

Abstract
We delineated and analyzed directly oriented paralogous low-copy repeats (DP-LCRs) in the most recent version of the human haploid reference genome. The computationally defined DP-LCRs were cross-referenced with our chromosomal microarray analysis (CMA) database of 25,144 patients subjected to genome-wide assays. This computationally guided approach to the empirically derived large data set allowed us to investigate genomic rearrangement relative frequencies and identify new loci for recurrent nonallelic homologous recombination (NAHR)-mediated copy-number variants (CNVs). The most commonly observed recurrent CNVs were NPHP1 duplications (233), CHRNA7 duplications (175), and 22q11.21 deletions (DiGeorge/velocardiofacial syndrome, 166). In the ∼25% of CMA cases for which parental studies were available, we identified 190 de novo recurrent CNVs. In this group, the most frequently observed events were deletions of 22q11.21 (48), 16p11.2 (autism, 34), and 7q11.23 (Williams-Beuren syndrome, 11). Several features of DP-LCRs, including length, distance between NAHR substrate elements, DNA sequence identity (fraction matching), GC content, and concentration of the homologous recombination (HR) hot spot motif 5′-CCNCCNTNNCCNC-3′, correlate with the frequencies of the recurrent CNVs events. Four novel adjacent DP-LCR-flanked and NAHR-prone regions, involving 2q12.2q13, were elucidated in association with novel genomic disorders. Our study quantitates genome architectural features responsible for NAHR-mediated genomic instability and further elucidates the role of NAHR in human disease.
Copy-number variants (CNVs) are an important cause of multiple genomic disorders (Stankiewicz and Lupski 2010; Girirajan et al. 2011). One major mechanism responsible for CNV formation is nonallelic homologous recombination (NAHR) (Stankiewicz and Lupski 2002), which occurs between two paralogous low-copy repeats (LCRs) or segmental duplications (Bailey et al. 2002). Utilizing directly oriented paralogous LCR (DP-LCR) copies in cis as recombination substrates for ectopic crossovers, NAHR can lead to recurrent genomic deletions and reciprocal duplications. Recent evidence suggests a greater than twofold genome-wide enrichment for CNVs between DP-LCRs (Li et al. 2012). NAHR events in trans between LCRs on nonhomologous chromosomes can cause recurrent constitutional translocations (Giglio et al. 2002; Ou et al. 2011). For LCRs in inverted orientation, Dittwald et al. (2013) showed that 12.0% of the human genome is potentially susceptible to NAHR-mediated inversions between inverse paralogous LCRs, with 942 genes (99 of which are on the X chromosome) predicted to be disrupted secondary to such an inversion. Locus-specific studies have shown that LCR size is correlated with NAHR frequency, suggesting that ectopic synapsis precedes ectopic crossing-over (Liu et al. 2012).
To date, ∼40 nonoverlapping genomic loci with deletion and/or reciprocal duplication associated with known syndromes have been identified as genomic disorders (Lupski 1998, 2009; Mefford 2009; Liu et al. 2012; Vissers and Stankiewicz 2012). Bioinformatic analyses have revealed many more regions of genomic instability in the human genome that are potentially prone to recurrent DNA rearrangements via NAHR; some of them may be pathogenic, but their phenotypic consequences remain to be elucidated.
Using genome-wide bioinformatic analyses in the human genome build hg16 (July 2003), Sharp et al. (2005) predicted 130 genomic intervals flanked by DP-LCRs >10 kb in size, of >95% DNA sequence identity, with the distance between the DP-LCRs ranging from 0.05–10 Mb. Using the same parameters for bioinformatic analyses of the genome build hg19 (February 2009), Liu et al. (2012) identified 608 intervals that collapsed into 89 regions prone to DP-LCR/NAHR. Most of the differences between these data sets result from the different DP-LCRs identified in these genome builds as well as various methods for collapsing the overlapping regions.
Here, we constructed bioinformatically a new genome-wide map of the DP-LCR-flanked regions in human genome build hg19 using a concept of LCR clusters. We then queried and cross-referenced our database of 25,144 high-resolution genomic analyses performed on patients referred for chromosomal microarray analysis (CMA) (Cheung et al. 2005). This approach enabled us to determine the relative frequencies in this clinical population of known recurrent genomic disorders and also to quantitate genome-wide genomic architectural features that are associated with individual locus events, to gain insights into the parameters rendering genomic instability. The frequency for ascertaining these genomic disorders varies dramatically and, as predicted previously, may reflect genome architecture and mechanism. We report the computationally determined genomic features that correlate with the empirically observed frequency of de novo recurrent rearrangements and further test, on a genome-wide scale, the “ectopic synapsis precedes ectopic crossing-over” hypothesis.
Results
To investigate genomic regions prone to NAHR instability, we used the following approaches. (1) We applied bioinformatic genome-wide analyses of genomic architecture for features/parameters derived from empirical locus-specific studies. (2) We queried a clinical population manifesting phenotypes due to genomic rearrangements, the CMA database at the Medical Genetics Laboratories (MGL) of Baylor College of Medicine (BCM), for genomic instability regions. Such intervals were indicated by genome-wide analyses of architectural features lending susceptibility to rearrangements and the quantitative characteristics of such structural features as well as the quantitative frequencies of rearrangements at a given locus. (3) We performed statistical modeling of the correlation between genomic architecture and clinical laboratory data for the molecular bases of recurrent rearrangements (the term “recurrent” in this manuscript refers to the common-sized rearrangements that arise de novo in the population two or more times at the same locus). (4) We used “wet bench” region-specific molecular analysis for confirmation of predicted NAHR events. Such an integrated interdisciplinary approach enabled us to glean crucial relationships between the human genome structural features and genomic instability manifested in a clinical population to provide mechanistic insights.
Bioinformatic genome-wide analyses
Genome-wide map of the DP-LCRs delineates LCR cluster-flanked/NAHR-prone regions
In the current genome build (hg19), we found 653 pairs of DP-LCRs (parameters defined in Methods; DP-LCRs refer to this computationally defined set). Using hierarchical LCR clustering, we defined 198 potential NAHR-prone genomic regions (Fig. 1; Supplemental Table S1; Supplemental Notes), 105 of which were flanked by DP-LCRs or DP-LCR clusters with intervening unique sequence. The remaining 93 mapped within the LCR clusters themselves (e.g., the 12q14.2 region responsible for globozoospermia, MIM# 613958) (Koscinski et al. 2011; Elinati et al. 2012).
Figure 1.

Schematic representation of LCR clustering. Horizontal arrows indicate LCR elements and their orientation; the same color represents a pair of paralogous LCRs. A hierarchical clustering tree is depicted above; the dashed horizontal line (violet) shows the height threshold for cutting this tree. Directly oriented paralogous LCRs (DP-LCRs) can potentially mediate NAHR events. The structure of LCR clusters (subunit structure, orientation, etc.) as well as the DNA sequence homology between LCR clusters flanking NAHR-prone regions often revealed extensive complexity, in contradistinction to the concept of a “segmental duplication” and more consistent with “complex LCR clusters” and with current accepted models for generating duplications and complex genomic rearrangements; e.g., FoSTeS (Lee et al. 2007) or MMBIR (Hastings et al. 2009) (Supplemental Fig. S1).
The computationally identified regions, as expected, showed sequence homology of the flanking regions, but rather than simple direct repeats or segmental duplications, these flanking regions were often represented by complex LCR clusters (Fig. 1; Supplemental Fig. S1). Fifty-three regions containing 193 pairs of DP-LCRs were associated with the known NAHR-mediated deletions and reciprocal duplications on autosomes and chromosome X (Supplemental Table S2; Liu et al. 2012; Vissers and Stankiewicz 2012). The genomic regions with high DP-LCRs pair density include 16p11.2p12.1 (22 pairs), 10q11.21q11.23 (18 pairs), 5q13.2 (spinal muscular atrophy, 13 pairs), and 15q25.2 (deletion A-C) (12 pairs).
