement of endothelial NO and peroxynitrite in hyperoxia mediated endothelial cell death (Brooks et al., 2001; Gu et al., 2002). Upon hyperoxia treatment for two days, from P7-P9, a significant decrease in the vaso-obliteration area was observed in eNOS deficient mice, as well as in wild type mice treated with the NOS inhibitor L-NNA. Further, the eNOS deficient OIR retina showed significant reduction in nitrotyrosine levels compared to control OIR retinas (Brooks et al., 2001). This demonstrates that NO is an important factor in the vascular injury associated with ROP. Another study using iNOS knockout mice and the iNOS inhibitor 1400W showed that iNOS is expressed in the ischemic retina and that iNOS blockade improved physiological vascular repair in the capillary dropout zone (Sennlaub et al., 2001). The investigators further showed that adverse effects of iNOS on vascular repair are mediated at least in part by a downregulation of VEGF receptor 2 in cells adjacent to iNOS-expressing cells.
Even though the role of NO in vascular injury is well discussed, so far no one has investigated whether arginase, another regulator of NO, plays any role in the vascular injury associated with oxygen induced retinopathy. An ongoing study from our laboratory has demonstrated that arginase II pays a key role in the hyperoxia-mediated vascular injury through a mechanism involving superoxide and peroxynitrite formation, and activation of microglia/macrophages (Suwanpradid et al., unpublished). Studies using arginase II deficient mice in the OIR model showed significant decreases in vaso-obliteration compared to the wild type OIR retina, suggesting that vascular survival is improved with a deficiency in arginase II. Interestingly, after the mice were returned to room air at P12, regrowth of vessels into the area of vaso-obliteration was faster in arginase II knockout mice than in the wild type mice. Deletion of arginase II gene in combination with haploid deficiency of arginase I did not show any additional protection compared with wild type mice at P12, and there was no difference in the vaso-obliteration area between the two knockout mice lines. Additionally there was a significant decrease in the levels of superoxide and peroxynitrite formation in the arginase II deficient OIR retina relative to the wild type OIR retina. This suggests that the observed vascular protection is mediated by reduction in nitrative stress in the OIR retina. Changes in NO signaling are expected in the retina during oxygen-induced retinopathy. Hence, it is possible that bioavailable NO levels are reduced in wild type retinas during OIR, causing an increase in peroxynitrite formation. Normalization of NO levels is expected in arginase II knockout retina due to increased availaibity of L-arginine. This should result in reduced formation of both superoxide and peroxynitrite. The ratio of activated microglia to resting microglia during hyperoxia was also significantly reduced in arginase II knock out OIR retina compared to OIR controls, suggesting a role for microglia in this vasoprotection. Previous work has shown increased numbers of actively phagocytizing microglia in TNFα knockout OIR retina (Stevenson et al., 2010). The investigators suggested this as a mechanism contributing to reduced vascular injury in the TNFα-deficient OIR retina.
In the second phase of ROP, the avascular retina becomes hypoxic and attempts to reinstate adequate levels of oxygen and nutrients to the vaso-obliterated retina by upregulation of angiogenic factors. The increase in growth factors results in uncontrolled proliferation and leads to pathological neovascularization. In contrast to normal developmental growth, this neovascularization is excessive, disorganized, and misdirected toward the vitreous, which is devoid of vessels under physiological conditions. Neovascularization is maximal at P17 and regresses thereafter (Sapieha et al., 2010). Our analyses of the area of retina covered by vitreo-retinal neovascular tufts showed no significant difference between wild type and arginase knockout retinas following the OIR treatment. However, at P17, the neovascular tufts were smaller in size in the arginase II knockout retina as compared with the wild type retinas. In the previous study using iNOS-deficient mice, iNOS deletion was vasoprotective during the later stages of OIR and resulted in a significant reduction in neovascularization (Sennlaub et al., 2001). Activated glial cells could be a major source of this enhanced retinal iNOS and reactive nitrogen species formation. Our studies have shown that glial activation is minimal in arginase II knockout OIR retina along with reduced expression of iNOS (Fig. 15, 16). However, pathological neovascularization does occur in our OIR model with arginase II deletion suggesting the involvement of additional factors in the regulation of neovascularization in OIR retina. Further work is required to elucidate the specific mechanisms by which arginase II modulates vascular injury and repair. Given our findings implicating arginase II in retinal neuronal degeneration and the well-established relationship between neuronal and vascular injury, it is tempting to speculate that arginase II mediates vascular injury by a mechanism involving neuronal cell toxicity.
5. Conclusions and perspective
Ischemic retinopathies are characterized by oxidative stress and inflammatory reactions leading to vascular and neuronal dysfunction and injury, altered blood flow, breakdown of the blood-retinal barrier and death of vascular and neuronal cells [for review, please see (Adamis and Berman, 2008; Antonetti et al., 2012; Tang and Kern, 2011)]. If unchecked, the vascular injury can progress to pathological neovascularization with fibrovascular scarring, leading to retinal detachment and blindness (Friedlander, 2007). Extensive literature from studies of diseases and disease models that affect vessels and organs outside the CNS support the concept that dysregulated arginase activity could be involved in all of these events. Excessive arginase activity has been shown to contribute to progression of diabetic vascular dysfunction by uncoupling NOS and also by activating the ornithine pathway (Romero et al., 2012; Romero et al., 2008). Each of these can have multiple adverse effects. Uncoupled NOS uses molecular oxygen to produce superoxide which will react rapidly with any available NO to form peroxynitrite a highly reactive inflammatory and toxic oxidant. This further reduces NO availability and increases oxidative stress. The decreases in NO will cause a decrease in endothelial cell-dependent vasorelaxation which will decrease blood flow, leading to ischemia. Decreases in endothelial cell-derived NO can also contribute to vascular inflammation because an adequate supply of NO is needed to block platelet aggregation and prevent leukocyte rolling and attachment to the vessel wall (Cooke and Dzau, 1997). Activation of the arginase/ornithine pathway may contribute to tissue fibrosis due to increases in formation of proline which promotes collagen formation or polyamines which enhance cell growth. Also, polyamine metabolism and products of polyamine oxidation have been shown to be involved in the pathogenesis of ischemic brain damage (Ivanova et al., 2002; Takano et al., 2005; Wood et al., 2006), gyrate atrophy of the retina and choroid (Takki, 1994), injury of the retinal pigment epithelium (Hayasaka et al, 2011) and diabetes-induced alterations in the function of potassium channels in the retinal microvasculature (Matsushita and Puro, 2006).
Based on these observations implicating the arginase/ornithine pathway in inflammation and injury in peripheral tissues and brain, we postulated that dysregulation of this pathway is involved in retinal vascular inflammation. Our study using a model of endotoxin-induced uveitis provided the first evidence that arginase is critically involved in acute retinal inflammation induced by endotoxin (Zhang et al, 2009a). We found that endotoxin treatment induces a prominent increase in arginase I expression and activity in the retina and retinal cells in vivo and in vitro. We used selective inhibition of arginase by gene deletion or pharmacologic inhibitor to demonstrate that arginase is an indispensible component in retinal inflammation during uveitis. Blockade of arginase prevented the production of inflammatory cytokines in retinal neuroglia and macrophage/microglia, inhibited leukocyte adhesion to the vessel wall, reduced signs of uveitis and prevented retina damage. Considering that 10% to 15% of blindness in the United States is due to uveitis (http://www.uveitissociety.org/pages/faq.html), reduction of arginase activity may provide a beneficial therapeutic approach.
Because vascular inflammation is also a predominant feature of diabetic retinopathy, we have evaluated the involvement of arginase in vascular dysfunction and injury in diabetic mice and retinal endothelial cells treated with high glucose. Our studies have provided the first evidence that arginase is critically involved in diabetes-induced vascular inflammation and impairment of endothelial cell-dependent vascular relaxation (Elms et al., 2013; Patel et al., In Press). We found that blockade of arginase activity by selective inhibitor or gene deletion increased bioavailable NO levels, reduced formation of superoxide and peroxynitrite, improved retinal endothelial dependent vasorelxation and decreased leukostasis as compared with the controls, which highlights the important role of arginase expression in diabetes-induced vascular dysfunction and injury. These results indicate that arginase is a potential therapeutic target for preserving bioavailability of NO, limiting oxidative stress and preventing early signs of vascular inflammation and injury during diabetic retinopathy. Whereas the role of overactive arginase in peripheral vascular dysfunction and injury has been a topic of active research, our studies are the first to mechanistically link overactive arginase to retinal vascular injury in diabetes.
