

Abstract
MGH2.1 is a herpes simplex virus type 1 (HSV1) oncolytic virus that expresses two prodrug-activating transgenes: the cyclophosphamide (CPA)-activating cytochrome P4502B1 (CYP2B1) and the CPT11-activating secreted human intestinal carboxylesterase (shiCE). Toxicology and biodistribution of MGH2.1 in the presence/absence of prodrugs was evaluated in mice. MGH2.1 ± prodrugs was cytotoxic to human glioma cells, but not to normal cells. Pharmacokinetically, intracranial MGH2.1 did not significantly alter the metabolism of intraperitoneally (i.p.) administered prodrugs in mouse plasma, brain, or liver. MGH2.1 did not induce an acute inflammatory reaction. MGH2.1 DNA was detected in brains of mice inoculated with 108 pfus for up to 60 days. However, only one animal showed evidence of viral gene expression at this time. Expression of virally encoded genes was restricted to brain. Intracranial inoculation of MGH2.1 did not induce lethality at 108 pfus in the absence of prodrugs and at 106 pfus in the presence of prodrugs. This study provides safety and toxicology data justifying a possible clinical trial of intratumoral injection of MGH2.1 with peripheral administration of CPA and/or CPT11 prodrugs in humans with malignant gliomas.
Keywords: carboxylesterase, CPT11, cyclophosphamide, cytochrome P450, herpes simplex virus, irinotecan
Introduction
Glioblastoma multiforme (GBM) is the most common and aggressive type of brain tumor with the median survival of 14.6 months for newly diagnosed patients.1 Considering that the median age of onset is 64 years, it is a sobering that the 5-year survival rate is reported to be under 5% among patients of 65 years of age or older.2 The current standard therapy for newly diagnosed glioma patients consists of maximum safe surgical resection of tumor mass followed by concomitant or sequential radiation and chemotherapy with temozolomide.3 Despite this aggressive multimodal treatment, the prognosis of most GBM patients remains poor and recurrence is almost inevitable with median survival of 5.0 months.4 Treating recurrent GBM is even more difficult than the newly diagnosed cases since the second surgical resection is not always feasible and in many cases, the tumor develops resistance to radiation and chemotherapy.
GBM is a disease caused by the accumulation of genetic mutations resulting in uncontrollable proliferation of cells, which are also highly heterogeneous. Recent advances in genomics, proteomics, and bioinformatics provide a vast amount of information underlying molecular mechanisms of GBM initiation and progression, leading to development of gene-targeted therapies.5,6,7 The three critical pathways involved in GBM are the RTK/RAS/PI3K pathway, the p53 pathway, and the CDK/cyclin/CDK inhibitor/RB pathway. These have been found to be disrupted in more than 75% of GBM samples.8 Considering the complex and compensatory nature of the signaling, therapeutic targeting of a single molecule, or even of an entire pathway, is not likely to achieve clinically significant or meaningful anticancer effects.