Comparison with the 130 DP-LCRs/NAHR regions reported by Sharp et al. (2005, 2006) revealed a relatively poor overlap; only 92 regions (71%) were successfully lifted over by the UCSC LiftOver tool to the current haploid human genome build hg19. Conversely, we observed a high rate of overlap with the 89 regions reported by Liu et al. (2012) (unpublished coordinates of these 89 regions, courtesy of Dr. Pengfei Liu) (Supplemental Notes; Supplemental Figs. S2, S3). Our approach also allowed us to segregate overlapping or adjacent DP-LCR-flanked fragments into distinct regions. For example, the thrombocytopenia-absent radius syndrome (TAR, MIM# 274000) region on 1q21 (Klopocki et al. 2007; Albers et al. 2012) and the 1q21.1 deletion/duplication syndrome region (MIM# 612474, 612475) (Brunetti-Pierri et al. 2008; Mefford et al. 2008) found in neuropsychiatric traits such as schizophrenia and autism (The International Schizophrenia Consortium 2008; Stefansson et al. 2008), in addition to three adjacent regions on chromosome 2q12.2q13 (Liu et al. 2012), were collapsed in previous reports but were separated by our analyses. Moreover, using the less stringent criterion for the length of flanking DP-LCRs copies, we have identified the STS deletions and duplications on Xp22.31 (MIM# 308100) (Hernández-Martín et al. 1999; Liu et al. 2011) that were not included in the analysis by Cooper et al. (2011) and CNVs in Xq28 (El-Hattab et al. 2011) that were not detected by the approach used by Liu et al. (2012).
As anticipated, due to the structural differences between the specific inversion haplotypes and the reference haploid genome, we did not detect DP-LCRs mediating two known recurrent CNVs: small CHRNA7 deletion/duplication in 15q13.3 (MIM# 612001) (Sharp et al. 2006, 2008; Shinawi et al. 2009; Szafranski et al. 2010) and 17q21.31 deletion/duplication (MIM# 610443/613533) (Koolen et al. 2006; Sharp et al. 2006; Shaw-Smith et al. 2006; Grisart et al. 2009; Itsara et al. 2012). Moreover, some known pathology-associated variants observed in patients with the 15q24 deletion syndrome (MIM# 613406), 15q24 A-D, 15q24 B-D, 15q24 B-E, and 15q24 D-E, were not detected since they are flanked by LCRs with DNA fraction matching <95%.
Potential disease-causing genes
We identified 2145 RefSeq genes overlapping or between the DP-LCRs (Supplemental Table S3). Among them, we found 39 known dosage-sensitive genes that could potentially manifest haploinsufficiency phenotypes with heterozygous deletions (Huang et al. 2010), nine of them not associated with known pathogenic NAHR-associated regions (see Discussion). In addition, we have identified 232 disease-causing (MIM, www.omim.org) genes with associated phenotypes (Supplemental Table S3).
CMA database analyses
Prevalence of the known pathogenic recurrent regions
From genome analyses performed on 25,144 patients referred for CMA in MGL at BCM, we identified 2129 known pathogenic recurrent NAHR-mediated CNVs (Fig. 2; Supplemental Table S4). In total, 1053 deletions versus 1076 duplications were observed; notably, in this clinical population we observed that deletions outnumbered duplications at most (28 of 52; 55.5%) of the loci studied.
Figure 2.

Site frequency spectrum of known pathogenic (de novo, inherited, or unknown origin) deletions and duplications in the MGL BCM CMA database. The most commonly observed regions of genomic instability are NPHP1 duplications (233), CHRNA7 duplications (175), and 22q11.21 deletions (DGS/VCFS, 166).
We identified and isolated the de novo (190 CNVs) from the inherited events of known parental origin (355 CNVs; parental genomic assay studies were available) (Fig.3) and from the events of unknown parental origin (1584, e.g., lack of available information about the parental studies). For de novo events, deletions outweigh duplications 159 to 31.
Figure 3.
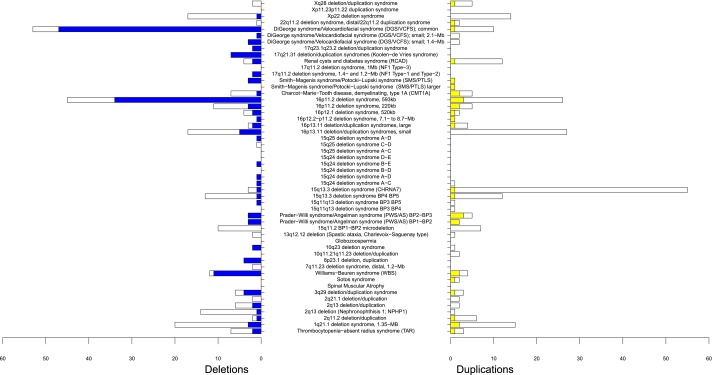
Known recurrent CNVs found in the MGL BCM CMA database divided into de novo (colored) and inherited (white), excluding ∼75% of events of unknown parental origin (i.e., no parental study was performed). Among de novo events, more deletions were found than reciprocal duplications.
We also identified one homozygous deletion of CHRNA7 in 15q13.3, one homozygous deletion of NPHP1 in 2q13, 24 hemizygous deletions of STS in Xp22.31, four homozygous duplications (or triplications) of NPHP1 in 2q13, one homozygous duplication (or triplication) of BP1/BP2 in 15q11.2, two homozygous duplications (or triplications) of CHRNA7 in 15q13.3, three homozygous duplications of the DiGeorge/velocardiofacial syndrome (DGS/VCFS) region in 22q11.21 (Bi et al. 2012), and one Prader-Willi/Angelman syndromes (PWS/AS) interstitial triplication in 15q11.2q13.
We have not found in our clinical cohort database any CNVs in the very LCR-rich regions on chr12:63,923,419-64,218,133 (globozoospermia) (Koscinski et al. 2011; Elinati et al. 2012), chr5:68,829,717-70,863,644 (spinal muscular atrophy; MIM# 253300) (Lefebvre et al. 1995), or chrX:153409725-153462352 (blue cone monochromacy, MIM# 303700; colorblindness, MIM# 303800). CNVs in these regions (not reported by Cooper et al. 2011) are likely underrepresented and underestimated due to both ascertainment biases from our selected study population (e.g., no males with infertility referred) and technical problems in detecting CNVs in short unique sequences.
We also identified somatic mosaicism events (FISH-verified) in three DP-LCR-flanked regions: one 8p23.1 deletion (60% mosaic), one 16p11.2 deletion (58%), and one 17q11.2 (NF1) deletion in 37% of cells examined, suggesting mitotic NAHR events. In addition, we found a mosaic deletion (58%) in the 16p11.2 autism region in one patient's mother; this event is distinct from a previously reported case (Shinawi et al. 2010).
