

Abstract
Congenital heart defect (CHD) occurs in 40% of Down syndrome (DS) cases. While carrying three copies of chromosome 21 increases the risk for CHD, trisomy 21 itself is not sufficient to cause CHD. Thus, additional genetic variation and/or environmental factors could contribute to the CHD risk. Here we report genomic variations that in concert with trisomy 21, determine the risk for CHD in DS. This case-control GWAS includes 187 DS with CHD (AVSD = 69, ASD = 53, VSD = 65) as cases, and 151 DS without CHD as controls. Chromosome 21–specific association studies revealed rs2832616 and rs1943950 as CHD risk alleles (adjusted genotypic P-values <0.05). These signals were confirmed in a replication cohort of 92 DS-CHD cases and 80 DS-without CHD (nominal P-value 0.0022). Furthermore, CNV analyses using a customized chromosome 21 aCGH of 135K probes in 55 DS-AVSD and 53 DS-without CHD revealed three CNV regions associated with AVSD risk (FDR ≤ 0.05). Two of these regions that are located within the previously identified CHD region on chromosome 21 were further confirmed in a replication study of 49 DS-AVSD and 45 DS- without CHD (FDR ≤ 0.05). One of these CNVs maps near the RIPK4 gene, and the second includes the ZBTB21 (previously ZNF295) gene, highlighting the potential role of these genes in the pathogenesis of CHD in DS. We propose that the genetic architecture of the CHD risk of DS is complex and includes trisomy 21, and SNP and CNV variations in chromosome 21. In addition, a yet-unidentified genetic variation in the rest of the genome may contribute to this complex genetic architecture.
Down syndrome (DS) is a common genomic disorder caused by trisomy of human chromosome 21 (Antonarakis 1998; Antonarakis et al. 2002, 2004; Antonarakis and Epstein 2006). Some of its phenotypes (e.g., cognitive impairment) are consistently present in all DS individuals albeit in variable severity, while others show incomplete penetrance (Antonarakis et al. 2002, 2004; Antonarakis and Epstein 2006). Among the most notable phenotypes with reduced penetrance are the congenital heart defects (CHD), since ∼40% of DS individuals present some form of CHD (Ferencz et al. 1989). The most frequent forms of CHD in DS cases are atrioventricular septal defects (AVSD), comprising 43% of the CHD cases, while ventricular septal defects (VSD), atrial septal defects (ASD), and tetralogy of fallot (TOF) comprise 32%, 19%, and 6% of the CHD in DS, respectively. In fact, AVSDs are almost exclusively seen in DS (Ferencz et al. 1989; Roizen and Patterson 2003). The complete underlying genomic or gene expression variation that contributes to the presence of a CHD in DS is unknown. Extensive efforts over the past years to gain a better understanding of the genetic basis of CHD in DS using rare cases of partial trisomy 21 have led to identification of genomic regions on chromosome 21 that, when triplicated, are consistently associated with CHD. An initial study of rare partial trisomy 21 cases suggested a minimal CHD candidate region on 21q22.3 of ∼5.27 Mb between markers D21S3 and PFKL (Barlow et al. 2001); this region was later narrowed down to 1.77 Mb (DSCAM-ZBTB21) (Korbel et al. 2009). In another similar study of partial trisomy 21 cases, the CHD region was mapped to a larger telomeric genomic segment of 15.4 Mb that overlaps with the segment described above (Lyle et al. 2009). An association study in the mid 1990s using a few microsatellite markers on chromosome 21 hinted at a potential association between variation of the COL6A1 gene region with CHD in Down syndrome (Davies et al. 1995). Also, Grossman et al. (2011) used Drosophila, mouse transgenesis, and a cardiac myoblast H9C2 cell model to show that the overexpression of DSCAM and COL6A2 genes cooperatively contributes to ASD in mice, increased abnormalities of heart rhythm, and failure in Drosophila, and promoted substrate adhesion in the H9C2 cell line. Recent studies also suggest the potential contribution of VEGFA (Ackerman et al. 2012), ciliome and Hedgehog (Ripoll et al. 2012), and folate (Locke et al. 2010) pathways to the pathogenicity of CHD in DS. There are also several mouse models for partial or complete trisomy syntenic to human chromosome 21 (Sago et al. 1998; Shinohara et al. 2001; Dunlevy et al. 2010; Yu et al. 2010). CHDs have been observed in the full “trisomy 21” homologous mice Tc1 (Dunlevy et al. 2010) and Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+ mice (Yu et al. 2010). In addition, CHDs have been observed in the partial “trisomy 21” model Ts65Dn that is trisomic for 13.4 Mb of the 22.9-Mb chromosome 21 syntenic regions (Moore 2006; Williams et al. 2008). Also, recently engineered duplication of a 5.43-Mb region of Mmu16 from Tiam1 to Kcnj6 in the mouse model, Dp(16)2Yey, was reported to cause CHD (Liu et al. 2011).
Candidate non–chromosome 21 genes have also been identified for susceptibility to several CHDs and AVSD in particular (not related to DS). Pathogenic mutations in the CRELD1 gene (on 3p25) have been found in 6% of individuals with non–trisomy 21– related AVSD (Robinson et al. 2003). Also, GATA4 mutations (on 8p23) have been found in families with cardiac malformations that included AVSD, VSD, insufficiency of cardiac valves, ASD, and thickening of the pulmonary valve; the data in these families suggest that the same pathogenic mutation could predispose to different types of heart defects in different individuals (Garg et al. 2003). The creation of transgenic mouse strains using cardiac-specific gene inactivation of hypomorphic Bmp4 alleles resulted in AVSDs that correlate with the level of Bmp4 expression (Jiao et al. 2003). Thus, it is conceivable that gene expression variation of certain loci could contribute to the phenotypic variation of heart defects in DS. Also, CNVs are important components of the overall genomic variability among individual genomes (Sharp et al. 2005; Beckmann et al. 2007). Rare and common CNVs have been associated with various phenotypes (Redon et al. 2006; Beckmann et al. 2007; Conrad et al. 2010; Craddock et al. 2010; Priest et al. 2012), and possibly they could also be one cause of the CHD risk in DS.
Our present hypothesis for the CHD phenotypes in DS individuals is that three copies of functional genomic elements on chromosome 21 and genetic variation of chromosome 21 and non–chromosome 21 loci predispose to abnormal heart development. Additional variables for these phenotypes could include unknown environmental factors and stochastic events. Thus, the CHD phenotypes are likely to be multifactorial, caused both by variation at multiple loci and interactions among them and with nongenetic factors. Here we aimed to contribute to the description of the genetic architecture of CHD in DS, and report the results of genome-wide and chromosome 21–specific SNP and CNV association studies using samples from DS individuals with and without CHD. We have also used SNPs that are associated with cis-eQTLs as a subset of the SNP space to test the hypothesis that sites controlling the quantitative difference in gene expression contribute to the development of CHD. Furthermore, genome-wide SNP-based interaction studies (GWIS) were attempted to further explore the complex genetic architecture of CHD in DS.