In this context, oncolytic virus-based gene therapy emerges as a potentially attractive platform by combining the viral replicative lytic capacity for tumors with additional transfer of genes that mediate anticancer effects.9 For GBM therapy, herpes simplex virus (HSV), adenovirus, measles, and reovirus have been extensively studied and their safety and efficacy have been evaluated in Phase 1 clinical trials.10,11 HSV type 1 (HSV1) oncolytic viruses possess several advantages for brain tumor treatment, namely, its capacity to accommodate several transgenes, its natural neurotropism, its high infectivity for and rapid lysis of cancer cells, the relative ease of genetic manipulation, and the availability of antiviral drugs to prevent unintentional viral spread.12,13,14 Very attenuated oncolytic HSVs (oHSVs) have been well tolerated in Phase I clinical trials for GBM. However, clinically significant efficacy remains elusive, likely because the current clinical generation of oHSV is attenuated and compromised in immune evasion ability and in the degree of replicative cytotoxicity.13,15,16
Newer generations of viral vectors have been developed to improve efficacy without compromising safety. MGH2 was originally engineered from wild HSV1 virus F strain:17 in addition to a deletion of the viral UL39 transgene that provides replicative selectivity for cells with p16 tumor suppressor defects18 and to deletions of both copies of the ICP34.5 neurovirulence genes (i.e., thereby rendering it genetically similar to G207, an oHSV tested in several clinical trials, refs. 19,20), MGH2 also contains two transgene transcriptional units encoding two different prodrug-activating genes: rat cytochrome P4502B1 (CYP2B1)21 and secreted human intestinal carboxylesterase (shiCE).22,23 CYP2B1 converts cyclophosphamide (CPA) into the active anticancer DNA-alkylating metabolite, phosphoramide mustard (PM), whereas shiCE converts irinotecan (CPT11) into the active topoisomerase I inhibitor, SN-38. MGH2 exhibited significant activity against human glioma cells both in vitro and in vivo that was enhanced by the addition of CPA and CPT11. CPA is an alkylating agent used in cancer treatment with dose-dependent biological activity as a cytotoxic and immunosuppressive agent at high dose and antiangiogenic and immunostimulatory agent at low dose.24 CYP2B1 encodes hepatic CYP2B1, an extensively studied prodrug-activating enzyme, which converts CPA to its anticancer metabolite PM.25 PM acts as a DNA cross-linking agent,26 altering DNA structure, and resulting in apoptotic cell death. CPA can also function as an immunomodulator that enhances oHSV replication through inhibition of antiviral natural killer cell and mononuclear cell responses.27,28,29,30,31,32,33,34,35 Irinotecan is also widely used in cancer treatment and activated by carboxylesterase (CE) into SN-38, a potent DNA topoisomerase I inhibitor.36 The efficacy of irinotecan has been reported to be enhanced when combined with other anticancer drugs in patients with glioma.37 The human intestinal form of CE expresses a truncated carboxyl terminus to enable the extracellular secretion of the drug on the surrounding noninfected cells (P. Potter, unpublished results). MGH2.1 in combination with CPA/CPT11 exerts its anticancer effects through four distinct modes of action: (i) immunomodulation by CPA improves oHSV replication; (ii) transgene-mediated activation of CPA and CPT11; (iii) direct oHSV replication and cytotoxicity; and (iv) bystander effect of cytotoxic metabolites released from infected/lysed cells. We have previously shown that oncolytic virus-mediated activation of the prodrugs, CPA and/or CPT11, produced more in vitro cytotoxicity against glioma cells and led to significantly increased survivorship of mice harboring brain glioma xenografts, when compared with treatment with prodrugs alone.17 In order to provide data related to this strategy's toxicology, safety, and biodistribution, we report experiments designed to show that mice tolerate the combination of oHSV and two prodrugs well. As part of the effort to proceed into clinical trials, MGH2 was genetically modified to MGH2.1 by removing a green fluorescent protein (GFP) expression cassette from its genome, as described in the Materials and Methods section. These data, thus, justify a possible clinical trial of MGH2.1 in combination with CPA and CPT11 in patients with malignant glioma.