Our studies using the OIR mouse model for ROP have demonstrated a novel role of arginase II in retinal neuro-vascular damage associated with ROP. Abnormal vasculature characterized by vaso-obliteration and pathological angiogenesis is the most recognized characteristic of ROP. From the clinical perspective, ROP is still considered as a vascular disease. However, recent studies have shown that retinal neuronal dysfunction is also an important component of the injury condition. Hence retinal neurons also need to be considered as targets when developing new therapies for ROP. Our studies in the OIR model demonstrate the involvement of arginase signaling in both neuronal dysfunction and vascular injury, suggesting that modulating the arginase/ornithine pathway is an important target for protecting the premature retina from oxygen-mediated injury.
6. Future directions
Targeting specific components of the arginase/ornithine pathway holds great promise as a therapy for multiple forms of ischemic retinopathy. However, there are many research questions that need to be addressed in order to better understand the role of arginase in retinopathy and effectively target potential therapies. Fruitful research areas that need to be addressed by future basic science and clinical studies are numerous. Research questions and topics of particular interest are outlined below.
6.1. Basic research
Which cell types are responsible for arginase-induced vascular dysfunction in diabetic retinopathy? We have evidence that arginase I is increased in retinal vascular cells, glia and macrophage/microglial cells. Which of these sources are responsible for the early signs of diabetic retinopathy observed in our animal models? Does activation of the arginase/ornithine pathway have a role in metabolic memory? If so, what is the underlying mechanism?
What is the role of polyamine oxidation in diabetic retinopathy? Does the arginase/polyamine pathway also have a role in the formation of acellular capillaries during the later stages of diabetic retinopathy? What role does the arginase/polyamine pathway play in diabetes-induced neuronal degeneration or RPE-photoreceptor dysfunction and breakdown of the blood retinal barrier in the diabetic retina?
What are the mechanisms responsible for increasing arginase expression/activity in the diabetic retina? Increased arginase exression/activity is associated with inflammatory cytokines and oxidative stress, both of which are elevated during diabetes. Peroxynitrite and H2O2 have been shown to increase arginase expression/activity in aortic endothelial cells, through PKC mediated activation of RhoA/Rho kinase pathway. Is this mechanism operative in the diabetic retina? What role do post-translational modifications of arginase have in increasing its activity?
What is the role of polyamine oxidation in retinal vascular injury, neuronal dysfunction and glial activation in the OIR model? Our data show a close correlation between neuronal and vascular degeneration in the OIR model. Are these pathologies causally related? If so, which insult is primary? Does arginase-induced alteration in NO signaling have a role in neurovascular injury during OIR?
6.2. Clinical/translational research
Which cells express arginase I and arginase II in the human retina? Is arginase expression, distribution or activity altered in retinas or surgical specimens for patients with diabetic retinopathy or other forms of ischemic retinopathy or inherited retinal degeneration?
The involvement of mutations in the ornithine aminotransferase (OAT) gene has been well established in gyrate atrophy of the retina and choroid. Do alterations in OAT or other components of the arginase/ornithine pathway have a role in AMD?
The impact of increases in polyamine formation and polyamine catabolism in brain injury and disease has been well established, and our studies have demonstated a role for this process in retinal neurodegeneration in mice. Could this component of the arginase/polyamine pathway also have a role in ganglion cell and optic nerve injury during glaucoma?
Inhibitors or blockers of the arginase/ornithine pathway are already in use for the treatment of patients with resistant parasitic infections and some forms of cancer. Can the progression of ischemic retinopathy be slowed or reversed by treatment with such compounds?
Could circulating levels of arginase or its metabolic products be used as biomarkers or predictors of retinopathy progression in patients with diabetes or other forms of retinopathy?
Highlights.
New therapies are needed for ischemic retinopathies
Arginase signaling is associated with neurovascular injury in ischemic retina
Arginase blockade reduces signs of inflammation and vascular dysfunction in the diabetic retina
Arginase II deletion reduces neuro-glial injury and limits retinal degeneration during oxygen-induced retinopathy.
Strategies for limiting arginase activity promise new therapy for ischemic retinopathies
Acknowledgements
We thank Chintan Patel, Esraa Shosha, Tahira Lemtalsi, Zhimin Xu, Shawn Elms and Wenbo Zhang for their contributions to the work described in this manuscript. This material is based on work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development (RBC) and by research grants from the Culver Vision Discovery Institute at Georgia Regents University (RBC), VA Research Career Scientist Award (RBC), VA Merit Review Award (RBC); PHS grants EY011766 (RBC & RWC), HL070215 (RWC), AHA 11SDG7440088 (SPN). The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributions
SPN, RWC and RBC prepared and edited the manuscript. MR, SPN, HFT and JS generated data for figures and contributed to writing of the paper.
None of the authors have any competing interests.
No ethical or Institutional Review Board (IRB) approval was required for this study.
All authors have completed the Unified Competing Interest form (available on request from the corresponding author) and declare: no support from any organization fo the submitted work; no financial relationships with any organizations that might have an interes in the submitted work, no relationships or activities that could appear to have influenced the submitted work.
Contributor Information
S. Priya Narayanan, Email: pnarayanan@gru.edu.
Modesto Rojas, Email: mrojas@gru.edu.
Jutamas Suwanpradid, Email: jsuwanpradid@gru.edu.
Haroldo A. Toque, Email: htoque@gru.edu.
R. William Caldwell, Email: wcaldwel@gru.edu.
References
- DCCT. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- UKPDS. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- Adamis AP, Berman AJ. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol. 2008;30:65–84. doi: 10.1007/s00281-008-0111-x. [DOI] [PubMed] [Google Scholar]
- Aguilar E, et al. Chapter 6. Ocular models of angiogenesis. Methods Enzymol. 2008;444:115–158. doi: 10.1016/S0076-6879(08)02806-1. [DOI] [PubMed] [Google Scholar]
- Aiello LP. Diabetic retinopathy. Diabetes Care. 1998;21:143–156. doi: 10.2337/diacare.21.1.143. [DOI] [PubMed] [Google Scholar]
- Akula JD, et al. Visual cycle modulation in neurovascular retinopathy. Exp Eye Res. 2010 doi: 10.1016/j.exer.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Akula JD, et al. The neurovascular relation in oxygen-induced retinopathy. Mol Vis. 2008;14:2499–2508. [PMC free article] [PubMed] [Google Scholar]
- Al-Shabrawey M, et al. Role of NADPH Oxidase and STAT3 in Statin-mediated Protection Against Diabetic Retinopathy. Invest Ophthalmol Vis Sci. 2008a;49:3231–3238. doi: 10.1167/iovs.08-1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shabrawey M, et al. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167:599–607. doi: 10.1016/S0002-9440(10)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shabrawey M, et al. Role of NADPH Oxidase in Retinal Vascular Inflammation. Invest. Ophthalmol. Vis. Sci. 2008b;49:3239–3244. doi: 10.1167/iovs.08-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon T, et al. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med. 1995;1:1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- Amin HJ, et al. Arginine supplementation prevents necrotizing enterocolitis in the premature infant. J Pediatr. 2002;140:425–431. doi: 10.1067/mpd.2002.123289. [DOI] [PubMed] [Google Scholar]
