

Abstract
Recent advancements in sequencing-based DNA methylation profiling methods provide an unprecedented opportunity to map complete DNA methylomes. These include whole-genome bisulfite sequencing (WGBS, MethylC-seq, or BS-seq), reduced-representation bisulfite sequencing (RRBS), and enrichment-based methods such as MeDIP-seq, MBD-seq, and MRE-seq. These methods yield largely comparable results but differ significantly in extent of genomic CpG coverage, resolution, quantitative accuracy, and cost, at least while using current algorithms to interrogate the data. None of these existing methods provides single-CpG resolution, comprehensive genome-wide coverage, and cost feasibility for a typical laboratory. We introduce methylCRF, a novel conditional random fields–based algorithm that integrates methylated DNA immunoprecipitation (MeDIP-seq) and methylation-sensitive restriction enzyme (MRE-seq) sequencing data to predict DNA methylation levels at single-CpG resolution. Our method is a combined computational and experimental strategy to produce DNA methylomes of all 28 million CpGs in the human genome for a fraction (<10%) of the cost of whole-genome bisulfite sequencing methods. methylCRF was benchmarked for accuracy against Infinium arrays, RRBS, WGBS sequencing, and locus-specific bisulfite sequencing performed on the same human embryonic stem cell line. methylCRF transformation of MeDIP-seq/MRE-seq was equivalent to a biological replicate of WGBS in quantification, coverage, and resolution. We used conventional bisulfite conversion, PCR, cloning, and sequencing to validate loci where our predictions do not agree with whole-genome bisulfite data, and in 11 out of 12 cases, methylCRF predictions of methylation level agree better with validated results than does whole-genome bisulfite sequencing. Therefore, methylCRF transformation of MeDIP-seq/MRE-seq data provides an accurate, inexpensive, and widely accessible strategy to create full DNA methylomes.
The haploid human genome contains ∼28 million CpGs that exist in methylated, hydroxymethylated, or unmethylated states. The methylation status of cytosines in CpGs influences protein–DNA interactions, gene expression, and chromatin structure and stability; and plays a vital role in the regulation of cellular processes including host defense against endogenous parasitic sequences, embryonic development, transcription, X-chromosome inactivation, and genomic imprinting, as well as possibly playing a role in learning and memory (Robertson 2005; Suzuki and Bird 2008; Laird 2010; Jones 2012). Understanding the role of DNA methylation in development and disease requires accurate assessment of the genomic distribution of these modifications (Laird 2010). Recent advancements in sequencing-based DNA methylation profiling methods provide an unprecedented opportunity to map complete DNA methylomes. Techniques for high-throughput detection of cytosine methylation include bisulfite conversion of unmethylated cytosines to uracil, immunoprecipitation with antibodies specific for methylated DNA, and cleavage of CpG-containing restriction sites by methylation-sensitive or methylation-dependent restriction endonucleases followed by sequencing or microarray hybridization (Bock 2012).
The most comprehensive method, bisulfite treatment followed by sequencing (whole-genome bisulfite sequencing [WGBS], including MethylC-seq [Lister et al. 2009] and BS-seq [Cokus et al. 2008; Laurent et al. 2010]), measures single-cytosine methylation levels genome-wide and directly estimates the ratio of molecules methylated rather than enrichment levels. However, this method requires essentially resequencing the entire genome multiple times for every experiment (with up to half the reads not even covering CpG sites). To obtain a complete DNA methylome, the total sequencing depth required for adequate coverage of each strand is equivalent to 30× of the human genome (90 Gb), which remains an expensive experiment. In addition to its high cost, bisulfite-converted genomes have lower sequence complexity and reduced GC content. Therefore, the performance of WGBS-based methods is also influenced by potential differences in the efficiency of amplification of methylated and unmethylated DNA copies of the same locus, and the ability to accurately align bisulfite-converted sequencing reads to the genome, which is more challenging than alignment of conventional reads (Krueger et al. 2012). As noted, 10% of CpGs in the mammalian genome remain refractory to alignment of bisulfite-converted reads (Laird 2010).
Reduced-representation bisulfite sequencing (RRBS) (Meissner et al. 2008) addresses the cost issue by measuring single-CpG methylation only in CpG-dense regions. For the human genome, it requires only ∼3 Gb of sequencing to achieve the same degree of sequencing depth for most regions of interest. However, RRBS's ability to interrogate a locus is dependent on its MspI-cut-site (CCGG) density and consequently measures 10%–15% of the CpGs in the human genome (Bock et al. 2010; Harris et al. 2010).
Restriction enzyme methods (e.g., MRE-seq) (Maunakea et al. 2010), on the other hand, typically incorporate parallel digestions with three to five restriction endonucleases. Using multiple cut sites, MRE-seq can cover close to 30% of the genome and saturates at ∼3 Gb of sequencing (Nair et al. 2011). These methylation-sensitive enzymes cut only restriction sites with unmethylated CpGs, and each read indicates the status of a single CpG. While methylated CpGs could be inferred by the absence of reads at cutting sites, this would require assuming perfect digestion, which is not typically done in practice.
In contrast to MRE-seq, methods using monoclonal antibodies against 5-methylcytosine (MeDIP-seq) (Weber et al. 2005; Maunakea et al. 2010) or methylated DNA-binding proteins (MBD-seq, domains of MBD2 alone, or in combination with MBD3L) (Serre et al. 2009), to enrich for methylated DNA independent of DNA sequence have been estimated to saturate at 5 Gb of sequencing (Nair et al. 2011). An important advantage of MeDIP over restriction enzyme methods is a lack of bias for a specific nucleotide sequence, other than CpGs. However, the relationship of enrichment to absolute methylation levels is confounded by variables such as CpG density (Pelizzola et al. 2008). Another inherent limitation of MeDIP-seq in its current form is its lower resolution (∼150 bp) compared with MRE-seq or bisulfite-based methods in that one or more of the CpGs in the immunoprecipitated DNA fragment could be responsible for the antibody binding.
Finally, array-based platforms are widely used. Approaches that couple bisulfite conversions with hybridization-based arrays (e.g., Illumina's HumanMethylation450 BeadChip arrays), while having single-base-pair resolution, are limited to a priori–targeted regions. For example, the Illumina BeadChip array assesses methylation at ∼485,000 targeted CpGs, averaging 17 CpGs per gene spread across CpG islands and gene loci. (For this analysis, we also used the previous BeadChip version, which contains 27,000 CpGs.)
As sequencing costs drop, the number of complete single-nucleotide DNA methylome maps is increasing; however, still only a few are publicly available for human. Barring a disruptive technological advance, the need for DNA methylome maps to address fundamental biological questions will likely continue to far outpace the production of new maps for years. In contrast, many more lower-cost DNA methylomes of either lower resolution or lower coverage have been generated across diverse biological and disease states. For example, the NIH Roadmap Epigenomics Project's current release of the Human Epigenome Atlas (Bernstein et al. 2010) contains eight WGBS data sets, 119 RRBS data sets, 49 MeDIP-seq, and 45 MRE-seq data sets. These methods yield largely comparable results but differ significantly in extent of genomic CpG coverage, resolution, quantitative accuracy, and cost, at least using current algorithms to interrogate the data (Bock et al. 2010; Harris et al. 2010). None of these existing methods provides single-CpG resolution, comprehensive genome-wide coverage, and a cost that is affordable for a typical laboratory, particularly when many samples are assayed. To address these needs, we describe here methylCRF, a novel conditional random fields–based algorithm that integrates methylated DNA immunoprecipitation (MeDIP-seq) and methylation-sensitive restriction enzyme (MRE-seq) sequencing data to predict DNA methylation levels at single-CpG resolution.
