

Abstract
Although more than thirty mammalian genomes have been sequenced to draft quality, very few of these include the Y chromosome. This has limited our understanding of the evolutionary dynamics of gene persistence and loss, our ability to identify conserved regulatory elements, as well our knowledge of the extent to which different types of selection act to maintain genes within this unique genomic environment. Here, we present the first MSY (male-specific region of the Y chromosome) sequences from two carnivores, the domestic dog and cat. By combining these with other available MSY data, our multiordinal comparison allows for the first accounting of levels of selection constraining the evolution of eutherian Y chromosomes. Despite gene gain and loss across the phylogeny, we show the eutherian ancestor retained a core set of 17 MSY genes, most being constrained by negative selection for nearly 100 million years. The X-degenerate and ampliconic gene classes are partitioned into distinct chromosomal domains in most mammals, but were radically restructured on the human lineage. We identified multiple conserved noncoding elements that potentially regulate eutherian MSY genes. The acquisition of novel ampliconic gene families was accompanied by signatures of positive selection and has differentially impacted the degeneration and expansion of MSY gene repertoires in different species.
Y chromosomes have arisen independently in divergent evolutionary lineages across the eukaryotic tree of life (Rice 1996; Marin and Baker 1998; Liu et al. 2004; Graves 2006; Koerich et al. 2008; Carvalho et al. 2009; Kaiser and Bachtrog 2010). While many genes are known to be Y-linked, the actual number of Y chromosomes that have been sequenced is extremely small (Skaletsky et al. 2003; Hughes et al. 2005; Kuroki et al. 2006; Clark et al. 2007; Koerich et al. 2008; Carvalho et al. 2009; Hughes et al. 2010, 2012) in relation to the rapid rate at which whole genomes are presently being sequenced. This reduced emphasis on sequencing Y chromosomes can be primarily attributed to their presumed low gene content and large amounts of repetitive DNA, that is often arrayed into long stretches of nearly identical sequence which precludes the use of shotgun sequencing approaches to assemble Y chromosome sequence into large, contiguous scaffolds. These assembly problems are further exacerbated when applying short-read next-generation sequence methods (Alkan et al. 2011).
The most completely sequenced and annotated Y chromosomes are from three recently diverged catarrhine primates: human, chimpanzee, and rhesus macaque (Skaletsky et al. 2003; Hughes et al. 2005, 2010, 2012; Kuroki et al. 2006 ). Comparisons between these species offer an important glimpse into the immense structural variation and complexity that can emerge on the Y within a very short period of evolutionary time. However, these three primate species last shared a common ancestor ∼21 million years ago (Mya) (Meredith et al. 2011) and thus offer limited comparative breadth to discriminate characteristics present across most mammalian Y chromosomes from idiosyncratic features reflective of a small evolutionary sample. This lack of phylogenetic scope has (1) hampered identification of the ancestral properties of eutherian Y chromosomes, (2) obscured broader patterns of evolutionary constraint and selection in a nonrecombining environment, and (3) hidden the frequency with which novel genes arise and/or acquire new functions in species with diverse phenotypes and reproductive strategies.
To expand our knowledge of eutherian Y chromosome structure and gene function, we generated the first extensive MSY (male-specific Y chromosome) sequence from two members of the Carnivora: the domestic cat, Felis silvestris catus, and domestic dog, Canis lupus familiaris. These two carnivore genomes provide a phylogenetically distinct vantage point with which to interpret the evolutionary patterns observed in the primate MSY comparisons, diverging from each other ∼55 Mya, and from primates ∼92 Mya (Meredith et al. 2011). Our combined analysis of two carnivore and three primate MSY sequences, together with physical mapping and functional sequence data from the mouse MSY, represent the first multiordinal comparison assessing deeper levels of evolutionary constraint. We also present a complete analysis of patterns of negative and positive selection to assess their effects on MSY degeneration and expansion.
Results and Discussion
MSY sequence assembly and annotation
Given the unique architecture of mammalian MSYs, and our sequencing strategy based upon 454-sequencing of pooled BAC clones (average read depth 25–30× coverage with 300-bp average read lengths), a combination of approaches was used to assemble the cat and dog MSY sequences. Previous studies show that eutherian Y chromosome sequence is divided primarily into two major sequence classes outside of the pseudoautosomal region(s) (PAR) (Skaletsky et al. 2003; Graves 2006). The first class, the X-degenerate sequences, primarily contains single-copy genes with homologous counterparts on the X chromosome. The carnivore X-degenerate regions were relatively straightforward to assemble using standard de novo assemblers and resulted in large contigs and scaffolds (N50 = 10 kb and 109 kb, respectively), aided by our physical mapping data.
The other class of sequences, referred to as ampliconic, contain both protein-coding and noncoding multicopy gene families that are almost exclusively expressed in testes and play an important role in spermatogenesis. Some ampliconic genes are formerly single-copy X-degenerate genes, while the remainder are derived from autosomal transposition events (Skaletsky et al. 2003). The ampliconic sequences found in sequenced catarrhine primate MSYs contain long stretches of nearly identical sequence that undergo frequent gene conversion, a mechanism which retards their genetic decay (Rozen et al. 2003; Skaletsky et al. 2003; Hughes et al. 2012). Given this structure and our use of shorter 454 read lengths (300-bp average), the resolution of the ampliconic gene regions required more meticulous manual assembly using read depth and paired-end information and was extensively validated (see Methods and Supplemental Material).