Statistical modeling quantitates genome architectural features rendering NAHR susceptibility
Genomic features related to the frequency of de novo recurrent rearrangements
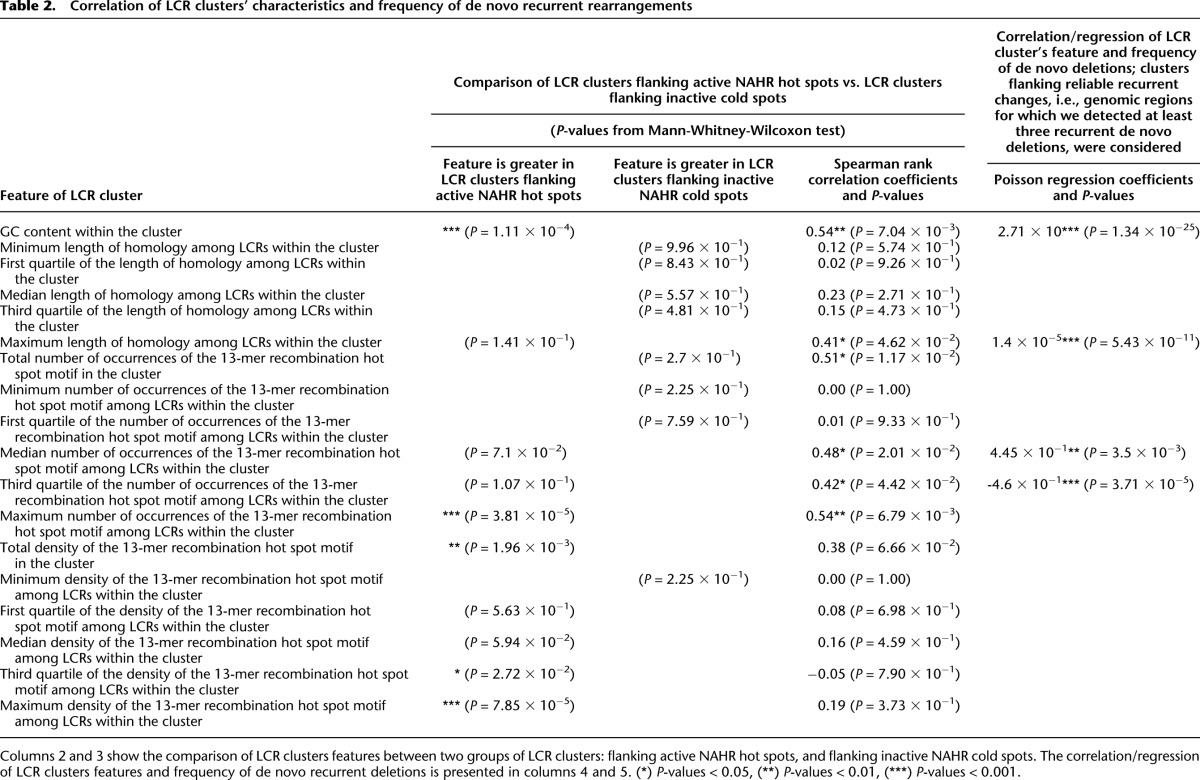
We performed genome-wide computational studies to delineate and quantitate genome architectural features rendering genome instability. We first determined the P-values from the Mann-Whitney-Wilcoxon tests, in which we compared DP-LCRs flanking the active NAHR hot spots, as determined by clinical population locus-specific frequencies, and DP-LCRs flanking the inactive cold spots (see Methods for details). We report herein the factors characterizing DP-LCRs that show a statistically significant outcome (Tables 1, 2, columns 2 and 3). For the same factors, we also computed the Spearman rank correlation coefficients on the set of DP-LCRs flanking the regions with at least three recurrent NAHR events detected (Tables 1, 2, column 4), as well as the factors that contribute significantly to the Poisson regression model (Tables 1, 2, column 5).
Table 1.
Correlation of DP-LCRs characteristics and frequency of de novo recurrent rearrangements
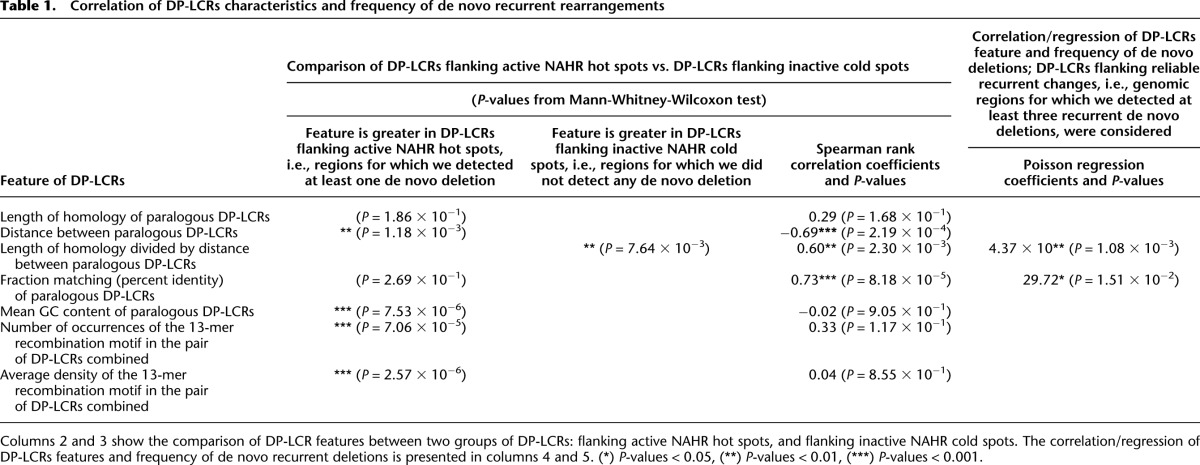
Table 2.
Correlation of LCR clusters' characteristics and frequency of de novo recurrent rearrangements
On a genome-wide scale, we found that the following properties of DP-LCRs correlate with NAHR frequency: (1) length of homology (weak association, Spearman correlation, P = 1.68 × 10−1); (2) distance between homologous pair; inverse relationship—the further the DP-LCRs are apart, the less frequent (Spearman correlation, P = 2.19 × 10−4); and (3) percent DNA sequence identity (i.e., fraction matching of DP-LCRs, P = 8.18 × 10−5). Notably, all DP-LCRs that flank frequent recurrent de novo deletions (i.e., for each we found at least four events in our CMA database) show a very high (>98%) level of fraction matching. Moreover, we found that a subset of DP-LCRs flanking active NAHR hot spots is characterized by an increased GC content (Mann-Whitney-Wilcoxon test, P = 7.53 × 10−6) and a density of the recombination hot spot motif 5′-CCNCCNTNNCCNC-3′ (Mann-Whitney-Wilcoxon test, P = 2.57 × 10−6) (Myers et al. 2008).
We also found significant correlations between the frequencies of NAHR events and the factors characterizing the LCR clusters: (1) the maximum length of homology among LCRs within a cluster (Spearman correlation, P = 4.62 × 10−2); (2) GC content within the cluster (Spearman correlation, P = 7.04 × 10−3); and (3) the maximum occurrences of the hot spot motif 5′-CCNCCNTNNCCNC-3′ among LCRs assigned to the cluster (Spearman correlation, P = 6.79 × 10−3). Finally, we observed that LCR clusters flanking active NAHR hot spots have a significantly greater GC content (Mann-Whitney-Wilcoxon test, P = 1.11 × 10−4) and an increased total density of the homologous recombination hot spot motif 5′-CCNCCNTNNCCNC-3′ (Mann-Whitney-Wilcoxon test, P = 1.96 × 10−3) when compared to other LCR clusters.
NAHR and crossover site predictions
Using the knowledge gained regarding NAHR sites or ectopic crossovers (Supplemental Table S5), we analyzed the distribution of the recombination hot spot motif 5′-CCNCCNTNNCCNC-3′ around the NAHR sites. As expected, we observed a significant enrichment of this recombination hot spot motif in the nearest vicinity of breakpoint locations, especially at the distance of up to 2 kb from breakpoints (Supplemental Fig. S4). The median distance from the breakpoint to the closest recombination hot spot motif was 2.1 kb (the mean was 5.8 kb, and the standard deviation was 6 kb). However, note that for 24 experimentally determined breakpoints (over one-third of all cases), the closest recombination hot spot motif was found <400 bp from the breakpoint location. Analysis of the distribution of other motifs not related to recombination showed no evidence for enrichment in the proximity to known NAHR sites.
Identification of novel genomic disorders in 2q12.2q13
We found three DP-LCR-flanked genomic regions on chromosome 2q12.2q13 mapping proximal and adjacent to NPHP1. Using CMA, we identified four differently sized recurrent deletions involving this region: an ∼1.7-Mb deletion in 2q12.2q12.3 in patients 1–3, an ∼0.6-Mb deletion of 2q12.3 in patients 4 and 5, an ∼1.2-Mb deletion in 2q12.3q13 in patient 8, and an ∼1.9-Mb deletion in patients 6 and 7 (Supplemental Table S3; Fig. 4). We also identified six individuals in the MGL BCM CMA database with the reciprocal duplications involving 2q12.2q13.