Results
Effects of MGH2.1 with and without CPA/CPT11 toward human glioma and normal cells
We first sought to establish the in vitro cytotoxicity of MGH2.1, CPA, and CPT11 at various dose levels in human astrocytes and three human glioma cell lines (Gli36, U87, and U251). MGH2.1 alone reduced the survival of all three glioma cell lines in a dose-dependent manner, but not that of human astrocytes, even at a multiplicity of infection (MOI) of 10 (Figure 1a). Each of the two prodrugs, CPA and CPT11, also reduced the survival of glioma cell lines but not that of human astrocytes (Figure 1b,c, respectively), in spite of their “prodrug” status perhaps because of incubation at 39.8 °C, when compared to controls. Because there was selective glioma cell cytotoxicity from the prodrugs alone at this high temperature, we next sought to determine if expression of the MGH2.1-encoded transgenes, CYP2B1 and shiCE, respectively converted the prodrugs CPA and CPT11 in glioma and normal cells to provide additional cytotoxicity (Figure 1d). For glioma cells, doses of MGH2.1, CPA, and CPT11 were selected at MOI of 0.1, 250 µmol/l and 0.05 µmol/l, respectively. For human astrocytes, doses of reagents were increased to MOI = 10, 1,000 µmol/l of CPA, and 0.2 µmol/l of CPT11. In order to study the effect of prodrug conversion without the confounding variable of MGH2.1 replicative cytotoxicity, the next set of experiments were conducted utilizing the temperature shift method,38 where 4 hours after infection of glioma cells with MGH2.1, viral replication is stopped by raising the temperature from 37 to 39.8 °C, in the presence or absence of prodrugs. Five days later, cells were counted. In spite of the temperature-mediated viral replicative block, the 4-hour replicative infection of MGH2.1 at MOI = 0.1 still reduced the survival of the three established glioma cell lines compared with control by 20–30% (Figure 1d). The combination of CPA and CPT11 also reduced the survival of all three glioma cells. Finally, the combination of initial infection with MGH2.1 that was replicatively blocked and of CPA and CPT11 significantly reduced survival when compared with CPA and CPT11 alone for Gli36 and U251 but not U87 glioma cells (Figure 1d). In order to minimize unconverted prodrug cytotoxicity and to maximize the degree of infectivity of cells, we repeated the experiment by reducing the dose of prodrugs by half and increasing the dose of MGH2.1 to a MOI of 1 in the Gli36 and U251 glioma cells only. The data of Figure 1e show that the increased MOI of MGH2.1 also increased the relative cytotoxicity in spite of the temperature-mediated replicative block, when added alone. Again, we detected a significant increase (of about 70%) in cytotoxicity when replicatively blocked MGH2.1 was combined with both prodrugs when compared with prodrugs alone (Figure 1e). In agreement with our previously published findings, this provided evidence that MGH2.1 did lead to additional conversion of the two prodrugs in infected cells.17 We then tested the cytotoxicity of MGH2.1 (MOI = 1) ± CPA and/or CPT11 against five primary glioma cells. While the temperature shift-mediated replicative block of MGH2.1 infection at MOI = 1 did not significantly reduce the cell number of primary glioma cells other than an approximate 10% decrease in X12, its combination with either or both prodrugs significantly reduced cell numbers of all glioma cells (P < 0.001) (Figure 1f). We next sought to determine if MGH2.1 replication in the presence or absence of prodrugs was cytotoxic to normal cells. These experiments were conducted at 37 °C to allow for viral replication. Infection of MGH2.1 at MOI = 10 in the presence of the both prodrugs led to a very small reduction in survival rates in some (human fibroblasts and skeletal muscle cells) but not all human primary normal cells (astrocytes, hepatic, human umbilical venous endothelial cell, renal, and smooth muscle) and also did not affect mouse neurons (Figure 1g). These data thus showed that MGH2.1 in combination with prodrugs was well tolerated by normal cells.
Figure 1.