Results
Genome-wide association study of CHD and its subphenotypes in DS
To identify genome-wide risk variants for the CHD in DS, we performed a case-control association study using 431,962 non–chromosome 21 SNPs. The cases were 187 samples of DS with various forms of CHD; the controls were 151 samples of DS without detectable CHD (see Methods). Figure 1 shows the “Manhattan plots” of the GWAS P-values for all combinations of heart defects (all CHD, AVSD alone, VSD alone, ASD alone). Our intent was to potentially detect GWAS significant signals for the different CHD subclasses. Supplemental Table S1 summarizes the top P-values from all of these association studies. Only one signal remains marginally significant after Bonferroni correction (Bonferroni adjusted P-value 0.03), with marker rs160890 on chromosome 5 when only the ASD phenotypic class was considered. The Q–Q plots of SNP association test of P-values are shown in Supplemental Figure S1.
Figure 1.

Genome-wide Manhattan plots for CHD in DS and its different subphenotypes across 431,962 SNPs based on allelic associations. −log10 P-values of SNP association tests are plotted relative to their position on each chromosome (alternating black and gray). Chromosome 21 trisomic SNPs are not included here (for details, see text and Fig. 2). The results shown are for DS-CHD (A), DS-AVSD (B), DS-ASD (C), and DS-VSD (D).
Association study using chromosome 21 SNP genotypes
To identify genomic risk variants on chromosome 21 for CHD in DS, we performed an association study using 7238 chromosome 21 trisomic SNPs. For this special trisomic association test, we calculated allelic and genotypic P-values (comparing the frequency of the four genotypic classes AAA, AAB, ABB, and BBB) in cases versus controls. The Bonferroni correction for multiple testing was used based on 7238 trisomic SNPs tested. The single-locus association test for 187 DS with CHD and 151 DS without CHD showed rs2832616 (nominal genotypic, P = 3.08 × 10−6) and rs1943950 (nominal genotypic, P = 6.83 × 10−6) as CHD risk alleles (Figure 2; Table 1). Both SNPs are located in the same LD block (r2 = 1) in the intergenic region between GRIK1 and CLDN17. Interestingly, both SNPs are cis-eQTLs for the KRTAP7-1 (Dimas et al. 2009; Yang et al. 2010).
Figure 2.

(A) Chromosome 21–wide Manhattan plot and (B) Q–Q plot of SNP genotypic association test P-values for 187 DS-CHD and 151 DS-without CHD using 7238 SNPs on chromosome 21. (Red line) The Bonferroni threshold for chromosome 21–wide α = 0.05. SNPs are plotted in megabases relative to their position on chromosome 21. Two SNPs within the same LD block (r2 = 1) reached chromosome 21–wide significance (P ≤ 0.05) (for details, see Table 1). (C) Regional association plot for the region identified to associate with DS-CHD. The panel shows the recombination rate in the region estimated from HapMap CEU data (http://hapmap.ncbi.nlm.nih.gov/), pairwise LD between SNPs in the region and the SNP identified (purple), and P-values for strength of associations and genes in the region. The r2 values are color-coded according to the scale on the panel.
Table 1.
Significant chromosome 21 trisomic SNPs association test results for DS-CHD and DS -ASD
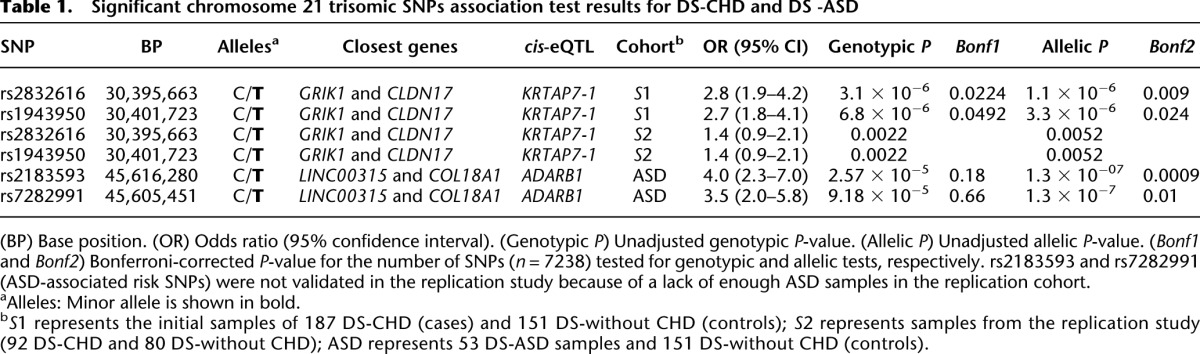
To validate these results, we genotyped the two risk CHD SNPs in a replication sample of 92 DS-CHD and 80 DS without CHD. The nominal genotypic P-value was 0.0022 for both SNPs (Table 1). Thus, we consider that these two SNPs were validated in the replication sample. When the separate phenotypic classes of CHD were considered, no statistically significant associations were found, except for DS-ASD using allelic P-values (Table 1). However, for this analysis, genotypic P-values were not significant (Table 1). This result was not further investigated.
Two-locus interactions
To test the hypothesis that there exist two loci interactions for CHD risk in DS, we used logistic regression to model the interaction between any two SNPs. Three different models were studied: (1) interactions among SNPs on the diploid fraction of the genome (i.e., excluding the chromosome 21); (2) interactions among SNPs on the trisomic fraction of the genome; and (3) interactions between trisomic SNPs on chromosome 21 and non–chromosome 21 cis-eQTLs.
Model 1: Interactions among SNPs on the diploid fraction of the genome
Since DS is likely to be a disorder of gene expression (Prandini et al. 2007), a two-locus interaction study was first performed using only cis-eQTL markers that are functionally related to gene expression variation. We used 8900 SNPs with strong association with gene expression (empirical P-value < 1.0 × 10−4) selected from the Geneva GenCord (Dimas et al. 2009) and HapMap3 CEU data sets (Stranger et al. 2012). After pruning the SNPs as described in Methods, 8126 SNPs remained. The interaction analysis was done by the fast-epistasis option in PLINK. The results are summarized in Table 2. In total, 33,011,875 SNP × SNP tests were done, and an uncorrected P-value of 6.0 × 10−9 was used to establish significance in our genome-wide cis-eQTL interaction study at the 0.05 level. Correction for multiple testing is done based on n × (n − 1)/8, where n is the number of SNPs tested (Becker et al. 2011). This cis-eQTL-SNP based interaction study for DS-CHD risk showed a significant interaction signal between rs972372 on chromosome 2 and rs681418 on chromosome 11 (P-value = 8.7 × 10−10) (Supplemental Fig. 2; Table 2). rs972372 is an intronic variant in the MAP4K4 gene and is associated with expression of the CNOT11 gene, while rs681418 is an intronic variant in the SPA17 gene and is associated with expression of the NRGN gene. This SNP–SNP interaction was tested in a replication study of 83 DS-CHD and 71 DS-without CHD, and the nominal P-value is 0.047 (Table 2). This marginal significance does not justify a clear declaration of validation of the SNP–SNP interaction result.
Table 2.
P-values of the top three pairs of potentially interacting cis-eQTLs for DS CHD

Furthermore, a two-locus interaction analysis was performed on the entire set of 431,962 SNPs genome-wide. An uncorrected P-value of 2.1 × 10−12 (Becker et al. 2011) was used to establish genome-wide significance at the 0.05 level. Supplemental Table S3 shows a summary of these analyses. None of the interacting pairs of SNPs become significant after correcting for multiple testing. Performing the same analysis after SNP LD pruning did not yield any significant signal (data not shown).