Results
Motivation for integrating MeDIP-seq and MRE-seq data
MeDIP-seq and MRE-seq provide complementary readouts of DNA methylation (Maunakea et al. 2010). Their protocols are simple and differ in important ways from bisulfite treatment methods (Harris et al. 2010; Maunakea et al. 2010). By using simple heuristics, the combination of these two methods gave promising results in identifying differentially methylated regions (DMRs) and intermediate or monoallelic methylation (Harris et al. 2010). Here we further explore the complementary nature of MeDIP-seq and MRE-seq. All genome-wide DNA methylation profiling methods have their own unique biases, which can lead to errors in assessing methylation states. Observed genome-wide measurements (MeDIP-seq, MRE-seq, WGBS, etc.) are derived from the actual methylation states of the sample, which is unknown or “hidden” from the investigators. These hidden methylation states are often inferred from the observed data, which are usually sequencing reads aligned to the reference genome. Because MeDIP-seq and MRE-seq are independent, complementary measurements of the same methylation state, our confidence in inferring the methylation state can be significantly increased when results from these two methods are integrated (Zhang et al. 2013). For example, a decrease in MeDIP-seq signal could reflect a biological event (we infer that this region is unmethylated) or could be a methodological artifact; but if the inferred unmethylated state is corroborated by an increase of MRE-seq signal, then the inference of unmethylation is stronger. Thus, integrating MeDIP-seq and MRE-seq is expected to significantly improve our ability to predict methylation levels accurately.
While MeDIP-seq and MRE-seq data correlate to a certain degree with WGBS measurements (Fig. 1A,B), their relationship is not well represented by a simple linear translation. It has also remained technically challenging to infer absolute methylation levels from enrichment measurements alone (Down et al. 2008; Pelizzola et al. 2008; Chavez et al. 2010; Harris et al. 2010). Importantly, existing high-resolution methylomes and prior regional analyses reveal that CpG methylation levels are highly nonrandom throughout the genome (Lister et al. 2009). The levels vary strongly with local CpG density, display distinct genomic feature-specific characteristics, and show strong correlation between neighboring CpG sites (Fig. 2A–C). These properties motivated us to use a formal statistical model to explore these complex relationships with the goal of making an accurate, comprehensive, high-resolution prediction of DNA methylation levels from MeDIP-seq and MRE-seq data.
Figure 1.

Relationship between MeDIP-seq and MRE-seq and MethylC-seq. (A) A kernelized density plot of per CpG MeDIP-seq normalized read count values as a function of MethylC-seq methylation levels shows a complex, approximately proportional relationship. (B) MRE-seq normalized read counts as a function of MethylC-seq methylation levels shows a complex, approximately inversely proportional relationship.
Figure 2.

CpG methylation levels are nonrandom throughout the genome. (A) A kernelized density plot of MethylC-seq methylation levels as a function of CpG density. Methylation varies in a CpG density–dependent manner with the majority of CpGs at 0–0.75 density with 75%–100% methylation and a smaller group at 0.75–1.25 density with almost 0% methylation. (B) CpG methylation levels as a function of their immediately 5′-CpG methylation level (up to 750 bp). (C) Distribution of MethylC-seq methylation levels at CpG islands, exons, and introns.
Summary of the methylCRF algorithm
We chose a conditional random field (CRF) model to integrate MeDIP-seq and MRE-seq data to predict genome-wide single-CpG methylation levels. Like the hidden Markov model (HMM), which has been extensively adopted by the computational biology field (Durbin et al. 1998), CRFs were initially developed for natural language processing (Lafferty et al. 2001), but their application in biological research has been limited (Wallach 2004). However, CRFs have several distinct advantages when modeling data with complex interdependencies, which is a common feature of biological data.
The primary advantage is due to the fact that CRFs model the conditional probability of the variable of interest (CpG methylation states) given observed variables (e.g., MRE scores and MeDIP scores), whereas HMMs model the joint probability of all model variables (Fig. 3A). By conditioning on the observed variables, CRFs are not confounded by correlation between them. This then allows CRFs to efficiently model more complex relationships between CpG methylation levels and larger numbers of potentially correlated and distant observations. In contrast, modeling the full joint probability either requires modeling the interdependencies between the observations (for example, between MeDIP-seq read count and CpG density) or making the assumption that they are conditionally independent (Fig. 3B)—which, in this case, they are not (Pelizzola et al. 2008). Since very little is actually known about possible additional confounding factors in these assays, this construction gives us significant freedom in choosing what data to use in predicting methylation levels. Considering the number of features that may influence CpG methylation, we believe that this is a critical benefit. This also allows the dependency of the methylation ratio of a CpG on any single observation or groups of observations anywhere in the genome to be accounted for with the model complexity growing only by feature number and not by distance of the dependency as would happen in an HMM. Long-range interactions are a common problem (DeCaprio et al. 2007; Yin and Li 2010). Of note, a CpG's methylation ratio can depend on observations in both directions, which would introduce loops in an HMM. Additionally, by not having to consider dependencies between observations, the addition of agglomerative and derivative features is trivial. In our case, this was extremely useful because we could define features incorporating large windows of MeDIP-seq and MRE-seq scores at each CpG without increasing the complexity of the inference and without considering their complicated dependence on individual MeDIP-seq and MRE-seq scores.
Figure 3.

CRF versus HMM. (A) In an HMM, the labels generate the observations, while in a CRF, co-occurrences of the label and observations are associated. (B) HMMs model the joint probability and must consequently model the dependencies in the observations, while CRFs only model the dependencies of the label on the data.
An additional, practical benefit of using CRFs is that the specification of the model is created by defining functions in a way that offers great flexibility. In a typical implementation, these functions are then instantiated with every combination of assignments of values for its parameter variables. However, one could instantiate with only one or a subset of the values that a variable can take without requiring the addition of a full probability distribution over all of the values. In a large model, this can provide a significant reduction in model complexity. Feature functions can also overlap or use a subset of variables of another. While subsetting does not add any expressiveness to the model, it provides an elegant and automated way to handle missing observations as well as to take advantage of a more detailed feature for some configurations of the variables while having a simpler representation for other configurations.
Lastly, since we are only interested in predicting correct methylation levels given our experimental data and the experimental data are fixed at test time, we do not believe there is any sacrifice in power by not modeling the full joint probability distribution. Also note that methylation ratios can be interpreted as a maximum likelihood estimate of the probability of a particular CpG being methylated, and, as our results indicate, this probabilistic interpretation appears effective.
Our complete CRF model, methylCRF, is described in detail in the Methods section. Features include MeDIP-seq and MRE-seq measurements covering individual CpGs, distance between neighboring CpGs, distribution of MRE sites, and genomic annotations including CpG islands, genes, repeats, and evolutionary conservation of DNA sequences. We also generated a variety of derived scores representing averaged experimental measurements in genomic windows of different sizes. We trained a separate CRF for each genomic feature, and for the final methylation estimates, we averaged the predictions for any CRF whose predictions overlapped. Training was performed using MethylC-seq (Lister et al. 2009)–measured methylation levels in randomly chosen regions representing 20% of the genome (Supplemental Table 1). Methylation levels were predicted genome-wide, and performance was evaluated using CpGs that were not used for training.
High concordance between methylCRF and WGBS predictions
Using methylCRF, we predicted individual methylation levels of 28 million CpGs for human H1 embryonic stem cells (ESC) with combined MeDIP-seq and MRE-seq data. Our predictions are in high concordance with MethylC-seq predictions on the same H1 cells, with a genome-wide correlation of 0.77 (Fig. 4A). methylCRF recapitulates the bimodal distribution of methylation levels identified by MethylC-seq (Fig. 4A). Using a previously developed concordance measurement (defined as the percent of CpGs with a methylation proportion difference less than 0.1 or 0.25) (Harris et al. 2010), methylCRF and MethylC-seq are 91% concordant within a 0.25 difference (Fig. 4B). This high concordance is illustrated by a genome-browser comparison between methylCRF and MethylC-seq of a representative genomic locus (Fig. 4C).
Figure 4.