Figure 4.

Four novel NAHR-prone regions on chromosome 2q12.2q13. (Top) Schematic representation of paralogous DP-LCRs (colored arrows) with their sequence homology and distance in between. UCSC display of LCR clusters and deletion CNVs found in patients 1–8 (middle) and deletion (red) and duplication (blue) CNVs from the DECIPHER and ISCA databases (bottom). Green arrows indicate the ST6GAL2, SLC5A7, EDAR, and RANBP2 genes proposed to contribute to the patients' phenotypes.
Crossover mapping by long-range polymerase chain reaction and DNA sequencing
Using long-range PCR primers specific for the proximal ∼25-kb and ∼29-kb DP-LCR subunits and to their distal paralogous copies within chromosome regions 2q12.2q12.3 and 2q12.3q13, respectively, we have obtained the patient-specific junction fragments anticipated from crossover that occurred within the predicted interval (Supplemental Table S7). We then sequenced and mapped the corresponding NAHR sites within: chr2:106,870,492-106,870,888 and chr2:108,538,023-108,538,419 (397 bp, 2q12.2q12.3) and chr2:109,138,102-109,138,135 and chr2:110,627,445-110,627,478 (34 bp, 2q12.3q13) (Supplemental Fig. S5). The crossovers occurred within 2362 bp of the nearest recombination hot spot motif 5′-CCNCCNTNNCCNC-3′. This coincides with our previous observation of the enrichment of this motif in the vicinity of the NAHR sites.
Other potential pathogenic syndromes
Our CMA database query revealed 13 additional DP-LCR-flanked genomic regions (Supplemental Table S8) with 80 CNVs (48 losses and 32 gains). Some of these CNVs represent atypical variants of known pathogenic NAHR-prone regions, i.e., Smith-Magenis/Potocki-Lupski syndromes (SMS/PTLS) or DGS/VCFS.
A patient with an atypical 22q11.21 deletion (0.692 Mb) distal to the TBX1 gene within the common DGS/VCFS region also had a NPHP1 duplication in 2q13, and a patient with epilepsy had an inherited deletion in chr7:55,731,114-56,507,219 (0.549–0.711 Mb).
Discussion
Bioinformatic analyses of the current (hg19) version of the human genome grouped DP-LCRs into LCR clusters using a hierarchical arranging of LCRs flanking the empirically defined NAHR-prone regions. Moreover, we analyzed the overlapping DP-LCRs/NAHR-prone regions independently (e.g., common and small DGS/VCFS deletions in 22q11.2, or 16p11.2p12.1 and 16p12.1 regions) (Supplemental Notes; Supplemental Figs. S6–S10), enabling a better classification of the NAHR-prone regions and identification of genomic instability prone regions, potentially revealing regions that could frequently undergo rearrangement in association with new genomic disorders.
The major differences between the DP-LCR-flanked/NAHR-prone genomic regions identified by Sharp et al. (2005, 2006) and those we now report are due to variations in the LCR content of different versions of the human genome as well as the parameters used to define the LCR clusters (Supplemental Notes). Additionally, in our analyses we intersected this new genome-wide map of the DP-LCR-flanked regions in the human genome to empirically derived mutational frequency data by query of the database with high-resolution genome assays performed on 25,144 patients referred for CMA. Thus, our approach enables an assessment of the relative frequencies of known recurrent genomic disorder rearrangements. This database was uniquely suited for this analysis because the arrays used in this patient cohort were specifically designed with genome-wide coverage of all the DP-LCR-flanked regions (Stankiewicz and Lupski 2002).
Genomic architecture and features rendering genomic instability
Frequencies of known NAHR-mediated deletion and duplication syndromes
Recently, Cooper et al. (2011) reported a whole-genome morbidity map of developmental delay (DD) for both recurrent and nonrecurrent CNVs derived from studies including >15,000 genome analyses of subjects. These samples were obtained from children ascertained with DD/intellectual disability (ID), who were referred for CMA at Signature Genomics Laboratories (SGL). The most prevalent identified recurrent genomic deletions were 22q11.21 (DGS/VCFS; common and small variants not distinguished), 15q11.2 (BP1/BP2), 2q13 (NPHP1), 16p11.2 (autism), 7q11.23 (Williams-Beuren syndrome [WBS]), and 15q13.3 (BP4-BP5). Of note, this order is consistent with the six most common deletions in our data set that included a more broadly ascertained clinical population.
To determine which DP-LCR-flanked recurrent CNVs arise most frequently (i.e., potentially have a higher NAHR rate) and thus provide insight into the mechanistic origin of the recurrent rearrangements, we examined the CMA database for the de novo CNVs. We then used these frequency data to identify the genomic features that may facilitate the NAHR events. Recently, the distribution of recurrent de novo CNVs was also presented by Girirajan et al. (2012) in a study of 2312 children with ID and congenital abnormalities and a known genomic disorder. Similar to our results (Fig. 3), the DiGeorge syndrome-critical region in 22q11.21 and the 16p11.2 autism region occur with relatively high frequency. In contrast to Girirajan et al. (2012), who focused on phenotypic consequences of the second large CNVs (the “second hit” hypothesis), we investigated the mechanistic underpinnings of NAHR by applying a bottom-up unbiased approach in bioinformatics analyses (from single elements through hierarchically derived clusters) to identify the structural genomic features of LCRs that correlate with the frequency of NAHR events.
Distribution of NAHR-mediated events in the CMA databases does not represent the prevalence of these events in the whole population, e.g., benign CNVs are underrepresented (we also have not considered in our analysis the NAHR-mediated 7q11.21 deletion that is considered as benign) (Rudd et al. 2009), and parental tests are usually performed in families with more severe disorders, thus influencing the calculations for de novo rates for genomic disorders with milder phenotypes. To overcome this ascertainment bias, Turner et al. (2008) calculated NAHR events for four genomic disorders: Charcot-Marie-Tooth disease type 1A (CMT1A), azoospermia factor a (AZFa, MIM# 415000), WBS, and SMS in spermatogenesis and determined that autosomal deletions occur approximately two times as often as their reciprocal duplications in male gametes. Consistent with these results, we have observed much fewer de novo duplications than deletions (31 vs. 159). In the individual loci/regions with a greater or even number of duplications vs. deletions, there were too few events (maximum six per region) to draw statistically significant conclusions.
Finally, a patient may have multiple recurrent rearrangements, which can potentially be associated with the phenotypic heterogeneity of the associated syndromes (Girirajan et al. 2012) or perhaps represent two discreet pathogenic mutations and the phenotypic consequences of a blending of phenotypes. In our cohort, we identified 75 patients with two known recurrent NAHR events (Supplemental Table S9): Among them, two patients have both CNVs occurring de novo and seven patients were observed to have one inherited CNV and one de novo CNV, a phenomenon reported 14 yr ago (Potocki et al. 1999). Six patients have two inherited CNVs; in one case, each CNV was inherited from a different parent. However, it is not clear whether this truly represents a more severe phenotype due to “two hits,” or that the patient has two rare phenotypes whose combined clinical features suggest a distinctly different disease. Further studies may help to better understand the phenotypic consequences of such combinations of CNVs and whether epistasis, digenic inheritance, or mutational load alone are responsible for the phenotype observed.
LCR features influencing NAHR rate
By assessing the recurrent deletions and duplications in patients with SMS and PTLS syndromes, respectively, and specifically investigating three different pairs of DP-LCRs with nearly identical fraction matching homologies of ∼98.6%, Liu et al. (2011) found that the natural logarithm (ln) frequency of the crossover positively correlates with the flanking DP-LCRs' length and is inversely influenced by the inter-LCR distance. From these data, they hypothesized that the probability of ectopic crossing-over increases with increased LCR length and that ectopic synapsis is a necessary precursor to ectopic crossing-over.
