

Abstract
Over 1200 recessive disease genes have been described in humans. The prevalence, allelic architecture, and per-genome load of pathogenic alleles in these genes remain to be fully elucidated, as does the contribution of DNA copy-number variants (CNVs) to carrier status and recessive disease. We mined CNV data from 21,470 individuals obtained by array-comparative genomic hybridization in a clinical diagnostic setting to identify deletions encompassing or disrupting recessive disease genes. We identified 3212 heterozygous potential carrier deletions affecting 419 unique recessive disease genes. Deletion frequency of these genes ranged from one occurrence to 1.5%. When compared with recessive disease genes never deleted in our cohort, the 419 recessive disease genes affected by at least one carrier deletion were longer and located farther from known dominant disease genes, suggesting that the formation and/or prevalence of carrier CNVs may be affected by both local and adjacent genomic features and by selection. Some subjects had multiple carrier CNVs (307 subjects) and/or carrier deletions encompassing more than one recessive disease gene (206 deletions). Heterozygous deletions spanning multiple recessive disease genes may confer carrier status for multiple single-gene disorders, for complex syndromes resulting from the combination of two or more recessive conditions, or may potentially cause clinical phenotypes due to a multiply heterozygous state. In addition to carrier mutations, we identified homozygous and hemizygous deletions potentially causative for recessive disease. We provide further evidence that CNVs contribute to the allelic architecture of both carrier and recessive disease-causing mutations. Thus, a complete recessive carrier screening method or diagnostic test should detect CNV alleles.
Over 1000 recessive genetic disorders have been described, and many of their corresponding disease genes identified (http://www.omim.org). While most of these conditions are individually rare (Srinivasan et al. 2010), their collective burden on health is noteworthy (Kumar et al. 2001; McCandless et al. 2004; Dye et al. 2011a,b). However, for many recessive diseases, the overall mutational spectrum, prevalence, and carrier frequency in the population remain obscure, as does an accurate estimate of the total per-genome load of recessive carrier mutations.
Identifying disease-causing mutations in individuals affected with recessive disease can provide a molecular diagnosis that not only brings an end to an often long diagnostic odyssey (Lupski et al. 2010; Field and Boat 2011), but can also potentially enable therapeutic options (Bainbridge et al. 2011; van Karnebeek and Stockler 2012). Carrier testing for heterozygous mutations in recessive disease genes also has clinical utility. An approach to multigene carrier screening in a clinical setting was described recently by Lazarin and colleagues who screened 23,453 individuals for selected, previously reported alleles (mostly single nucleotide variants, or SNVs) associated with 108 recessive disorders (Srinivasan et al. 2010; Lazarin et al. 2013). Twenty-four percent of individuals were carriers for at least one condition, and 5.2% were carriers for two or more recessive traits. These statistics represent lower bounds, as only 417 alleles (average of approximately four alleles per gene) were assessed. Notably, only five of the 417 genotyped alleles (∼1%) were DNA copy-number variants (CNVs) (Srinivasan et al. 2010).
Incorporating next-generation sequencing into carrier screening, Bell et al. (2011) recently demonstrated that capture sequencing of 437 recessive disease genes could identify SNVs in a group of 104 individuals, most of whom were known carriers or patients with a recessive disease. A few gross deletions were assayed for and detected, although custom capture baits were designed for each based on a priori knowledge of their presence and location. The method of Bell et al. (2011) is not yet clinically available. Furthermore, the gene list did not include many recessive disease genes, for example genes for recessive deafness, intellectual disability, or adult-onset cancers. An estimation of carrier load was made (average of 2.4 mutations per individual, range 0–7), although cell lines were the source of DNA, potentially affecting the accuracy of this value (Epeldegui et al. 2007).
Whole-genome and whole-exome sequences of a number of individuals have yielded some additional information about per-genome load of carrier mutations (Gonzaga-Jauregui et al. 2012). For example, Ashley et al. (2010) analyzed the whole-genome sequence of a single individual, identifying five carrier SNVs previously reported as pathogenic, and two novel, likely damaging carrier variants. Berg et al. (2013) analyzed 80 whole-genome sequences and found an average of 5.5 potential SNV carrier variants per genome, with a range of 0–12. MacArthur et al. (2012) analyzed data from the 1000 Genomes Project (1000GP) pilot phase, identifying 26 known and 21 predicted disease-causing alleles; however, the per-person load of variants was not enumerated. The 1000GP pilot phase data were further mined by Xue et al. (2012) to more accurately identify likely damaging mutations; however, the predicted consequence of each variant (i.e., disease-causing vs. carrier) was not systematically addressed in light of the inheritance pattern(s) of any associated Mendelian disease(s). Carrier alleles were not addressed in the recent initial publication of the Phase 1 1000GP data (The 1000 Genomes Project Consortium 2012).
CNVs encompassing recessive disease genes have been described as carrier alleles and disease-causing mutations for a number of conditions, and are occasionally among the most common causative mutations for a condition (Luzi et al. 1995; Rafi et al. 1995). While some reports of genome-wide copy-number analyses have mentioned carrier CNVs (e.g., Pang et al. 2010, which examined losses affecting any of 472 recessive disease genes in data from the Venter genome), this information is absent from others (Mills et al. 2011). Thus, the genome-wide contribution of CNVs to carrier states and recessive disease-causing mutations, and the diagnostic yield of aCGH to detect these variants, are not yet clear.
We sought to describe the contribution of CNVs to recessive carrier states and recessive disease. To this end, we examined the genomes of a clinical cohort of 21,470 subjects analyzed by aCGH (also known as chromosomal microarray analysis, or CMA) (Cheung et al. 2005) for CNVs deleting part or all of at least one known recessive disease gene.
Results
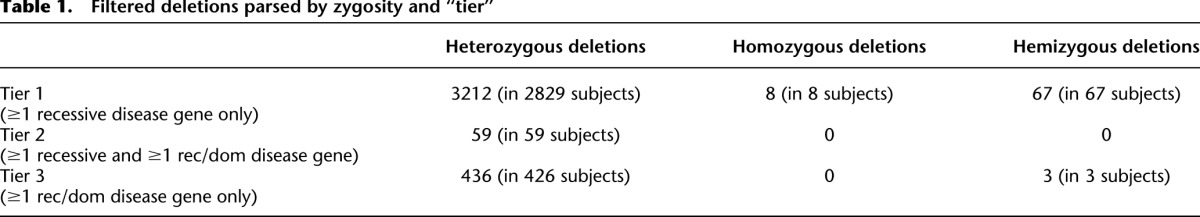
Computational analysis identified 165,595 CNVs in our cohort of 21,470 individuals using a nontargeted, genome-wide CGH array (“V7” array; 19,707 CNVs in 4928 subjects, avg. 4.0/subject) and a genome-wide CGH array with supplemental exon coverage (“V8” array; 145,888 CNVs in 16,542 subjects; avg. 8.8/subject). Initial filtering steps yielded 6372 deletions affecting at least one exon of a Mendelian disease gene and not affecting any genes associated solely with dominant diseases or diseases with complex inheritance patterns (Supplemental Fig. S1). While each deletion contained one or more genes associated with a recessive phenotype, some of these genes are solely associated with a recessive phenotype (“recessive disease genes”), whereas others are associated with both recessive and dominant phenotypes (“rec/dom disease genes”). As CNVs deleting rec/dom disease genes could contribute to either recessive or dominant disease, these CNVs were ordered in lower priority “tiers” (Tiers 2 and 3; Supplemental Fig. S1) than were CNVs affecting only recessive disease genes (Tier 1). Deletions were then parsed by zygosity and subjected to final quality control (Supplemental Methods; Supplemental Fig. S1), yielding the number of heterozygous, homozygous, and hemizygous deletions listed in Table 1. Detailed CNV data are shown as Supplemental Table S2.