We used a combination of molecular cytogenetic data, cDNA capture sequences, and RNA-seq data to annotate and estimate the overall sequence coverage in both of our MSY assemblies. Nearly half of the ∼20-Mb metacentric dog Y chromosome is comprised of a nucleolar organizer region (NOR) that covers Yp, while the MSY is restricted to the proximal portion of Yq (Figs. 1, 2; Supplemental Table 1). The majority of Yq is comprised of the 6.6-Mb PAR, indicating that the functional canine MSY sequence (∼2.5 Mb) may represent a minimal Y chromosome. Similarly, we estimated that the nearly 2-Mb cat X-degenerate sequence spanned by our physical map (Pearks Wilkerson et al. 2008) is largely complete. We did not attempt to sequence the cat ampliconic region because previous and unpublished cytogenetic and sequence data indicate this >30-Mb region covering the long arm and pericentromeric region of the Y is comprised of a limited set of highly repetitive gene families which are difficult to assemble with next-generation sequence data, thus requiring further mapping and sequencing efforts (Supplemental Fig. 1; Supplemental Table 1; Murphy et al. 2006).
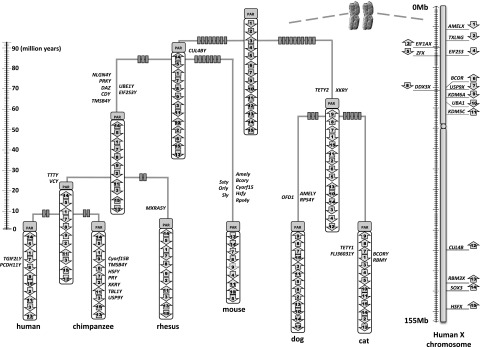
Figure 1.

Schematic representation of the MSY chromosome gene content of six mammals. Different gene/sequence classes are displayed in different colors (roughly following Skaletsky et al. 2003; Hughes et al. 2005, 2010, 2012 for clarity). Mouse physical mapping data are from Mazeyrat et al. (1998). Each sequence schematic is drawn to a different scale to emphasize differences in single-copy X-degenerate gene compaction.
Figure 2.

Fluorescent in situ hybridization and qPCR of dog NOR, PAR, and ampliconic genes. (A) Dog has six autosomal NORs (CFA7, 17, 20; arrows), and one on Y. A total of seven NORs are observed in males and six in females (not shown). (Red) NOR probe. (Green) PAR probe (represented by BAC clone RPCI-183L18). (B) NOR probe (green on Yp+Ycen) and PAR probe (red on Yq) are at opposite ends of the dog Y. (C) Zoomed dog Y showing the location of NOR and the PAR in relation to the centromere. Dog Y is sub-metacentric. (D) KAL1 and TBL1 containing BAC clones hybridize to distal Xp and Yq. (E) TSPY and CUL4BY cDNA probes localize to Ycen-proximal Yq. (F) Quantitative PCR results for SRY and TSPY/TSPYL in three male domestic dogs.
cDNA capture and de novo assembly of testis RNA-seq data allowed us to thoroughly characterize the cat and dog single and multicopy/ampliconic transcribed gene repertoire, which facilitated comparisons with other Y chromosomes. We found clear evidence for a canine ortholog of EIF2S3Y in the de novo assembled RNA-seq transcriptome that was not present in our BAC-based assembly. This is likely caused by lack of coverage (∼4× haploid Y coverage) in the canine BAC library we screened, or inadequate probe design used for the filter hybridizations. We found no orthologs of other eutherian X-degenerate MSY genes in the cat transcriptome assembly that were not already present in our assembly.
Using these approaches, we identified a total of 18 and 20 genes/gene families in the dog and cat MSY sequences, respectively (Fig. 1). We combined our carnivore gene lists with data from the mouse (Mazeyrat et al. 1998; Vernet et al. 2011) and primate (Skaletsky et al. 2003; Hughes et al. 2010, 2012) MSYs, to identify a set of 17 core genes putatively present on the ancestral eutherian Y chromosome (Fig. 3; Table 1). Most of these ancestral genes are broadly transcribed in diverse tissue types and have functional counterparts on the X chromosome (Supplemental Fig. 2; Skaletsky et al. 2003; Pearks Wilkerson et al. 2008). Notably, the two carnivore MSY sequences showed the lowest rates of X-degenerate gene loss when compared to sequenced primate MSYs and mouse mapping and functional data. Taken together, our analysis provides a more accurate estimate of the size and gene composition of the ancestral X-degenerate region (Table 1; Fig. 3).
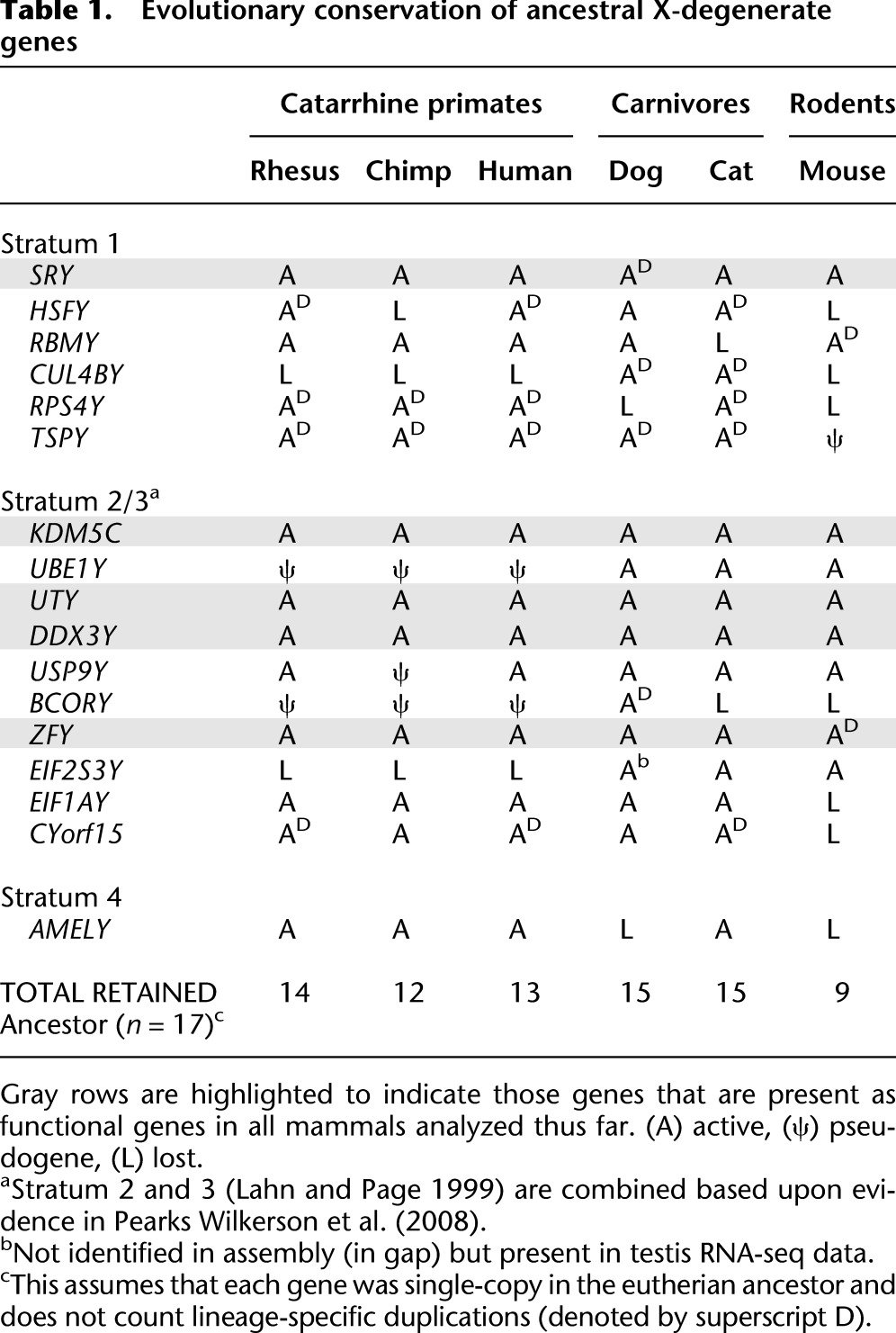
Figure 3.