Our analyses using the Spearman rank correlation (exploratory phase) and the Poisson regression (appropriate and recommended for count-type data), even when not controlling for fraction matching of the flanking LCR, confirm this phenomenon on a genome-wide scale. Although we detected only a weak association between the de novo CNV frequency and length of homology of DP-LCRs, we found a clearly significant correlation with the length of homology of DP-LCRs divided by the distance between them (Table 1). Cooper et al. (2011) have shown that the LCRs flanking active hot spots are larger and show higher sequence identity compared to the inactive spots. However, our study is the first statistically rigorous genome-wide analysis showing nontrivial correlations between the recurrent rearrangement relative frequencies, presumably reflecting mutational rates, and the various LCR architectural features. In addition, we studied the largest number of uniformly ascertained samples using a sensitive and comprehensive (in terms of genes covered) genomic assay.
Importantly, we also found that DNA fraction matching of the DP-LCRs flanking the NAHR hot spots strongly correlates with the de novo deletion/duplication frequency (Table 1). Although this phenomenon was previously suggested in the literature (Redon et al. 2006; Cooper et al. 2011; Girirajan et al. 2011), it has not been statistically confirmed until now.
Finally, we have shown that our definition of LCR clusters may enable better elucidation of the structural characteristics of the NAHR flanking regions. In particular, we have found that NAHR hot spots are characterized by increased GC content and increased saturation of the hot spot motif 5′-CCNCCNTNNCCNC-3′ (Table 2).
NAHR hot spots and crossover site predictions
Our data revealed that DP-LCRs mediating recurrent CNVs are characterized by greater GC content and increased saturation of the 13-mer recombination hot spot motif 5′-CCNCCNTNNCCNC-3′ (Table 1) when compared to other DP-LCRs. In the “ectopic synapsis precedes ectopic crossovers” model proposed by Liu et al. (2012), whereas the length and fraction matching (i.e., % identity) between flanking DP-LCR may assist in ectopic synapsis formation, perhaps the effective concentration of hot spot motifs within the paired DP-LCR helps determine whether a crossover occurs within the ectopic synapsis. The latter findings are consistent with the experimental observations of the frequency of NAHR-mediated recurrent triplications due to double crossover at the STS locus, given that the HR hot spot motif is contained within a minisatellite repeat at that locus (Liu et al. 2011); although two independent crossovers could be identified, it is not clear whether they occurred in one generation or in serial inter-generational passages since the de novo event was not available for study.
Interestingly, we also observed a significant enrichment of the recombination hot spot motif 5′-CCNCCNTNNCCNC-3′ in the vicinity of the NAHR sites, consistent with both NAHR and allelic homologous recombination (AHR) using the identical HR hot spot motif (Lupski 2004; Lindsay et al. 2006; Myers and McCarroll 2006) and observable difference in saturation of the 13-mer recombination hot spot motif between the DP-LCRs flanking NAHR sites and the DP-LCRs flanking inactive cold spots. These data confirm the previous observations that NAHR and AHR hot spots share common features (Lupski 2004) and can overlap at some loci (Lindsay et al. 2006; Myers and McCarroll 2006) and confirm assumptions that NAHR breakpoints may colocalize with some of the homologous recombination hot spots (Myers et al. 2008). These data also further support the “ectopic synapsis precedes ectopic crossing-over” model of Liu et al. (2011).
Haploinsufficient genes in NAHR-prone regions
Dang et al. (2008) suggested that haploinsufficient genes are less likely than other genes to map within the regions flanked by LCRs. We re-did this analysis for the regions flanked by DP-LCRs and found the opposite relationship (Fisher exact test, P = 0.0486) between the proportions of the haploinsufficient genes (13%) (Huang et al. 2010) and RefSeq genes (9.2%) that are contained within the DP-LCRs-flanked regions. Interestingly, this discrepancy is even higher if we consider a subset of the genome associated with known pathogenic NAHR-prone regions (Supplemental Table S2)—10% of haploinsufficient genes versus 6% RefSeq genes (Fisher exact test, P = 0.012). This may be caused by the fact that many dosage-sensitive genes outside the disease-associated regions are not yet known, and vice versa, these regions are better explored due to robust phenotypic consequences of deletion/duplication of these genes. On the other hand, the overrepresentation of dosage-sensitive genes in unstable regions may stimulate differentiation between organisms.
Moreover, we found nine known dosage-sensitive genes not previously associated with NAHR regions (BECN1, BRCA1, GRN, KLHL10, PCGF2, SMARCB1, STAT5A, STAT5B, and TP53BP2) (Supplemental Table S3); thus, DNA rearrangements can make a significant contribution to genomic disorders potentially involving these genes. However, some of the regions occupied by these genes may never be disrupted by NAHR due to unknown mechanisms that prevent recurrent rearrangements.
Gene conversions
Two paralogous genes that harbor NAHR sites may be also more prone to gene conversion events. To date, a number of such gene conversion events have been reported for genes mapping in paralogous LCRs (Chuzhanova et al. 2009), e.g., SMN1 and its very highly similar (99.99%) copy SMN2 (Lefebvre et al. 1995), responsible for autosomal recessive spinal muscular atrophy (SMA, MIM# 253300), GYPE and GYPA genes in chromosome 4q31, associated with blood group MN (MIM# 111300) (Huang et al. 2000), and NCF1B and NCF1, mutated in patients with chronic granulomatous disease (MIM# 233700) (Vázquez et al. 2001). For pachyonychia congenita type 2 (MIM# 167210) (Hashiguchi et al. 2002), we found DP-LCRs with fraction matching 94.99% (chr17:28,894,052-28,902,101 and chr17:39,776,063-39784174, separated by 10.874 Mb) and harboring the KRT17P3 and KRT17 genes. Of note, a few other gene conversion events reported by Chuzhanova et al. (2009) overlap DP-LCRs but with sequence identity lower than 95% or separated by <50 kb, suggesting potential different mechanism(s) mediating these gene conversions (Chen et al. 2010).
Novel genomic disorders
Deletions in 2q12.2q13
The proximal chromosome 2q11.2q21.1 is very LCR-rich (Fig. 5). Homozygous recurrent deletions involving NPHP1 in 2q13 result in the kidney disorder nephronopthisis. An ∼1.71-Mb recurrent deletion in the more distal region on 2q13 has been associated with ID and dysmorphism (Yu et al. 2012). Chromosome 2q13q14.1 encompasses the evolutionary breakpoint of the ancestral centric fusion of two chromosomes in nonhuman primates (Fan et al. 2002). Dharmadhikari et al. (2012) recently described small recurrent 2q21.1 deletions in patients with DD/ID, attention-deficit hyperactivity disorder, epilepsy, and other neurobehavioral abnormalities. Liu et al. (2012) and Sharp et al. (2005, 2006) (chr2:106475604-113302597, hg16; unsuccessful lift over to hg19) considered 2q12.2q13 as a single region. Our unbiased bottom-up approach enabled us to subdivide this genomic interval into four adjacent and overlapping regions (see Supplemental Notes for clinical discussion). We sequenced the 2q12.2q13 deletion breakpoints within the directly oriented paralogous subunits of the flanking LCR clusters, demonstrating NAHR as a mechanism of formation.
Figure 5.

DNA sequence homology between four LCR clusters in the 2q12.2q13 region (chr2:106,985,338-110,870,754) for paralogous subunits larger than 1 kb in size (hg19). (Top and bottom) UCSC Segmental Duplications (segdup) track representing the 2q12.2q13 region. (Middle) Results of Miropeats program analysis among all four clusters.