Table 1.
Filtered deletions parsed by zygosity and “tier”
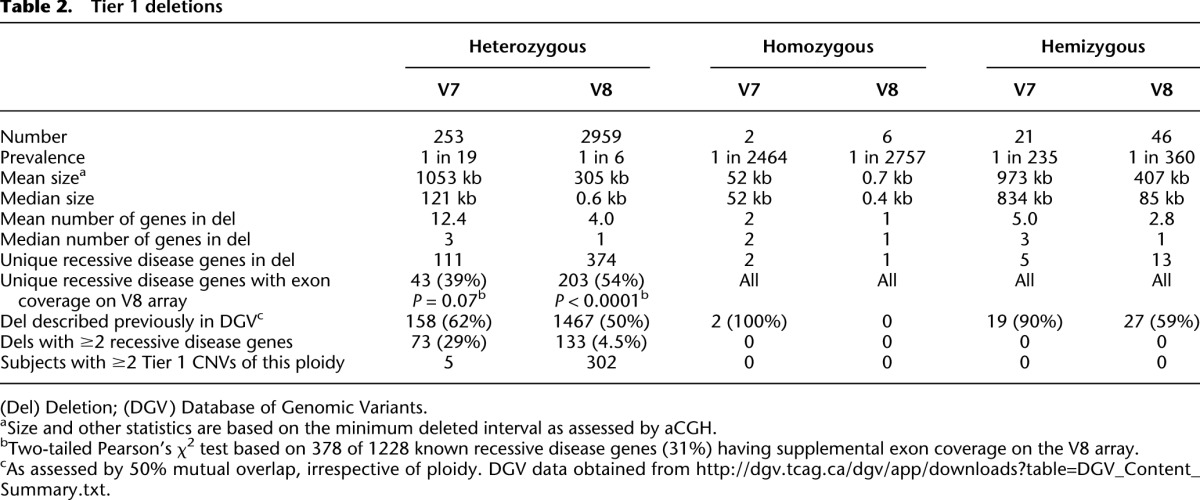
The percentage of subjects with a CNV passing the above filtering steps is displayed as Supplemental Figure S2. As Tiers 2 and 3 represent a small proportion of the total CNVs (Supplemental Figure S2) and the CNVs within them are less readily interpretable, subsequent data and analyses focus on Tier 1 deletions. Statistics concerning Tier 1 deletions are presented in depth in Table 2.
Table 2.
Tier 1 deletions
Heterozygous deletions of recessive disease genes: potential carrier CNVs
The 3212 heterozygous Tier 1 deletions (Table 2; Supplemental Table S2) represent potential carrier alleles. These were identified at an overall prevalence of approximately one per 19 subjects for V7 cases and one per six subjects for V8 cases (Table 2). The distribution of Tier 1 heterozygous deletion sizes is shown in Figure 1A, and the distribution of RefSeq genes per CNV in Figure 1B. These data demonstrate the predominance of small, single-gene events among V8 heterozygous Tier 1 deletions, likely detected because of the exon-focused design of the V8 array. The distribution of recessive disease genes per CNV is shown in Figure 1C.
Figure 1.

Attributes of the 3212 Tier 1 heterozygous deletions (potential carrier CNVs). Data are divided by array version (V7, blue; V8, plum) and based on the minimum deleted interval of each CNV. (A–C) The distributions of (A) deletion size, (B) number of RefSeq genes contained within each deletion, and (C) recessive disease genes per deletion. The spectrum of deletions identified by the V8 (exon-focused) array contains proportionally more small, single-gene events. (D) Distribution of heterozygous Tier 1 deletions per subject. A total of 18,641 subjects had no heterozygous Tier 1 deletion and are not shown. (E) Distribution of total recessive disease genes deleted per individual. This is an estimate of the distribution of per-person recessive carrier load attributable to copy-number variation. Individuals with no heterozygous Tier 1 deletion are omitted.
In total, 419 of 1228 known recessive disease genes (34%) were encompassed or disrupted by at least one Tier 1 heterozygous deletion. Of the 359 genes screened by capture sequencing by Bell et al. (2011) and designated as recessive disease genes in Supplemental Table S1, 140 were deleted by at least one Tier 1 heterozygous CNV (Supplemental Fig. S3). These copy-number variants or a proportion thereof may have evaded a capture sequencing screen as in Bell et al. (2011). The remaining 279 recessive disease genes deleted by Tier 1 heterozygous CNVs were not screened for mutations by Bell and colleagues.
Many apparent carrier CNV alleles (1625/3212, 51% of heterozygous Tier 1 deletions) matched intervals in the Database of Genomic Variants (DGV; http://dgv.tcag.ca/), a database of CNV data from control individuals, by 50% mutual overlap (Table 2; Supplemental Table S2). This serves as a positive control of our assay and analysis and suggests that this subset of deletions we report may represent nonprivate variants derived from founder events and/or recurrent CNV formation. Deletions not previously cataloged in DGV may be novel.
Of the heterozygous Tier 1 deletions described above, 490 (15.3%) affected X-linked genes in females, a special type of carrier state.
Carrier frequency
The frequency with which each of the 419 unique recessive disease genes is deleted among Tier 1 heterozygous CNVs ranges from one occurrence to 314 (1.5%; V7 and V8 combined), with a frequency distribution shown in Figure 2A and Supplemental Table S3. There are a few commonly affected genes and many rarely affected genes. We investigated whether our ascertainment of affected recessive disease genes was “saturated,” i.e., whether testing additional individuals would identify additional unique mutated recessive disease genes. Figure 2B plots our chronological ascertainment of unique recessive disease genes and indicates that it continues to rise even after assessing 2829 subjects with a Tier 1 heterozygous deletion, consistent with genomic observations of rare variant SNV alleles (Lupski et al. 2011; Marth et al. 2011).
Figure 2.