In vitro cytotoxicity assays. (a) Dose effect of MGH2.1 alone. HA and human glioma cell lines (Gli36, U87, and U251) were treated with increasing concentrations of MGH2.1. Three hours after infection with MGH2.1 at 37 °C, cells were washed to remove unattached viral particles and fresh medium was added back. Five days later, surviving cells were measured colorimetrically with CytoTox 96 Non-Radioactive Cytotoxicity Assay and confirmed numerically with a Coulter counter. Plotted values represent the mean ratio of surviving cells after treatment with MGH2.1 versus mock compared with mock, from experiments performed in triplicate and are defined as the fractional cell ratio. Standard deviations are indicated by error bars. While MGH2.1 did not show statistically significant cytotoxicity on HA, MGH2.1 at MOI = 1 and 10 caused significant reduction of cell survival of glioma cell lines (P < 0.0001). (b) Dose effect of CPA alone. Cells were incubated in medium containing CPA at 37 °C for 3 hours and then transferred to a 39.8 °C incubator. Four days later, surviving cells were measured. CPA caused significant cytotoxicity on Gli36 at 250 µmol/l (P = 0.0041), 500 µmol/l (P = 0.0050), and 1,000 µmol/l (P < 0.0001); U87 at 250 µmol/l (P = 0.0020), 500 µmol/l (P = 0.0391), and 1,000 µmol/l (P = 0.0084); and U251 at 500 µmol/l (P = 0.0043) and 1,000 µmol/l (P < 0.0001). (c) Dose effect of CPT11 alone. Cells were treated similarly to cells treated in b. CPT11 caused significant cytotoxicity to Gli36 at 0.05 µmol/l (P = 0.0195), 0.1 µmol/l (P = 0.0355), and 0.2 µmol/l (P = 0.0002); U87 at 0.1 µmol/l (P = 0.0324) and 0.2 µmol/l (P = 0.0054); and U251 at 0.05 µmol/l (P = 0036), 0.1 µmol/l (P = 0.0005), and 0.2 µmol/l (P < 0.0001). (d) Effect of TS MGH2.1 in combination with CPA and/or CPT11 in HA (MOI = 10) and glioma cell lines (MOI = 0.1). Cells were incubated in medium containing CPA at 37 °C for 3 hours and then transferred to a 39.8 °C incubator to stop MGH2.1 replication. By stopping viral replication, cytotoxicity mediated by CYP2B1 and shiCE conversion of CPA and CPT11, respectively, into their active anticancer metabolites can be assayed without the confounding variable of cytotoxicity mediated by the replicating oncolytic virus, as detailed in Aghi et al. (1999).38 Four days later, surviving cells were measured. Compared with no-treatment controls, the combination of MGH2.1 and prodrugs caused significant cytotoxicity to Gli36 (P < 0.001), U87 (P = 0.003), and U251 (P < 0.001), while no effect was observed for HA. (e) Effect of CPA and CPT11 versus TS MGH2.1 + CPA and CPT11 in Gli36 and U251 cells. The MOI of MGH2.1 was increased to 1 and the dose of prodrugs was halved compared with that in d, in order to maximize cell infectivity and minimize effects of prodrugs alone. (f) Effect of TS MGH2.1 and CPA and/or CPT11 on glioma “stem-like” cells, G97, G68, OG02, and X12. (g) Effect of replicative MGH2.1 with or without prodrugs against a panel of normal cells: R. Epi., Hepato., SM, HUVEC, HA, SkM, Fibro., and MN cells. Cells were infected with MGH2.1 for 1 hour at 37 °C and then fresh medium containing 1,000 µmol/l CPA and/or CPT11 was added. Unlike above experiments, incubation was carried out at 37 °C for the full 5 days, before measuring surviving cells. Statistical analyses were conducted by Dunnet's method to evaluate (a) MGH2.1 dose effect, linear models (analysis of variance) with Bonferroni correction to control for type I error to evaluate dose effects of (b) CPA, or (c) CPT11, and Student's t test was used to evaluate combination effects of (e–g) MGH2.1 ± CPA ± CPT11. *P < 0.05; #P < 0.01. All comparisons are made between the treatment effect and the mock control. CPA, cyclophosphamide; Fibro., pulmonary fibroblast; HA, human astrocyte; Hepato., hepatocyte; HUVEC, human umbilical venous endothelial cell; MN, mouse neural cells; MOI, multiplicity of infection; R.Epi., renal epithelial cells; SkM, skeletal muscle cells; SM, smooth muscle cells; TS, temperature-shifted.