MSY chromosome rearrangements. Estimate of chromosome rearrangement and gain/loss events within the MSY of six mammalian Y chromosomes. Fifteen of 17 MSY chromosome genes present on the ancestral eutherian MSY are indicated by numbered arrows, with the name and location of the X chromosome gametologs displayed on the right. TSPY and RPS4Y are both dispersed multicopy gene families in multiple species and cannot be confidently assigned to a single position in the terminal taxa, nor in the ancestor. Arrows indicate the gene orientation within each chromosome. Rectangles on each branch represent the minimum number of rearrangements. Inferred gene losses are shown at the right side of each branch, while inferred gene gains are shown on the left side of each branch. Mouse physical mapping data are from Mazeyrat et al. (1998).
Table 1.
Evolutionary conservation of ancestral X-degenerate genes
We characterized a number of lineage-specific changes in the carnivore MSY sequences. We found that the canine ortholog of the therian sex determining gene, SRY, expanded to seven copies within a 700-kb tandem repeat structure (Supplemental Figs. 3, 4), the largest number reported for a mammal, classifying it as an ampliconic gene. The canine MSY also retained two diverged, yet functional copies of an ancestral X-degenerate gene, BCORY, which has pseudogene relics on the primate MSYs. The most remarkable gene identified was a novel testis-specific protein-coding gene (termed DYNG for Dog Y chromosome Novel Gene) (Supplemental Fig. 5). DYNG encodes a 3887-aa protein that includes an ∼1300 residue domain composed of an array of divergent repeats. Neither the domain nor the remaining coding sequence showed any similarity to known transcripts or proteins in public databases, indicating that the gene evolved de novo. Based on its restricted gene expression profile, we speculate that DYNG plays a novel role in canine testis development or spermatogenesis.
Despite preserving a similar number of ancestral X-degenerate genes as the dog, the cat MSY has also evolved extensively since it diverged from the caniform lineage ∼55 Mya. We identified the first example of a novel X-degenerate fusion gene that incorporates parts of the ancestral EIF1AY and CYorf15 genes (Supplemental Fig. 6). The greatest architectural change in the felid lineage, however, was the acquisition and expansion of several novel, testis-specific gene families across Yq: FLJ36031Y and TETY1 were both derived from autosomal genes, and CUL4BY, TSPY, and TETY2 derived from genes shared ancestrally with the X chromosome (Fig. 1; Supplemental Fig. 1; Murphy et al. 2006). These lineage-specific cat Y chromosome gene families range in size from 30 to greater than 100 gene copies and are copy-number-variable within members of the same species (Supplemental Fig. 1). FISH analysis using domestic cat cDNA probes revealed that these same gene families are greatly expanded on the snow leopard MSY, suggesting that they arose prior to the radiation of the cat family (Supplemental Fig. 1).
Gene novelty via X transposition and pseudoautosomal region movement
An additional mechanism, X-to-Y chromosome transposition, can give rise to a third class of MSY sequence. A 4.5-Mb transposed block harboring two genes (Fig. 1) distinguishes the human MSY from chimpanzee and rhesus (Skaletsky et al. 2003; Hughes et al. 2005, 2010, 2012; Kuroki et al. 2006). Here, we found novel evidence for recurrent X-Y transposition events that gave rise to OFD1Y-like genes in dog and cattle (Chang et al. 2011), and pseudogenes on the human and chimp MSY regions (Supplemental Fig. 7; Hughes et al. 2010). While previous studies provide evidence for ongoing gene conversion between X-Y gene pairs (Skaletsky et al. 2003), statistical tests reject a similar hypothesis for OFD1Y-like genes (Supplemental Fig. 8). Further, we determined that the dog X-Y transposition encompasses a total of 120 kb, includes a pseudogene relic of the adjacent TRAPPC2 gene, and exhibits uniform synonymous substitution rates and levels of intronic divergence that are considerably reduced relative to other canine X-degenerate genes (Supplemental Fig. 3; Supplemental Table 2). The acquisition and retention of canine OFD1Y was concomitant with signatures of positive selection and evolution of testis-specific function (Supplemental Figs. 2, 8).