- Antonetti DA, et al. Diabetic retinopathy. N Engl J Med. 2012;366:1227–1239. doi: 10.1056/NEJMra1005073. [DOI] [PubMed] [Google Scholar]
- Ash DE. Structure and function of arginases. J Nutr. 2004;134:2760S–2764S. doi: 10.1093/jn/134.10.2760S. discussion 2765S–2767S. [DOI] [PubMed] [Google Scholar]
- Ashton H, et al. Therapeutics X. Topical antifungal agents. Br J Dermatol. 1970;82:539–541. doi: 10.1111/j.1365-2133.1970.tb02219.x. [DOI] [PubMed] [Google Scholar]
- Ashton N, et al. Effect of oxygen on developing retinal vessels with particular reference to the problem of retrolental fibroplasia. Br J Ophthalmol. 1954;38:397–432. doi: 10.1136/bjo.38.7.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulak KS, et al. Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proc Natl Acad Sci U S A. 2001;98:12056–12061. doi: 10.1073/pnas.221269198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward GW. The scotopic threshold response in diabetic retinopathy. Eye (Lond) 1989;3(Pt 5):626–637. doi: 10.1038/eye.1989.97. [DOI] [PubMed] [Google Scholar]
- Bachetti T, et al. Arginase pathway in human endothelial cells in pathophysiological conditions. J Mol Cell Cardiol. 2004;37:515–523. doi: 10.1016/j.yjmcc.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Bagnost T, et al. Cardiovascular effects of arginase inhibition in spontaneously hypertensive rats with fully developed hypertension. Cardiovasc Res. 2010 doi: 10.1093/cvr/cvq081. [DOI] [PubMed] [Google Scholar]
- Barber AJ, et al. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52:1156–1163. doi: 10.1167/iovs.10-6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber AJ, et al. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783–791. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnaby AM, et al. Development of scotopic visual thresholds in retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2007;48:4854–4860. doi: 10.1167/iovs.07-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp MH, et al. Role of thromboxane in retinal microvascular degeneration in oxygen-induced retinopathy. J Appl Physiol. 2001;90:2279–2288. doi: 10.1152/jappl.2001.90.6.2279. [DOI] [PubMed] [Google Scholar]
- Beckman JS, et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:620–624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz DE, et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108:2000–2006. doi: 10.1161/01.CIR.0000092948.04444.C7. [DOI] [PubMed] [Google Scholar]
- Bhatia S, et al. Antioxidant status, lipid peroxidation and nitric oxide end products in patients of type 2 diabetes mellitus with nephropathy. Clin Biochem. 2003;36:557–562. doi: 10.1016/s0009-9120(03)00094-8. [DOI] [PubMed] [Google Scholar]
- Bivalacqua TJ, et al. Overexpression of arginase in the aged mouse penis impairs erectile function and decreases eNOS activity: influence of in vivo gene therapy of antiarginase. Am J Physiol Heart Circ Physiol. 2007;292:H1340–H1351. doi: 10.1152/ajpheart.00121.2005. [DOI] [PubMed] [Google Scholar]
- Boor P, et al. Treatment targets in renal fibrosis. Nephrol Dial Transplant. 2007;22:3391–3407. doi: 10.1093/ndt/gfm393. [DOI] [PubMed] [Google Scholar]
- Brafman A, et al. Inhibition of oxygen-induced retinopathy in RTP801-deficient mice. Invest Ophthalmol Vis Sci. 2004;45:3796–3805. doi: 10.1167/iovs.04-0052. [DOI] [PubMed] [Google Scholar]
- Braissant O, et al. L-arginine uptake, the citrulline-NO cycle and arginase II in the rat brain: an in situ hybridization study. Brain Res Mol Brain Res. 1999;70:231–241. doi: 10.1016/s0169-328x(99)00151-5. [DOI] [PubMed] [Google Scholar]
- Brooks SE, et al. Reduced severity of oxygen-induced retinopathy in eNOS-deficient mice. Invest Ophthalmol Vis Sci. 2001;42:222–228. [PubMed] [Google Scholar]
- Buga GM, et al. NG-hydroxy-L-arginine and nitric oxide inhibit Caco-2 tumor cell proliferation by distinct mechanisms. Am J Physiol. 1998;275:R1256–R1264. doi: 10.1152/ajpregu.1998.275.4.R1256. [DOI] [PubMed] [Google Scholar]
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762–769. doi: 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- Caldwell RB, et al. Vascular dysfunction in retinopathy-an emerging role for arginase. Brain Res Bull. 2009;81:303–309. doi: 10.1016/j.brainresbull.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, et al. Hypoxia-induced retinal angiogenesis in zebrafish as a model to study retinopathy. PLoS One. 2008;3:e2748. doi: 10.1371/journal.pone.0002748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceriello A. The emerging challenge in diabetes: the "metabolic memory". Vascul Pharmacol. 2012;57:133–138. doi: 10.1016/j.vph.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Chandra S, et al. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br J Pharmacol. 2012;165:506–519. doi: 10.1111/j.1476-5381.2011.01584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CI, et al. The involvement of tyrosine kinases, cyclic AMP/protein kinase A, and p38 mitogen-activated protein kinase in IL-13-mediated arginase I induction in macrophages: its implications in IL-13-inhibited nitric oxide production. J Immunol. 2000;165:2134–2141. doi: 10.4049/jimmunol.165.4.2134. [DOI] [PubMed] [Google Scholar]
- Chen B, et al. Hypoxia promotes human pulmonary artery smooth muscle cell proliferation through induction of arginase. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1151–L1159. doi: 10.1152/ajplung.00183.2009. [DOI] [PubMed] [Google Scholar]
- Cho NC, et al. Selective loss of S-cones in diabetic retinopathy. Arch Ophthalmol. 2000;118:1393–1400. doi: 10.1001/archopht.118.10.1393. [DOI] [PubMed] [Google Scholar]
- Clermont AC, Bursell SE. Retinal blood flow in diabetes. Microcirculation. 2007;14:49–61. doi: 10.1080/10739680601072164. [DOI] [PubMed] [Google Scholar]
- Cohen RA, Tong X. Vascular oxidative stress: the common link in hypertensive and diabetic vascular disease. Journal of cardiovascular pharmacology. 2010;55:308–316. doi: 10.1097/fjc.0b013e3181d89670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke JP, Dzau VJ. Nitric oxide synthase: role in the genesis of vascular disease. Annu Rev Med. 1997;48:489–509. doi: 10.1146/annurev.med.48.1.489. [DOI] [PubMed] [Google Scholar]
- Cooke JP, et al. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. The Journal of clinical investigation. 1991;88:1663–1671. doi: 10.1172/JCI115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JD, et al. Arginase-boronic acid complex highlights a physiological role in erectile function. Nat Struct Biol. 1999;6:1043–1047. doi: 10.1038/14929. [DOI] [PubMed] [Google Scholar]
- Curtis TM, et al. Microvascular lesions of diabetic retinopathy: clues towards understanding pathogenesis? Eye. 2009;23:1496–1508. doi: 10.1038/eye.2009.108. [DOI] [PubMed] [Google Scholar]
- Curtis TM, Scholfield CN. The role of lipids and protein kinase Cs in the pathogenesis of diabetic retinopathy. Diabetes Metab Res Rev. 2004;20:28–43. doi: 10.1002/dmrr.431. [DOI] [PubMed] [Google Scholar]
- Darley-Usmar V, et al. Nitric oxide and oxygen radicals: a question of balance. FEBS Lett. 1995;369:131–135. doi: 10.1016/0014-5793(95)00764-z. [DOI] [PubMed] [Google Scholar]
- Davel LE, et al. Arginine metabolic pathways involved in the modulation of tumor-induced angiogenesis by macrophages. FEBS Lett. 2002;532:216–220. doi: 10.1016/s0014-5793(02)03682-7. [DOI] [PubMed] [Google Scholar]