Conclusions
In summary, we used empirically derived patient data and mechanistic-guided bioinformatic analyses of the human genome to study the disease-associated genomic instability caused by DP-LCRs. Systematic screening of a large clinical database allowed us not only to detect and experimentally confirm novel NAHR regions but also to statistically investigate genome architectural features that correlate with genome instability and disease susceptibility. Our data show that LCRs represent complex structure with subunits revealing differences in both orientations and percent sequence identity. Architectural features rendering susceptibility to genomic rearrangements include: LCR length, percent fraction matching of paralogous segments, and the density of the HR hot spot motif. The novelty of this study is the statistical investigation and elucidation of genomic characteristics of the instability of NAHR recombination hot spots and the integration with genomic analyses done on a large patient cohort to yield mechanistic insights.
It should be noted that our research was based on a bottom-up approach (from the LCR pairs through LCR clusters to NAHR-prone regions) that is unbiased and uniform. We show that such comprehensive analyses constitute an effective way of elucidating human genome function and basic studies of genomic instability and its consequences for human health.
Methods
Patient ascertainment
Individuals with 2q12.2q13 deletions and duplications reported here were identified after referral for CMA to clinical laboratories, including BCM (patients 1, 2, 4, 5, 7, and 8), SGL (patient 6), and Murdoch Children's Research Institute, Parkville VIC, Australia (patient 3). Clinical information was obtained for patients 1, 4, and 5 following informed consent under a protocol approved by the Institutional Review Board (IRB) for Human Subject Research at BCM. The patients' clinical descriptions are provided in the Supplemental Notes.
Bioinformatic genome analyses
Definition and identification of DP-LCRs
The reference DNA sequences were downloaded from the UCSC Genome Browser (NCBI build 37/hg19, www.genome.ucsc.edu). From the Segmental Dups track (Bailey et al. 2002), a subset of DP-LCRs longer than 8 kb were selected (see Supplemental Notes; Supplemental Fig. S11), that map between 50 kb and 10 Mb from each other (including length of the smaller copy), with fraction matching >95%, not spanning centromeres (criteria from the literature, e.g., Sharp et al. 2005; Liu et al. 2012).
LCR clusters
After identifying DP-LCRs, we collapsed them into the LCR seeds (regions with 100% LCRs/Gaps content). We subsequently organized these LCR seeds hierarchically into clusters. The distances between the seeds were measured as the number of base pairs between the closest ends of the LCR seeds using a single linkage method (Supplemental Notes; Fig. 1).
We elected to use one threshold for the maximal distance between the LCR clusters with the same criteria (i.e., we have cut the hierarchical cluster tree at the same height across its width); however, certain genomic regions (e.g., 10q11.21q11.23) (Stankiewicz et al. 2012) encompass much larger LCR blocks, suggesting the hierarchical cluster tree should be trimmed at a higher level.
Other bioinformatic tools
DNA sequence similarities were analyzed using BLAT (http://genome.ucsc.edu) and assembled using Sequencher v4.8 (GeneCodes, Ann Arbor, MI, USA). Bioinformatic analyses used R software (www.r-project.org). Approved gene symbols were used according to HUGO Gene Nomenclature Committee resources (http://www.genenames.org). Transferring coordinates between genome builds was performed using UCSC LiftOver tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver). Automatic processing of OMIM was performed using OMIM API (http://omim.org/help/api).
To better visualize the chromosome architecture in the DP-LCR-flanked regions, we used the ICAass (v 2.5) algorithm. The graphical display was performed using Miropeats (v 2.01) (The Genome Institute at Washington University, St Louis, Missouri). The program was run using two thresholds of 1000 bp (http://www.genome.ou.edu/miropeats.html) (Parsons 1995).
CMA database analyses
Frequency of the known pathogenic syndromes
We calculated the frequencies of 52 known NAHR-mediated pathogenic deletions and duplications (we excluded chromosome Y from our analyses and modeling) in the CMA database in the MGL at BCM. Using oligonucleotide coordinates and data from parental studies, we classified them in one of three groups: de novo (dn), parental (par), and unknown, based on the reported inheritance. It should be noted that all but one (i.e., NPHP1 on 2q13) of the known autosomal recurrent CNVs manifest as dominant disorders.
Novel potentially pathogenic recurrent CNVs
We also analyzed 436 DP-LCR pairs that have not been associated with known pathogenic NAHR-mediated genomic deletions and/or duplications (excluding those on chromosome Y). We used oligonucleotide coordinates and data from parental studies for processing CMA rearrangements that were reported and interpreted.
Statistical modeling based on genome-wide analyses and CMA data
Genomic features related to the frequency of de novo recurrent rearrangements
For this aim, we selected from the CMA database the set of deletions that are most likely to be de novo events. This set was then filtered for CNVs that are flanked by at least one pair of DP-LCRs (i.e., left and right breakpoints are located within left and right paralogous copies, respectively) overlapping with known pathogenic NAHR-prone regions (Supplemental Table S2). Using this set, for each DP-LCR we assign the number of de novo deletion events that are flanked by this DP-LCR. In our study, we use this number as an estimation of the frequency of recurrent de novo deletions in the given region (frequencies of de novo deletions are plotted in Fig.3).
Regions flanked by DP-LCRs, for which we found evidence for at least one NAHR event, we denoted as “active NAHR hot spots.” Remaining regions surrounded by DP-LCRs we marked as “inactive NAHR cold spots.” To analyze the architectural differences between two groups of flanking DP-LCRs—active NAHR hot spots and inactive cold spots—we performed a series of nonparametric Mann-Whitney-Wilcoxon tests.
Subsequently, we focused on the genomic regions with at least three recurrent NAHR events detected. Our analyses of the correlations between the frequencies of the recurrent NAHR-mediated deletion and their specific genomic architectural features were performed in two steps. First, we used exploratory analysis with the Spearman rank correlation, in which we identified factors that statistically significantly correlated with the NAHR frequency. Second, we applied a Poisson regression, the most adequate method for analysis of the count data. This kind of regression analysis builds the model that explains the response variable assuming that it has a Poisson distribution, i.e., the logarithm of its expected value can be modeled by a linear combination of parameters. The advantage of the regression approach over standard hypotheses testing was discussed by McElduff et al. (2010).
Utilizing the model parameters, we analyzed the genomic features that characterize the regions prone to recurrent de novo CNVs. First, we focused on DP-LCRs by investigating their length of homology, the distances, fraction matching scores between paralogous copies, average GC content, and the presence of the 13-mer recombination hot spot motif 5′-CCNCCNTNNCCNC-3′ (the histone methyltransferase PRDM9 binding site). Next, we analyzed different features of the LCR clusters (e.g., length of LCRs, GC content within the cluster, or concentration of the recombination hot spot motif) flanking the NAHR-prone regions. In particular, we studied the distributions of three parameters (i.e., LCR lengths, number of hot spot motifs in LCRs, and density of motifs in LCRs) by means of their robust statistics (median, first, and third quartile, minimum and maximum). We then calculated the above-mentioned statistics for all LCR clusters, taking into account all direct and inverse paralogous LCR copies. Moreover, we determined the total number of occurrences of the 13-mer recombination hot spot motif 5′-CCNCCNTNNCCNC-3′ and its saturation, as well as the GC content inside the clusters.
NAHR junction prediction
We analyzed the reported NAHR junctions (Supplemental Table S5) for evidence of enrichment of the 13-mer recombination hot spot motif 5′-CCNCCNTNNCCNC-3′ by comparing the frequency of this motif within 20 kb of the NAHR with the frequency of other 13-mers.
2q12.2q13 region-specific molecular analyses
DNA isolation
Genomic DNA was extracted from peripheral blood using the Puregene DNA isolation kit (Gentra System).