An additional distinguishing characteristic of carnivore Y chromosomes is that they possess substantially larger PARs (∼6.6 Mb) than the ∼2.7- and 0.7-Mb homologous regions in catarrhine primates and mouse, respectively. This difference is due to the carnivore pseudoautosomal boundary (PAB) extending to the terminus of the SHROOM2 gene (Fig. 1; Supplemental Figs. 3, 9), immediately proximal to the carnivore-specific, testis-specific gene TETY2 (Murphy et al. 2006) that arose from PAB movement (Supplemental Figs. 10, 11). Indeed, physical mapping data indicate that most eutherian PARs are markedly longer than human and mouse PARs (Raudsepp et al. 2012) and that the larger primate X-degenerate regions are merely remnants of recent PAB movement and incorporation of ancestral PAR sequence into these three MSYs. When these differences in PAR sequence size are taken into account, we conclude that the ancestral eutherian X-degenerate sequence was small, probably less than 2 Mb (Fig. 1; Supplemental Fig. 3).
Conserved noncoding elements
The phylogenetic breadth provided by our analysis of divergent eutherian MSYs allowed us to identify extreme conservation of MSY microsynteny and conserved noncoding elements. Pairwise comparisons revealed highly diverged chromosome structure and gene orders (Fig. 3; Supplemental Fig. 3). However, one gene cluster containing USP9Y, DDX3Y, and UTY has been preserved in over 340 million years of independently sampled evolutionary history (Figs. 1, 3; Supplemental Fig. 12). Phylogenomic evidence demonstrates microsynteny conservation is favored to maintain gene coregulation (Engstrom et al. 2007; Irimia et al. 2012). Therefore, we searched multispecies MSY alignments for evolutionarily conserved sequences, reasoning that these would correspond to elements that regulate MSY gene transcription. Given the high degree of structural rearrangement (Hughes et al. 2010, 2012), accelerated sequence evolution (Repping et al. 2006; Pearks Wilkerson et al. 2008), and lack of recombination, we hypothesized that retention of ancestral Y chromosome sequence would be extremely rare outside of gene boundaries. Accordingly, we found that the vast majority of highly conserved alignments corresponded to known gene exons (Fig. 4; Supplemental Fig. 3). However, we identified many conserved elements outside of known coding regions, in addition to 47 candidate noncoding regions that had significant predicted RNA secondary structure (Fig. 4; Supplemental Fig. 3). Most predicted elements had supporting data for potential regulatory function based on ChIP-seq and expression data (Supplemental Table 4).
Figure 4.
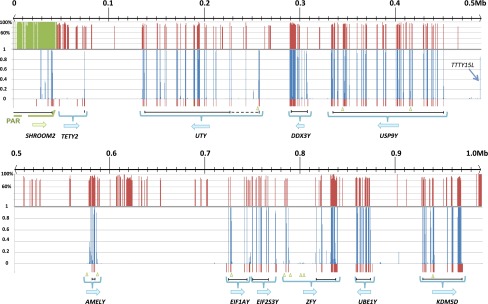
Sequence conservation across five mammalian Y chromosomes. The cat Y chromosome sequence is shown as the reference. Five-way (human, chimpanzee, rhesus, dog, and cat) sequence conservation is calculated and shown by blue columns (bottom panel). The conservation between cat X and Y chromosome sequences is shown by red/green columns (top panel). Gene boundaries are shown below each panel with blue brackets, with arrows defining orientation. Vertical red bars at the bottom of each panel indicate known gene exons. Green triangles along the bottom indicate the locations of conserved elements with potential secondary structure based on significant RNAz scores. Black lines at the bottom indicate the coding region span of each gene (dashed lines indicate conserved coding sequences identified in other mammals but not cat).
One notable evolutionarily conserved element corresponds to the 5′ end of the human TTTY15 gene, which encodes a testis-specific, long noncoding RNA previously identified only in human and chimpanzee (Skaletsky et al. 2003; Kuroki et al. 2006). We speculate that this noncoding element may be associated with coregulation of the conserved USP9Y, DDX3Y, and UTY gene cluster, to which it has remained physically proximal in divergent mammal lineages despite rampant chromosomal rearrangement. Exactly which protein-coding genes are regulated by TTTY15 and other conserved noncoding elements identified here remains a fertile area of future investigation.
The effects of selection on MSY degeneration and expansion
The complete sequence of the human Y chromosome revealed a complex interleaving of X-degenerate and ampliconic sequences distributed across the chromosome (Skaletsky et al. 2003). In contrast, the chimpanzee and, to a greater extent, rhesus X-degenerate and ampliconic regions are partitioned into two functionally diverged and physically delineated compartments (Hughes et al. 2010, 2012). Similarly, the dog and cat Y chromosomes show a dichotomous architecture comparable to other MSYs (Mazeyrat et al. 1998; Vernet et al. 2011), with the vast majority of the conserved X-degenerate genes compressed into a small physical space (Fig. 1). When considered in a phylogenetic context, we propose the ancestral X-degenerate gene region was reduced to as little as 2 Mb prior to the radiation of modern eutherian orders ∼100 Mya and has remained physically partitioned from ampliconic sequences in most mammals, possibly due to constraints imposed by gene regulation. This pattern of rapid size reduction and selection for a core set of MSY genes, followed by a long period of evolutionary retention (Supplemental Fig. 13), provides additional evidence for the exponential decay model (Hughes et al. 2012). In support of this model, we analyzed patterns of selection on all MSY protein-coding genes (Table 2). Our results demonstrate that nearly all single-copy X-degenerate protein-coding genes are under strong purifying selection across mammalian orders (Hughes et al. 2005, 2012).