- Davoodi J, et al. Overexpression and characterization of the human mitochondrial and cytosolic branched-chain aminotransferases. J Biol Chem. 1998;273:4982–4989. doi: 10.1074/jbc.273.9.4982. [DOI] [PubMed] [Google Scholar]
- de Boer J, et al. Role of nitric oxide and superoxide in allergen-induced airway hyperreactivity after the late asthmatic reaction in guinea-pigs. Br J Pharmacol. 2001;133:1235–1242. doi: 10.1038/sj.bjp.0704191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Caterina R, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. The Journal of clinical investigation. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa MS, et al. Arginase 2 and Nitric Oxide Synthase: Pathways associated with the pathogenesis of thyroid tumors. Free Radic Biol Med. 2010 doi: 10.1016/j.freeradbiomed.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Deignan JL, et al. Contrasting features of urea cycle disorders in human patients and knockout mouse models. Mol Genet Metab. 2008;93:7–14. doi: 10.1016/j.ymgme.2007.08.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delage B, et al. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer. 2010;126:2762–2772. doi: 10.1002/ijc.25202. [DOI] [PubMed] [Google Scholar]
- Dembinska O, et al. Graded contribution of retinal maturation to the development of oxygen-induced retinopathy in rats. Invest Ophthalmol Vis Sci. 2001;42:1111–1118. [PubMed] [Google Scholar]
- Dizikes GJ, et al. Isolation of human liver arginase cDNA and demonstration of nonhomology between the two human arginase genes. Biochem Biophys Res Commun. 1986;141:53–59. doi: 10.1016/s0006-291x(86)80333-3. [DOI] [PubMed] [Google Scholar]
- Doughan AK, et al. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- Downie LE, et al. Neuronal and glial cell changes are determined by retinal vascularization in retinopathy of prematurity. J Comp Neurol. 2007;504:404–417. doi: 10.1002/cne.21449. [DOI] [PubMed] [Google Scholar]
- Du Y, et al. Diabetes-induced nitrative stress in the retina, and correction by aminoguanidine. J. Neurochem. 2002;80:771–779. doi: 10.1046/j.0022-3042.2001.00737.x. [DOI] [PubMed] [Google Scholar]
- Durante W, et al. Thrombin stimulates vascular smooth muscle cell polyamine synthesis by inducing cationic amino acid transporter and ornithine decarboxylase gene expression. Circ Res. 1998;83:217–223. doi: 10.1161/01.res.83.2.217. [DOI] [PubMed] [Google Scholar]
- Durante W, et al. Arginase: a critical regulator of nitric oxide synthesis and vascular function. Clin Exp Pharmacol Physiol. 2007;34:906–911. doi: 10.1111/j.1440-1681.2007.04638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Remessy A, Behzadian MA, Abou-Mohamed G, Franklin T, Caldwell RW, Caldwell RB. Peroxynitrite increases vascular permeability in experimental diabetes by a mechanism involving increased expression of VEGF and urokinase plasminogen activator receptor (uPAR) Am J Pathol. 2003;162:1995–2004. doi: 10.1016/S0002-9440(10)64332-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis EA, et al. Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/Wor diabetic rat. Free Radic Biol Med. 2000;28:91–101. doi: 10.1016/s0891-5849(99)00216-6. [DOI] [PubMed] [Google Scholar]
- Elms SC, et al. The role of arginase I in diabetes-induced retinal vascular dysfunction in mouse and rat models of diabetes. Diabetologia. 2013;56:654–662. doi: 10.1007/s00125-012-2789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esch F, et al. Purification of a multipotent antideath activity from bovine liver and its identification as arginase: nitric oxide-independent inhibition of neuronal apoptosis. J Neurosci. 1998;18:4083–4095. doi: 10.1523/JNEUROSCI.18-11-04083.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasipe FR, et al. Arginine supplementation improves rotorod performance in sickle transgenic mice. Hematology. 2004;9:301–305. doi: 10.1080/10245330410001714185. [DOI] [PubMed] [Google Scholar]
- Filbin MT. The Muddle with MAG. Mol Cell Neurosci. 1996;8:84–92. doi: 10.1006/mcne.1996.0047. [DOI] [PubMed] [Google Scholar]
- Fletcher EL, et al. The significance of neuronal and glial cell changes in the rat retina during oxygen-induced retinopathy. Doc Ophthalmol. 2010;120:67–86. doi: 10.1007/s10633-009-9193-6. [DOI] [PubMed] [Google Scholar]
- Friedlander M. Fibrosis and diseases of the eye. J Clin Invest. 2007;117:576–586. doi: 10.1172/JCI31030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton AB, Hansen RM. Electroretinogram responses and refractive errors in patients with a history of retinopathy prematurity. Doc Ophthalmol. 1995;91:87–100. doi: 10.1007/BF01203688. [DOI] [PubMed] [Google Scholar]
- Fulton AB, Hansen RM. Photoreceptor function in infants and children with a history of mild retinopathy of prematurity. J Opt Soc Am A Opt Image Sci Vis. 1996;13:566–571. doi: 10.1364/josaa.13.000566. [DOI] [PubMed] [Google Scholar]
- Fulton AB, et al. The neurovascular retina in retinopathy of prematurity. Prog Retin Eye Res. 2009;28:452–482. doi: 10.1016/j.preteyeres.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton AB, et al. The rod photoreceptors in retinopathy of prematurity: an electroretinographic study. Arch Ophthalmol. 2001;119:499–505. doi: 10.1001/archopht.119.4.499. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- Glazer ES, et al. Bioengineered human arginase I with enhanced activity and stability controls hepatocellular and pancreatic carcinoma xenografts. Transl Oncol. 2011;4:138–146. doi: 10.1593/tlo.10265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-Trolin C, et al. Increased red blood cell polyamines in ALS and Parkinson's disease. Exp Neurol. 2002;177:515–520. doi: 10.1006/exnr.2002.7952. [DOI] [PubMed] [Google Scholar]
- Good WV, Gendron RL. Retinopathy of prematurity: gone today, here tomorrow? Clin Experiment Ophthalmol. 2005;33:339–340. doi: 10.1111/j.1442-9071.2005.01057.x. [DOI] [PubMed] [Google Scholar]
- Gotoh T, et al. Chromosomal localization of the human arginase II gene and tissue distribution of its mRNA. Biochem Biophys Res Commun. 1997;233:487–491. doi: 10.1006/bbrc.1997.6473. [DOI] [PubMed] [Google Scholar]
- Gotoh T, et al. Molecular cloning of cDNA for nonhepatic mitochondrial arginase (arginase II) and comparison of its induction with nitric oxide synthase in a murine macrophage-like cell line. FEBS Lett. 1996;395:119–122. doi: 10.1016/0014-5793(96)01015-0. [DOI] [PubMed] [Google Scholar]
- Griendling KK, et al. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- Gruetter CA, et al. Relaxation of bovine coronary artery and activation of coronary arterial guanylate cyclase by nitric oxide, nitroprusside and a carcinogenic nitrosoamine. Journal of cyclic nucleotide research. 1979;5:211–224. [PubMed] [Google Scholar]
- Gu X, et al. Effects of sustained hyperoxia on revascularization in experimental retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2002;43:496–502. [PubMed] [Google Scholar]
- Gualtieri M, et al. Contrast sensitivity mediated by inferred magno- and parvocellular pathways in type 2 diabetics with and without nonproliferative retinopathy. Invest Ophthalmol Vis Sci. 2011;52:1151–1155. doi: 10.1167/iovs.09-3705. [DOI] [PubMed] [Google Scholar]
- Guo X, et al. ERM protein moesin is phosphorylated by advanced glycation end products and modulates endothelial permeability. Am J Physiol Heart Circ Physiol. 2009;297:H238–H246. doi: 10.1152/ajpheart.00196.2009. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Harrison DG. Vascular NADPH oxidases as drug targets for novel antioxidant strategies. Drug Discov Today. 2006;11:524–533. doi: 10.1016/j.drudis.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, et al. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- Haffner I, et al. Regulation of arginase-1 expression in macrophages by a protein kinase A type I and histone deacetylase dependent pathway. J Cell Biochem. 2008;103:520–527. doi: 10.1002/jcb.21422. [DOI] [PubMed] [Google Scholar]