Model 2: Interactions among SNPs on the trisomic chromosome 21
For the study of interactions between SNPs on chromosome 21 contributing to CHD, three different tests were performed. First, all 7238 genotyped SNPs on chromosome 21 were used, performing 26,190,703 interaction tests (P-value cutoff for 0.05-level genome-wide significance: 7.5 × 10−9). Then, after LD SNP pruning, only 2950 SNPs were used for interaction analysis, performing 4,349,775 tests (P-value cutoff for 0.05-level genome-wide significance: 4.5 × 10−8). Finally, all chromosome 21 cis-eQTLs (166 cis-eQTLs) available from the Geneva GenCord and HapMap3 CEU data sets were used for interaction analysis based on cis-eQTLs. A total of 13,695 tests were performed (P-value cutoff for 0.05-level significance: 1.4 × 10−5). None of these tests showed a significant result after correcting for multiple testing (data not shown).
Model 3: Interactions between SNPs on chromosome 21 and SNPs on the diploid fraction of the genome
An interaction analysis was performed between all chromosome 21 SNPs (7238 trisomic SNPs) and cis-eQTLs present in the non–chromosome 21 fraction of the genome (8126 SNPs) in order to identify possible interactions between trisomic and disomic regions of the genome contributing to CHD. None of the tested 32,209,100 SNP pair comparisons remained significant after adjusting for multiple testing (an uncorrected P-value of 6.2 × 10−9 was used to establish genome-wide significance at the 0.05 level) (data not shown).
Chromosome 21 CNV association analyses
Copy number variation on the trisomic chromosome 21 could also contribute to the CHD risk in DS and particularly for the AVSD risk (Beckmann et al. 2007; de Smith et al. 2010). After calling CNVs on chromosome 21 (see Methods), association tests using 55 DS-AVSD as cases and 53 DS-without CHD as controls were performed on 4401 CNV tests obtained after performing intersection across all the samples. The P-values of association tests are shown in Figure 3. CNV association tests with FDR ≤0.05 or simpleM P-value ≤0.05 were considered to be significant. Twenty-one CNV tests out of 4401 passed this threshold, constituting three different CNV regions (Table 3). Both FDR and simpleM-based multiple testing correction identified the same CNV regions as significantly associated. These CNV regions, termed here as CNV1, CNV2, and CNV3, are covered by 42, 20, and 62 consecutive probes in our customized NimbleGen CGH array, respectively. All of these consecutive probes for each CNV region showed the same copy number state.
Figure 3.

(A) Chromosome 21–wide Manhattan plot of P-values for DS-AVSD across 4401 consecutive chromosomal regions for CNV association. The P-values are calculated by a two-by-three Fisher's exact test. (Horizontal line) The FDR threshold for chromosome 21–wide α ≤ 0.05; CNV tests are plotted in equidistance. Three CNV regions reached genome-wide significance (FDR ≤ 0.05). (B) Overview of the CNV1 region (Chr21: 42,066,443–42,071,313) 6 kb upstream of the RIPK4 gene. This 4870-bp CNV region (in gray) is defined by merging six contiguous CNV tests (in black) (for details, see text). CTCF, REST, and other transcription factor binding sites are present in this region as well as the histone mark H3K4me1 (data from http://genome.ucsc.edu/ENCODE/). Additionally, an inversion (in pink), reported in the database of genomic variation, overlaps with this CNV region. (C) Overview of the 1820-bp CNV2 region (Chr21: 42,284,480–42,286,300) defined by merging two contiguous CNV tests overlapping with the last exon of the ZBTB21 gene.
Table 3.
Candidate chromosome 21 CNV regions and the frequency of deletion (Del), duplication (Dup), and copy neutral among DS-AVSD (n = 55) and DS without CHD (n = 53)
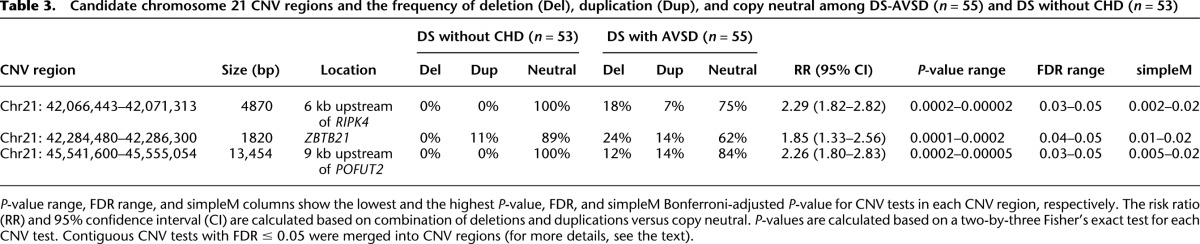
CNV1 (Chr21: 42,066,443–42,071,313) (P-value = 2.5 × 10−4, FDR = 0.05) is a 4.9-kb CNV region located 6 kb upstream of RIPK4 (Fig. 3B; Table 3). It contains PBX3 and BCL3 transcription factor binding sites detected in different cell lines (Fig. 3B). Whereas deletions and duplications were found in 25% of DS-AVSDs, no such events were observed in the controls (Table 3). The risk ratio for this CNV is 2.29 (95% CI: 1.82–2.82) (Table 3). Moreover, CNV1 overlaps with an inversion reported in the Database of Genomic Variants (Variation_37237) (DGV, http://dgv.tcag.ca/) (Fig. 3B). Also for CNV1, there is a SNP probe for rs13048349 that resides within this CNV region and shows the presence of variation in copy number in this region in GenCord cohort (Dimas et al. 2009) that are 215 healthy individuals genotyped by Illumina 550K SNP beadchips, and in our collection of Down syndrome individuals (Supplemental Fig. S3C,D), while the adjacent SNP probe for rs13048349 outside of this CNV region did not show any copy number variation (Supplemental Fig. S3A,B).
CNV2 (Chr21: 42,284,480–42,286,300) (P-value = 1.9 × 10−4, FDR = 0.05) is a 1.8-kb CNV region located within ZBTB21 including its final exon. Whereas deletions were found in 24% of DS-AVSDs, no deletion was observed in the controls (Table 3). The risk ratio for this CNV region is 1.85 (95% CI: 1.33–2.56) (Table 3). Interestingly, both CNV1 and CNV2 fall within the 1.7-Mb CHD critical region defined before (Korbel et al. 2009). CNV3 (Chr21: 45,541,600–45,555,054) (P-value = 4.8 × 10−5, FDR = 0.05) is a 13.5-kb region located 9 kb upstream of the POFUT2 gene. CNV3 includes many transcription factor binding sites identified in a variety of cell lines (EP300, CTCF, BCL3, PBX3, TAF1, CEBPB, HEY1) and a long noncoding RNA (LINC00315). While the frequency of deletion and duplication in DS-AVSDs is 26%, no such events were observed in the controls (Table 3). The risk ratio for this CNV is 2.26 (95% CI: 1.80–2.83) (Table 3). Moreover, CNV3 overlaps with a duplicated fragment (Variation_53268) reported in the DGV. In addition, all three detected CNV regions overlap with much larger deletions and duplications reported in the DECIPHER database (https://decipher.sanger.ac.uk) associated with developmental delay (Fig. 3).
To detect the presence of these CNVs in the general (apparently healthy) population, we used qPCR to see if CNV1, CNV2, and CNV3 are present in the Geneva GenCord cohort (62 samples). We observed duplication of CNV1, CNV2, and CNV3 with 9%, 14%, and 16% frequencies, respectively (Supplemental Fig. S4), while no deletions were detected.