Table 2.
Results of dN/dS analyses using coding sequences matching the longest human transcript
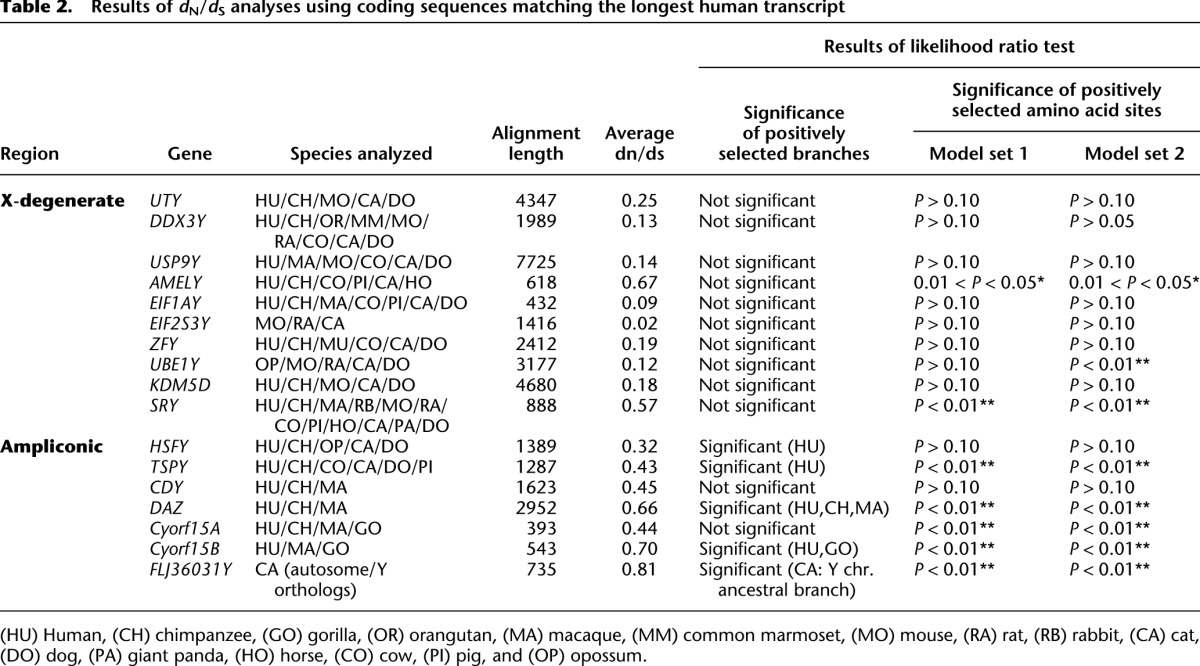
While purifying selection has promoted retention of a small suite of housekeeping genes that are shared with the X, each eutherian mammal MSY has acquired, to varying degrees, repertoires of male-benefit genes with testis-limited expression (Graves 2006). In cat and mouse, the MSY ampliconic regions have expanded into massive chromosome arms, tens of megabases in length, while by comparison the rhesus and dog MSYs have comparatively little ampliconic sequence (Fig. 1). The mouse ampliconic region contains a small number of highly dispersed, lineage-specific gene families that regulate sperm head morphology and development (Mazeyrat et al. 1998; Toure et al. 2004; Vernet et al. 2011; Cocquet et al. 2012). Similarly, cat Yq is comprised of a limited number of highly duplicated testis-specific transcripts (Supplemental Fig. 4) (Murphy et al. 2006). With few exceptions, we observed widespread signatures of positive selection among lineage-specific ampliconic gene families in carnivores (e.g., FLJ36031Y), mouse (e.g., Ssty and Sly), cattle (HSFY) (Hamilton et al. 2011), and primates (DAZ). By contrast, evidence of positive selection was virtually absent in the analyses of single-copy, ubiquitously expressed X degenerate genes (Table 2; Supplemental Fig. 14). Ampliconic genes were frequently characterized by novel exon origination (Supplemental Figs. 6, 10), in some cases resulting in sequences that aligned poorly with large portions of their autosomal or X-linked homologs (e.g., TETY2, DAZ, CDY). These characteristics are hallmarks of rapid evolution observed in reproduction-related proteins (Swanson and Vacquier 2002).
The mouse MSY contains the fewest intact X-degenerate genes of those surveyed here (Table 1), while simultaneously harboring a very large ampliconic gene repertoire evolving by positive selection. By contrast, the cat MSY contains several large multicopy gene families under positive selection (Table 1; Supplemental Fig. 1), yet retains one of the largest functional X-degenerate gene repertoires, thus failing to support predictions that strong positive selection should promote Y chromosome degeneration (Rice 1987; Bachtrog 2004). Rather than continue to degrade, many mammalian MSYs have maintained intact ancestral gene repertoires over tens of millions of years, and in some cases have dramatically expanded their gene content as a consequence of positive selection acting on testis-specific gene families. Recent studies suggest that rapid evolution of mouse X-Y multicopy genes may be driven by post-meiotic conflict (Cocquet et al. 2012), and we hypothesize that gene expansions observed in the cat family (Supplemental Fig. 1) are driven by similar mechanisms.
In conclusion, we find that multiordinal comparisons of eutherian Y chromosome sequences have provided a much improved perspective on lineage-specific gene loss and expansion and have allowed for the first sequence-based reconstruction of ancestral eutherian Y chromosome gene content, as well as the identification of putative MSY regulatory elements. Furthermore, our analysis suggests that many Y chromosomes have rapidly expanding gene repertoires, and that a fine balance between negative and positive selection shapes the gene content of eutherian Y chromosomes. Future sequencing of Y chromosomes with diverse structural variation and sequence class compositions will provide a more thorough understanding of the role that life history traits, behavior, and mating strategies play in Y chromosome amplification and degeneration. Finally, we show that next-generation sequencing of BAC clones combined with careful manual assembly can produce accurate draft assemblies of ampliconic MSY regions.