Prodrug metabolism
CPA is metabolized by the hepatic cytochrome 450 system,25,39,40,41,42 whereas CPT11 is metabolized by serum CEs.23,43 To determine if there were alterations in the normal metabolism of both prodrugs when administered systemically in the presence of intracranial MGH2.1, prodrug metabolite levels, SN-38 (CPT11 metabolite) and PM (CPA metabolite), were assayed in plasma, brain, and liver of Balb/C mice intracranially injected with 106 pfus of MGH2.1 or mock (heat-inactivated MGH2.1). The day after virus injection, both CPA and CPT11 were administered intraperitoneally (i.p.) at a dose of 2 mg in 100 µl phosphate-buffered saline (PBS). Animals were killed at 10 minutes, 30 minutes, 1, 2, 4, and 6 hours after prodrug injection, and blood, brain, and liver were immediately harvested and processed for measurements of metabolites by mass spectrometry (detailed procedure in Supplementary Materials and Methods). As expected, there were higher concentrations of both prodrugs and their metabolites in liver compared with plasma and brain (Figure 2). No significant difference in the levels of CPA, CPT11, PM, and SN-38 in plasma, brain, and livers, except for SN-38 levels in plasma (P = 0.0213, two-tailed paired t test) was observed between MGH2.1 and mock infection groups. Taken together, these results indicated that intracranial injection of MGH2.1 did not significantly alter the metabolism of systemic CPA and CPT11.
Figure 2.

Prodrug metabolite assay. Concentrations of the prodrug, (a) CPA, and its metabolite, (b) PM, and of the prodrug, (c) CPT11, and its metabolite, (d) SN-38, were measured in plasma, brain, and liver of mice injected with MGH2.1 or mock in the presence of systemic CPA and CPT11. Viable MGH2.1 (1 × 106 pfus) or heat-inactivated mock was inoculated into mouse brains, stereotactically. The next day, 2 mg of CPA and 2 mg of CPT11 was injected intraperitoneally, and blood, brain, and liver were collected at 10 minutes and 30 minutes, 1, 2, 4 and 6 hours after dosing. The concentrations of analytes are shown as ng/ml for the plasma samples and ng/g for brain or liver samples. Each plot represents the average of duplicate samples and standard deviations were indicated by error bars. Student's t test was used for comparison between viable MGH2.1 and mock groups. CPA, cyclophosphamide; PM, phosphoramide mustard.
Detection of viral DNA and transcripts in brain and trigeminal ganglia
We then sought to determine presence of viral DNA and transcripts in brains and trigeminal ganglia (TG). Seven days after virus inoculation at a dose of 5 × 107 pfus, viral DNA polymerase was detected in all brain samples regardless of prodrugs. Interestingly, viral genomic DNA was also detected in TG in two of three animals treated with MGH2.1 + CPA, one of three animals treated with MGH2.1 + CPT11, and none of the animals treated with MGH2.1 alone (Figure 3a). We then analyzed for presence of MGH2.1 at 60 days. After injection of MGH2.1 (1 × 107 or 1 × 108 pfus), viral DNA polymerase was still present in five out of five brains inoculated with 1 × 108 pfus of MGH2.1 and in three out of five brains inoculated with 1 × 107 pfus of MGH2.1 (Figure 3b). However, no viral DNA was detected in TG. Expression of viral latency-associated transcript (LAT) and of the two encoded transgenes (rat CYP2B1 and human shiCE) was evaluated by reverse transcription-PCR (RT-PCR) of brain and TG obtained from the mice, 60 days after MGH2.1 intracranial injection. Expression of LAT was detected in none of the five brains and one of five TGs from the lower MGH2.1 dose group, and one of five brains and two of five TG samples from the higher dose group (Figure 3c). The transgene shiCE transcript was expressed in one of the TGs that also expressed LAT in the higher dose group. However, no CYP2B1 transgene expression was observed in any group.
Figure 3.

Detection of viral genomic DNA and LATs. (a) MGH2.1 (5 × 107 pfus) ± CPA (intraperitoneally (i.p.) at 1, 3, 5, and 7 days) or CPT11 (i.p. at 1 day) was intracerebrally injected in mice and viral genomic DNA was isolated from brain and TG 7 days after virus inoculation. (b) MGH2.1 (107 and 108 pfus) was intracerebrally injected in mice and viral genomic DNA was isolated from brain and TG 60 days after virus inoculation. PCR for viral DNA pol was carried out by 35-cycle amplification, and amplified products were separated by agarose gel electrophoresis. Mouse β-actin was used as an internal control for genomic DNA. (c) Expression of transgenes (rat CYP2B1 and human shiCE) and LAT were analyzed by reverse transcription-PCR 60 days after intracerebral inoculation of MGH2.1 or PBS. Mouse GAPDH was used as an internal control for mRNA. CPA, cyclophosphamide; CYP2B1, cytochrome P4502B1; LAT, latency-associated transcript; PBS, phosphate-buffered saline; pol, polymerase; shiCE, secreted human intestinal carboxylesterase; TG, trigeminal ganglia.