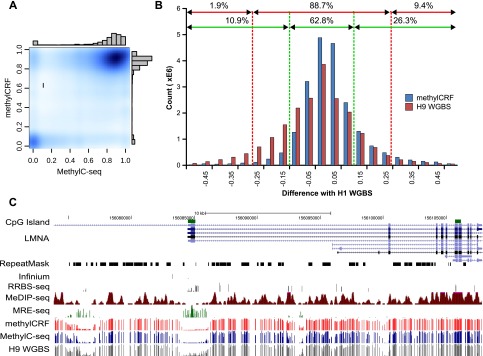
Concordance between MethylC-seq and methylCRF. (A) Kernelized density plot comparing H1 ESC (male) MethylC-seq and methylCRF methylation levels at each CpG with at least one MethylC-seq read. MethylCRF recapitulates the bimodal distribution of MethylC-seq. (B) The number of CpGs as a function of the difference between MethylC-seq and methylCRF methylation levels—the two agree within 25% for 91% of the CpGs and within 10% for 70% of the CpGs. The difference between BS-seq (H9 ESC, female) and MethylC-seq (H1 ESC) on common CpGs is also plotted for comparison. (C) Genome Browser view of per CpG methylation levels across a representative test region on chromosome 1.
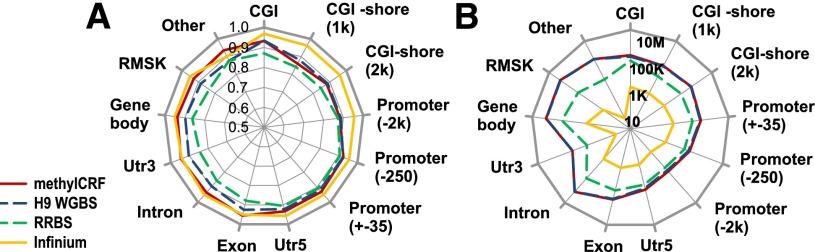
We next compared methylCRF and MethylC-seq on various genomic features (Fig. 5A). methylCRF and MethylC-seq agreed at an exceptionally high level for CpGs within CpG islands, promoters, 5′ UTRs, and exons with 93%, 93%, 93%, and 96% respective concordances. The concordance decreased in RepeatMasker-annotated regions and regions with no annotation (Fig. 5A), possibly reflecting higher mapping errors in these regions, particularly for the reduced complexity reads from bisulfite conversion.
Figure 5.
Comparison between MethylC-seq and methylCRF and other methylation assays. (A) Concordance of methylCRF, BS-seq, RRBS, and Infinium array with MethylC-seq within a 25% window broken out by annotated genomic features. Note that BS-seq (H9) is a female sample, while MethylC-seq (H1) is a male sample. Only CpGs in common were compared. (B) The number of CpGs used for each comparison on a log10 scale.
Benchmarking against other experimental methods
Several additional DNA methylation data sets exist for the H1 ESC line, including data obtained with RRBS and an Infinium methylation array. In addition, a WGBS data set was generated for the H9 human embryonic stem cell line (BS-seq) (Laurent et al. 2010). Data from this closely related ESC line provide the closest “biological replicate” of the MethylC-seq ESC H1 WGBS data set.
When compared with MethylC-seq, methylCRF's performance is almost indistinguishable in the comparison between MethylC-seq and BS-seq on these ESC cell lines (Figs. 4B,C, 5A). Specifically, within a 28% difference window, per-CpG methylation levels between the MethylC-seq (H1) and BS-seq (H9) are 90% concordant, while methylCRF (H1) predictions reach the same concordance with a window of 23%. These windows decrease to 26% for H9 and 18% for methylCRF when we limit the comparison to CpGs with high MethylC-seq (H1) read coverage (e.g., >10 reads) and not in repetitive regions.
RRBS has comparable concordance levels to methylCRF when compared with MethylC-seq. The Infinium array data appear to have slightly higher concordance, which might be a result of having many fewer (∼28,000) CpGs for comparison and/or nonrandom selection of CpGs on the Infinium platform (Figs. 4C, 5A). The high concordance among these popular methods is consistent with previous comparisons (Bock et al. 2010; Harris et al. 2010). However, these methods clearly interrogate very different fractions of the DNA methylomes, as evidenced by the Genome Browser view (Fig. 4C) and CpG coverage comparison (Fig. 5B).
Robust performance across a variety of measurements
The strength of WGBS predictions is significantly influenced by sequencing coverage. Previous analyses suggest that the methylation level of individual CpGs can only be confidently estimated when sequencing depth is at least 10 (Harris et al. 2010). Therefore, typically the minimum requirement for a WGBS experiment is to sequence the bisulfite-converted genome to a depth of 30× (Krueger et al. 2012). However, even at this sequencing depth, a significant number of CpGs still are not covered by enough reads (Fig. 6A). Indeed, we observe increased concordance with increasing MethylC-seq coverage. For example, with a minimum 10-read coverage level, the concordance within a 0.25-threshold window between methylCRF and MethylC-seq increased to 93% (from 91%, minimum one-read coverage) (Fig. 6A).
Figure 6.

Factors affecting concordance between MethylC-seq and methylCRF. (A) Concordance with MethylC-seq as a function of MethylC-seq read count (CpG coverage) at 10% and 25% windows for both methylCRF and BS-seq. The right y-axis (blue bars) indicates the number of CpGs with that coverage. (B) Concordance with MethylC-seq as a function of CpG density at 25% windows for both methylCRF and BS-seq. (C) Concordance of methylCRF within a 25% window broken out by annotated genomic features when only MeDIP-seq, MRE-seq, or genomic features are used. Concordance of BATMAN using MeDIP-seq is also plotted for comparison. (D) methylCRF accuracy on CGIs with high or low methylation (as defined by MethylC-seq). The Lo set of CGIs are those with an average CpG methylation ≤0.2, while the Hi set are those with an average methylation ≥0.8.
CpG density is a major confounding factor in analyzing methyl-cytosine enrichment-based methods (Down et al. 2008; Pelizzola et al. 2008). For example, inferring methylation levels in CpG-poor regions is thought to be highly inaccurate or impossible using MeDIP-seq (Pelizzola et al. 2008). Therefore, we examined methylCRF's performance across regions with differing CpG density and found that the concordance between methylCRF and MethylC-seq does not vary significantly based on CpG density (Fig. 6B).
We also compared methylCRF with Batman (Down et al. 2008), a popular method for analyzing MeDIP-seq data. Since Batman predicts methylation levels in windows of fixed size and not of single CpGs, we assigned each CpG the methylation level of its window. methylCRF consistently outperforms BATMAN in all categories (Fig. 6D).
Since our model learns separate CRFs for each genomic feature, we asked if it is possible that the high correlations between methylCRF and MethylC-seq could be explained by each CRF capturing the a priori methylation distributions of genomic features instead of using the experimental data. To examine this relationship, we applied methylCRF (1) without MeDIP-seq data, (2) without MRE-seq data, and (3) with neither MeDIP-seq nor MRE-seq data, i.e., with only genomic features (Fig. 6C). The experimental data do, indeed, make a large difference in our predictions. Interestingly, MRE-seq alone performs slightly better than MeDIP-seq alone. This may be due to the ability of a CRF to incorporate a priori knowledge that most CpGs are methylated, thus making some MeDIP-seq information redundant. However, it is important to note that the combination of MeDIP-seq and MRE-seq improves performance significantly.