Allele frequency spectrum and assortment of heterozygous Tier 1 deletions (potential carrier CNVs). (A) Histogram of the prevalence with which each of 419 recessive disease genes is deleted by heterozygous Tier 1 CNVs, demonstrating a predominance of rarely affected genes and a few more commonly deleted genes. (B) Chronological ascertainment of unique recessive disease genes affected by heterozygous Tier 1 deletions. As more individuals with Tier 1 heterozygous deletions are analyzed (x-axis), additional recessive disease genes are identified that were not previously found to be deleted in our cohort, up to a total of 419 of 1228 known recessive disease genes (34%). The ascertainment of unique, deleted recessive disease genes continues to rise even after assessing 2829 subjects with a Tier 1 heterozygous deletion. (C) To determine whether Tier 1 heterozygous CNVs are distributed randomly among subjects, we compared the number of V8 individuals with two or three Tier 1 heterozygous CNVs deleting a single recessive disease gene (279 subjects; red line) to that expected by chance (black probability distribution; see text and Supplemental Methods). There was no statistically significant enrichment of individuals with multiple potential carrier deletions (P = 0.312), suggesting that carrier CNVs, numerically, are distributed randomly among our cohort. (D) Co-occurrence (or absence of co-occurrence) of heterozygous deletions in all pairs of 374 recessive disease genes among V8 cases displayed as a correlation matrix. Genes are plotted along each axis consecutively by genomic position. (Blue) Relative enrichment of codeletion; (red) relative paucity of codeletion.
The most frequently deleted genes in our cohort are listed in Supplemental Table S5. Some display a variety of deletion alleles, while others are the substrate of only one or a few ancestral and/or recurrent deletions. Among CNVs affecting commonly deleted genes, some very likely represent carrier alleles (e.g., deletions of NPHP1), while others appear to be too common in our cohort to be deleterious, given the incidence of the associated recessive disease(s) (e.g., deletions of a noncoding, alternative exon of LEPREL1); these common CNV alleles likely represent benign polymorphisms. Some entries in Supplemental Table S5 have intermediate frequencies, a diversity of alleles, or an unknown disease incidence in the population, rendering their roles as carrier alleles less clear.
The LEPREL1 noncoding exon deletions (above) led us to assess the overall contribution of CNVs affecting noncoding exons in our data set. We compared a test set of 2803 Tier 1 heterozygous deletions overlapping RefSeq exons to the UCSC Genome Browser CCDS track (a consensus track of the protein-coding portions of the genome; data not shown). Five hundred eighty-eight of the 2803 CNVs (21%) did not delete a coding exon; however, 238 of these 588 (40%) were the LEPREL1 deletion mentioned above, and another 240/588 (40%) were NKX2-6 deletions, also featured in Supplemental Table S5 (the RefSeq and UCSC gene models disagree concerning whether NKX2-6 is coding, and it is not part of CCDS). When the LEPREL1 and NKX2-6 CNVs are removed from the test set, the remaining 110 CNVs deleting noncoding exons make up a small proportion of the total (110/2325, 4.7%).
We compared SNV carrier frequencies in recessive disease genes screened by Lazarin et al. (2013) to deletion carrier frequencies in our cohort (Fig. 3; Supplemental Table S10). We limited this analysis to genes that were assigned purely recessive status (i.e., not rec/dom, etc.) and that have exonic probe coverage on the V8 (exon-focused) CGH array, and used only V8 Tier 1 deletion data. For the 49 genes fulfilling these criteria, SNV carrier frequency was 13.5 times higher than deletion carrier frequency; however, for five of the 49 genes (CTNS, LAMC2, SACS, CLN8, and ALDH3A2) deletion carrier frequency was higher than that of SNVs. CTNS is notable in that a single 57-kb deletion eliminating the first 10 exons of the gene is present in either the heterozygous or homozygous state in 76% of European patients with nephropathic cystinosis (OMIM #219800), making it the predominant mutation for this condition (Forestier et al. 1999). Twenty-five V8 patients were carriers for this allele, and five for larger deletions affecting CTNS. This deletion was not screened for by Lazarin et al. (2013). Figure 3 compares the SNV and deletion carrier frequencies in Supplemental Table S10, and demonstrates a lack of correlation between the two (Spearman correlation coefficient = 0.122, P[estimated] = 0.399), in support of the idea that each recessive disease locus may differ in the frequency contribution of SNV versus CNV alleles.
Figure 3.

Comparison of deletion carrier frequency in our cohort to point mutation carrier frequency reported by Lazarin et al. (2013). Values are derived from Supplemental Table S10. SNV and deletion carrier frequencies for a given gene are poorly correlated (Spearman correlation coefficient = 0.122; P[estimated] = 0.399). Genes for which deletion carrier frequency is higher than SNV carrier frequency are labeled and in green.
CNVs spanning two or more recessive disease genes
Two hundred six Tier 1 heterozygous CNVs deleted multiple recessive disease genes, with a range of two to six of such genes in each deletion (Table 2; Fig. 1C; Supplemental Table S2). These deletions contributed to the difference between the number of CNVs per individual (Fig. 1D) and the total carrier load in that individual (Fig. 1E). In contrast to a carrier point mutation, a single heterozygous deletion containing two or more recessive disease genes confers carrier status for multiple recessive conditions, each of which could manifest by a mutation on the remaining allele. In addition, if such a deletion is homozygous or hemizygous, it could lead to a complex recessive phenotype; for example, the autosomal recessive hypotonia-cystinuria syndrome (OMIM #606407) and X-linked deletions of Xp11.4-p21.2 leading to combinations of Duchenne muscular dystrophy, ornithine transcarbamylase deficiency, McLeod syndrome, and chronic granulomatous disease in males (Peng et al. 2007). Furthermore, the multiply heterozygous state could potentially itself manifest disease (i.e., digenic or oligogenic inheritance) if the genes involved encode proteins in the same pathway and contribute to a mutational load that surpasses a threshold for disease (Lupski 2012).
We investigated how many genomic regions exist in the human genome in which two or more recessive disease genes in cis are not separated by a known dominant or rec/dom disease gene or centromere, as each of these regions may predict a locus for which deletion may eliminate or disrupt two or more recessive disease genes, with potential consequences as described above. We analyzed our gene list (Supplemental Table S1) and found that 294 such genomic regions exist (Fig. 4; Supplemental Table S4; Supplemental Methods), containing between two and 10 recessive disease genes and including 19 regions on the X chromosome.
Figure 4.

Two hundred ninety-four chromosomal regions contain “consecutive” recessive disease genes. A homozygous or hemizygous deletion containing two or more recessive disease genes not interrupted by a dominant or rec/dom disease gene could lead to a complex recessive phenotype (i.e., a recessive contiguous gene syndrome); heterozygous deletion of the same region may render an individual a carrier for two or more recessive conditions. Such “consecutive” recessive disease genes are indicated by the purple bars above each chromosome, which span from the start of the first recessive disease gene in the series to the end of the last one. Selected chromosomal bands are numbered for locational reference.
Individuals with multiple carrier deletions
Three hundred seven subjects had multiple Tier 1 heterozygous deletions (range 2–7) (Fig. 1D), contributing to the total CNV carrier load per individual shown in Figure 1E. We examined whether these CNVs (specifically, the gene deletions they cause) are distributed independently among the V8 subjects in our cohort. To do this, we modeled the expected number of V8 individuals with multiple recessive disease genes deleted in trans using a binomial distribution (Fig. 2C; Supplemental Methods). The number of individuals in our cohort with multiple potential carrier deletions does not significantly deviate from the modeled expectation (P = 0.312) (Fig. 2C), suggesting that the number of CNV carrier alleles is distributed randomly among V8 subjects in our cohort.