Methods
Mapping, fluorescence in situ hybridization (FISH) and sequencing
Canine Y chromosome BAC clones were identified by screening filter sets of the RPCI-81 male dog (Doberman breed) BAC library. We used probes designed from known genes and cDNAs captured with a probe made from flow-sorted dog Y chromosome DNA (Murphy et al. 2006; Pearks Wilkerson et al. 2008). BAC clones were grown, DNA extracted, end sequenced, and fingerprinted to build contigs with FPC as described (Pearks Wilkerson et al. 2008). End sequences were used to design additional probes for library screening. STS content mapping was also performed and used to order and orient the BAC clones. DNA from 25 canine BAC clones and 18 feline BAC clones (Pearks Wilkerson et al. 2008) selected from minimum tiling paths were sheared to ∼3-kb insert length and pooled in equimolar ratios to prepare 454 Life Sciences (Roche) FLX paired-end libraries. Pooled libraries for dog and cat were each sequenced to ∼21× and 27× coverage, respectively. FISH was performed with biotin- and dioxigenin-labeled BAC clone and cDNA probes, to help confirm order and relative chromosomal coverage of BAC contigs. A combination of Sanger and Illumina GAII/HiSeq reads were used for analysis of the cDNA capture and tissue-specific RNA-seq libraries.
De novo assembly of cat and dog Y chromosome sequences
De novo assembly of 454-sequencing reads was performed with Newbler 2.5p1 (Margulies et al. 2006) and MIRA 3 (Chevreux et al. 2004). The initial sequence assembly was subjected to custom manual assembly in problematic regions (Supplemental Material).
Repeatmasking
RepeatMasker3.3 (http://www.repeatmasker.org/) was used to detect the type and location of repetitive sequences within each Y chromosome sequence assembly. We also downloaded the draft Felis catus cat genome assembly (felCat6.2) and ran RepeatModeler1.0.5 (http://www.repeatmasker.org/RepeatModeler.html) to identify novel cat-specific repetitive elements not present within the current Repeatmasker library. These new repeats were added, and the repeatmasking analysis was repeated.
Gene annotation and gene expression data analysis
For the dog MSY annotation, we used four sources of expressed sequences: (1) Sanger-sequencing reads from testis cDNA captured using flow-sorted Y chromosome DNA as a probe; (2) the canine EST database; (3) published RNA-seq data from multiple tissues (NCBI RNA-seq data accession no. SRP009687); and (4) Illumina sequences derived from three cDNA capture libraries (testis, brain, and kidney) using MSY BAC clone probe pools. BLAST, Bowtie 2 (Langmead and Salzberg 2012), and TopHat (Trapnell et al. 2009) were used for transcript assembly alignment and detection of alternative transcript splicing variants.
To annotate the domestic cat MSY assembly, we used two sources of expressed sequences: (1) previously published full-length cat Y chromosome mRNA sequences, as well as novel reads identified using cDNA capture with flow-sorted Y chromosome probes (Murphy et al. 2006, 2008); and (2) Illumina RNA-seq reads from domestic cat testis mRNA. Full-length cDNAs were assembled to the reference MSY sequence to identify intron-exon boundaries using Spidey (http://www.ncbi.nlm.nih.gov/spidey/). SOAPdenovo-Trans (http://soap.genomics.org.cn/SOAPdenovo-Trans.html) was used to perform de novo assembly of transcriptome sequences using RNA-seq data. Assembled transcripts were realigned to our MSY genomic sequences to modify the gene annotations, as well as identify potential splicing variants.
To evaluate the potential of missing genes in the cat and dog X-degenerate assemblies caused by gaps of BAC clone overlap and coverage, we applied several steps to check the integrity of our data. First, we used dog and cat RNA-seq data to generate de novo transcript assemblies in SOAPdenovo-Trans. Then, we compiled a list of available mammalian MSY mRNA sequences from all available species (human, chimp, rhesus macaque, mouse, rat, cattle, horse) to query the assembled transcriptomes, employing a relaxed BLAST threshold setting (e < 10−5). All retrieved sequences were then queried against the published domestic dog (canFam3) and cat (felCat6.2) genome assemblies using BLAST and BLAT to identify potential orthologs, reasoning that transcripts of autosomal or X chromosome paralogs can be clearly distinguished from true MSY genes. All identified MSY gene transcripts were then compared to our Y chromosome assemblies to evaluate the integrity of the estimated gene content.
Intrachromosomal sequence comparison and conservation tests
Genomic sequences for human (hg19), chimpanzee (panTro3), mouse (mm9), and dog (canFam3) were obtained from NCBI. The domestic cat draft genome assembly (felCat6.2) was used for cat inter-chromosomal sequence similarity tests. Published MSY sequences of human (Skaletsky et al. 2003), chimpanzee (Hughes et al. 2010), and rhesus (Hughes et al. 2012) were compared to the carnivore MSY sequences using custom Perl code applying the BLAST search engine. Y chromosome sequence alignments were performed with BLASTZ (Schwartz et al. 2004), Multiz, and TBA (Miller et al. 2004). Conservation scores from multiple sequence alignments were obtained with phyloFit/phastCons in the PHAST package (http://compgen.bscb.cornell.edu/phast/). Self dot-plot analyses of cat and dog Y chromosomes were performed using published Perl scripts (Hughes et al. 2005). MGR (Bourque and Pevzner 2002) was used to estimate the minimum number of rearrangements among 15 orthologous MSY synteny blocks shared between the ancestral X and Y chromosomes. We applied the software RNAz (Washietl et al. 2005) to predict potential noncoding RNAs or other regulatory elements among all conserved alignments.