To further demonstrate that experimental data, but not the a priori methylation status of genomic features, drive our prediction, we compared the rates of concordance for methylated and unmethylated CpG islands. Using MethylC-seq scores for H1 ES cells, we defined 17,189 unmethylated and 6728 methylated CpG islands with an average methylation level of ≤0.2 and ≥0.8, respectively. We compared these with the average methylCRF scores for H1 ES cells for each of these CpG islands (Fig. 6D). Clearly, methylCRF predicts similar sets of methylated and unmethylated CpG islands as MethylC-seq, and it does both equally well. Furthermore, on a per-CpG level, unmethylated CpG islands concordance is 0.98, while methylated CpG islands concordance is 0.96. This analysis strongly suggests that while we take advantage of a priori information like genomic features, the algorithm clearly integrates experimental data, relationships within the data, and between data and genomic features to make accurate predictions. We performed a similar analysis on the subset of CpG islands located in promoters—that is, a partitioning of CpG islands independent of the model of the CpG island–specific CRF—and obtained similar results (Supplemental Fig. S1). Similar results were also obtained when we restricted our analysis to intergenic CpG islands (Supplemental Fig. S2).
methylCRF accuracy is robust when applied to a second sample
Having demonstrated that methylCRF can accurately predict differential DNA methylation of CpGs independent of the characteristic methylation status of their genomic feature, we tested whether our model, trained on data from H1 embryonic stem cells, would generalize to data of other samples. This includes testing whether methylCRF can predict differential DNA methylation at a genomic locus between different samples. We reason that if methylCRF is completely dependent on genomic features or is overtrained with ESC data, we would not be able to distinguish between data sets generated from other cell or tissue types.
We generated WGBS, Infinium HumanMethylation450 BeadChip, MeDIP-seq, and MRE-seq data profiles of a human fetal neural stem cells (NSCs) culture (Hu-F-NSC-02, neurosphere cultured cells, ganglionic eminence derived, fetal age of 21 wk) (Supplemental Table 1). We generated a single-CpG-resolution DNA methylome of this sample using methylCRF. We performed similar concordance analysis between predictions of methylCRF and that of WGBS, and between methylCRF and Infinium arrays. The overall concordance is consistently high for comparison of methylCRF against either WGBS or Infinium arrays (Fig. 7A). Specifically, methylCRF and WGBS were 88% concordance within a 0.25 difference window, and 65% concordance within a 0.10 difference window. Additionally, we defined differentially methylated CpG islands between the H1 ESCs and the fetal NSCs using the WGBS data. Out of 26,845 CpG islands, we identified 233 that have significantly different methylation status between H1 ESCs and fetal NSCs, such that their average methylation levels are less than 0.2 in one sample but greater than 0.8 in the other. These WGBS-defined, cell-type-specific differences in CpG island methylation were mirrored by similar differences between H1 ESCs and fetal NSCs estimated by methylCRF (Fig. 7B), suggesting that methylCRF can faithfully predict differential DNA methylation between two samples.
Figure 7.

Applying methylCRF to fetal NSCs. (A) Concordances between methylCRF and WGBS data and between methylCRF and Infinium array broken out by annotated genomic features. (B) CpG islands were grouped as “indifferent” and “different” based on their methylation levels in H1 ESC and fetal NSC (Hu-F-NSC-02) assessed by WGBS data. Actual difference distributions were plotted between H1 ESC (WGBS, red), or H9 ESC (WGBS, blue), or H1 ESC (methylCRF, green) and fetal NSCs (WGBS).
Finally, we evaluated the ability of methylCRF to predict intermediate methylation levels. We did not include imprinted control regions (ICRs) as a genomic feature in training. However, when we examined the methylation status of known ICRs (obtained from https://atlas.genetics.kcl.ac.uk and summarized in Supplemental Table 2), we found that the majority of the ICRs exhibited intermediate methylation levels based on methylCRF prediction in both H1 ESCs and fetal NSCs (Fig. 8A,B), and the levels were consistent with those determined by WGBS-based methods (Fig. 8A,B). Genome Browser views of the data were provided for two exemplar ICRs (Fig. 8C,D).
Figure 8.

Comparing methylCRF and WGBS predictions in imprinted control regions. (A) Known imprinted control regions (ICRs; https://atlas.genetics.kcl.ac.uk) (Supplemental Table 2) were grouped based on WGBS data (H1 ESC, MethylC-seq), as “Lo” (average methylation ≤0.2), “Mid” (average methylation between 0.2 and 0.8), and “Hi” (average methylation ≥0.8). Boxplots represent average methylation levels of these ICRs based on MethylC-seq and methylCRF. (B) Same as A, except for fetal NSCs. (C) A Genome Browser view of ICR near gene MEST (mesoderm specific transcript, chr7). (D) A Genome Browser view of ICR near gene NDN (necdin, chr15).
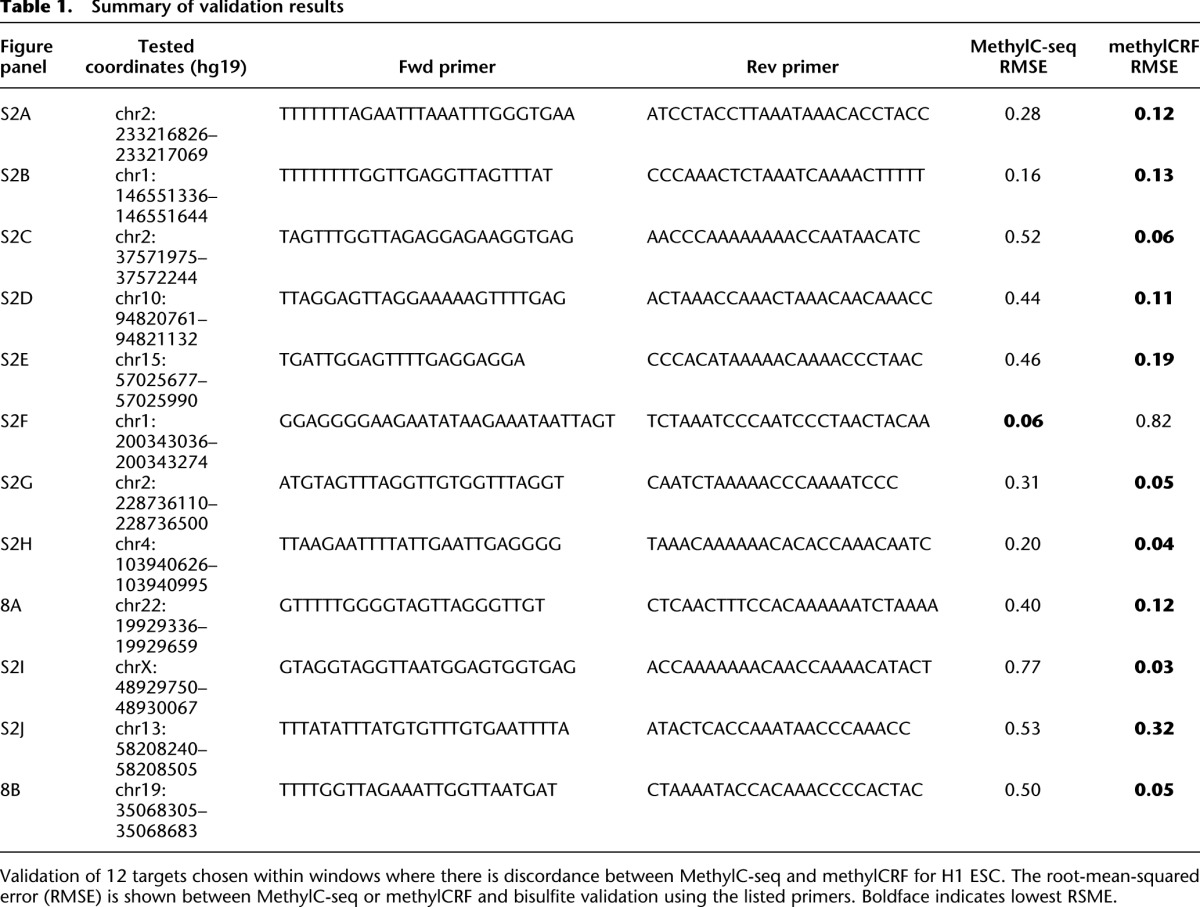
Experimental validation
For regions where methylCRF and MethylC-seq results were discordant in H1 ESCs, we experimentally validated methylation status by performing PCR amplification of bisulfite-converted DNA, followed by Sanger sequencing of cloned amplicon DNA. Out of 12 regions that show disagreement, bisulfite validation agreed better with methylCRF in 11 cases and agreed better with MethylC-seq in only one case. Two of the tested loci are shown in Figure 9, while the remaining sites are summarized in Table 1 and Supplemental Figure S3.