Furthermore, we correlated these CNV states detected by qPCR in the GenCord cohort and gene expression of nearby genes based on mRNA-seq data available from the same data set (Supplemental Fig. S5; Gutierrez-Arcelus et al. 2013). CNV2, which resides in the final exon of ZBTB21, correlated with increased expression of this gene compared with copy neutral state (one-sided Mann-Whitney U-test, P = 0.024) (Supplemental Fig. S5). CNV2 is also associated with expression of the C2CD2 gene (one-sided Mann-Whitney U-test, P = 0.0029), which lies 108 kb upstream. CNV3 also is associated with an increase in expression of the nearby gene COL18A1 (one-sided Mann-Whitney U-test, P = 0.045), which lies 158 kb upstream (Supplemental Fig. S5).
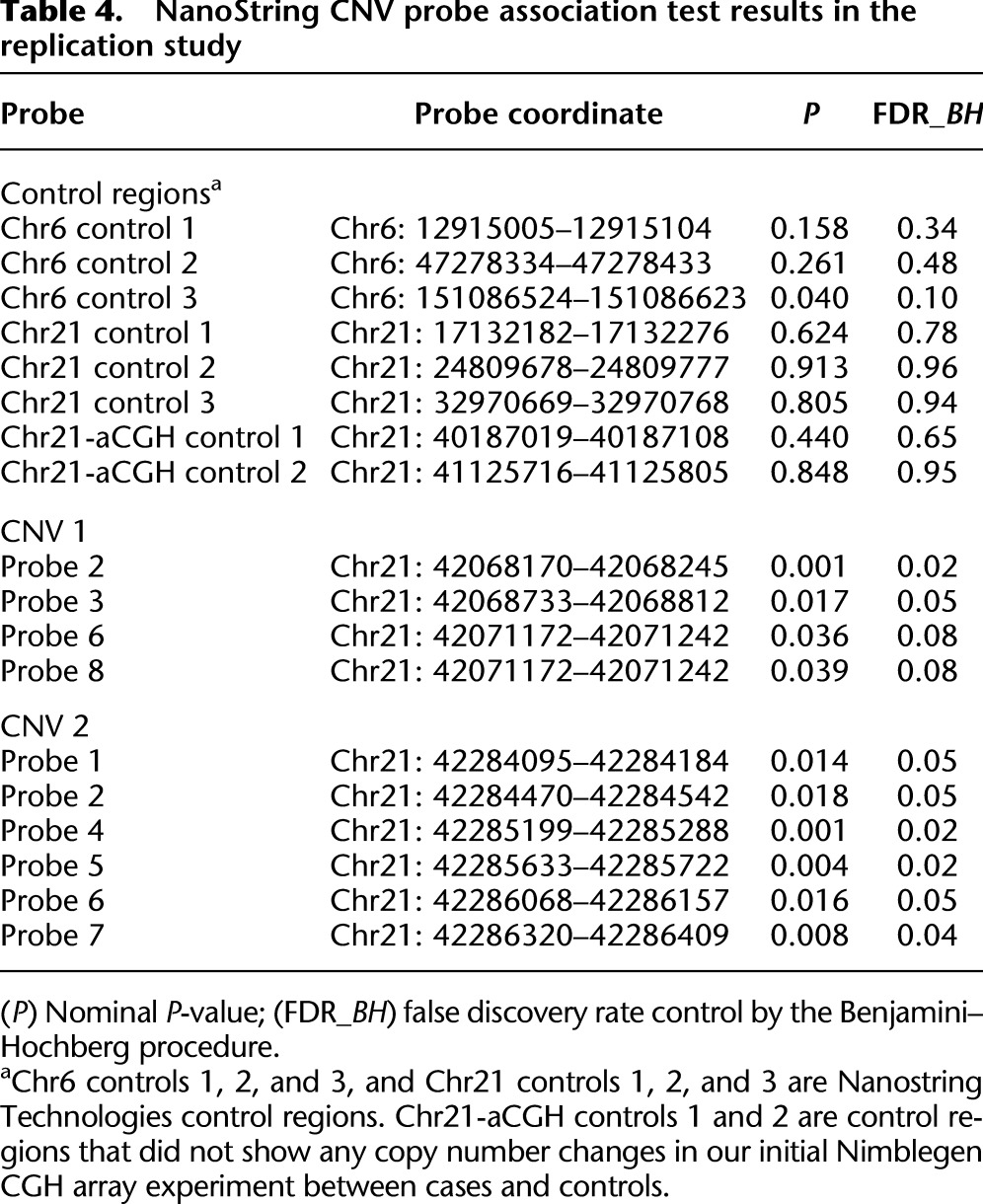
We subsequently tested the significance of these risk CNVs in a replication study of 49 DS-AVSD cases and 45 DS-without CHD controls using NanoString nCounter technology (see Methods). The one-sided Mann-Whitney U-test was used to assess the presence of copy number differences between cases and controls for the three detected AVSD-DS risk CNVs. The presence of less copies for CNV1 in DS-AVSD cases as compared with DS-without CHD is shown by two probes (FDR ≤ 0.05) and marginally by an additional two probes (FDR < 0.1) out of eight probes tested (Table 4). Also, the presence of less copies for CNV2 in DS-AVSD as compared with DS-without CHD is shown by six probes (FDR ≤ 0.05) out of seven probes tested (Table 4). Thus, we conclude that the risk CNV1 and CNV2 were validated in a replication sample. For CNV3, none of the 11 probes tested show any significant signal.
Table 4.
NanoString CNV probe association test results in the replication study
Discussion
Recently, significant progress has been made in the definition of regulatory pathways that control normal and abnormal cardiac valve development involving well-regulated mechanisms for cell movements and cell–cell interactions among distinct populations of heart precursor cells (Gumbiner 1996; Santiago-Martinez et al. 2006; Totong et al. 2011). This suggests that molecules that regulate cell migration and cell–cell adhesion may contribute to CHD. The fact that the frequency of CHD in DS is much higher than in normal euploid individuals raises the hypothesis that dosage-sensitive genes on chromosome 21 greatly increase the risk for CHD. However, the underlying genomic or gene expression variation and the allelic architecture that contributes to the manifestation of a CHD in DS are unknown.
In this study, our hypothesis is that three copies of functional genomic elements on chromosome 21 and genetic variation of chromosome 21 and non–chromosome 21 loci predispose to CHD in DS. Moreover, to test the hypothesis that quantitative difference in gene expression contributes to the development of CHD, cis-eQTLs were used as a subset of the genomic variation space. Genome-wide interaction studies (GWIS) were attempted to further reveal the complex genetic architecture of CHD in DS. Furthermore, we hypothesized that chromosome 21 CNVs could also contribute to the increased risk of the CHD in DS.
These GWAS and GWIS were performed on a population of 187 DS individuals with CHD and 151 DS individuals without CHD. Although the sample size of this study is not large, power calculations (Purcell et al. 2003) suggest that we could likely identify loci with odds ratio >2. PCA analyses did not reveal any considerable population stratification (cases and controls showed similar distributions in the space defined by the first two Principal Components), and we did not formally detect any significant inflation factor (lambda = 1.01) (Supplemental Fig. S7).
The most notable results of this study regarding identifying CHD risk loci in DS, that have been verified in our replication study as well, were as follows:
rs2832616 and rs1943950 within the same LD block on chromosome 21 (both cis-eQTLs for KRTAP7-1 gene) are CHD risk alleles (odds ratios of 2.8 and 2.7, respectively).