To determine whether any specific genes in trans are more or less commonly codeleted, we performed pairwise comparisons of codeletion frequency for all pairs of 374 genes deleted by V8 Tier 1 heterozygous deletions (Fig. 2D). In addition to the expected cis interactions, we found pairs of genes in trans that showed some evidence for enriched co-occurrence of deletion.
While many recessive disease genes codeleted in a single individual have unrelated functions, a few cases exist in which these genes appear to be in related pathways. An example is subject 1399, in whom a CNV at 6p21.32 (CNV 1080) deletes three recessive immune genes: PSMB8, the disease gene for autoinflammation, lipodystrophy, and dermatosis syndrome (OMIM #256040), and TAP1 and TAP2, both disease genes for type I bare lymphocyte syndrome (OMIM #604571). In the same individual, a CNV at 16p11.2 (CNV 2211) deletes one recessive immune gene (CD19, the disease gene for common variable immunodeficiency 3 [OMIM #613493]) and two recessive disease genes unrelated to immune function. Thus, this patient is heterozygously deleted for four genes related to recessive immunological conditions due to both multiple carrier CNVs and a CNV spanning multiple recessive disease genes. Owing to the anonymized fashion in which our study was conducted, it is unknown whether this multiply heterozygous state contributes to an immune phenotype.
Genomic signatures of CNV carrier mutations
We investigated whether particular features of recessive disease genes correlated with their deletion in our cohort. To do this, we compared the 419 recessive disease genes deleted at least once by Tier 1 heterozygous CNVs with all remaining recessive disease genes. Recessive disease genes deleted at least once were farther from the nearest dominant disease gene than were the remaining recessive disease genes (median distance 1.55 Mb vs. 0.66 Mb, respectively; P = 3.3 × 10−15, Wilcoxon rank sum test with continuity correction) (Fig. 5A). Additionally, these genes were larger (median genomic size 45.1 kb vs. 24.0 kb; P = 9.3 × 10−16) and had lower fractional content of Alu elements (see Supplemental Methods) (median 11.9% vs. 18.0%; P = 2.33 × 10−11) (Fig. 5B,C).
Figure 5.

Features of recessive disease genes deleted in our cohort. The 419 recessive disease genes deleted at least once by a Tier 1 heterozygous CNV were compared with all other recessive disease genes. Those deleted in our cohort are (A) farther from the nearest dominant disease gene (P = 3.3 × 10−15); (B) larger, genomically (P = 9.3 × 10−16); and (C) had a lower fractional Alu content (P = 2.3 × 10−11) than the remaining recessive disease genes. All P-values result from the Wilcoxon rank sum test with continuity correction.
CNV validation and robustness
To investigate the possibility of false-positive CNV calls, particularly among deletions encompassing only a few (e.g., nine or fewer array probes, we examined the average probe log2 value across each heterozygous Tier 1 deletion, parsed by the number of array probes within each deletion (Supplemental Table S8; Supplemental Fig. S4). Additionally, a subset of CNVs was assessed by PCR using primers spanning the hypothesized deletion breakpoints and/or by FISH. Of 56 total PCRs, seven had equivocal results, and of the remaining 49 cases, 39 (80%) confirmed the deletion (Supplemental Fig. S4; Supplemental Table S2; data not shown). All 187 deletions assayed by FISH were confirmed, including 20 (11%) with nine or fewer array probes (Supplemental Table S2; Supplemental Fig. S4; data not shown). FISH confirmed one CNV for which PCR was equivocal (CNV 315).
Despite the above data suggesting that most small CNV calls in our data set are robust, we repeated several analyses (Figs. 1A, 2B, 5) using only data from CNVs comprised of ≥10 probes. This analysis demonstrates that our findings persist even without small CNVs (Supplemental Table S9; Supplemental Fig. S5):
Homozygous deletions
Eight homozygous deletions were identified (Fig. 6; Supplemental Table S6): A homozygous deletion at 2p21 (subject 163) deletes the recessive disease gene SLC3A1 (mutated in cystinuria, OMIM #220100) as well as much of PREPL (Fig. 6A). Homozygous deletions spanning both SLC3A1 and PREPL have been described as causing hypotonia-cystinuria syndrome (HCS; OMIM #606407); larger homozygous losses at this locus including additional genes cause the so-called 2p21 deletion syndrome, which has a more severe phenotype (Parvari et al. 2001). A spectrum of HCS deletions at this locus have been described in patients and carriers (Martens et al. 2007), consistent with the variety of heterozygous deletions at this locus among subjects in our cohort (Fig. 6A). It is unknown whether subject 163 suffers from neonatal hypotonia, failure to thrive, growth retardation, and/or type I cystinuria. If so, exon-targeted aCGH has potentially provided a molecular diagnosis for this rare, phenotypically complex recessive condition.
Figure 6.

Homozygous Tier 1 deletions. Each illustrates a unique feature of homozygous deletions. (A) The hypotonia-cystinuria syndrome (HCS) region (top) is homozygously deleted in subject 163 (middle, light gray shading on probe log2 plot). Twelve heterozygous Tier 1 deletions affect SLC3A1 (bottom) in our cohort, demonstrating diverse sizes and locations of carrier alleles. Each is plotted by minimum (thick line) and maximum (thin line) boundaries. No Tier 1 CNV affected solely PREPL. (B) NPHP1, the disease gene for juvenile nephronophthisis 1 and other conditions, is homozygously deleted as part of a compound heterozygous deletion, likely mediated by the complex repeat architecture of this region (colored bars above RefSeq genes track). Arrow indicates heterozygously deleted region. (C) Six homozygous deletions of a noncoding alternative exon 1 of LEPREL1. This CNV is mostly likely not pathogenic (see text). Database of Genomic Variant CNVs are shown (“table browser query on dgv”).
The deletion at 2q13 in subject 133 results in the complete elimination of NPHP1, LINC00116, and exon 1 of MALL (Fig. 6B). Homozygous deletion of NPHP1 is the most common cause of juvenile nephronophthisis 1 (OMIM #256100) (Konrad et al. 1996). Homozygous or compound heterozygous mutations in NPHP1 are also associated with Joubert syndrome 4 (OMIM #609583) and Senior-Løken syndrome 1 (OMIM #266900). The NPHP1 region is characterized by complex repeat structure (Fig. 6B), predisposing to various rearrangements by nonallelic homologous recombination (Saunier et al. 2000). The deletion in subject 133 appears to be a compound heterozygous deletion, as indicated by the heterozygous loss of the rest of MALL and a few microRNA genes (Fig. 6B). Neither subject 133 nor subject 163 have other likely pathogenic CNVs in addition to the homozygous deletions described (data not shown).
Six homozygous deletions eliminated a noncoding alternative exon 1 of the LEPREL1 gene at 3q28 (Fig. 6C). A homozygous missense mutation in this gene has been described in a single consanguineous pedigree segregating with high myopia with cataract and vitreoretinal degeneration (OMIM #614292) (Mordechai et al. 2011). The high frequency of homozygotes and carriers in our cohort suggests that this is most likely not a disease-causing variant for this condition.