Figure 9.

Experimental validation of regions where there is discordance between MethylC-seq and methylCRF. Genome Browser view and site-specific bisulfite sequencing validation for each region (○ unmethylated CpG; • methylated CpG). The line graph shows the methylation levels estimated by MethylC-seq, RRBS, bisulfite validation, and methylCRF. (A) chr22: 19929336–19929659; MethylC-seq predicted on average a methylation level of 80% methylated, while methylCRF and bisulfite validation agreed on a level of 40% methylated. (B) chr19: 35068305–35068683; MethylC-seq predicted on average a methylation level of 60% methylated, while methylCRF and bisulfite validation agreed that the region is more than 90% to completely methylated.
Table 1.
Summary of validation results
Discussion
DNA methylation is an epigenetic mark that has important regulatory roles in a broad range of biological processes and diseases (Jones 2012). Understanding the role of DNA methylation in development and disease requires knowledge of the distribution of these modifications in the genome. The technology is now available for studying DNA methylation genome-wide, at high resolution and in a large number of samples (Bock 2012). Previous comparisons suggest that many popular methods yield largely comparable results, but they differ significantly in extent of genomic CpG coverage, resolution, quantitative accuracy, and cost (Bock et al. 2010; Harris et al. 2010), at least using current algorithms to interrogate the data.
We introduce a combined computational and experimental strategy to produce single-CpG-resolution DNA methylomes of all 28 million CpGs in the human genome at a fraction of the cost of whole-genome bisulfite sequencing methods. Our computational model, methylCRF, is based on conditional random fields, a model similar to the well-known hidden Markov model, initially developed for natural language processing but less applied in biomedicine. Using this model, we integrated data from two complementary DNA methylation assays (MeDIP-seq and MRE-seq) to predict methylation at single-CpG resolution that were similar to the results from WGBS on the DNA of the same cell line. However, the cost of our two assays combined is <10% of a whole-genome bisulfite sequencing methylome. We showed that methylation levels assessed by methylCRF from MeDIP-seq/MRE-seq data are indistinguishable from a biological replicate of whole-genome bisulfite sequencing.
A complete genome-wide DNA methylome of a given sample will describe methylation levels of every CpG in the genome, ∼28 million in humans. The WGBS-based method is considered the only approach capable of producing such single-CpG-resolution DNA methylomes. It is perhaps the most celebrated method in DNA methylomics to date and generally considered superior to enrichment-based methods (Krueger et al. 2012). One important reason that WGBS appears conceptually superior to enrichment-based methods is that transformation of sequencing results to direct estimates of methylation levels of individual CpGs is straightforward—once data are aligned to the reference genome, one can simply count converted and unconverted Cs to infer methylation levels. Although WGBS does not directly measure single-CpG methylation levels of a sample, investigators can easily infer methylation levels based on experimental data (by sequencing alleles from multiple cells) derived from the true methylation states, i.e., observed counts of converted and unconverted Cs. Such intuitive heuristics makes WGBS seem straightforward.
Similarly, enrichment-based data are also derived from true methylation states. However, current analytical methods for enrichment-based data usually calculate enrichment scores that are indicative of regional DNA methylation levels corrected by local CpG distribution (Down et al. 2008; Chavez et al. 2010) but do not predict single-CpG methylation levels. Our novel algorithm closes this gap—we can predict single-CpG methylation levels based on MeDIP-seq and MRE-seq data, two fundamentally different methods. The algorithm represents a fundamental advancement in statistical modeling over methods currently applied to enrichment-based methods.
There are still significant barriers to individual laboratories adopting WGBS as a routine assay, mainly the high production cost. Our method costs only a fraction of that of WGBS, yet can achieve comparable results. Importantly, the cost saving is scalable; any anticipated reduction in sequencing cost will reduce the cost of WGBS and our method in equal proportions. We performed saturation analysis of MeDIP and MRE and concluded that ∼30M MRE reads and 50M MeDIP reads are required to reach saturation for measuring a human DNA methylome (Supplemental Fig. S4; Supplemental Notes). This translates to 1×–1.5× coverage of the human genome. For WGBS, the requirement is at least 20×–30× coverage. This striking 20-fold difference in required coverage will remain unchanged across different next-generation sequencing platforms. To generate 30× coverage for a human sample is still expensive or even prohibitive for most laboratories. Very often investigators need to assay multiple samples to identify biologically interesting differences with reasonable statistical significance.
Additionally, bisulfite-converted genomes have lower sequence complexity. This not only causes problems in library construction and cluster formation on a sequencing plate, but also more profoundly affects alignment of bisulfite reads to the genome, i.e., mapping. Several algorithms have been developed to improve mapping of WGBS data (Xi and Li 2009; Coarfa et al. 2010; Krueger and Andrews 2011; Frith et al. 2012; Otto et al. 2012), but the problem remains not entirely solved. The confidence of mapping WGBS reads is generally lower than mapping standard, non-bisulfite-converted reads. Since CpG methylation calls are predicted based on aligned reads, how accuracy of methylation calls relates to mapping quality remains uncharacterized. For example, while the effect of biased (C does not match T) versus unbiased (C matches T) alignment has been analyzed (Krueger et al. 2012), no one, to our knowledge, has examined the possibly more critical, alignment biases based on the number of Cs in a read in either type of alignment.
In contrast, MeDIP and MRE protocols produce standard sequencing reads for which existing statistics were designed. Although two libraries are constructed, the total cost (reagents and labor) is comparable to constructing one WGBS, according to published protocols (http://www.roadmapepigenomics.org/protocols). Mapping of MeDIP and MRE reads uses standard mapping tools and is more accurate than mapping of reads from WGBS.
In our experimental validation, we examined 12 loci where MethylC-seq and methylCRF predictions do not agree in H1 ESC. We used bisulfite conversion, PCR, cloning, and sequencing as our validation method because it is considered the gold standard for targeted DNA methylation prediction, and we could exclude the possibility that differences are caused by bisulfite conversion. Nevertheless, in 11 out of 12 loci, the gold-standard approach gave methylation levels that were closer to those from methylCRF predictions than from WGBS. Our interpretation of this result is that most errors made by WGBS might be due to misalignment; however, a comprehensive analysis of WGBS mapping is needed to be certain. Alternatively, these differences may reflect true biological variation. This raises the profound question again: How much of the WGBS-predicted DNA methylome is actually incorrect due to challenges in mapping bisulfite-converted reads? We are eager to explore this question in future studies.
When compared with RRBS and Illumina arrays, our method is obviously much more comprehensive. Our method provides 10-fold to 20-fold more coverage than RRBS or available methylation arrays. Investigators may want to use array-based assays when their target CpG sites are directly interrogated by the array. However, many regions of interest, for example, repeats or cryptic promoters, will not be assessed. There are also a number of examples in which the specific CpG site interrogated by an array does not reflect the true methylation status of the genomic feature (e.g., a promoter) and may lead to false conclusions. Our method not only provides a comprehensive method for exploratory studies, but as the cost of WGBS drops sufficiently for exploratory analysis, the concomitant drop in cost of methylCRF application on MeDIP-seq and MRE-seq will provide investigators a platform to comprehensively address biological questions by comparing multiple samples, conditions, or variances genome-wide.