CMA
A total of 25,144 patients referred for CMA in MGL at BCM were screened using custom-designed exon-targeted aCGH oligonucleotide microarrays V7 (105K, total 5950), V8 (180K, total 16,639) (Boone et al. (2010), V8.3 (400K, total 2061), and V9 (400K, total 494) OLIGO designed in MGL at BCM (http://www.bcm.edu/geneticlabs/) and manufactured by Agilent Technology as previously described (Szafranski et al. 2010). The most common reasons for testing in these patients were: DD/ID (∼26.7%), autism spectrum disorders (ASDs; ∼9.3%), seizures (∼7.6%), dysmorphic features (6.3%), heart defects (2.9%), speech delay (∼2.1%), attention deficit hyperactivity disorder (ADHD; ∼1.9%), and others (∼26.8%). In ∼16.4% of cases, no indication was provided. Additional subjects with deletions within the 2q12.2q13 region were identified using bacterial artificial chromosome (BAC)-based (SignatureChip version 4) (Bejjani et al. 2005) and oligonucleotide-based aCGH (SignatureChipOS, custom-designed by Signature Genomics, version 3.1, 135K from RocheNimbleGen) (Duker et al. 2010) (patient 6) and by Illumina SNP array HumanCytoSNP-12 300K (patient 3 and the mother).
FISH analyses
Confirmatory and parental FISH analyses with the BAC clones were performed using standard procedures.
Allele-specific long-range PCR and DNA sequencing
Deletion junctions were amplified using long-range PCR primers designed to harbor at least three nucleotide mismatches (cis-morphisms) based on the comparison of the paralogous LCR sequence variants. Forward primers were specific to the directly oriented LCR subunit in the proximal LCR cluster, and reverse primers were located in the paralogous copy in the distal LCR cluster. This strategy allowed preferential amplification of the predicted junction fragment of the deletion generated by the recombination of LCRs. Primers (Supplemental Table S7) were designed using the Primer 3 software (http://frodo.wi.mit.edu/primer3/). Amplification of the breakpoint junction fragments, marking the crossover, was performed using Takara LA Taq Polymerase (Takara Bio Inc.) according to the manufacturer's protocol. The following PCR conditions were used: 94°C for 1 min, followed by 30 cycles of 94°C for 30 s, and 68°C for 12 min, and 72°C for 10 min. PCR products were treated with ExoSAP-IT (USB) to remove unconsumed dNTPs and primers, and directly Sanger-sequenced using BigDye Terminator Cycle Sequencing performed according to the manufacturer's protocol (Applied Biosystems).
Data access
The aCGH data sets from BCM CMA can be accessed through the NCBI dbVar database (http://www.ncbi.nlm.nih.gov/dbvar/) under accession number nstd79. The NAHR site sequences have been deposited in the DNA Data Bank of Japan (DDBJ; http://www.ddbj.nig.ac.jp/) under accession numbers AB817973 and AB817974.
Competing interest statement
J.R.L. is a consultant for Athena Diagnostics, owns stock in 23andMe and Ion Torrent Systems Inc., and is a coinventor on multiple U.S. and European patents for DNA diagnostics. J.A.R. is an employee of SGL, a subsidiary of PerkinElmer, Inc. Furthermore, the Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular diagnostic testing (MGL; http://www.bcm.edu/geneticlabs/). S.A. served on the national advisory board of Questcor Pharmaceuticals and received an honorarium for serving in this position.
Acknowledgments
We thank Dr. Pengfei Liu, Ian M. Campbell, Amber N. Pursley, and Kristen T. Maliszewski for helpful discussions. This work was supported in part by the Intellectual and Developmental Disabilities Research Center (IDDRC) (grant number P30 HD024064) and the National Institute of Neurological Disorders and Stroke (National Institutes of Health) (grant number R01 NS058529) to J.R.L., the Polish National Science Center (grant number 2011/01/B/NZ2/00864) to A.G. and P.D., the EU through the European Social Fund (grant number UDA-POKL.04.01.01-00-072/09-00) to P.D. C.P.S. is a recipient of a Doris Duke Clinical Scientist Development Award. P.D. is supported by a START fellowship from the Foundation for Polish Science.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.152454.112.
References
- Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, Jolley JD, Cvejic A, Kostadima M, Bertone P, et al. 2012. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 44: 435–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE 2002. Recent segmental duplications in the human genome. Science 297: 1003–1007 [DOI] [PubMed] [Google Scholar]
- Bejjani BA, Saleki R, Ballif BC, Rorem EA, Sundin K, Theisen A, Kashork CD, Shaffer LG 2005. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: Is less more? Am J Med Genet A 134: 259–267 [DOI] [PubMed] [Google Scholar]
- Bi W, Probst FJ, Wiszniewska J, Plunkett K, Roney EK, Carter BS, Williams MD, Stankiewicz P, Patel A, Stevens CA, et al. 2012. Co-occurrence of recurrent duplications of the DiGeorge syndrome region on both chromosome 22 homologues due to inherited and de novo events. J Med Genet 49: 681–688 [DOI] [PubMed] [Google Scholar]
- Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, Kang SH, Yang Y, Wiszniewska J, Nowakowska BA, et al. 2010. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat 31: 1326–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, et al. 2008. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet 40: 1466–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JM, Férec C, Cooper DN 2010. Gene conversion in human genetic disease. Genes 1: 550–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, Yatsenko SA, Cooper ML, Furman P, Stankiewicz P, et al. 2005. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med 7: 422–432 [DOI] [PubMed] [Google Scholar]
- Chuzhanova N, Chen JM, Bacolla A, Patrinos GP, Férec C, Wells RD, Cooper DN 2009. Gene conversion causing human inherited disease: Evidence for involvement of non-B-DNA-forming sequences and recombination-promoting motifs in DNA breakage and repair. Hum Mutat 30: 1189–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. 2011. A copy number variation morbidity map of developmental delay. Nat Genet 43: 838–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VT, Kassahn KS, Marcos AE, Ragan MA 2008. Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur J Hum Genet 16: 1350–1357 [DOI] [PubMed] [Google Scholar]
- Dharmadhikari AV, Kang SH, Szafranski P, Person RE, Sampath S, Prakash SK, Bader PI, Phillips JA, Hannig V, Williams M, et al. 2012. Small rare recurrent deletions and reciprocal duplications in 2q21.1, including brain-specific ARHGEF4 and GPR148. Hum Mol Genet 21: 3345–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittwald P, Gambin T, Gonzaga-Jauregui C, Carvalho CM, Lupski JR, Stankiewicz P, Gambin A 2013. Inverted low-copy repeats and genome instability—a genome-wide analysis. Hum Mutat 34: 210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duker AL, Ballif BC, Bawle EV, Person RE, Mahadevan S, Alliman S, Thompson R, Traylor R, Bejjani BA, Shaffer LG, et al. 2010. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet 18: 1196–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hattab AW, Fang P, Jin W, Hughes JR, Gibson JB, Patel GS, Grange DK, Manwaring LP, Patel A, Stankiewicz P, et al. 2011. Int22h-1/int22h-2-mediated Xq28 rearrangements: Intellectual disability associated with duplications and in utero male lethality with deletions. J Med Genet 48: 840–850 [DOI] [PubMed] [Google Scholar]
- Elinati E, Kuentz P, Redin C, Jaber S, Vanden Meerschaut F, Makarian J, Koscinski I, Nasr-Esfahani MH, Demirol A, Gurgan T, et al. 2012. Globozoospermia is mainly due to DPY19L2 deletion via non-allelic homologous recombination involving two recombination hot spots. Hum Mol Genet 21: 3695–3702 [DOI] [PubMed] [Google Scholar]
- Fan Y, Linardopoulou E, Friedman C, Williams E, Trask BJ 2002. Genomic structure and evolution of the ancestral chromosome fusion site in 2q13-2q14.1 and paralogous regions on other human chromosomes. Genome Res 12: 1651–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio S, Calvari V, Gregato G, Gimelli G, Camanini S, Giorda R, Ragusa A, Guerneri S, Selicorni A, Stumm M, et al. 2002. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am J Hum Genet 71: 276–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Campbell CD, Eichler EE 2011. Human copy number variation and complex genetic disease. Annu Rev Genet 45: 203–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, Filipink RA, McConnell JS, Angle B, Meschino WS, et al. 2012. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med 367: 1321–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisart B, Willatt L, Destrée A, Fryns JP, Rack K, de Ravel T, Rosenfeld J, Vermeesch JR, Verellen-Dumoulin C, Sandford R 2009. 17q21.31 microduplication patients are characterised by behavioural problems and poor social interaction. J Med Genet 46: 524–530 [DOI] [PubMed] [Google Scholar]