Neither consanguinity nor absence of heterozygosity (AOH) were assessed overall in our cohort. However, SNP array data were available for a single subject (2930) with a homozygous LEPREL1 deletion (Supplemental Fig. S6; Methods available upon request). These indicated >665 Mb of AOH, consistent with consanguinity. LEPREL1 was located within one region of AOH, both explaining the origin of this subject's homozygous deletion and indicating that our algorithm correctly called the zygosity of this CNV.
Hemizygous deletions
Sixty-seven males in our cohort had hemizygous Tier 1 deletions (Table 2; Supplemental Tables S2, S7). In total, hemizygous Tier 1 deletions encompass 14 unique, recessive, X-linked genes (Supplemental Table S7). The most commonly deleted genes are DMD (16 hemizygous deletions; one per 1034 V8 subjects), STS (10 hemizygous deletions; one per 1654 V8 subjects), and OPN1LW (eight hemizygous deletions; one per 2068 V8 subjects).
Discussion
Recessive genetic diseases, while individually rare, number in the thousands and contribute substantially to the burden of disease with a genetic basis. The extent to which DNA copy-number variations (CNVs) contribute to recessive disease or recessive carrier mutations has not been investigated on a genome-wide scale in a large cohort. We sought to identify and describe CNV carrier and recessive disease-causing alleles using genomic copy-number data from a clinical cohort of 21,470 individuals.
Heterozygous deletions of recessive disease genes: potential carrier CNVs
Potential carrier CNVs were identified by mining our data set for heterozygous deletions encompassing or disrupting recessive disease genes. We calculated the prevalence of such events using two different CGH array platforms. These values provide provisional estimates of the CNV contribution to recessive carrier load upon which future investigations can improve. The V8 array, with both backbone probes and supplemental probes localized to the exons of selected genes, identified more CNVs and CNVs of markedly smaller minimum size than did the V7 (backbone probes only) array. Thus, aCGH using a higher-resolution, exon-focused design enables the detection of additional potential carrier alleles, particularly small CNVs encompassing or disrupting single genes.
An abundance of small carrier deletions, many of which encompass only a few array probes, raises the possibility of false-positive CNV calls (Haraksingh et al. 2011). We took multiple measures to filter out low-quality CNVs and found that over 50% of filtered CNVs matched alleles in the Database of Genomic Variants (DGV), and a high percentage of filtered CNVs tested by PCR or FISH were validated. Additionally, when we repeated several of our analyses using only CNVs spanning ≥10 probes, each of our original conclusions was upheld (Supplemental Fig. S5). Moreover, an abundance of small CNVs is consistent with the overall allele frequency spectrum of CNVs in personal genomes (e.g., Wheeler et al. 2008; Conrad et al. 2010; Mills et al. 2011).
Carrier frequency
We estimated gene-specific CNV carrier frequencies for the 419 recessive disease genes deleted heterozygously in our cohort; these are characterized by a few commonly mutated genes and a “long tail” of more rarely variant loci, consistent with observations of SNV carrier alleles (Srinivasan et al. 2010). The ascertainment of unique recessive disease genes deleted lessened over time, but did not appear to reach saturation, reminiscent of the continued identification of novel SNPs in personal genomes despite a multitude of previously sequenced genomes (Lupski et al. 2011; Marth et al. 2011; Gonzaga-Jauregui et al. 2012). Abundant novel alleles are also consistent with low-frequency technical error; however, our finding of continued novelty persists even among our highest confidence (≥10 probe) CNVs, indicating that the observed copious diversity of CNV carrier alleles exists and extends even to large CNVs.
We compared the gene-specific CNV carrier frequencies in our cohort to SNV carrier frequencies recently published by Lazarin et al. (2013) and found that the two are not correlated. Our study also identified CNVs affecting hundreds of recessive disease genes not assayed by either Lazarin et al. (2013) or Bell et al. (2011) in their approaches to carrier screening. This indicates that while an SNV-centric carrier screen of genes with high SNV carrier frequency in the population may be ideal for efficient SNV carrier screening, the list of priority genes may be different if carrier states caused by CNVs are considered.
CNVs spanning two or more recessive disease genes
By identifying heterozygous CNVs in our cohort that span two or more recessive disease genes, we identified variants that, in a single allele, may render the affected individual a carrier for multiple recessive diseases. Such CNVs were present in ∼1% of subjects in our cohort and encompassed between two and six recessive disease genes each. Our genome-wide prediction of loci susceptible to this kind of mutation (Fig. 4; Supplemental Table S4) indicates that up to 10 recessive disease genes can be deleted in cis without deleting a known dominant or rec/dom disease gene. We hypothesize that the carrier load from CNVs may be particularly skewed in individuals with deletions spanning multiple recessive disease genes, and may even exceed the SNV carrier load.
Analogous to dominant contiguous gene syndromes, in which two or more discrete dominant disease genes may contribute to a complex phenotype (Campbell et al. 2012), recessive contiguous gene syndromes have also been described (e.g., hypotonia-cystinuria syndrome; OMIM #606407). By identifying all contiguous recessive disease genes in the genome, we delineate hundreds of potential complex recessive phenotypes (recessive contiguous gene syndromes) that could result from homozygous or hemizygous deletion of these loci, further developing the concept of oligogenic inheritance and cis-genetics, in contrast to the trans-genetics in which Mendelism is so rooted (Lupski et al. 2011). Such phenotypes may challenge diagnostic acumen; thus, a laboratory method that assays for CNVs at high resolution in these predicted genomic regions may facilitate a molecular diagnosis.
Individuals with multiple carrier deletions
In addition to CNVs affecting multiple recessive disease genes, multiple carrier CNVs in the same individual could contribute to an overall load of carrier mutations. Indeed, a subset of subjects in our cohort (>1%) has multiple potential CNV carrier alleles. Estimates of the recessive carrier load per individual have been proposed for some time (Muller 1950; Morton et al. 1956; Morton 1960) and, recently, empirical estimates have been made from genome-wide or targeted analyses (Bell et al. 2011; Berg et al. 2013; Lazarin et al. 2013), suggesting that the average per-person SNV carrier load is likely <10 carrier mutations. Thus, our data suggest that multiple CNVs may contribute appreciably to the overall carrier load in some individuals.
Genomic signatures of CNV carrier mutations
We found that recessive disease genes deleted at least once in our cohort tended to be located, in terms of median distance, two to three times farther from the nearest dominant disease gene than were never-deleted recessive disease genes. This correlation may suggest a mechanism whereby an adjacent locus (dominant gene) can affect population allele frequency at another locus (CNV frequency in a recessive disease gene), since a deletion encompassing both genes would potentially lead to dominant disease and be selected against (Fig. 7A,B). This finding persists among CNVs ≥10 probes, the most robust group of CNVs in our analyses (Supplemental Fig. S5C).
Figure 7.