The accuracy of methylCRF was benchmarked against WGBS, RRBS, Infinium array, and locus-specific bisufite sequencing on H1 human ESCs. In addition, we determined that the high concordance between methylCRF and WGBS is consistent across most genomic feature sets and across all CpG density levels. The power of the method stems from its integrative nature—methylCRF is able to integrate a priori information about the expected methylation states of various types of genomic features, two complementary and independent experimental measurements of methylation states, and hidden relationships among neighboring CpG sites. Genomic sequences and features provide a default expectation of their methylation status. Indeed, the CpG content of the genome reflects germ-cell methylation states during the course of evolution (Li et al. 2012) and has been used to estimate methylation levels directly (Das et al. 2006). This is reflected by the overall concordance of 0.66 when methylCRF makes predictions based on genomic features alone, which represents an expectation of methylation of a majority of CpGs in a normal somatic cell. The concordance is significantly improved when either MeDIP-seq or MRE-seq data are integrated, and the highest concordance is obtained when the data sets are combined. Importantly, methylation predictions made by methylCRF are conditioned on both genomic features and experimental data and are not driven by genomic features alone. This is supported by the accurate separation of methylated CpG islands from unmethylated ones (Fig. 6D), even when focusing on promoter regions and/or intergenic CpG islands (Supplemental Figs. 1, 2).
The accuracy of methylCRF was further benchmarked on WGBS and Infinium array data from a second sample. Here methylCRF trained on H1 ESC data was applied to MeDIP-seq and MRE-seq of a fetal-brain NSC sample. The concordances between methylCRF and WGBS, and between methylCRF and Infinium array, were at similarly high levels as those obtained on analyzing H1 ESC data. Moreover, methylCRF can reliably identify differentially methylated regions between the two samples. This strongly suggests that our model trained on ESC data can be applied to data of other samples.
In the present implementation of methylCRF, we only consider CpG methylation and assume that all signals obtained from MeDIP-seq and MRE-seq are results of CpG methylation. We also assume WGBS-produced methylation signal and ignore complications caused by hydroxymethylation. We note that methylations of cytosines in the context of other than CpG (i.e., CHG and CHH) are rare in somatic cells but are, indeed, present in embryonic stem cells, usually in low levels, and are associated with highly methylated CpGs (Lister et al. 2009). The biological significance of CHG and CHH methylation in mammalian cells is yet to be determined. Our statistical model is general enough to incorporate non-CpG cytosine methylation, but we focused on CpG methylation in this study. Our statistical model is also general enough to incorporate data on hydroxymethylation when they become more and more available.
Our study has several limitations. Because the cell line we used to train, H1 ESC, is male, it is possible that the additional X chromosomes in female samples may not be as accurate. This is because males will only have one allele aligning the reference X chromosome, whereas a female will have two, resulting in twice as many reads. In fact, this may also affect WGBS accuracy. The concordance within the 0.25 difference window between H1 and H9 on chrX alone drops to 81%, whereas excluding chrX raises the concordance 92%. The concordance of methylCRF with H9 on chrX is 79%, whereas without chrX it is 90%. Note that this also provides a natural experiment that suggests how methylCRF will perform in cases of large-scale genomic aberrations such as segmental or even chromosomal duplication or deletion, which is frequently found in cancer. Both WGBS and methylCRF seem to be proportionally less accurate when alleles and possibly segments are added or deleted. Nevertheless, once more WBGS data become available, we can trivially extend methylCRF to include a field for structural variations that could be estimated by standard means. Additionally, since MeDIP-seq and MRE-seq are sequencing based, we can make use of existing tools to add SNP-based features and to include input (unenriched sequences). We note that WGBS is at a disadvantage when considering SNPs; in particular, C → T SNPs will either be reported as an unmethylated C unless strand-specific alignment is available or even worse will align to cause false-positive alignments in other locations.
Another potential limitation is that the H1 WGBS (MethylC-seq) and MeDIP-seq and MRE-seq were performed on separate passages of the H1 ESC line and assayed in separate laboratories. This may explain why methylCRF was consistently validated by the bisulfate cloning method over WGBS. However, if this were true, two important inferences can be drawn. First, notable changes in methylation can be seen even between passage numbers. Second, these validations show methylCRF's sensitivity in detecting DMRs based on experimental data—even in very similar biological contexts. Additionally, the accuracy on fetal NSCs is slightly lower than on H1 ESCs. This may suggest that H1 ESCs may have differences in their global methylation than other cell types. While this does not stop methylCRF from detecting tissue-specific DMRs, it suggests that the accuracy may improve further if we retrain methylCRF simultaneously on WGBS from multiple cell types.
Finally, because of our use of genomic feature-specific predictions, methylCRF accuracy may suffer when some part of the methylation machinery breaks down or behaves differently, for instance, as in some cancers. We have indirectly tested this in a sample case, however. Figure 2C shows CpG islands to have an extremely biased distribution of low methylation. However, Figure 6D shows equivalent accuracy in CpG islands that represent a genomic feature with, in the statistical view of methylCRF, aberrant methylation.
Despite the promise of WGBS-based methods, the number of publicly available, complete, single-CpG-resolution DNA methylomes is still small in contrast to the number of lower-resolution and/or lower-coverage DNA methylomes generated by less-expensive methods (e.g., MeDIP-seq and MRE-seq generated by individual laboratories and by the Roadmap Epigenomics Project). Our method can convert these data into single-base resolution, complete DNA methylomes, and thus significantly increase the value of such existing data sets.
In summary, our results suggest that methylCRF is an effective statistical framework capable of integrating two fundamentally different sequencing-based DNA methylation assays—MeDIP-seq and MRE-seq—to predict genome-wide, single-CpG-resolution methylome maps. The concordance of our methylCRF predictions with WGBS falls within the range of concordance between two WGBS experiments on similar cells. methylCRF will thus significantly increase the value of high-coverage DNA methylomes produced using much less expensive methods and provide a general statistical framework for integrating contributions from various types of DNA methylation data regardless of their coverage, resolution, and the nature of their readout.
Methods
methylCRF implementation
methylCRF is implemented using the theoretical framework of conditional random fields (Lafferty et al. 2001). This general framework expresses the conditional probability Pr(Y|X) of a series of hidden states, the random variables Y, given the observed data X:
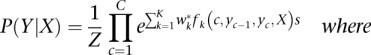 |
Y is the methylation level of every CpG and X is the observations (MeDIP-seq, MRE-seq, genomic context). The C CpGs are indexed by c, and the K feature functions f are indexed by k. The weights w1 … wk are learned via gradient ascent of the log likelihood. Z is the partition function, which provides the global normalization and is the sum of all sequences of methylation levels given X:
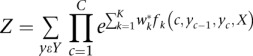 |
Our approach to data features was initially to include anything that we thought might in some way affect methylation. We then let an L1 normalization term during training determine which features were not important by pushing their weights to 0. Therefore, the choice of important features was learned from the data. We split the data into different ranges of effect. We included MeDIP and MRE scores both at the CpG (D0 and M0) and within windows of 20 bp, 200 bp, 2 kb, and 20 kb. We included whether a CpG was at an MRE restriction site (ER) and the distance in base pairs to the previous CpG (PC). From UCSC Genome Browser tracks (Kent et al. 2002), we included a 46-way mammalian phastCons conservation score. We included GC% in 20-bp, 150-bp, and 500-bp windows, and CpG density in a 150-bp window.
We defined one CRF feature for each one of these data features combined with the methylation level of the current and previous, 5′ CpG. We also added CRF features for the next two MeDIP and MRE scores on both the 5′ and 3′ sides. We then defined compound features. We included a feature combining D0 and PC to possibly address the nonlinear relationship between MeDIP and CpG density. We also included one large feature including factors that appeared to be interacting (data not shown), including both the current and previous CpG methylation as well as D0, M0, and PC for the current CpG as well as the two CpGs to upstream and downstream of the current. This feature also included MeDIP and MRE in 20-bp and 2-kb windows, ER, and GC in 20-bp and 150-bp windows for the current CpG as well as ER for the surrounding two CpGs. We additionally included four more features consisting of subsets of these as a fallback for rare combinations of values. A diagram of the complete model is illustrated in Supplemental Figure S5.