- Hashiguchi T, Yotsumoto S, Shimada H, Terasaki K, Setoyama M, Kobayashi K, Saheki T, Kanzaki T 2002. A novel point mutation in the keratin 17 gene in a Japanese case of pachyonychia congenita type 2. J Invest Dermatol 118: 545–547 [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR 2009. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Martín A, González-Sarmiento R, De Unamuno P 1999. X-linked ichthyosis: An update. Br J Dermatol 141: 617–627 [DOI] [PubMed] [Google Scholar]
- Huang CH, Chen Y, Blumenfeld OO 2000. A novel Sta glycophorin produced via gene conversion of pseudoexon III from glycophorin E to glycophorin A gene. Hum Mutat 15: 533–540 [DOI] [PubMed] [Google Scholar]
- Huang N, Lee I, Marcotte EM, Hurles ME 2010. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6: e1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Schizophrenia Consortium 2008. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455: 237–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsara A, Vissers LE, Steinberg KM, Meyer KJ, Zody MC, Koolen DA, de Ligt J, Cuppen E, Baker C, Lee C, et al. 2012. Resolving the breakpoints of the 17q21.31 microdeletion syndrome with next-generation sequencing. Am J Hum Genet 90: 599–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopocki E, Schulze H, Strauss G, Ott CE, Hall J, Trotier F, Fleischhauer S, Greenhalgh L, Newbury-Ecob RA, Neumann LM, et al. 2007. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am J Hum Genet 80: 232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R, Kooy RF, Reyniers E, Romano C, Fichera M, et al. 2006. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet 38: 999–1001 [DOI] [PubMed] [Google Scholar]
- Koscinski I, Elinati E, Fossard C, Redin C, Muller J, Velez de la Calle J, Schmitt F, Ben Khelifa M, Ray PF, Ray P, et al. 2011. DPY19L2 deletion as a major cause of globozoospermia. Am J Hum Genet 88: 344–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247 [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. 1995. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80: 155–165 [DOI] [PubMed] [Google Scholar]
- Li J, Harris RA, Cheung SW, Coarfa C, Joeng M, Goodell MA, White LD, Patel A, Kang S-H, Shaw C, et al. 2012. Genomic hypomethylation in the human germline associates with selective structural mutability in the human genome. PLoS Genet 8: e1002692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay SJ, Khajavi M, Lupski JR, Hurles ME 2006. A chromosomal rearrangement hot spot can be identified from population genetic variation and is coincident with a hot spot for allelic recombination. Am J Hum Genet 79: 890–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Lacaria M, Zhang F, Withers M, Hastings PJ, Lupski JR 2011. Frequency of nonallelic homologous recombination is correlated with length of homology: Evidence that ectopic synapsis precedes ectopic crossing-over. Am J Hum Genet 89: 580–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Carvalho CM, Hastings P, Lupski JR 2012. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev 22: 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR 1998. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet 14: 417–422 [DOI] [PubMed] [Google Scholar]
- Lupski JR 2004. Hot spots of homologous recombination in the human genome: Not all homologous sequences are equal. Genome Biol 5: 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR 2009. Genomic disorders ten years on. Genome Med 1: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElduff F, Cortina-Bojra M, Chan SK, Wade A 2010. When t-tests or Wilcoxon-Mann-Whitney tests won't do. Adv Physiol Educ 34: 128–133 [DOI] [PubMed] [Google Scholar]
- Mefford HC 2009. Genotype to phenotype-discovery and characterization of novel genomic disorders in a “genotype-first” era. Genet Med 11: 836–842 [DOI] [PubMed] [Google Scholar]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, et al. 2008. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med 359: 1685–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SR, McCarroll SA 2006. New insights into the biological basis of genomic disorders. Nat Genet 38: 1363–1364 [DOI] [PubMed] [Google Scholar]
- Myers S, Freeman C, Auton A, Donnely P, McVean G 2008. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet 40: 1124–1129 [DOI] [PubMed] [Google Scholar]
- Ou Z, Stankiewicz P, Xia Z, Breman AM, Dawson B, Wiszniewska J, Szafranski P, Cooper ML, Rao M, Shao L, et al. 2011. Observation and prediction of recurrent human translocations mediated by NAHR between nonhomologous chromosomes. Genome Res 21: 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JD 1995. Miropeats: Graphical DNA sequence comparisons. Comput Appl Biosci 11: 615–619 [DOI] [PubMed] [Google Scholar]
- Potocki L, Chen KS, Koeuth T, Killian J, Iannaccone ST, Shapira SK, Kashork CD, Spikes AS, Shaffer LG, Lupski JR 1999. DNA rearrangements on both homologues of chromosome 17 in a mildly delayed individual with a family history of autosomal dominant carpal tunnel syndrome. Am J Hum Genet 64: 471–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. 2006. Global variation in copy number in the human genome. Nature 444: 444–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd MK, Keene J, Bunke B, Kaminsky EB, Adam MP, Mulle JG, Ledbetter DH, Martin CL 2009. Segmental duplications mediate novel, clinically relevant chromosome rearrangements. Hum Mol Genet 18: 2957–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, et al. 2005. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 77: 78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, et al. 2006. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet 38: 1038–1042 [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, Stevenson RE, Schroer RJ, Novara F, De Gregori M, Ciccone R, et al. 2008. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet 40: 322–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, Curley R, Cumming S, Dunn C, Kalaitzopoulos D, et al. 2006. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet 38: 1032–1037 [DOI] [PubMed] [Google Scholar]
- Shinawi M, Schaaf CP, Bhatt SS, Xia Z, Patel A, Cheung SW, Lanpher B, Nagl S, Herding HS, Nevinny-Stickel C, et al. 2009. A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat Genet 41: 1269–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, et al. 2010. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet 47: 332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR 2002. Genome architecture, rearrangements and genomic disorders. Trends Genet 18: 74–82 [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR 2010. Structural variation in the human genome and its role in disease. Annu Rev Med 61: 437–455 [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Kulkarni S, Dharmadhikari AV, Sampath S, Bhatt SS, Shaikh TH, Xia Z, Pursley AN, Cooper ML, Shinawi M, et al. 2012. Recurrent deletions and reciprocal duplications of 10q11.21q11.23 including CHAT and SLC18A3 are likely mediated by complex low-copy repeats. Hum Mutat 33: 165–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietiläinen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, et al. 2008. Large recurrent microdeletions associated with schizophrenia. Nature 455: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafranski P, Schaaf CP, Person RE, Gibson IB, Xia Z, Mahadevan S, Wiszniewska J, Bacino CA, Lalani S, Potocki L, et al. 2010. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: Benign or pathological? Hum Mutat 31: 840–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, Blayney ML, Beck S, Hurles ME 2008. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet 40: 90–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez N, Lehrnbecher T, Chen R, Christensen BL, Gallin JI, Malech H, Holland S, Zhu S, Chanock SJ 2001. Mutational analysis of patients with p47-phox-deficient chronic granulomatous disease: The significance of recombination events between the p47-phox gene (NCF1) and its highly homologous pseudogenes. Exp Hematol 29: 234–243 [DOI] [PubMed] [Google Scholar]
- Vissers LE, Stankiewicz P 2012. Microdeletion and microduplication syndromes. Methods Mol Biol 838: 29–75 [DOI] [PubMed] [Google Scholar]
- Yu HE, Hawash K, Picker J, Stoler J, Urion D, Wu BL, Shen Y 2012. A recurrent 1.71 Mb genomic imbalance at 2q13 increases the risk of developmental delay and dysmorphism. Clin Genet 81: 257–264 [DOI] [PubMed] [Google Scholar]