Phylogenetic tree reconstruction and gene conversion tests
Multiple sequence (nucleotide and amino acid sequences) alignments were performed using MAFFT (Katoh and Toh 2010). We used MODELTEST (Posada and Crandall 1998) to estimate the best fitting substitution models for each gene. RAxML 7.0 (Stamatakis 2006) was used for maximum likelihood (ML) tree searching and bootstrap tests. We used GENECONV (http://www.math.wustl.edu/∼sawyer/geneconv/) to identify probable regions that had undergone gene conversion between or among different genes. To confirm Geneconv results, we used a sliding window approach to identify regions within the gene alignment that departed significantly from the assumed species tree (Meredith et al. 2011). In this step, sequences were aligned with MAFFT using stringent parameter settings. A series of 600-bp windows (with a 10-nt step between each window) was used to extract sequence data from the sequence alignments, from the start of the 5′ UTR to the end of the 3′ UTR. ML trees were constructed for each sequence window alignment using RAxML, with 100 bootstrap replicates performed to assess nodal support. These trees were compared with a tree built using a constraint topology derived from the whole gene alignment.
Selection tests
To evaluate signatures of natural selection on each MSY coding gene, we used PAML (Yang 2007) and a published phylogenetic tree for mammals (Meredith et al. 2011). For branch-specific tests, two model sets are used: (1) a free-ratio model with one ratio model; and (2) a two ratio model with one ratio model (tested branches suggested by the results of free-ratio and one-ratio comparison). Likelihood ratio tests are applied to results of the two ratio/one ratio comparison to evaluate statistical significance. In order to test for positively selected amino acid sites within each gene alignment, we used two pairs of models: (1) model M1 and model M2a (model set 1 in Table 2); and (2) model M7 and model M8 (model set 2 in Table 2). To test relative synonymous and nonsynonymous rates on specific branches of the phylogeny, branch-site tests were employed. For this test, the null hypothesis and alternative hypotheses are set with fixed dN/dS ratios (equal to 1) and variable dN/dS ratios to calculate maximum likelihood values independently. Accession numbers of published sequences involved in tests described above are shown in Supplemental Table 3.
Quantitative real-time PCR
To estimate the relative copy number of genes within the MSY, genomic DNA from three domestic cats and three domestic dogs was isolated using the Invitrogen PureGene kit, and RNA was removed using RNase A. Primers pairs were designed to produce amplicons of 195–205 bp from genomic DNA for each gene, in most cases spanning intron/exon boundaries. Three known single-copy genes for cat (DDX3Y, USP9Y, UTY), seven putative multicopy genes for cat (TSPY, TETY1, TETY2, CUL4BY, RPS4YL, HSFY, FLJ36031Y), and three putative multicopy genes for dog (SRY, TSPY, TSPY-like) were evaluated using the Roche 480 SYBR-Green protocol. Reactions contained 50 ng genomic DNA in a 10-μl reaction volume in 384-microwell plates. All qPCR reactions were performed in triplicate on a Roche Applied Science LightCycler 480 instrument with thermal cycling conditions of 95°C for 10 min, followed by 45 cycles of 95°C for 15 sec, and 60°C for 1 min. Primer efficiency standardization using a twofold genomic DNA dilution series over three orders of magnitude was performed for each primer pair. Copy number was calculated using the 2-ΔΔct method (Schmittgen and Livak 2008), with the known single-copy gene UBE1Y used as the control for quantification in both cat and dog.
Data access
Raw 454 sequence reads have been deposited in the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession numbers SRS375726 and SRS375675. Illumina RNA-seq reads from domestic cat testis mRNA have been deposited in SRA under accession number SRA059462. The dog Y chromosome transcript sequences have been deposited in the NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession numbers JX964851–JX964871.
Acknowledgments
We thank Wes Warren and Tina Graves (The Genome Institute at Washington University) for performing BAC fingerprinting, contig construction, and 454-DNA sequencing, and Natalia Serdukova (Institute of Molecular and Cellular Biology, Novosibirsk, Russia) for providing the NOR probe. This work was supported by grants from the Morris Animal Foundation (D06FE-063 and D12FE-019 to W.J.M.) and the American Kennel Club, Canine Health Foundation (01262 to W.J.M. and 779 to W.J.M. and T.R.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.154286.112.