The distributions of methylation levels are genomic feature specific (Fig. 2C), so we reasoned that the methylation level transitions between neighboring CpGs are also genomic feature specific. To address this, we trained a separate CRF for each genomic feature: one for each of the genomic annotations in RefGene (5′ UTR, gene body, exon, intron, 3′ UTR, CGI), for the derived types (distal promoter, TSS-2 kb; proximal promoter, TSS-250 bp; core promoter, TSS ± 35 bp; 1 kb flanking each CpG Island; and 2 kb flanking each CpG Island), one for each Repeat class (DNA, LINE, LTR, RNA, SINE, low complexity sequence and simple repeats, and other), and one for the remainder of the genome not covered by any of the previous CRFs.
Training was performed using MethylC-seq (Lister et al. 2009)–measured methylation levels in randomly chosen regions representing 20% of the genome. We used only CpGs with at least 10-read coverage. We performed separate discretization for each CRF. Each of the CRFs was trained using crfasgd (Bottou and Bousquet 2008) using default settings.
CRFs are typically discriminately trained by iteratively ascending their gradient. While the function is convex and thus converges to a global maximum, the whole CRF must be evaluated once for every iteration in the ascent that poses performance issues. However, CRFs have been shown to handily model millions of features (Sha and Pereira 2003). Additionally, the ascent can be performed online providing two benefits: (1) potentially less overfitting due to the less optimal solution and (2) speed of analysis (Bottou and Bousquet 2008). Being discriminately trained, however, CRFs need to be handled carefully so as to ensure their generalizability to future data.
For CpGs that are annotated with multiple features, we combine the methylation predictions by averaging the predictions of the corresponding CRFs and giving each CRF an equal vote. Performance was evaluated using CpGs that were not used for training.
Discretization heuristic
CRFs are rooted in the Natural Language Processing (NLP) community and thus model discrete rather than continuous variables. There has been at least one paper extending CRFs to rankings that require developing a continuous CRF (Qin et al. 2008). Additionally, in the derivation of CRFs, there is no restriction on the form of the random variables, and thus continuous predictors are also an option. However, the theoretical and practical work on modeling and training of discrete CRFs is very extensive. Additionally, the relationship between the predictors and the methylation ratios are complex. Finally, Naïve Bayes is known to perform better with discretized variables (Dougherty et al. 1995). We, therefore, decided to follow in this line of work by representing the relationship between the predictors and methylation ratios as piecewise constant. The trade-off in avoiding the choice of the correct family of continuous distributions for the methylation ratios as well as the predictors in the continuous case is that we must determine where to cut the range of a predictor into pieces. This is equivalent to discretization for which considerable work has been done.
While equal-range or equal-size discretization is straightforward, they did not perform very well (data not shown). We instead chose to use supervised discretization using the methylation ratios to guide the discretization. While good entropy-based methods do exist, they would require the methylation ratios to already be discretized, posing a chicken-and-the-egg problem for which we could not find an existing solution. This led us to develop a two-step heuristic consisting of clustering to discretize the methylation ratios followed by supervised discretization of each predictor. In the first step, which we term “order-preserving clustering,” we cluster the predictors and the methylation ratios together. We iteratively up-weight the methylation ratios and recluster until there is an order between clusters such that all of the methylation values in one cluster are larger than the previous (Supplemental Fig. 6). Given this as a data model–driven discretization of the methylation ratios, we then use supervised discretization on each predictor individually. Note that this heuristic can use any pairwise distance metric, clustering method, or supervised discretization method. We used k-means (Macqueen 1967) and Euclidean distance for clustering and CAIM (Kurgan and Cios 2004) for discretization.
MeDIP-seq, MRE-seq, and WGBS data
All data were obtained from the NIH Roadmap Epigenomics Mapping Centers' repository for human reference epigenome atlas (Bernstein et al. 2010). Experiments were performed under the guidelines of the Roadmap Epigenomics Project (http://www.roadmapepigenomics.org/protocols). Specifically, MeDIP-seq and MRE-seq experiments were performed as described previously (Maunakea et al. 2010). All data have been previously submitted to NCBI and are listed in Supplemental Table 1.
The reads were aligned with bowtie (Langmead et al. 2009) to HG19. The MRE reads were normalized to account for differences in enzyme efficiency and scoring consisted of tabulating reads with ends at each CpG (Maunakea et al. 2010). To allow for comparison between experiments, the CpG read counts for MeDIP were scaled so that the 75th percentile of CpGs with at least one read is 10. Since for each MeDIP read, the CpG that was bound by the antibody cannot be determined, a fractional count was added to each CpG for the read. The final MeDIP score is the sum of CpG scores within the specified window.
Genomic features
RepeatMasker annotations, CpG islands, genomic super duplications, 46-way phastCons, and refGene coding loci features were all downloaded from the UCSC Genome Browser (Kent et al. 2002). The GC percent, CpG density, and MRE sites were calculated using HG19.
Training and prediction
For training, we randomly selected both the location and size of genomic fragments of 75 kb to 750 kb in length comprising ∼20% of the genome. We used only CpGs with at least 10 BS reads. We performed separate discretization for each CRF. For the k-means step in the discretization of BS methylation values, we arbitrarily chose 10 clusters because this seemed a reasonable cutoff for the granularity for the measurement of CpG methylation that we would be interested in. We used Euclidean distance as our metric. Further details on our discretization method are discussed above. Each of the CRFs was trained using crfasgd (Bottou and Bousquet 2008) using default settings. The data for each CRF were split on gaps of >750 bp between consecutive CpGs.
For prediction, the data were created as for the training data. For the final methylCRF predictions, we combined the predicted methylation levels of all the CRFs by averaging the predictions for CpGs that were shared by multiple CRFs.
Genome Browser tracks are available as part of Roadmap Epigenomics Project's data visualization hub: http://VizHub.wustl.edu.
Bisulfite treatment and library construction for WGBS
One to five micrograms of gDNA was sonicated to an approximate size range of 200–400 bp. Size selection is performed on a PAGE gel to obtain DNA fragments of 200–300 bp. DNA are quantified by fluorescent incorporation (Qubit, Invitrogen). The library preparation includes end-repair and phosphorylation with NEBNextTM or Illumina Sample Prep Kit reagents, and addition of an A base to the 3′ end of the DNA fragments. Methylated adaptors are ligated, and size selection is performed to remove excess free adaptors. The ligated DNA is quantified by Qubit, and ∼100 ng of DNA is used for bisulfite conversion. A methylated adaptor ligated to unmethylated lambda-phage DNA (NEB) is used as an internal control for assessing the rate of bisulfite conversion. The ratio of target library to λ is 1600:1. Bisulfite conversion of the methylated adaptor-ligated DNA fragments follows the FFPE Tissue Samples Protocol from QIAGEN's EpiTect Bisulfite Kit. Cleanup of the bisulfite-converted DNA is performed, and a second round of conversion is applied. Enrichment of adaptor-ligated DNA fragments is accomplished by dividing the template into five aliquots followed by eight cycles of PCR with adaptor primers. Post-PCR size selection of the PCR products from the five reactions is performed on a PAGE gel. Following 100-bp paired-end sequencing on an HiSeq2000, sequence reads were aligned and processed through the Bismark pipeline.
Infinium assay
Bisulfite conversion was performed on 1 μg of genomic DNA using the EZ DNA methylation kit (Zymo Research) as per the manufacturer's alternative incubation conditions protocol. The bisulfite-converted DNA was amplified and hybridized to an Infinium HumanMethylation450 beadchip (Illumina) following the Infinium HD methylation assay protocol at the UCSF Genomics Core Facility. Methylation levels (beta values) were determined using the Methylation Module of the Illumina GenomeStudio software.