- Hagenfeldt L, et al. Plasma amino acids in relation to metabolic control in insulin-dependent diabetic children. Acta Paediatr Scand. 1989;78:278–282. doi: 10.1111/j.1651-2227.1989.tb11070.x. [DOI] [PubMed] [Google Scholar]
- Hammes HP, et al. Nerve growth factor prevents both neuroretinal programmed cell death and capillary pathology in experimental diabetes. Mol Med. 1995;1:527–534. [PMC free article] [PubMed] [Google Scholar]
- Hansen RM, Fulton AB. Background adaptation in children with a history of mild retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2000;41:320–324. [PubMed] [Google Scholar]
- Harrison WW, et al. Prediction, by retinal location, of the onset of diabetic edema in patients with nonproliferative diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52:6825–6831. doi: 10.1167/iovs.11-7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartnett ME. Studies on the pathogenesis of avascular retina and neovascularization into the vitreous in peripheral severe retinopathy of prematurity (an american ophthalmological society thesis) Trans Am Ophthalmol Soc. 2010;108:96–119. [PMC free article] [PubMed] [Google Scholar]
- Hayasaka S, et al. Retinal risks of high-dose ornithine supplements: a review. Br J Nutr. 2011;106:801–811. doi: 10.1017/S0007114511003291. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Iguchi A. Possibility of the regression of atherosclerosis through the prevention of endothelial senescence by the regulation of nitric oxide and free radical scavengers. Geriatr Gerontol Int. 2010;10:115–130. doi: 10.1111/j.1447-0594.2009.00581.x. [DOI] [PubMed] [Google Scholar]
- He T, et al. Interaction between iNOS and COX-2 in hypoxia-induced retinal neovascularization in mice. Arch Med Res. 2007;38:807–815. doi: 10.1016/j.arcmed.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Hein TW, et al. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: counteracting role of arginase. Faseb J. 2003;17:2328–2330. doi: 10.1096/fj.03-0115fje. [DOI] [PubMed] [Google Scholar]
- Horowitz S, et al. Increased arginase activity and endothelial dysfunction in human inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1323–G1336. doi: 10.1152/ajpgi.00499.2006. [DOI] [PubMed] [Google Scholar]
- Huynh NN, et al. Arginase II knockout mouse displays a hypertensive phenotype despite a decreased vasoconstrictory profile. Hypertension. 2009a;54:294–301. doi: 10.1161/HYPERTENSIONAHA.108.121731. [DOI] [PubMed] [Google Scholar]
- Huynh NN, et al. The vascular effects of different arginase inhibitors in rat isolated aorta and mesenteric arteries. Br J Pharmacol. 2009b;156:84–93. doi: 10.1111/j.1476-5381.2008.00036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoguchi T, et al. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J Am Soc Nephrol. 2003;14:S227–S232. doi: 10.1097/01.asn.0000077407.90309.65. [DOI] [PubMed] [Google Scholar]
- Ishizaki E, et al. Functional K(ATP) channels in the rat retinal microvasculature: topographical distribution, redox regulation, spermine modulation and diabetic alteration. J Physiol. 2009;587:2233–2253. doi: 10.1113/jphysiol.2009.169003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova S, et al. Neuroprotection in cerebral ischemia by neutralization of 3-aminopropanal. Proc Natl Acad Sci U S A. 2002;99:5579–5584. doi: 10.1073/pnas.082609299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova S, et al. Cerebral ischemia enhances polyamine oxidation: identification of enzymatically formed 3-aminopropanal as an endogenous mediator of neuronal and glial cell death. J Exp Med. 1998;188:327–340. doi: 10.1084/jem.188.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer RK, et al. Mouse model for human arginase deficiency. Mol Cell Biol. 2002;22:4491–4498. doi: 10.1128/MCB.22.13.4491-4498.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson GR, et al. Inner retinal visual dysfunction is a sensitive marker of non-proliferative diabetic retinopathy. Br J Ophthalmol. 2012;96:699–703. doi: 10.1136/bjophthalmol-2011-300467. [DOI] [PubMed] [Google Scholar]
- Jeglinski W, et al. Difluoromethylornithine counteracts lesion-induced astrogliosis in rat hippocampus: enhancement of inhibitory effect by combined treatment with GM1 ganglioside. Acta Neurobiol Exp (Wars) 1996;56:549–553. doi: 10.55782/ane-1996-1159. [DOI] [PubMed] [Google Scholar]
- Jeyabalan G, et al. Arginase blockade protects against hepatic damage in warm ischemia-reperfusion. Nitric Oxide. 2008;19:29–35. doi: 10.1016/j.niox.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, et al. Protein disregulation in red blood cell membranes of type 2 diabetic patients. Biochem Biophys Res Commun. 2003;309:196–200. doi: 10.1016/s0006-291x(03)01559-6. [DOI] [PubMed] [Google Scholar]
- Jin Y, et al. Mice deficient in Mkp-1 develop more severe pulmonary hypertension and greater lung protein levels of arginase in response to chronic hypoxia. Am J Physiol Heart Circ Physiol. 2010;298:H1518–H1528. doi: 10.1152/ajpheart.00813.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joussen AM, editor. Retinal Vascular Diseases. Berlin, Germany: Springer-Verlag; 2007. [Google Scholar]
- Jung C, et al. Arginase inhibition mediates cardioprotection during ischaemiareperfusion. Cardiovasc Res. 2010;85:147–154. doi: 10.1093/cvr/cvp303. [DOI] [PubMed] [Google Scholar]
- Kampfer H, et al. Expression and activity of arginase isoenzymes during normal and diabetes-impaired skin repair. J Invest Dermatol. 2003;121:1544–1551. doi: 10.1046/j.1523-1747.2003.12610.x. [DOI] [PubMed] [Google Scholar]
- Kapoor M, et al. Effects of epicatechin gallate on wound healing and scar formation in a full thickness incisional wound healing model in rats. Am J Pathol. 2004;165:299–307. doi: 10.1016/S0002-9440(10)63297-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur C, et al. Cellular and vascular changes in the retina of neonatal rats after an acute exposure to hypoxia. Invest Ophthalmol Vis Sci. 2009;50:5364–5374. doi: 10.1167/iovs.09-3552. [DOI] [PubMed] [Google Scholar]
- Kenyon NJ, et al. Arginases I and II in lungs of ovalbumin-sensitized mice exposed to ovalbumin: sources and consequences. Toxicol Appl Pharmacol. 2008;230:269–275. doi: 10.1016/j.taap.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, et al. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol. 2009;107:1249–1257. doi: 10.1152/japplphysiol.91393.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Klein BE. Diabetic eye disease. Lancet. 1997;350:197–204. doi: 10.1016/S0140-6736(97)04195-0. [DOI] [PubMed] [Google Scholar]
- Koga T, et al. Coinduction of nitric oxide synthase and arginine metabolic enzymes in endotoxin-induced uveitis rats. Exp Eye Res. 2002;75:659–667. doi: 10.1006/exer.2002.2062. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, et al. Abnormalities of retinal metabolism in diabetes or experimental galactosemia VIII. Prevention by aminoguanidine. Curr Eye Res. 2000;21:814–819. doi: 10.1076/ceyr.21.4.814.5545. [DOI] [PubMed] [Google Scholar]
- Kruman I, et al. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo HS, et al. The combination of peripheral nerve grafts and acidic fibroblast growth factor enhances arginase I and polyamine spermine expression in transected rat spinal cords. Biochem Biophys Res Commun. 2007;357:1–7. doi: 10.1016/j.bbrc.2007.02.167. [DOI] [PubMed] [Google Scholar]
- Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174:4880–4891. doi: 10.4049/jimmunol.174.8.4880. [DOI] [PubMed] [Google Scholar]