A 4.9-kb CNV upstream of the RIPK4 gene (CNV1) in the previously reported CHD region of chromosome 21 with a risk ratio of 2.29.
A 1.8-kb CNV within the ZBTB21 gene (CNV2) in the previously reported CHD region of chromosome 21 with a risk ratio of 1.85.
A pair of interacting cis-eQTLs involving CNOT11 on chromosome 2 and NRGN on chromosome 11 (Bonferroni-adjusted P-value < 0.05).
The chromosome 21 CNV analyses revealed three CNV regions, two of which (CNV1 and CNV2) were verified in an independent replication study and map within the previously reported CHD region of chromosome 21 in DS (Fig. 3; Tables 3, 4). The combined effect of trisomy 21 and the presence of less copies than the neutral state for CNV1 and CNV2 seem to be associated with the AVSD risk in DS. This highlights the importance of local copy number variation on chromosome 21 in the context of trisomy in the pathogenicity of AVSD in DS. CNV1, which resides 6 kb upstream of RIPK4, contains two transcription factor binding sites (PBX3 and BCL3) and shows enrichment for H3K4me1 detected in different cell lines (Fig. 3B), suggesting that this CNV region may contain regulatory elements for the nearby gene RIPK4. Previously, our laboratory has shown an exceptional overexpression of RIPK4 in the heart tissue of the Ts65Dn DS mouse model (Lyle et al. 2004). This, combined with the fact that RIPK4 resides in the minimal critical CHD region on chromosome 21 (Korbel et al. 2009), suggests a possible important role in the pathogenicity of CHD in DS. CNV2, which also maps within the critical chromosome 21 CHD region, involves the last and largest exon of ZBTB21, which encodes the functional portion of the protein (Fig. 3C). ZBTB21, which acts as a transcription repressor (Wang et al. 2005), has been shown to interact with PPP2R2B (Glatter et al. 2009), which in Drosophila regulates the WNT/beta-catenin signaling pathway. This pathway is required for cardiac differentiation in human embryonic stem cells (Paige et al. 2010).
Furthermore, our genome-wide two-locus interaction analysis based on whole-genome cis-eQTLs for CHD shows a significant pair of interacting SNPs located in the MAP4K4 and SPA17 genes (Table 2). Interaction analyses based on whole-genome markers revealed no significant interacting pair of SNPs after adjusting for multiple testing (Supplemental Table S3). Analysis of Gene Ontology (GO) categories (Huang et al. 2009) on the affected genes showed no significant enrichment (Supplemental Table S7).
In summary, the findings of this study reveal several candidate risk loci for CHD in DS. It is reasonable that many genes or other functional genomic elements may contribute to the development of CHD, since a very large number of genes and signaling pathways regulate heart development, and variation in all of these genes may have a contribution (minor or major) to the risk of CHD in DS. Our results support a multifactorial model for the development of CHD, and a complex architecture of risk alleles with cumulative effects that explain the developmental phenotypes.
Methods
Sample collection and genotyping
To identify the genetic components that confer susceptibility to the CHD phenotypes, a cohort of 187 DS patients with well-characterized CHD phenotypes (through ultrasonography, echocardiography, and/or surgery) (AVSD, VSD, ASD) and 151 ethnically matched DS controls (DS without a CHD) were genotyped to perform genetic association studies. Informed consent was obtained for all samples, and the study was approved by the Geneva University Ethics Committee. All DS individuals (cases and controls) are fully trisomic for the entire chromosome 21. The distribution of phenotypical attributes of 187 DS-CHD individuals is as follows: AVSD = 69 (37.0%), ASD = 51 (27.2%), VSD = 67 (35.8%) (Supplemental Table S2).
The genomic DNA used in our GWAS was extracted from blood. Genotyping of 750 ng of genomic DNA was performed using Illumina 550K and 610K BeadChips according to the manufacturer's Infinium II protocol (Illumina).
The genomic DNA used in our replication study was extracted from blood or lymphoblastoid cell lines. The distribution of phenotypical attributes of our replication samples of 92 DS-CHD individuals collected at the Cardiology Department of Hôpital Necker-Enfants malades (Paris) after obtaining informed consent is as follows: AVSD = 64, ASD = 7, VSD = 16, and others = 5. The 80 DS without CHD controls of the replication study come from the Jérôme Lejeune Foundation (Paris).
For quality-control analyses, data from samples were used if a minimum of a 98% call rate was observed for the sample in GenomeStudio data analysis software (Illumina) across all the SNPs common to both HumanHap550 and HumanHap610 BeadChips. Other criteria for SNP filtering were as follows: cluster separation > 0.3, MAF > 0.04, HWE P-value > 0.05, HET − 0.2 < 0 > 0.2. Due to the complexity of SNP calling in trisomic regions of the genome, we removed chromosome 21 SNPs (trisomic SNPs) from this phase of analysis and called and analyzed them separately (see below). After filtering, subsequent analyses were performed on a data set of 431,962 SNPs with the overall call rate of 99.997%. For linkage disequilibrium (LD)–based SNP pruning, we used the option based on recursively removing SNPs within a sliding window of 50 SNPs that shifts 5 SNPs along with each step, and a variance inflation factor (VIF) threshold of 2 (as recommended in the PLINK toolset).
Chromosome 21 SNPs calling
Clustering programs such as GenomeStudio (Illumina) assume a diploid state for each SNP. Since there is no standard algorithm for calling triploid genomes, chromosome 21 SNPs were not adequately called by the software; thus, they were investigated separately and genotypes were called by an in-house procedure. In GenomeStudio software, we used the filter option to choose only chromosome 21 SNPs (trisomic SNPs). Subsequently, we used the sample graph option in GenomeStudio to discriminate four clusters of SNPs by plotting intensity (R) versus allele ratio (theta) for each individual. These four clusters represented the homozygous AAA genotypes (samples with a low theta value), the heterozygous AAB genotypes (samples with an intermediate low theta value), the ABB genotypes (samples with an intermediate high theta value), and the homozygous BBB genotypes (samples with high theta values) (Supplemental Fig. S6). The boundaries for each cluster were defined visually, and we avoid including SNPs located between the boundaries of any two adjacent clusters. Then the SNP data for each cluster were imported in an Excel spreadsheet and were assigned to the respective genotype (AAA, AAB, ABB, and BBB) manually. This manual procedure was performed for calling chromosome 21 trisomic SNPs for all the DS samples used in this study. All the DS samples are fully trisomic for chromosome 21 and clearly showed four clusters of genotypes in the GenomeStudio software for chromosome 21 SNPs. To evaluate the quality of trisomic SNPs called by Illumina beadchips, we genotyped rs7282991 and rs2183593 on chromosome 21 by pyrosequencing for 96 DS samples that were also genotyped by Illumina BeadChips. We found 100% concordance for the genotypes called by both methods. To check for trisomic SNPs that violate Hardy-Weinberg equilibrium (HWE) in Down syndrome individuals, we used a method previously developed (Kerstann et al. 2004). HWE was tested using a two-by-four Fisher's exact test (HWE P-value > 0.05). A total of 7238 SNPs out of 8251 chromosome 21 SNPs were retained.