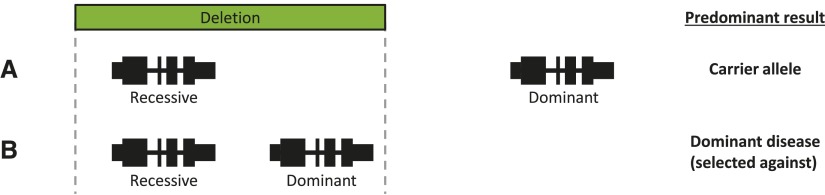
Model explaining the potential impact of adjacent genomic features on gene-specific deletion carrier frequency. (A) Recessive disease gene without a nearby dominant disease gene (e.g., NPHP1). Most deletions encompassing this recessive disease gene do not also delete a dominant disease gene and are thus solely carrier deletions. (B) Recessive disease gene near to a haploinsufficient dominant disease gene (e.g., MYO15A and RAI1, respectively). Many deletions of the recessive disease gene also delete the dominant disease gene, rendering the individual a carrier and affected with dominant disease. This mutation is selected out of the population.
We also found that recessive disease genes deleted at least once in our cohort tended to be larger, genomically, than those never deleted. This correlation may be the result of a confounding variable, for example number of exons or likelihood of exon coverage on the V8 array. However, it may also suggest that, for large recessive disease genes, many deletions do not span beyond the boundary of the gene. Thus, irrespective of this recessive disease gene's proximity to the nearest dominant disease gene, many deletions in this gene are solely carrier deletions.
Furthermore, recessive disease genes deleted at least once in our cohort tended to have a lower Alu content than recessive disease genes never deleted. It has been proposed that Alu elements play a role in the generation of genome rearrangements by various mechanisms (Lehrman et al. 1985; Boone et al. 2011). Thus, loci with high Alu density may be predisposed to copy-number change, and as such, these loci might be expected to be enriched for variants contributing to dominant (Boone et al. 2011) or semi-dominant (Lehrman et al. 1985) phenotypes, for which new mutation is important. In our cohort, Alu density is not associated with recessive disease genes affected by CNV carrier states, potentially because of the minor importance of new mutation, as compared with inherited variation, for recessive alleles. The reason for a lower Alu concentration (as opposed to statistically equivalent) among recessive disease genes deleted at least once is unknown.
Homozygous deletions
Three homozygously deleted regions illustrate the ability of screening aCGH to provide a molecular diagnosis for recessive disease, including one case of a two-gene homozygous deletion potentially leading to HCS, a recessive contiguous gene syndrome. The total number of homozygotes in our cohort appears small, given that this is a population screened because of a clinical phenotype. The subjects studied are largely from a diverse urban population with no historical evidence for a high percentage of consanguineous unions, although frequency of consanguinity in our clinical cohort has not been experimentally assessed. The possibility of an overly stringent log2 cutoff for homozygous deletions (−2) can likely be excluded (Supplemental Fig. S7). One factor that may contribute to the small number of homozygous CNVs is that our CNV-calling algorithm does not incorporate dim probes (probes with very low fluorescence) into CNV calls.
Hemizygous deletions
The V8 hemizygous deletion frequency and V8 carrier frequency are similar for most genes in Supplemental Table S7. This is consistent with the following: (1) For nonlethal X-linked recessive conditions, the prevalence in the population of carrier females and affected males should be somewhat similar; and (2) the mothers of boys identified by aCGH to have an X-linked potentially pathogenic CNV are often tested by aCGH in order to estimate recurrence risk; such carrier females are included in our cohort. Two genes, however, do not follow the above pattern: OCRL and OPN1LW.
Heterozygous OCRL deletions are present in 314 females in our cohort (Supplemental Tables S2, S5). All are the same, single-exon (exon 16 of 24) in-frame loss, and are likely benign based on the high frequency of this event in our cohort and the low incidence of Lowe syndrome (OMIM #309000) and Dent disease 2 (OMIM #300555). In contrast to the large number of female heterozygotes, there is only a single male with a hemizygous OCRL deletion (an 11-probe loss of exons 5 and 6). Whereas it is possible that the exon 16 OCRL losses are technical artifacts, it is perhaps more likely that they are not ascertained in the hemizygous state owing to dim probes that are excluded from the CNV-calling algorithm. Note also that no homozygous OCRL deletion was detected.
OPN1LW encodes red cone pigment, associated with red-green color vision defects (OMIM #303700 and #303900) present in ∼8% of northern European males (Deeb and Motulsky 1993). This gene was deleted in eight V8 males compared with no heterozygous deletions in V8 females. This may be explained by the homology shared between OPN1LW and the nearby OPN1MW (green cone pigment gene; often present in multiple iterative copies): A deletion resulting from recombination between OPN1LW and OPN1MW, the most common mutation causing red-green colorblindness (Nathans et al. 1986), leaves a remaining hybrid gene copy and may leave one or more intact gene copies. Thus, at this multiallelic CNV locus, CGH probe log2 ratios are expected to be attenuated; indeed, the average log2 in our cohort for hemizygous deletions of this locus was −1.13, compared with −2.97 for hemizygous deletions of all other genes. Log2 ratios of heterozygous deletions do not fall below the computational threshold (−0.415) defined by our algorithm (data not shown), explaining the lack of carrier CNVs in OPN1LW in Supplemental Tables S2 and S7.
Limitations
The following are potential limitations of our study, which suggest opportunities for future research: (1) We did not examine copy-number gains; (2) many deletions were not validated by an alternative molecular method in addition to aCGH; (3) we did not manually review each deletion, its relation to gene structure, and consider published disease-causing and benign variants to assign its potential pathogenicity. This requires an approach, for example in Boone et al. (2013) and de Leeuw et al. (2012), that has yet to be fully automated and can be difficult when the mutation is novel or has not been studied by case-control or experimental (i.e., model organism) approaches; (4) the minimum interval of copy-number change was used to determine the genes deleted by each CNV. Thus, some CNVs may encompass additional genes; (5) pseudogenes may prevent array probe coverage of some recessive disease genes (e.g., SMN1, which does not have exonic probe coverage on our array because of the pseudogene SMN2); (6) some exons are not unique or large enough to contain multiple probes; thus, exon coverage is not equal for all exon-covered genes; (7) our assignment of inheritance patterns to genes was largely computational and may benefit from manual review of these 2125 genes; (8) deletions falling within known genomic disorder regions were only eliminated if they overlapped a known, dominant disease gene; (9) deletions encompassing rec/dom disease genes were eliminated from most analyses because of the possibility that heterozygous mutations in these genes may cause dominant disease; however a proportion are likely true carrier mutations.
Final considerations
The CNVs found in our study that represent true carrier states and disease alleles should be assayed in any comprehensive recessive diagnostic or carrier screening test. An important avenue for future research will be to ascertain the yield of recessive carrier and disease-causing alleles (CNV, SNV, and other variant types) identified by clinical exome and whole-genome sequencing.
Methods
Subjects
Array CGH was performed clinically on DNA from 21,470 subjects, comprised of patients and prenatal cases referred to the Baylor College of Medicine Medical Genetics Laboratories between July 2008 and April 2012 for suspicion of a condition of genetic origin, and their parents. Subject data were anonymized for our analyses.