- Lange PS, et al. Novel roles for arginase in cell survival, regeneration, and translation in the central nervous system. J Nutr. 2004;134:2812S–2817S. doi: 10.1093/jn/134.10.2812S. discussion 2818S–2819S. [DOI] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Lechner M, et al. Inducible nitric oxide synthase (iNOS) in tumor biology: the two sides of the same coin. Semin Cancer Biol. 2005;15:277–289. doi: 10.1016/j.semcancer.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Li H, et al. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab. 2001;280:E75–E82. doi: 10.1152/ajpendo.2001.280.1.E75. [DOI] [PubMed] [Google Scholar]
- Li Q, Puro DG. Adenosine activates ATP-sensitive K(+) currents in pericytes of rat retinal microvessels: role of A1 and A2a receptors. Brain Res. 2001;907:93–99. doi: 10.1016/s0006-8993(01)02607-5. [DOI] [PubMed] [Google Scholar]
- Lima e Silva R, et al. Trans-scleral delivery of polyamine analogs for ocular neovascularization. Exp Eye Res. 2006;83:1260–1267. doi: 10.1016/j.exer.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Lima e Silva R, et al. Suppression and regression of choroidal neovascularization by polyamine analogues. Invest Ophthalmol Vis Sci. 2005;46:3323–3330. doi: 10.1167/iovs.04-1210. [DOI] [PubMed] [Google Scholar]
- Lindemann D, Racke K. Glucocorticoid inhibition of interleukin-4 (IL-4) and interleukin-13 (IL-13) induced up-regulation of arginase in rat airway fibroblasts. Naunyn Schmiedebergs Arch Pharmacol. 2003;368:546–550. doi: 10.1007/s00210-003-0839-8. [DOI] [PubMed] [Google Scholar]
- Liu K, et al. Development of the electroretinographic oscillatory potentials in normal and ROP rats. Invest Ophthalmol Vis Sci. 2006;47:5447–5452. doi: 10.1167/iovs.06-0702. [DOI] [PubMed] [Google Scholar]
- Loirand G, et al. Rho exchange factors in the cardiovascular system. Curr Opin Pharmacol. 2008;8:174–180. doi: 10.1016/j.coph.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez JG, et al. Diabetes induces pulmonary artery endothelial dysfunction by NADPH oxidase induction. American journal of physiology. Lung cellular and molecular physiology. 2008;295:L727–L732. doi: 10.1152/ajplung.90354.2008. [DOI] [PubMed] [Google Scholar]
- Lovett FA, et al. Convergence of Igf2 expression and adhesion signalling via RhoA and p38 MAPK enhances myogenic differentiation. J Cell Sci. 2006;119:4828–4840. doi: 10.1242/jcs.03278. [DOI] [PubMed] [Google Scholar]
- Maarsingh H, et al. Arginase strongly impairs neuronal nitric oxide-mediated airway smooth muscle relaxation in allergic asthma. Respir Res. 2006;7:6. doi: 10.1186/1465-9921-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarsingh H, et al. Arginase and pulmonary diseases. Naunyn Schmiedebergs Arch Pharmacol. 2008a;378:171–184. doi: 10.1007/s00210-008-0286-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarsingh H, et al. Arginine homeostasis in allergic asthma. Eur J Pharmacol. 2008b;585:375–384. doi: 10.1016/j.ejphar.2008.02.096. [DOI] [PubMed] [Google Scholar]
- Madan A, Penn JS. Animal models of oxygen-induced retinopathy. Front Biosci. 2003;8:d1030–d1043. doi: 10.2741/1056. [DOI] [PubMed] [Google Scholar]
- Marrero MB, et al. Role of Janus kinase/signal transducer and activator of transcription and mitogen-activated protein kinase cascades in angiotensin II- and platelet-derived growth factor-induced vascular smooth muscle cell proliferation. J Biol Chem. 1997;272:24684–24690. doi: 10.1074/jbc.272.39.24684. [DOI] [PubMed] [Google Scholar]
- Masuda H, et al. Modulation of intrinsic cavernous tone and nitric oxide production by arginase in rabbit corpus cavernosum. J Urol. 2004;171:490–494. doi: 10.1097/01.ju.0000088343.68746.65. [DOI] [PubMed] [Google Scholar]
- Matsushita K, et al. Diabetes-induced inhibition of voltage-dependent calcium channels in the retinal microvasculature: role of spermine. Invest Ophthalmol Vis Sci. 2010;51:5979–5990. doi: 10.1167/iovs.10-5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K, Puro DG. Topographical heterogeneity of K(IR) currents in pericyte-containing microvessels of the rat retina: effect of diabetes. J Physiol. 2006;573:483–495. doi: 10.1113/jphysiol.2006.107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken E, et al. The lipid peroxidation by-product 4-hydroxynonenal is toxic to axons and oligodendrocytes. J Cereb Blood Flow Metab. 2000;20:1529–1536. doi: 10.1097/00004647-200011000-00002. [DOI] [PubMed] [Google Scholar]
- McLeod DS, et al. Clinical and histopathologic features of canine oxygen-induced proliferative retinopathy. Invest Ophthalmol Vis Sci. 1998;39:1918–1932. [PubMed] [Google Scholar]
- Merzouk S, et al. Antioxidant status and levels of different vitamins determined by high performance liquid chromatography in diabetic subjects with multiple complications. Gen Physiol Biophys. 2003;22:15–27. [PubMed] [Google Scholar]
- Meurs H, et al. Airway hyperresponsiveness in asthma: lessons from in vitro model systems and animal models. Eur Respir J. 2008;32:487–502. doi: 10.1183/09031936.00023608. [DOI] [PubMed] [Google Scholar]
- Mills CD, et al. Macrophage arginine metabolism and the inhibition or stimulation of cancer. J Immunol. 1992;149:2709–2714. [PubMed] [Google Scholar]
- Ming XF, et al. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation. 2004;110:3708–3714. doi: 10.1161/01.CIR.0000142867.26182.32. [DOI] [PubMed] [Google Scholar]
- Mintz-Hittner HA, et al. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. 2011;364:603–615. doi: 10.1056/NEJMoa1007374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanaka K, et al. Immunohistochemical localization of arginase II and other enzymes of arginine metabolism in rat kidney and liver. Histochem J. 1998;30:741–751. doi: 10.1023/a:1003468726969. [DOI] [PubMed] [Google Scholar]
- Morris CR, et al. Nitric oxide and arginine dysregulation: a novel pathway to pulmonary hypertension in hemolytic disorders. Curr Mol Med. 2008;8:620–632. doi: 10.2174/156652408786241447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CR, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. Jama. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SM., Jr Regulation of enzymes of the urea cycle and arginine metabolism. Annu Rev Nutr. 2002;22:87–105. doi: 10.1146/annurev.nutr.22.110801.140547. [DOI] [PubMed] [Google Scholar]
- Morris SM., Jr Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol. 2009;157:922–930. doi: 10.1111/j.1476-5381.2009.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison LD, Kish SJ. Brain polyamine levels are altered in Alzheimer's disease. Neurosci Lett. 1995;197:5–8. doi: 10.1016/0304-3940(95)11881-v. [DOI] [PubMed] [Google Scholar]
- Mumenthaler SM, et al. Expression of arginase II in prostate cancer. Int J Oncol. 2008;32:357–365. [PubMed] [Google Scholar]
- Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol. 2009;158:638–651. doi: 10.1111/j.1476-5381.2009.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka T, et al. Impaired retinal circulation in patients with type 2 diabetes mellitus: retinal laser Doppler velocimetry study. Invest Ophthalmol Vis Sci. 2010;51:6729–6734. doi: 10.1167/iovs.10-5364. [DOI] [PubMed] [Google Scholar]
- Nakamura S, et al. Morphological and functional changes in the retina after chronic oxygen-induced retinopathy. PLoS One. 2012;7:e32167. doi: 10.1371/journal.pone.0032167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan SP, et al. Arginase 2 deletion reduces neuro-glial injury and improves retinal function in a model of retinopathy of prematurity. PLoS One. 2011;6:e22460. doi: 10.1371/journal.pone.0022460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita Y, et al. The key role of IL-6-arginase cascade for inducing dendritic cell-dependent CD4(+) T cell dysfunction in tumor-bearing mice. J Immunol. 2012;190:812–820. doi: 10.4049/jimmunol.1103797. [DOI] [PubMed] [Google Scholar]
- NEI. The Prevalence of Diabetic Retinopathy Among Adults in the United States. 2008 http://www.nei.nih.gov/eyedata/pbd3.asp.