Association analysis for non–chromosome 21 SNPs was performed by a Fisher's exact test of allele counts in DS individuals with CHD and its different classes (AVSD, ASD, and VSD) and DS individuals without CHD, as implemented in PLINK toolset v0.99q software (Purcell et al. 2007). Quantile-quantile plots (Q–Q plots) were generated by WGAViewer (Ge et al. 2008) to detect inflation of statistics due to population stratification. LocusZoom software was used for showing the regional association plot (Pruim et al. 2010). A logistic regression model provided in PLINK was applied for genome-wide interaction analyses based on the coefficient b3 in Y ∼ b0 + b1.A + b2.B + b3.AB + e, which is a model based on allele dosage for each SNP, A and B (Purcell et al. 2007).
For chromosome 21 trisomic SNPs association and interaction studies, custom scripts implemented in R programming language were used. To test for possible differences in disease susceptibility due to single- and two-locus interaction, a Fisher's exact test and a logistic regression model were applied, respectively. Logistic regression was also used to assess the statistical interaction of trisomic SNPs on chromosome 21 with the disomic SNPs (only cis-eQTLs) on the rest of the genome.
Population stratification
Population stratification for the GWAS data was examined by Principal Component Analysis (PCA) using the EIGENSTRAT software v3.0 (Price et al. 2006). We also estimated ancestry proportion by an identity-by-state-based (IBS) clustering method implemented in the PLINK toolset. Briefly, 104,338 SNPs were used by the EIGENSTRAT software and all available SNPs (n = 431,962) by the PLINK IBS clustering method to assess an underlying population structure (Supplemental Fig. S7). There was no evidence of population stratification, because the genomic inflation factor calculated by both EIGENSTRAT and PLINK is 1.01 and 1.02, respectively.
SNP genotyping by pyrosequencing and Sanger sequencing
Pyrosequencing was performed for genotyping rs2832616, rs2183593, rs7282991, and rs9723772. Sanger sequencing was used for genotyping rs1943950 and rs681418, which we were not able to genotype by pyrosequencing.
The PCR primers were designed using Pyrosequencing Assay Design version 1.0.6 (Qiagen; Supplemental Tables S5, S6). The pyrosequencing reactions were automatically performed with a PSQ 96MA system (Qiagen) according to the manufacturer's instructions. All reactions were constructed as recommended by the manufacturer's instructions. The PCR conditions for pyrosequencing were 94°C for 5 min; (94°C for 30 sec, 60°C for 30 sec; Tm reduces ½ °C per cycle, and 72°C for 20 sec) for nine cycles; (94°C for 30 sec, 55°C for 30 sec, and 72°C for 20 sec) for 29 cycles; 72°C for 5 min. The PCR conditions for Sanger sequencing were 94°C for 2 min; (94°C for 30 sec, 55°C for 30 sec, and 72°C for 45 sec) for 26 cycles; 72°C for 5 min.
Chromosome 21 CNVs analyses
To assess whether CNVs on chromosome 21 could predispose to CHD, we comprehensively searched both for known and novel CNVs on chromosome 21 using a custom CGH array. A subset of 55 DS with AVSD and 53 DS without CHD samples were compared in a case-control approach.
Array design
To detect CNVs potentially contributing to the CHD in DS, NimbleGen 12 × 135K custom arrays were used. These arrays were designed to screen human chromosome 21 for both known and de novo CNVs. High-density coverage (a mean of one probe per 100 bp) was used for a 9-Mb region (Chr21: 38,000,000–46,944,323) that is known to be associated with a CHD in DS. 89,443 probes were printed for this region. In addition, to genotype 95 known chromosome 21 CNVs (Conrad et al. 2010), a minimum of 30 probes per known CNV was printed on the arrays. Furthermore, 500 probes on the unique portions of chromosome X and chromosome Y were printed on each subarray to act as internal controls in sex-mismatched hybridizations and to allow detection of possible sample mixes in our cohort. The remaining euchromatic portion of chromosome 21 (Chr21: 13,260,001–37,999,999) was covered by the remaining 42,876 probes available on the NimbleGen 12 × 135K array. As a reference DNA, a lymphoblastoid cell line derived from a DS individual without CHD was used for all analyses. The reference DNA is fully trisomic for chromosome 21 (Supplemental Fig. S8). Dye swap experiments and technical replicates (between and within arrays and subarrays) were performed to measure dye bias effects and to remove random errors introduced in the experiment.
Hybridization signals were extracted using NimbleScan software v2.5 to generate General Feature Format (GFF) files, and SignalMap software v1.9.0.05 (NimbleGen) was used subsequently to visualize and analyze the data. Log2 ratios were obtained at each probe position and then were quantile-normalized to correct for interarray variability (Supplemental Fig. S9). Putative CNVs were called as chromosomal segments with unusually high or low log2 ratios of fluorescent intensity between the test DNA (DS with or without CHD) and reference DNA (a DS individual without CHD) using the genome alteration detection analysis (GADA) algorithm (Pique-Regi et al. 2008) using the options “-M 10 -T 12 -a 0.2.”
CNVs calling by GADA were followed by intersection across all the individuals to obtain CNV intersected regions (CNV tests) (Valsesia et al. 2011). A total of 4401 intersected CNV tests spanning the entire chromosome 21 were identified according to three classes (deletion, duplication, and copy neutral). We applied a two-by-three Fisher's exact test to detect any association signal for 4401 intersected CNV tests. CNV tests with FDR ≤ 0.05 were considered to be significant, and contiguous CNV tests with FDR ≤ 0.05 were merged into CNV regions. Moreover, we used simpleM (Gao et al. 2008, 2010), a data-driven approach based on PCA that estimates the actual number of effective tests (Meff) in association studies. Briefly, it computes eigenvalues from the pairwise 4401 × 4401 CNV correlation matrix and then derives the Meff using PCA. Meff is then used in a Bonferroni correction on the P-values resulting from two-by-three Fisher's exact test for 4401 CNV tests.
NanoString CodeSet design
NanoString nCounter technology was used to verify the detected risk CNVs in a replication study of 49 DS-AVSD and 45 DS-without CHD following NanoString's recommended protocol for CNV analyses. The three detected DS-AVSD-associated risk CNVs were included in the CodeSet in addition to six control regions selected by NanoString Technologies (three regions on chromosome 21 and three regions on chromosome 6) and an additional two control regions on chromosome 21 that, based on our previous NimbleGen CGH arrays, did not show any copy number variation between cases and controls. CNV1, CNV2, and CNV3 were covered by eight, seven, and 11 probes, respectively. The CodeSet also contains a set of 10 probes called invariant probes designed against autosomal genomic regions that are not copy number variant. The invariant probes were used for normalization purposes. Moreover, the CodeSet includes six positive dsDNA control probes, each targeting a unique DNA sequence present in every assay. The positive control probes were used for technical normalization. Probe sets for each CNV region in the CodeSet were designed and synthesized at NanoString Technologies. All procedures related to DNA quantification, hybridization, detection, and scanning were carried out as recommended by NanoString Technologies at the Genomics platform at the University of Geneva.
NanoString data processing started with a technical normalization of raw NanoString counts for each designed probe using the counts obtained for positive-control probe sets prior to normalization using an invariant probe set. All normalization procedures were performed by nSolver software v1.1 (NanoString Technologies). Finally, we estimated the copy number ratio for each probe relative to the reference sample. One-sided Mann–Whitney U-tests were used to compare the fold change ratio for each probe between cases and controls. The P-value related to each probe was FDR-corrected based on the total 35 probes tested.