Systemic biodistribution of virally encoded transcripts and viral LAT after MGH2.1 intracerebral injection
We next proceeded to assay for the presence of MGH2.1-encoded transcripts in multiple organs after MGH2.1 inoculation in mice brains. Brains, liver, lung, heart, intestines, testis, spleen, lymph nodes, and TG of Balb/C mice intracranially injected with MGH2.1 (5 × 107 pfus) in the presence or absence of systemic prodrugs were harvested and RT-PCR for CYP2B1, shiCE, and LAT was carried out, 7 days after MGH2.1 injection (Figure 4). Since CYP2B1 expression is under control of the HSV1 IE 4/5 promoter (Supplementary Figure S1a), its expression would indicate persistent viral infection in a tissue, unlike LAT which may indicate only latent virus. RNA extracted from U251 cells infected with MGH2.1 or wild-type F strained was utilized as positive and negative control, respectively. Distribution of transgenes and LAT was essentially restricted to the brain regardless of administration of CPA and/or CPT11. One out of three mice injected with MGH2.1 in the presence of CPA showed LAT expression in the TG, suggestive of possible viral latency or distribution due to immunomodulating effect of CPA.
Figure 4.

Biodistribution of intracerebrally injected MGH2.1. MGH2.1 (5 × 107 pfus) ± CPA (intraperitoneally (i.p.) at 1, 3, 5, and 7 days) or CPT11 (i.p. at 1 day) was intracerebrally injected in mice and viral mRNA was isolated after 7 days. Reverse transcription-PCR for the two encoded transgenes (rat CYP2B1 and human shiCE), for the viral LAT gene and, for mouse GAPDH as a control was carried out in brains, liver, lung, heart, intestines, testis, spleen, lymph nodes, and TG. RNA from U251 infected with MGH2.1 or wild-type F strain was used as control. RNA after reverse-transcription was amplified for 35 cycles, and amplified products were separated by agarose gel electrophoresis. CPA, cyclophosphamide; CYP2B1, cytochrome P4502B1; LAT, latency-associated transcript; LN, lymph node; shiCE, secreted human intestinal carboxylesterase; TG, trigeminal ganglia.
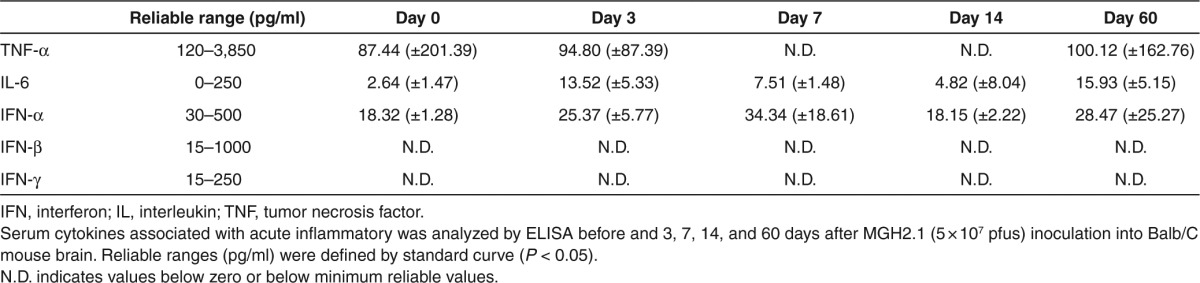
Serum cytokine profile in animals after intracranial injection with MGH2.1
We investigated whether there was a change in serum cytokines associated with acute inflammatory responses after intracranial injection with MGH2.1. Serum was prepared from Balb/C mice 3, 7, 14, and 60 days after intracranial injection of 5 × 107 pfus of MGH2.1. Serum from mice without virus injection was utilized as a negative control. There was a small increase in interleukin-6 (IL-6) after MGH2.1 intracranial inoculation that remained elevated for the duration of the experiment. There were no changes in tumor necrosis factor-α (TNF-α), interferon-α (IFN-α), IFN-β, or IFN-γ before or after MGH2.1 intracranial injection (Table 1). This suggested that intracerebral MGH2.1 was associated with minor increases in serum IL-6 without evidence of clinical significance.