- Neu J. Glutamine supplements in premature infants: why and how. J Pediatr Gastroenterol Nutr. 2003;37:533–535. doi: 10.1097/00005176-200311000-00004. [DOI] [PubMed] [Google Scholar]
- Neu J, et al. The dipeptide Arg-Gln inhibits retinal neovascularization in the mouse model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2006;47:3151–3155. doi: 10.1167/iovs.05-1473. [DOI] [PubMed] [Google Scholar]
- Neufeld AH, et al. Loss of retinal ganglion cells following retinal ischemia: the role of inducible nitric oxide synthase. Exp Eye Res. 2002;75:521–528. doi: 10.1006/exer.2002.2042. [DOI] [PubMed] [Google Scholar]
- Nicholson BP, Schachat AP. A review of clinical trials of anti-VEGF agents for diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2010;248:915–930. doi: 10.1007/s00417-010-1315-z. [DOI] [PubMed] [Google Scholar]
- Nicoletti R, et al. Vitreous polyamines spermidine, putrescine, and spermine in human proliferative disorders of the retina. Br J Ophthalmol. 2003;87:1038–1042. doi: 10.1136/bjo.87.8.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesman MR, et al. Therapeutic effect of liposomal superoxide dismutase in an animal model of retinopathy of prematurity. Neurochem Res. 1997;22:597–605. doi: 10.1023/a:1022474120512. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- O’Connor AR, et al. Long-term ophthalmic outcome of low birth weight children with and without retinopathy of prematurity. Pediatrics. 2002;109:12–18. doi: 10.1542/peds.109.1.12. [DOI] [PubMed] [Google Scholar]
- Oguni M, et al. Ontogeny of copper-zinc and manganese superoxide dismutase in the developing rat retina: immunohistochemical and immunochemical study. Ophthalmic Res. 1995;27:227–233. doi: 10.1159/000267710. [DOI] [PubMed] [Google Scholar]
- Ong WY, et al. Kainate-induced neuronal injury leads to persistent phosphorylation of cAMP response element-binding protein in glial and endothelial cells in the hippocampus. Exp Brain Res. 2000;131:178–186. doi: 10.1007/s002219900329. [DOI] [PubMed] [Google Scholar]
- Osborne NN, et al. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23:91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Osowska S, et al. Citrulline increases arginine pools and restores nitrogen balance after massive intestinal resection. Gut. 2004;53:1781–1786. doi: 10.1136/gut.2004.042317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki M, et al. Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J Clin Invest. 2002;110:331–340. doi: 10.1172/JCI15215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, et al. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HY, et al. Early diabetic changes in the nerve fibre layer at the macula detected by spectral domain optical coherence tomography. Br J Ophthalmol. 2011;95:1223–1228. doi: 10.1136/bjo.2010.191841. [DOI] [PubMed] [Google Scholar]
- Patel C, et al. Arginase as a mediator of diabetic retinopathy. Frontiers in Inflammation. doi: 10.3389/fimmu.2013.00173. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn JS, et al. Variable oxygen exposure causes preretinal neovascularization in the newborn rat. Invest Ophthalmol Vis Sci. 1993;34:576–585. [PubMed] [Google Scholar]
- Pernet V, et al. A role for polyamines in retinal ganglion cell excitotoxic death. J Neurochem. 2007;103:1481–1490. doi: 10.1111/j.1471-4159.2007.04843.x. [DOI] [PubMed] [Google Scholar]
- Pieper GM, Dondlinger LA. Plasma and vascular tissue arginine are decreased in diabetes: acute arginine supplementation restores endothelium-dependent relaxation by augmenting cGMP production. J Pharmacol Exp Ther. 1997;283:684–691. [PubMed] [Google Scholar]
- Pierce EA, et al. Regulation of vascular endothelial growth factor by oxygen in a model of retinopathy of prematurity. Arch Ophthalmol. 1996;114:1219–1228. doi: 10.1001/archopht.1996.01100140419009. [DOI] [PubMed] [Google Scholar]
- Pocernich CB, Butterfield DA. Acrolein inhibits NADH-linked mitochondrial enzyme activity: implications for Alzheimer's disease. Neurotox Res. 2003;5:515–520. doi: 10.1007/BF03033161. [DOI] [PubMed] [Google Scholar]
- Pollan C. Retinopathy of prematurity: an eye toward better outcomes. Neonatal Netw. 2009;28:93–101. doi: 10.1891/0730-0832.28.2.93. [DOI] [PubMed] [Google Scholar]
- Prager TC, et al. The pattern electroretinogram in diabetes. Am J Ophthalmol. 1990;109:279–284. doi: 10.1016/s0002-9394(14)74550-7. [DOI] [PubMed] [Google Scholar]
- Reid KM, et al. Liver I/R injury is improved by the arginase inhibitor, N(omega)-hydroxy-nor-L-arginine (nor-NOHA) Am J Physiol Gastrointest Liver Physiol. 2007;292:G512–G517. doi: 10.1152/ajpgi.00227.2006. [DOI] [PubMed] [Google Scholar]
- Retnakaran R, Zinman B. Type 1 diabetes, hyperglycaemia, and the heart. Lancet. 2008;371:1790–1799. doi: 10.1016/S0140-6736(08)60767-9. [DOI] [PubMed] [Google Scholar]
- Rivera JC, et al. Understanding retinopathy of prematurity: update on pathogenesis. Neonatology. 2011;100:343–353. doi: 10.1159/000330174. [DOI] [PubMed] [Google Scholar]
- Romero MJ, et al. Diabetes-induced vascular dysfunction involves arginase I. Am J Physiol Heart Circ Physiol. 2012;302:H159–H166. doi: 10.1152/ajpheart.00774.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero MJ, et al. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ Res. 2008;102:95–102. doi: 10.1161/CIRCRESAHA.107.155028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S, et al. Ischemia induces significant changes in purine nucleoside concentration in the retina-choroid in rats. Exp Eye Res. 1997;65:771–779. doi: 10.1006/exer.1997.0391. [DOI] [PubMed] [Google Scholar]
- Ryoo S, et al. Endothelial arginase II: a novel target for the treatment of atherosclerosis. Circ Res. 2008;102:923–932. doi: 10.1161/CIRCRESAHA.107.169573. [DOI] [PubMed] [Google Scholar]
- Ryoo S, et al. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ Res. 2006;99:951–960. doi: 10.1161/01.RES.0000247034.24662.b4. [DOI] [PubMed] [Google Scholar]
- Sabbatini M, et al. Arginase inhibition slows the progression of renal failure in rats with renal ablation. Am J Physiol Renal Physiol. 2003;284:F680–F687. doi: 10.1152/ajprenal.00270.2002. [DOI] [PubMed] [Google Scholar]
- Santhanam L, et al. Arginase and vascular aging. J Appl Physiol. 2008;105:1632–1642. doi: 10.1152/japplphysiol.90627.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JM, et al. Diabetic retinopathy, superoxide damage and antioxidants. Curr Pharm Biotechnol. 2011;12:352–361. doi: 10.2174/138920111794480507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapieha P, et al. Retinopathy of prematurity: understanding ischemic retinal vasculopathies at an extreme of life. J Clin Invest. 2010;120:3022–3032. doi: 10.1172/JCI42142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satriano J. Arginine pathways and the inflammatory response: interregulation of nitric oxide and polyamines: review article. Amino Acids. 2004;26:321–329. doi: 10.1007/s00726-004-0078-4. [DOI] [PubMed] [Google Scholar]
- Schieffer B, et al. Role of NAD(P)H oxidase in angiotensin II-induced JAK/STAT signaling and cytokine induction. Circ Res. 2000;87:1195–1201. doi: 10.1161/01.res.87.12.1195. [DOI] [PubMed] [Google Scholar]
- Schwartz IF, et al. Augmented arginine uptake, through modulation of cationic amino acid transporter-1, increases GFR in diabetic rats. Kidney Int. 2004;65:1311–1319. doi: 10.1111/j.1523-1755.2004.00508.x. [DOI] [PubMed] [Google Scholar]
- Seiler N. Oxidation of polyamines and brain injury. Neurochem Res. 2000;25:471–490. doi: 10.1023/a:1007508008731. [DOI] [PubMed] [Google Scholar]
- Sennlaub F, et al. Inducible nitric oxide synthase mediates the change from retinal to vitreal neovascularization in ischemic retinopathy. J Clin Invest. 2001;107:717–725. doi: 10.1172/JCI10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennlaub F, et al. Inducible nitric oxide synthase mediates retinal apoptosis in ischemic proliferative retinopathy. J Neurosci. 2002;22:3987–3993. doi: 10.1523/JNEUROSCI.22-10-03987.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shantz LM, Levin VA. Regulation of ornithine decarboxylase during oncogenic transformation: mechanisms and therapeutic potential. Amino Acids. 2007;33:213–223. doi: 10.1007/s00726-007-0531-2. [DOI] [PubMed] [Google Scholar]
- Shatanawi A, et al. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am J Physiol Cell Physiol. 2011;300:C1181–C1192. doi: 10.1152/ajpcell.00328.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer JD, et al. Differential regulation of macrophage arginine metabolism: a proposed role in wound healing. Am J Physiol. 1997;272:E181–E190. doi: 10.1152/ajpendo.1997.272.2.E181. [DOI] [PubMed] [Google Scholar]