Bisulfite validation
Total genomic DNA underwent bisulfite conversion following an established protocol (Grunau et al. 2001) with the following modifications: incubation at 95°C for 1 min and 50°C for 59 min for a total of 16 cycles. Regions of interest were amplified with PCR primers (Table 1) and subsequently cloned using pCR2.1/TOPO (Invitrogen). Individual bacterial colonies were subjected to PCR using vector-specific primers and sequenced (Quintara Biosciences). The data were analyzed with online software BISMA (Rohde et al. 2010). The results are summarized in Table 1 and Supplemental Figure 3.
Software availability
MethylCRF is completely open source software. The source code, parameter sets, and genomic data sets, as well as instructions are available at http://methylCRF.wustl.edu.
Acknowledgments
We thank collaborators in the Reference Epigenome Mapping Centers (REMC), Epigenome Data Analysis and Coordination Center, and NCBI who have generated and processed the data used in this project. We acknowledge support from the NIH Roadmap Epigenomics Program, sponsored by the National Institute on Drug Abuse (NIDA) and the National Institute of Environmental Health Sciences (NIEHS). J.B.C. is supported by a Career Development Award from the Dermatology Foundation. J.F.C., M.H., and T.W. are supported by NIH grant 5U01ES017154. T.W. is supported by the Basil O'Connor Starter Scholar Research Award Grant no. 5-FY10-491 from the March of Dimes Foundation, the Edward Jr. Mallinckrodt Foundation, P50CA134254, R01HG007354, and a generous startup package from the Department of Genetics, Washington University. K.L.L. and C.L.M. are supported by NIH Grants P01CA095616 and P01CA142536 and a grant from the Sontag Foundation. T.W. thanks David Haussler for insightful discussions during the early development of the project.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.152231.112.
References
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. 2010. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 28: 1045–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C 2012. Analysing and interpreting DNA methylation data. Nat Rev Genet 13: 705–719 [DOI] [PubMed] [Google Scholar]
- Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, Jager N, Gnirke A, Stunnenberg HG, Meissner A 2010. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 28: 1106–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottou L, Bousquet O 2008. The tradeoffs of large scale learning. Adv Neural Inf Process Syst 20: 161–168 [Google Scholar]
- Chavez L, Jozefczuk J, Grimm C, Dietrich J, Timmermann B, Lehrach H, Herwig R, Adjaye J 2010. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res 20: 1441–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coarfa C, Yu F, Miller C, Chen Z, Harris R, Milosavljevic A 2010. Pash 3.0: A versatile software package for read mapping and integrative analysis of genomic and epigenomic variation using massively parallel DNA sequencing. BMC Bioinformatics 11: 572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE 2008. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452: 215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Dimitrova N, Xuan Z, Rollins RA, Haghighi F, Edwards JR, Ju J, Bestor TH, Zhang MQ 2006. Computational prediction of methylation status in human genomic sequences. Proc Natl Acad Sci 103: 10713–10716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaprio D, Vinson JP, Pearson MD, Montgomery P, Doherty M, Galagan JE 2007. Conrad: Gene prediction using conditional random fields. Genome Res 17: 1389–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty J, Kohavi R, Sahami M 1995. Supervised and unsupervised discretization of continuous features. In Machine Learning: Proceedings of the Twelfth International Conference (ed. Prieditis A, Russell S), pp. 194–202. Morgan Kaufmann, San Francisco. http://ai.stanford.edu/∼ronnyk/disc.pdf [Google Scholar]
- Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Graf S, Johnson N, Herrero J, Tomazou EM, et al. 2008. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol 26: 779–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R, Eddy SR, Krogh A, Mitchison G 1998. Biological sequence analysis: Probabilistic models of proteins and nucleic acids. Cambridge University Press, Cambridge, UK [Google Scholar]
- Frith MC, Mori R, Asai K 2012. A mostly traditional approach improves alignment of bisulfite-converted DNA. Nucleic Acids Res 40: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunau C, Clark SJ, Rosenthal A 2001. Bisulfite genomic sequencing: Systematic investigation of critical experimental parameters. Nucleic Acids Res 29: E65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, et al. 2010. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 28: 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA 2012. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492 [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The human genome browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR 2011. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Kreck B, Franke A, Andrews SR 2012. DNA methylome analysis using short bisulfite sequencing data. Nat Methods 9: 145–151 [DOI] [PubMed] [Google Scholar]
- Kurgan LA, Cios KJ 2004. CAIM discretization algorithm. IEEE Trans Knowl Data Eng 16: 145–153 [Google Scholar]
- Lafferty J, McCallum A, Pereira F 2001. Conditional random fields: Probabilistic models for segmenting and labeling sequence data. In Proceedings of the Eighteenth International Conference on Machine Learning (ed. Brodley CE, Pohoreckyj Danyluk A), pp. 282–289. Morgan Kaufmann, San Francisco [Google Scholar]
- Laird PW 2010. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 11: 191–203 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. 2010. Dynamic changes in the human methylome during differentiation. Genome Res 20: 320–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Harris RA, Cheung SW, Coarfa C, Jeong M, Goodell MA, White LD, Patel A, Kang S-H, Shaw C, et al. 2012. Genomic hypomethylation in the human germline associates with selective structural mutability in the human genome. PLoS Genet 8: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macqueen J. 1967. Some methods of classification and analysis of multivariate observations. In Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability, Vol. 1: Statistics (ed. Le Cam LM, Neyman J), pp. 281–297. University of California Press, Berkeley, CA. [Google Scholar]
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. 2010. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466: 253–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SS, Coolen MW, Stirzaker C, Song JZ, Statham AL, Strbenac D, Robinson MD, Clark SJ 2011. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 6: 34–44 [DOI] [PubMed] [Google Scholar]
- Otto C, Stadler P, Hoffmann S 2012. Fast and sensitive mapping of bisulfite-treated sequencing data. Bioinformatics 28: 1698–1704 [DOI] [PubMed] [Google Scholar]
- Pelizzola M, Koga Y, Urban AE, Krauthammer M, Weissman S, Halaban R, Molinaro AM 2008. MEDME: An experimental and analytical methodology for the estimation of DNA methylation levels based on microarray derived MeDIP-enrichment. Genome Res 18: 1652–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin T, Liu T, Zhang X, Wang D, Li D 2008. Global ranking using continuous conditional random fields. NIPS 2008: 1281–1288 [Google Scholar]
- Robertson KD 2005. DNA methylation and human disease. Nat Rev Genet 6: 597–610 [DOI] [PubMed] [Google Scholar]
- Rohde C, Zhang Y, Reinhardt R, Jeltsch A 2010. BISMA—fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics 11: 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre D, Lee BH, Ting AH 2009. MBD-isolated genome sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res 38: 391–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha F, Pereira F 2003. Shallow parsing with conditional random fields. In Proceedings of the 2003 Conference of the North American Chapter of the Association for Computational Linguistics on Human Language Technology, Vol. 1, pp. 134–141. Association for Computational Linguistics, Stroudsburg, PA [Google Scholar]
- Suzuki MM, Bird A 2008. DNA methylation landscapes: Provocative insights from epigenomics. Nat Rev Genet 9: 465–476 [DOI] [PubMed] [Google Scholar]
- Wallach H. 2004. Conditional random fields: An introduction. Department of Computer and Information Science Technical Report MS-CIS-04-21, University of Pennsylvania. http://repository.upenn.edu/cis reports/22.
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D 2005. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37: 853–862 [DOI] [PubMed] [Google Scholar]
- Xi Y, Li W 2009. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Li J 2010. Detecting copy number variations from array CGH data based on a conditional random field model. J Bioinform Comput Biol 8: 295–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Zhou Y, Lin N, Lowdon RF, Hong C, Nagarajan RP, Cheng JB, Li D, Stevens M, Lee HJ, et al. 2013. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res (this issue). doi: 10.1101/gr.156539.113 [DOI] [PMC free article] [PubMed] [Google Scholar]