Quantitative polymerase chain reaction
Since CNV risk alleles for AVSD in DS should be also common CNVs in normal healthy individuals (healthy non–Down syndrome individuals), quantitative polymerase chain reaction (qPCR) was used to verify their frequency in the normal population. For this purpose, we used 62 DNA samples from the Geneva GenCord collection (Dimas et al. 2009). The primers designed for each risk CNV plus a control region are shown in Supplemental Table S4. Primer pairs were tested and efficiencies were measured using standard curves from serial dilutions of genomic DNA. qPCR was performed using the SYBR Green PCR Kit with a standard protocol, in conjunction with the 7900HT Real-Time PCR System from Applied Biosystems (ABI). Each reaction was carried out in triplicate in 384-well plates in a total volume of 10 μL containing 1× ABI CyberGreen Master mix, 0.9 mM each primer, and with ∼2 ng of DNA. Liquid handling for the 384-well plates was performed with a Biomek robot (Beckman Coulter). The thermal profile recommended by Applied Biosystems was used for amplification (50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 sec, and 60°C for 1 min). The generated data were analyzed with SDS 2.2 software (Applied Biosystems). The quantification was carried out using a pair of primers for the ADARB1 gene on chromosome 21 as a reference gene. For gene copy number assignment, the CT values for each set of triplicates were averaged and normalized against the control primers for the reference gene. The relative copy number for each CNV was calculated as described before (Livak and Schmittgen 2001). We applied a Gaussian Mixture Modeling (GMM) method (Valsesia et al. 2012) to detect copy number variation from the distribution of copy number ratios in an unsupervised approach. We also performed Mann–Whitney U-tests between these CNV states and gene expression of nearby genes (Supplemental Fig. S5). The gene expression data based on mRNA-seq for the Geneva GenCord cohort (Gutierrez-Arcelus et al. 2013) are available for primary fibroblast cells, T cells, and lymphoblastoid cell lines (LCLs).
Genome annotation
Human genome hg18/NCBI 36 was used as our reference.
Data access
The genotyping and chromosome 21 CNV data from this study have been submitted to the EMBL-EBI European Genome-phenome Archive (http://www.ebi.ac.uk/ega/) under accession number EGAS00000000129.
Acknowledgments
We gratefully acknowledge the individuals and families for their participation. We thank the Genomics Platform of the NCCR program “Frontiers in Genetics” for their help in performing qPCR, NanoString, and SNP genotyping experiments. We are also grateful to E.T. Dermitzakis and M. Gutierrez-Arcelus for making available the Geneva GenCord data. We also thank F. Bena, S. Gimelli, E. Stathaki, Y. Dupre, and M. Gagnebin, who assisted in our aCGH experiments. We also thank V. Salle at the Hôpital Necker-Enfants maladies for assisting in our replication sample recruitment. The computations were performed at the Vital-IT (http://www.vital-it.ch) Center for high-performance computing of the SIB Swiss Institute of Bioinformatics. Also our special thanks to the CRB-BioJel Center (http://www.crb-institutlejeune.com/) for their contribution in our sample collection. The study was supported by grants from the NCCR–Frontiers in Genetics, the European AnEuploidy project, the Fondation Child Care, the SNF 144082, the ERC 249968 to S.E.A., and the Spanish Ministry of Ecomomy and Competitivity to X.E. P.M. was supported by a grant from the Bodossaki foundation. K.P. was supported by the EMBO long-term fellowship program ALTF 527-2010.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.147991.112.
References
- Ackerman C, Locke AE, Feingold E, Reshey B, Espana K, Thusberg J, Mooney S, Bean LJ, Dooley KJ, Cua CL, et al. 2012. An excess of deleterious variants in VEGF-A pathway genes in Down-syndrome-associated atrioventricular septal defects. Am J Hum Genet 91: 646–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis SE 1998. 10 years of genomics, chromosome 21, and Down syndrome. Genomics 51: 1–16 [DOI] [PubMed] [Google Scholar]
- Antonarakis SE, Epstein CJ 2006. The challenge of Down syndrome. Trends Mol Med 12: 473–479 [DOI] [PubMed] [Google Scholar]
- Antonarakis SE, Lyle R, Deutsch S, Reymond A 2002. Chromosome 21: A small land of fascinating disorders with unknown pathophysiology. Int J Dev Biol 46: 89–96 [PubMed] [Google Scholar]
- Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S 2004. Chromosome 21 and Down syndrome: From genomics to pathophysiology. Nat Rev Genet 5: 725–738 [DOI] [PubMed] [Google Scholar]
- Barlow GM, Chen XN, Shi ZY, Lyons GE, Kurnit DM, Celle L, Spinner NB, Zackai E, Pettenati MJ, Van Riper AJ, et al. 2001. Down syndrome congenital heart disease: A narrowed region and a candidate gene. Genet Med 3: 91–101 [DOI] [PubMed] [Google Scholar]
- Becker T, Herold C, Meesters C, Mattheisen M, Baur MP 2011. Significance levels in genome-wide interaction analysis (GWIA). Ann Hum Genet 75: 29–35 [DOI] [PubMed] [Google Scholar]
- Beckmann JS, Estivill X, Antonarakis SE 2007. Copy number variants and genetic traits: Closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet 8: 639–646 [DOI] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. 2010. Origins and functional impact of copy number variation in the human genome. Nature 464: 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S, Vukcevic D, Barnes C, Conrad DF, Giannoulatou E, et al. 2010. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464: 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies GE, Howard CM, Farrer MJ, Coleman MM, Bennett LB, Cullen LM, Wyse RK, Burn J, Williamson R, Kessling AM 1995. Genetic variation in the COL6A1 region is associated with congenital heart defects in trisomy 21 (Down's syndrome). Ann Hum Genet 59: 253–269 [DOI] [PubMed] [Google Scholar]
- de Smith AJ, Trewick AL, Blakemore AI 2010. Implications of copy number variation in people with chromosomal abnormalities: Potential for greater variation in copy number state may contribute to variability of phenotype. HUGO J 4: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al. 2009. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 325: 1246–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlevy L, Bennett M, Slender A, Lana-Elola E, Tybulewicz VL, Fisher EM, Mohun T 2010. Down's syndrome-like cardiac developmental defects in embryos of the transchromosomic Tc1 mouse. Cardiovasc Res 88: 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferencz C, Neill CA, Boughman JA, Rubin JD, Brenner JI, Perry LW 1989. Congenital cardiovascular malformations associated with chromosome abnormalities: An epidemiologic study. J Pediatr 114: 79–86 [DOI] [PubMed] [Google Scholar]
- Gao X, Starmer J, Martin ER 2008. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet Epidemiol 32: 361–369 [DOI] [PubMed] [Google Scholar]
- Gao X, Becker LC, Becker DM, Starmer JD, Province MA 2010. Avoiding the high Bonferroni penalty in genome-wide association studies. Genet Epidemiol 34: 100–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, et al. 2003. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424: 443–447 [DOI] [PubMed] [Google Scholar]
- Ge D, Zhang K, Need AC, Martin O, Fellay J, Urban TJ, Telenti A, Goldstein DB 2008. WGAViewer: Software for genomic annotation of whole genome association studies. Genome Res 18: 640–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatter T, Wepf A, Aebersold R, Gstaiger M 2009. An integrated workflow for charting the human interaction proteome: Insights into the PP2A system. Mol Syst Biol 5: 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman TR, Gamliel A, Wessells RJ, Taghli-Lamallem O, Jepsen K, Ocorr K, Korenberg JR, Peterson KL, Rosenfeld MG, Bodmer R, et al. 2011. Over-expression of DSCAM and COL6A2 cooperatively generates congenital heart defects. PLoS Genet 7: e1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner BM 1996. Cell adhesion: The molecular basis of tissue architecture and morphogenesis. Cell 84: 345–357 [DOI] [PubMed] [Google Scholar]