- Singh R, et al. Arginase activity in human breast cancer cell lines: N(omega)-hydroxy-L-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 2000;60:3305–3312. [PubMed] [Google Scholar]
- Sinha P, et al. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–645. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- Smith G, et al. Immunolocalisation of the VEGF receptors FLT-1, KDR, and FLT-4 in diabetic retinopathy. Br J Ophthalmol. 1999;83:486–494. doi: 10.1136/bjo.83.4.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LE, et al. Essential role of growth hormone in ischemia-induced retinal neovascularization. Science. 1997;276:1706–1709. doi: 10.1126/science.276.5319.1706. [DOI] [PubMed] [Google Scholar]
- Smith LE, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- Sonta T, et al. Evidence for contribution of vascular NAD(P)H oxidase to increased oxidative stress in animal models of diabetes and obesity. Free Radic Biol Med. 2004;37:115–123. doi: 10.1016/j.freeradbiomed.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Sparkes RS, et al. The gene for human liver arginase (ARG1) is assigned to chromosome band 6q23. Am J Hum Genet. 1986;39:186–193. [PMC free article] [PubMed] [Google Scholar]
- Stempin CC, et al. Arginase induction promotes Trypanosoma cruzi intracellular replication in Cruzipain-treated J774 cells through the activation of multiple signaling pathways. Eur J Immunol. 2004;34:200–209. doi: 10.1002/eji.200324313. [DOI] [PubMed] [Google Scholar]
- Stevenson L, et al. Reduced nitro-oxidative stress and neural cell death suggests a protective role for microglial cells in TNFalpha−/− mice in ischemic retinopathy. Invest Ophthalmol Vis Sci. 2010;51:3291–3299. doi: 10.1167/iovs.09-4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano K, et al. Neuronal and glial responses to polyamines in the ischemic brain. Curr Neurovasc Res. 2005a;2:213–223. doi: 10.2174/1567202054368335. [DOI] [PubMed] [Google Scholar]
- Takano K, et al. Oxidative metabolites are involved in polyamine-induced microglial cell death. Neuroscience. 2005b;134:1123–1131. doi: 10.1016/j.neuroscience.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Takki K. Gyrate atrophy of the choroid and retina associated with hyperornithinaemia. Br J Ophthalmol. 1974;58:3–23. doi: 10.1136/bjo.58.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011;30:343–358. doi: 10.1016/j.preteyeres.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik HE, et al. Simvastatin improves diabetes-induced coronary endothelial dysfunction. J Pharmacol Exp Ther. 2006;319:386–395. doi: 10.1124/jpet.106.106823. [DOI] [PubMed] [Google Scholar]
- Thengchaisri N, et al. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arterioscler Thromb Vasc Biol. 2006;26:2035–2042. doi: 10.1161/01.ATV.0000233334.24805.62. [DOI] [PubMed] [Google Scholar]
- Tomitori H, et al. Polyamine oxidase and acrolein as novel biochemical markers for diagnosis of cerebral stroke. Stroke. 2005;36:2609–2613. doi: 10.1161/01.STR.0000190004.36793.2d. [DOI] [PubMed] [Google Scholar]
- Toque HAF, et al. Arginase II Deletion Increases Corpora Cavernosa Relaxation in Diabetic Mice. J Sex Med. 2010;8(3):722–733. doi: 10.1111/j.1743-6109.2010.02098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey KA, et al. Characterization of retinal function and glial cell response in a mouse model of oxygen-induced retinopathy. J Comp Neurol. 2011;519:506–527. doi: 10.1002/cne.22530. [DOI] [PubMed] [Google Scholar]
- Vlassara H, Palace MR. Diabetes and advanced glycation endproducts. J Intern Med. 2002;251:87–101. doi: 10.1046/j.1365-2796.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- Waddington SN, et al. Arginase activity is modulated by IL-4 and HOArg in nephritic glomeruli and mesangial cells. Am J Physiol. 1998;274:F473–F480. doi: 10.1152/ajprenal.1998.274.3.F473. [DOI] [PubMed] [Google Scholar]
- Waugh WH, et al. Oral citrulline as arginine precursor may be beneficial in sickle cell disease: early phase two results. J Natl Med Assoc. 2001;93:363–371. [PMC free article] [PubMed] [Google Scholar]
- Wei LH, et al. IL-4 and IL-13 upregulate arginase I expression by cAMP and JAK/STAT6 pathways in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2000;279:C248–C256. doi: 10.1152/ajpcell.2000.279.1.C248. [DOI] [PubMed] [Google Scholar]
- Weigert A, Brune B. Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide. 2008;19:95–102. doi: 10.1016/j.niox.2008.04.021. [DOI] [PubMed] [Google Scholar]
- White AR, et al. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension. 2006;47:245–251. doi: 10.1161/01.HYP.0000198543.34502.d7. [DOI] [PubMed] [Google Scholar]
- Witte MB, Barbul A. Arginine physiology and its implication for wound healing. Wound Repair Regen. 2003;11:419–423. doi: 10.1046/j.1524-475x.2003.11605.x. [DOI] [PubMed] [Google Scholar]
- Wong TY, Scott IU. Clinical practice. Retinal-vein occlusion. N Engl J Med. 2010;363:2135–2144. doi: 10.1056/NEJMcp1003934. [DOI] [PubMed] [Google Scholar]
- Wood PL, et al. Neurotoxicity of reactive aldehydes: the concept of "aldehyde load" as demonstrated by neuroprotection with hydroxylamines. Brain Res. 2006;1095:190–199. doi: 10.1016/j.brainres.2006.04.038. [DOI] [PubMed] [Google Scholar]
- Wu G, et al. Arginine deficiency in preterm infants: biochemical mechanisms and nutritional implications. J Nutr Biochem. 2004;15:442–451. doi: 10.1016/j.jnutbio.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Wu WC, et al. Visual acuity, optical components, and macular abnormalities in patients with a history of retinopathy of prematurity. Ophthalmology. 2012;119:1907–1916. doi: 10.1016/j.ophtha.2012.02.040. [DOI] [PubMed] [Google Scholar]
- Xia Y, Zweier JL. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6954–6958. doi: 10.1073/pnas.94.13.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. Faseb J. 2004;18:1746–1748. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- Yamakura F, et al. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J Biol Chem. 1998;273:14085–14089. doi: 10.1074/jbc.273.23.14085. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, et al. Selective reduction of the S cone electroretinogram in diabetes. Br J Ophthalmol. 1996;80:973–975. doi: 10.1136/bjo.80.11.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, et al. Inhibition of arginase I activity by RNA interference attenuates IL-13-induced airways hyperresponsiveness. J Immunol. 2006;177:5595–5603. doi: 10.4049/jimmunol.177.8.5595. [DOI] [PubMed] [Google Scholar]
- Yang Z, Ming XF. Endothelial arginase: a new target in atherosclerosis. Curr Hypertens Rep. 2006;8:54–59. doi: 10.1007/s11906-006-0041-8. [DOI] [PubMed] [Google Scholar]
- Yokota H, et al. Neuroprotection from retinal ischemia/reperfusion injury by NOX2 NADPH oxidase deletion. Invest Ophthalmol Vis Sci. 2011;52:8123–8131. doi: 10.1167/iovs.11-8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, et al. Acrolein, IL-6 and CRP as markers of silent brain infarction. Atherosclerosis. 2009;203:557–562. doi: 10.1016/j.atherosclerosis.2008.07.022. [DOI] [PubMed] [Google Scholar]
- Yu Z, et al. 3-Aminopropanal is a lysosomotropic aldehyde that causes oxidative stress and apoptosis by rupturing lysosomes. Apmis. 2003;111:643–652. doi: 10.1034/j.1600-0463.2003.1110607.x. [DOI] [PubMed] [Google Scholar]
- Yu Z, et al. Human neuroblastoma (SH-SY5Y) cells are highly sensitive to the lysosomotropic aldehyde 3-aminopropanal. Brain Res. 2004;1016:163–169. doi: 10.1016/j.brainres.2004.04.075. [DOI] [PubMed] [Google Scholar]
- Zahedi K, et al. Polyamine catabolism is enhanced after traumatic brain injury. J Neurotrauma. 2010;27:515–525. doi: 10.1089/neu.2009.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, et al. Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. Faseb J. 2001;15:1264–1266. doi: 10.1096/fj.00-0681fje. [DOI] [PubMed] [Google Scholar]
- Zhang W, et al. Arginase activity mediates retinal inflammation in endotoxin-induced uveitis. Am J Pathol. 2009a;175:891–902. doi: 10.2353/ajpath.2009.081115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, et al. NAD(P)H oxidase-dependent regulation of CCL2 production during retinal inflammation. Invest Ophthalmol Vis Sci. 2009b;50:3033–3040. doi: 10.1167/iovs.08-2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zharikov S, et al. Uric acid decreases NO production and increases arginase activity in cultured pulmonary artery endothelial cells. Am J Physiol Cell Physiol. 2008;295:C1183–C1190. doi: 10.1152/ajpcell.00075.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann N, et al. Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003;111:1863–1874. doi: 10.1172/JCI17912. [DOI] [PMC free article] [PubMed] [Google Scholar]