References
- Alkan C, Sajjadian S, Eichler EE 2011. Limitations of next-generation genome sequence assembly. Nat Methods 8: 61–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D 2004. Evidence that positive selection drives Y-chromosome degeneration in Drosophila miranda. Nat Genet 36: 518–522 [DOI] [PubMed] [Google Scholar]
- Bourque G, Pevzner PA 2002. Genome-scale evolution: Reconstructing gene orders in the ancestral species. Genome Res 12: 26–36 [PMC free article] [PubMed] [Google Scholar]
- Carvalho AB, Koerich LB, Clark AG 2009. Origin and evolution of Y chromosomes: Drosophila tales. Trends Genet 25: 270–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Klabnik JL, Liu WS 2011. Regional selection acting on the OFD1 gene family. PLoS ONE 6: e26195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WEG, Wetter T, Suhai S 2004. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 14: 1147–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, Iyer VN, et al. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218 [DOI] [PubMed] [Google Scholar]
- Cocquet J, Ellis PJ, Mahadevaiah SK, Affara NA, Vaiman D, Burgoyne PS 2012. A genetic basis for a postmeiotic X versus Y chromosome intragenomic conflict in the mouse. PLoS Genet 8: e1002900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom PG, Ho Sui SJ, Drivenes O, Becker TS, Lenhard B 2007. Genomic regulatory blocks underlie extensive microsynteny conservation in insects. Genome Res 17: 1898–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves JAM 2006. Sex chromosome specialization and degeneration in mammals. Cell 124: 901–914 [DOI] [PubMed] [Google Scholar]
- Hamilton CK, Revay T, Domander R, Favetta LA, King WA 2011. A large expansion of the HSFY gene family in cattle shows dispersion across Yq and testis-specific expression. PLoS ONE 6: e17790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Pyntikova T, Minx PJ, Graves T, Rozen S, Wilson RK, Page DC 2005. Conservation of Y-linked genes during human evolution revealed by comparative sequencing in chimpanzee. Nature 437: 100–103 [DOI] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Pyntikova T, Graves TA, van Daalen SK, Minx PJ, Fulton RS, McGrath SD, Locke DP, Friedman C, et al. 2010. Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature 463: 536–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Graves T, Fulton RS, Dugan S, Ding Y, Buhay CJ, Kremitzki C, et al. 2012. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature 483: 82–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M, Tena JJ, Alexis M, Fernandez-Minan A, Maeso I, Bogdanovic O, de la Calle-Mustienes E, Roy SW, Gomez-Skarmeta JL, Fraser HB 2012. Extensive conservation of ancient microsynteny across metazoans due to cis-regulatory constraints. Genome Res 22: 2356–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser VB, Bachtrog D 2010. Evolution of sex chromosomes in insects. Annu Rev Genet 44: 91–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Toh H 2010. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26: 1899–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerich LB, Wang XY, Clark AG, Carvalho AB 2008. Low conservation of gene content in the Drosophila Y chromosome. Nature 456: 949–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroki Y, Toyoda A, Noguchi H, Taylor TD, Itoh T, Kim DS, Kim DW, Choi SH, Kim IC, Choi HH, et al. 2006. Comparative analysis of chimpanzee and human Y chromosomes unveils complex evolutionary pathway. Nat Genet 38: 158–167 [DOI] [PubMed] [Google Scholar]
- Lahn BT, Page DC 1999. Four evolutionary strata on the human X chromosome. Science 286: 964–967 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZY, Moore PH, Ma H, Ackerman CM, Ragiba M, Yu QY, Pearl HM, Kim MS, Charlton JW, Stiles JI, et al. 2004. A primitive Y chromosome in papaya marks incipient sex chromosome evolution. Nature 427: 348–352 [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen ZT, et al. 2006. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin I, Baker BS 1998. The evolutionary dynamics of sex determination. Science 281: 1990–1994 [DOI] [PubMed] [Google Scholar]
- Mazeyrat S, Saut N, Sargent CA, Grimmond S, Longepied G, Ehrmann IE, Ellis PS, Greenfield A, Affara NA, Mitchell MJ 1998. The mouse Y chromosome interval necessary for spermatogonial proliferation is gene dense with syntenic homology to the human AZFa region. Hum Mol Genet 7: 1713–1724 [DOI] [PubMed] [Google Scholar]
- Meredith RW, Janecka JE, Gatesy J, Ryder OA, Fisher CA, Teeling EC, Goodbla A, Eizirik E, Simao TLL, Stadler T, et al. 2011. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science 334: 521–524 [DOI] [PubMed] [Google Scholar]
- Miller W, Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AFA, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14: 708–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy WJ, Wilkerson AJP, Raudsepp T, Agarwala R, Schaffer AA, Stanyon R, Chowdhary BP 2006. Novel gene acquisition on carnivore Y chromosomes. PLoS Genet 2: 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearks Wilkerson AJ, Raudsepp T, Graves T, Albracht D, Warren W, Chowdhary BP, Skow LC, Murphy WJ 2008. Gene discovery and comparative analysis of X-degenerate genes from the domestic cat Y chromosome. Genomics 92: 329–338 [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA 1998. MODELTEST: Testing the model of DNA substitution. Bioinformatics 14: 817–818 [DOI] [PubMed] [Google Scholar]
- Raudsepp T, Das PJ, Avila F, Chowdhary BP 2012. The pseudoautosomal region and sex chromosome aneuploidies in domestic species. Sex Dev 6: 72–83 [DOI] [PubMed] [Google Scholar]
- Repping S, van Daalen SKM, Brown LG, Korver CM, Lange J, Marszalek JD, Pyntikova T, van der Veen F, Skaletsky H, Page DC, et al. 2006. High mutation rates have driven extensive structural polymorphism among human Y chromosomes. Nat Genet 38: 463–467 [DOI] [PubMed] [Google Scholar]
- Rice WR 1987. Genetic hitchhiking and the evolution of reduced genetic-activity of the Y-sex chromosome. Genetics 116: 161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WR 1996. Evolution of the Y sex chromosome in animals. Bioscience 46: 331–343 [Google Scholar]
- Rozen S, Skaletsky H, Marszalek JD, Minx PJ, Cordum HS, Waterston RH, Wilson RK, Page DC 2003. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 423: 873–876 [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ 2008. Analyzing real-time PCR data by the comparative C-T method. Nat Protoc 3: 1101–1108 [DOI] [PubMed] [Google Scholar]
- Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W 2004. Human-mouse alignments with BLASTZ. Genome Res 13: 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J, Bieri T, et al. 2003. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423: 825–837 [DOI] [PubMed] [Google Scholar]
- Stamatakis A 2006. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690 [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Vacquier VD 2002. The rapid evolution of reproductive proteins. Nat Rev Genet 3: 137–144 [DOI] [PubMed] [Google Scholar]
- Toure A, Grigoriev V, Mahadevaiah SK, Rattigan A, Ojarikre OA, Burgoyne PS 2004. A protein encoded by a member of the multicopy Ssty gene family located on the long arm of the mouse Y chromosome is expressed during sperm development. Genomics 83: 140–147 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernet N, Mahadevaiah SK, Ojarikre OA, Longepied G, Prosser HM, Bradley A, Mitchell MJ, Burgoyne PS 2011. The Y-encoded gene Zfy2 acts to remove cells with unpaired chromosomes at the first meiotic metaphase in male mice. Curr Biol 21: 787–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Hofacker IL, Stadler PF 2005. Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci 102: 2454–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z 2007. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586–1591 [DOI] [PubMed] [Google Scholar]