- Gutierrez-Arcelus M, Lappalainen T, Montgomery SB, Buil A, Ongen H, Yurovsky A, Bryois J, Giger T, Romano L, Planchon A, et al. 2013. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife 2: e00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Jiao K, Kulessa H, Tompkins K, Zhou Y, Batts L, Baldwin HS, Hogan BL 2003. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev 17: 2362–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerstann KF, Feingold E, Freeman SB, Bean LJ, Pyatt R, Tinker S, Jewel AH, Capone G, Sherman SL 2004. Linkage disequilibrium mapping in trisomic populations: Analytical approaches and an application to congenital heart defects in Down syndrome. Genet Epidemiol 27: 240–251 [DOI] [PubMed] [Google Scholar]
- Korbel JO, Tirosh-Wagner T, Urban AE, Chen XN, Kasowski M, Dai L, Grubert F, Erdman C, Gao MC, Lange K, et al. 2009. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci 106: 12031–12036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Morishima M, Yu T, Matsui S, Zhang L, Fu D, Pao A, Costa AC, Gardiner KJ, Cowell JK, et al. 2011. Genetic analysis of Down syndrome-associated heart defects in mice. Hum Genet 130: 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Locke AE, Dooley KJ, Tinker SW, Cheong SY, Feingold E, Allen EG, Freeman SB, Torfs CP, Cua CL, Epstein MP, et al. 2010. Variation in folate pathway genes contributes to risk of congenital heart defects among individuals with Down syndrome. Genet Epidemiol 34: 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle R, Gehrig C, Neergaard-Henrichsen C, Deutsch S, Antonarakis SE 2004. Gene expression from the aneuploid chromosome in a trisomy mouse model of down syndrome. Genome Res 14: 1268–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle R, Bena F, Gagos S, Gehrig C, Lopez G, Schinzel A, Lespinasse J, Bottani A, Dahoun S, Taine L, et al. 2009. Genotype–phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur J Hum Genet 17: 454–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS 2006. Postnatal lethality and cardiac anomalies in the Ts65Dn Down syndrome mouse model. Mamm Genome 17: 1005–1012 [DOI] [PubMed] [Google Scholar]
- Paige SL, Osugi T, Afanasiev OK, Pabon L, Reinecke H, Murry CE 2010. Endogenous Wnt/β-catenin signaling is required for cardiac differentiation in human embryonic stem cells. PLoS ONE 5: e11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique-Regi R, Monso-Varona J, Ortega A, Seeger RC, Triche TJ, Asgharzadeh S 2008. Sparse representation and Bayesian detection of genome copy number alterations from microarray data. Bioinformatics 24: 309–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prandini P, Deutsch S, Lyle R, Gagnebin M, Delucinge Vivier C, Delorenzi M, Gehrig C, Descombes P, Sherman S, Dagna Bricarelli F, et al. 2007. Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance. Am J Hum Genet 81: 252–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D 2006. Principal Components Analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909 [DOI] [PubMed] [Google Scholar]
- Priest JR, Girirajan S, Vu TH, Olson A, Eichler EE, Portman MA 2012. Rare copy number variants in isolated sporadic and syndromic atrioventricular septal defects. Am J Med Genet A 158: 1279–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ 2010. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 26: 2336–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Cherny SS, Sham PC 2003. Genetic Power Calculator: Design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19: 149–150 [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. 2007. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. 2006. Global variation in copy number in the human genome. Nature 444: 444–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoll C, Rivals I, Ait Yahya-Graison E, Dauphinot L, Paly E, Mircher C, Ravel A, Grattau Y, Blehaut H, Megarbane A, et al. 2012. Molecular signatures of cardiac defects in Down syndrome lymphoblastoid cell lines suggest altered ciliome and Hedgehog pathways. PLoS ONE 7: e41616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SW, Morris CD, Goldmuntz E, Reller MD, Jones MA, Steiner RD, Maslen CL 2003. Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects. Am J Hum Genet 72: 1047–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizen NJ, Patterson D 2003. Down's syndrome. Lancet 361: 1281–1289 [DOI] [PubMed] [Google Scholar]
- Sago H, Carlson EJ, Smith DJ, Kilbridge J, Rubin EM, Mobley WC, Epstein CJ, Huang TT 1998. Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc Natl Acad Sci 95: 6256–6261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Martinez E, Soplop NH, Kramer SG 2006. Lateral positioning at the dorsal midline: Slit and roundabout receptors guide Drosophila heart cell migration. Proc Natl Acad Sci 103: 12441–12446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, et al. 2005. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 77: 78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara T, Tomizuka K, Miyabara S, Takehara S, Kazuki Y, Inoue J, Katoh M, Nakane H, Iino A, Ohguma A, et al. 2001. Mice containing a human chromosome 21 model behavioral impairment and cardiac anomalies of Down's syndrome. Hum Mol Genet 10: 1163–1175 [DOI] [PubMed] [Google Scholar]
- Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, Sekowska M, Smith GD, Evans D, Gutierrez-Arcelus M, et al. 2012. Patterns of cis regulatory variation in diverse human populations. PLoS Genet 8: e1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totong R, Schell T, Lescroart F, Ryckebusch L, Lin YF, Zygmunt T, Herwig L, Krudewig A, Gershoony D, Belting HG, et al. 2011. The novel transmembrane protein Tmem2 is essential for coordination of myocardial and endocardial morphogenesis. Development 138: 4199–4205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsesia A, Rimoldi D, Martinet D, Ibberson M, Benaglio P, Quadroni M, Waridel P, Gaillard M, Pidoux M, Rapin B, et al. 2011. Network-guided analysis of genes with altered somatic copy number and gene expression reveals pathways commonly perturbed in metastatic melanoma. PLoS ONE 6: e18369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsesia A, Stevenson BJ, Waterworth DM, Mooser V, Vollenweider P, Waeber G, Jongeneel CV, Beckmann JS, Kutalik Z, Bergmann S 2012. Identification and validation of copy number variants using SNP genotyping arrays from a large clinical cohort. BMC Genomics 13: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Kudoh J, Takayanagi A, Shimizu N 2005. Novel human BTB/POZ domain-containing zinc finger protein ZNF295 is directly associated with ZFP161. Biochem Biophys Res Commun 327: 615–627 [DOI] [PubMed] [Google Scholar]
- Williams AD, Mjaatvedt CH, Moore CS 2008. Characterization of the cardiac phenotype in neonatal Ts65Dn mice. Dev Dyn 237: 426–435 [DOI] [PubMed] [Google Scholar]
- Yang TP, Beazley C, Montgomery SB, Dimas AS, Gutierrez-Arcelus M, Stranger BE, Deloukas P, Dermitzakis ET 2010. Genevar: A database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics 26: 2474–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Li Z, Jia Z, Clapcote SJ, Liu C, Li S, Asrar S, Pao A, Chen R, Fan N, et al. 2010. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet 19: 2780–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]