Array comparative genomic hybridization (aCGH)
DNA from 4928 subjects was analyzed using the BCM version 7 (“V7”) CGH array, a custom, 105k-probe, nontargeted oligonucleotide array characterized primarily by “backbone” probes distributed with relatively even spacing throughout the genome (one probe per ∼30 kb); several dozen genomic disorder regions and disease loci were also interrogated with enhanced probe coverage (Kang et al. 2010). A total of 16,542 subjects were analyzed with the BCM version 8 (“V8”) CGH array, a custom, 180k-probe oligonucleotide array based on V7, but with supplemental exonic and intronic probe coverage of over 1700 known and candidate disease genes, including 378 known recessive disease genes, 420 known dominant disease genes, and 93 genes associated with recessive and dominant disease (rec/dom disease genes) (Supplemental Table S1). The V8 array and aCGH procedures have been described (Boone et al. 2010).
Assigning inheritance patterns to genes
We compiled a list of all known Mendelian disease genes as of May 2012 and assigned to each one or more inheritance patterns based on its association with Online Mendelian Inheritance in Man (OMIM; http://www.omim.org) disease phenotypes (Supplemental Methods). The resultant list contains 1228 recessive disease genes, 732 dominant genes, and 161 genes associated with both recessive and dominant genetic disease (rec/dom disease genes) (Supplemental Table S1).
Identifying potential carrier CNVs
To identify potential carrier variants, CNVs were filtered as described in the Supplemental Methods and in Supplemental Fig. S1. Briefly: (1) As the effect on gene function of duplications and higher order copy-number gains is challenging to predict (Bacino and Cheung 2010; Boone et al. 2013), only deletions were included in our analysis; (2) Only CNVs affecting at least one recessive disease gene and no dominant disease genes were included; this was done to exclude CNVs that, in the heterozygous state, may cause disease and thus not be representative of carrier variants found in the general population; (3) Only autosomal CNVs and X-linked CNVs in females with log2 values consistent with heterozygous deletion were included; (4) Three “tiers” of potential carrier mutations were assigned (Tiers 1, 2, and 3) based on the likelihood of association with recessive disease and nonassociation with dominant disease (see Supplemental Fig. S1; Supplemental Results); (5) Filtering was based on the minimum extent of each CNV. All genomic coordinates are based on the February 2009 assembly of the reference human genome (GRCh37/hg19), unless otherwise specified.
Identifying hemizygous and homozygous CNVs affecting recessive disease genes
Hemizygous CNVs were the X- and Y-linked CNVs in males remaining after filtering as described above for carrier CNVs. Homozygous CNVs were all other remaining CNVs with an average probe ratio log2 consistent with a homozygous loss (Supplemental Figs. S1, S7).
CNV validation
A subset of deletions was subjected to PCR (size range 3–114 array probes) and/or FISH (size range 3–703 probes) validation as described previously (Boone et al. 2010; Supplemental Table S2; Supplemental Fig. S4).
Data access
CNV data have been deposited at dbVar (http://www.ncbi.nlm.nih.gov/dbvar/studies/) under accession number nstd80.
Competing interest statement
J.R.L. is a paid consultant for Athena Diagnostics, holds stock ownership in 23andMe, Inc. and Ion Torrent Systems, Inc., and is a co-inventor on multiple United States and European patents related to molecular diagnostics. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered in the Medical Genetics Laboratories (http://www.bcm.edu/geneticlabs/).
Acknowledgments
We thank Drs. Jonathan Berg and Stephen Kingsmore for help in assembling the recessive disease gene list, and Joep de Ligt for commenting on the manuscript. P.M.B. and I.M.C. are fellows of the Baylor College of Medicine Medical Scientist Training Program (T32GM007330). P.M.B. is also supported by a National Eye Institute (NEI) Training Program grant (T32EY007102) from the United States National Institutes of Health (NIH) and grants from the Wintermann Foundation and the Baylor Research Advocates for Student Scientists. This work was supported in part by the National Institute of Neurological Disorders and Stroke (RO1NS058529 to J.R.L.) and the National Human Genome Research Institute (U54HG006542).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.156075.113.
References
- The 1000 Genomes Project Consortium 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley EA, Butte AJ, Wheeler MT, Chen R, Klein TE, Dewey FE, Dudley JT, Ormond KE, Pavlovic A, Morgan AA, et al. 2010. Clinical assessment incorporating a personal genome. Lancet 375: 1525–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacino CA, Cheung S-W 2010. Introductory comments on special section–genomic microduplications: When adding may equal subtracting. Am J Med Genet A 152A: 1063–1065 [DOI] [PubMed] [Google Scholar]
- Bainbridge MN, Wiszniewski W, Murdock DR, Friedman J, Gonzaga-Jauregui C, Newsham I, Reid JG, Fink JK, Morgan MB, Gingras M-C, et al. 2011. Whole-genome sequencing for optimized patient management. Sci Transl Med 3: 87re83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, et al. 2011. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med 3: 65ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JS, Adams M, Nassar N, Bizon C, Lee K, Schmitt CP, Wilhelmsen KC, Evans JP 2013. An informatics approach to analyzing the incidentalome. Genet Med 15: 36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, Kang S-HL, Yang Y, Wiszniewska J, Nowakowska BA, et al. 2010. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat 31: 1326–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Liu P, Zhang F, Carvalho CMB, Towne CF, Batish SD, Lupski JR 2011. Alu-specific microhomology-mediated deletion of the final exon of SPAST in three unrelated subjects with hereditary spastic paraplegia. Genet Med 13: 582–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Soens ZT, Campbell IM, Stankiewicz P, Cheung SW, Patel A, Beaudet AL, Plon SE, Shaw CA, McGuire AL, et al. 2013. Incidental copy-number variants identified by routine genome testing in a clinical population. Genet Med 15: 45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Yatsenko SA, Hixson P, Reimschisel T, Thomas M, Wilson W, Dayal U, Wheless JW, Crunk A, Curry C, et al. 2012. Novel 9q34.11 gene deletions encompassing combinations of four Mendelian disease genes: STXBP1, SPTAN1, ENG, and TOR1A. Genet Med 14: 868–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, Yatsenko SA, Cooper ML, Furman P, Stankiewicz P, et al. 2005. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med 7: 422–432 [DOI] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. 2010. Origins and functional impact of copy number variation in the human genome. Nature 464: 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw N, Dijkhuizen T, Hehir-Kwa JY, Carter NP, Feuk L, Firth HV, Kuhn RM, Ledbetter DH, Martin CL, van Ravenswaaij-Arts CMA, et al. 2012. Diagnostic interpretation of array data using public databases and internet sources. Hum Mutat 33: 930–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeb SS, Motulsky AG 1993. Red-green color vision defects. In GeneReviews (ed. Pagon RA). University of Washington, Seattle, WA. [Google Scholar]
- Dye DE, Brameld KJ, Maxwell S, Goldblatt J, Bower C, Leonard H, Bourke J, Glasson EJ, O'Leary P 2011a. The impact of single gene and chromosomal disorders on hospital admissions of children and adolescents: A population-based study. Public Health Genomics 14: 153–161 [DOI] [PubMed] [Google Scholar]
- Dye DE, Brameld KJ, Maxwell S, Goldblatt J, O'Leary P 2011b. The impact of single gene and chromosomal disorders on hospital admissions in an adult population. J Community Genet 2: 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epeldegui M, Hung YP, McQuay A, Ambinder RF, Martínez-Maza O 2007. Infection of human B cells with Epstein-Barr virus results in the expression of somatic hypermutation-inducing molecules and in the accrual of oncogene mutations. Mol Immunol 44: 934–942 [DOI] [PubMed] [Google Scholar]
- Field MJ, Boat TF, ed. 2011. Rare diseases and orphan products: Accelerating research and development National Academy of Sciences, Washington, D.C. [PubMed] [Google Scholar]
- Forestier L, Jean G, Attard M, Cherqui S, Lewis C, van't Hoff W, Broyer M, Town M, Antignac C 1999. Molecular characterization of CTNS deletions in nephropathic cystinosis: Development of a PCR-based detection assay. Am J Hum Genet 65: 353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C, Lupski JR, Gibbs RA 2012. Human genome sequencing in health and disease. Annu Rev Med 63: 35–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraksingh RR, Abyzov A, Gerstein M, Urban AE, Snyder M 2011. Genome-wide mapping of copy number variation in humans: Comparative analysis of high resolution array platforms. PLoS ONE 6: e27859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S-HL, Shaw C, Ou Z, Eng PA, Cooper ML, Pursley AN, Sahoo T, Bacino CA, Chinault AC, Stankiewicz P, et al. 2010. Insertional translocation detected using FISH confirmation of array-comparative genomic hybridization (aCGH) results. Am J Med Genet A 152A: 1111–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad M, Saunier S, Heidet L, Silbermann F, Benessy F, Calado J, Le Paslier D, Broyer M, Gubler M-C, Antignac C 1996. Large homozygous deletions of the 2q13 region are a major cause of juvenile nephronophthisis. Hum Mol Genet 5: 367–371 [DOI] [PubMed] [Google Scholar]
- Kumar P, Radhakrishnan J, Chowdhary MA, Giampietro PF 2001. Prevalence and patterns of presentation of genetic disorders in a pediatric emergency department. Mayo Clin Proc 76: 777–783 [DOI] [PubMed] [Google Scholar]
- Lazarin GA, Haque IS, Nazareth S, Iori K, Patterson AS, Jacobson JL, Marshall JR, Seltzer WK, Patrizio P, Evans EA, et al. 2013. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: Results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med 15: 178–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrman MA, Schneider WJ, Südhof TC, Brown MS, Goldstein JL, Russell DW 1985. Mutation in LDL receptor: Alu-Alu recombination deletes exons encoding transmembrane and cytoplasmic domains. Science 227: 140–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR 2012. Digenic inheritance and Mendelian disease. Nat Genet 44: 1291–1292 [DOI] [PubMed] [Google Scholar]
- Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DCY, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, et al. 2010. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med 362: 1181–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA 2011. Clan genomics and the complex architecture of human disease. Cell 147: 32–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzi P, Rafi MA, Wenger DA 1995. Characterization of the large deletion in the GALC gene found in patients with Krabbe disease. Hum Mol Genet 4: 2335–2338 [DOI] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, et al. 2012. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335: 823–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens K, Heulens I, Meulemans S, Zaffanello M, Tilstra D, Hes FJ, Rooman R, François I, de Zegher F, Jaeken J, et al. 2007. Global distribution of the most prevalent deletions causing hypotonia-cystinuria syndrome. Eur J Hum Genet 15: 1029–1033 [DOI] [PubMed] [Google Scholar]
- Marth GT, Yu F, Indap AR, Garimella K, Gravel S, Leong WF, Tyler-Smith C, Bainbridge M, Blackwell T, Zheng-Bradley X, et al. 2011. The functional spectrum of low-frequency coding variation. Genome Biol 12: R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCandless SE, Brunger JW, Cassidy SB 2004. The burden of genetic disease on inpatient care in a children's hospital. Am J Hum Genet 74: 121–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470: 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordechai S, Gradstein L, Pasanen A, Ofir R, El Amour K, Levy J, Belfair N, Lifshitz T, Joshua S, Narkis G, et al. 2011. High myopia caused by a mutation in LEPREL1, encoding prolyl 3-hydroxylase 2. Am J Hum Genet 89: 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton NE 1960. The mutational load due to detrimental genes in man. Am J Hum Genet 12: 348–364 [PMC free article] [PubMed] [Google Scholar]
- Morton NE, Crow JF, Muller HJ 1956. An estimate of the mutational damage in man from data on consanguineous marriages. Proc Natl Acad Sci 42: 855–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ 1950. Our load of mutations. Am J Hum Genet 2: 111–176 [PMC free article] [PubMed] [Google Scholar]
- Nathans J, Piantanida TP, Eddy RL, Shows TB, Hogness DS 1986. Molecular genetics of inherited variation in human color vision. Science 232: 203–210 [DOI] [PubMed] [Google Scholar]
- Pang AW, MacDonald JR, Pinto D, Wei J, Rafiq MA, Conrad DF, Park H, Hurles ME, Lee C, Venter JC, et al. 2010. Towards a comprehensive structural variation map of an individual human genome. Genome Biol 11: R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvari R, Brodyansky I, Elpeleg O, Moses S, Landau D, Hershkovitz E 2001. A recessive contiguous gene deletion of chromosome 2p16 associated with cystinuria and a mitochondrial disease. Am J Hum Genet 69: 869–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Redman CM, Wu X, Song X, Walker RH, Westhoff CM, Lee S 2007. Insights into extensive deletions around the XK locus associated with McLeod phenotype and characterization of two novel cases. Gene 392: 142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafi MA, Luzi P, Chen YQ, Wenger DA 1995. A large deletion together with a point mutation in the GALC gene is a common mutant allele in patients with infantile Krabbe disease. Hum Mol Genet 4: 1285–1289 [DOI] [PubMed] [Google Scholar]
- Saunier S, Calado J, Benessy F, Silbermann F, Heilig R, Weissenbach J, Antignac C 2000. Characterization of the NPHP1 locus: Mutational mechanism involved in deletions in familial juvenile nephronophthisis. Am J Hum Genet 66: 778–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan BS, Flannick J, Patterson AS, Chang CC, Pham T, Young S, Kaushal A, Lee J, Patrizio P, Evans EA 2010. A universal carrier test for the long tail of Mendelian disease. Reprod Biomed Online 21: 537–551 [DOI] [PubMed] [Google Scholar]
- van Karnebeek CDM, Stockler S 2012. Treatable inborn errors of metabolism causing intellectual disability: A systematic literature review. Mol Genet Metab 105: 368–381 [DOI] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Makhijani V, Roth GT, Gomes X, et al. 2008. The complete diploid genome of an individual by massively parallel sequencing. Nature 452: 872–876 [DOI] [PubMed] [Google Scholar]
- Xue Y, Chen Y, Ayub Q, Huang N, Ball EV, Mort M, Phillips AD, Shaw K, Stenson PD, Cooper DN, et al. 2012. Deleterious- and disease-allele prevalence in healthy individuals: Insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet 91: 1022–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]