Table 1. Serum chemokine levels in Balb/C after MGH2.1 intracerebral inoculation.
Lethality of MGH2.1 with and without prodrugs after intracranial injection
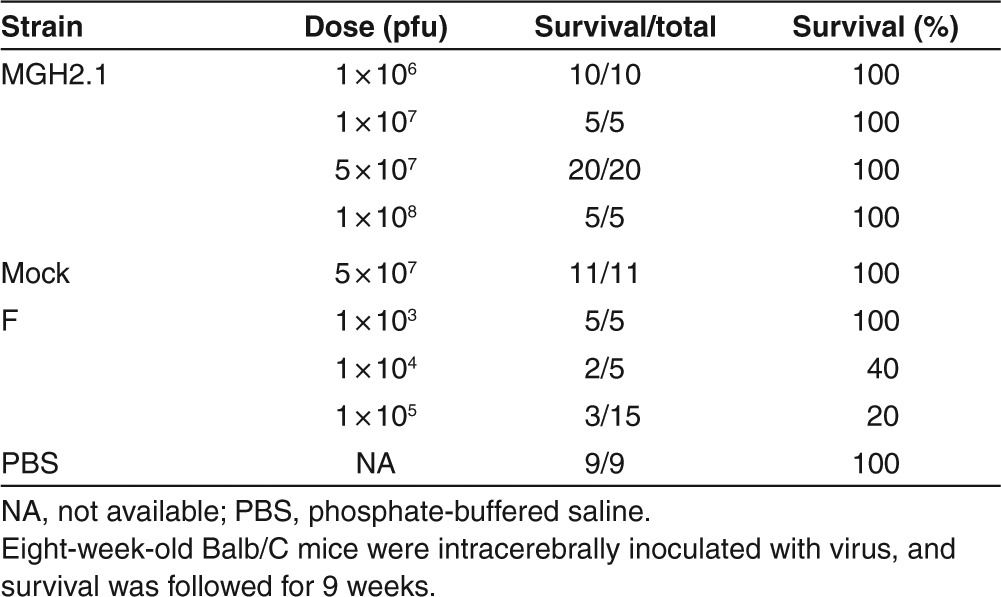
To further determine if there was evidence of severe clinical toxicity, a survival study was conducted. Intracerebral toxicity of MGH2.1 was compared with that of wild-type F strain in young (aged 8 weeks) Balb/C mice. Different doses of viruses (5 µl volume) were stereotactically injected into the white matter of the right hemisphere of mice. The body weight of animals was recorded weekly and the survival was followed for 9 weeks. MGH2.1 did not cause lethality at the highest dose tested (108 pfus), whereas lethality was evident with F strain at a dose as low as 104 pfus (Table 2). In fact, the calculated LD50 for F strain was 6.8 × 103 pfus, in agreement with published studies. There was some initial weight loss in mice inoculated with MGH2.1 at the higher inoculated doses, but weight returned to baseline within a few weeks after viral inoculation with weight gain ensuing thereafter (Supplementary Figure S2a). In fact, by the end of 9 weeks, there were no statistically significant differences in body weights between mice injected with 1 × 108 pfus of MGH2.1 versus those injected with mock.
Table 2. Lethality of MGH2.1 and wild-type strain F after intracerebral injection in Balb/C mice.
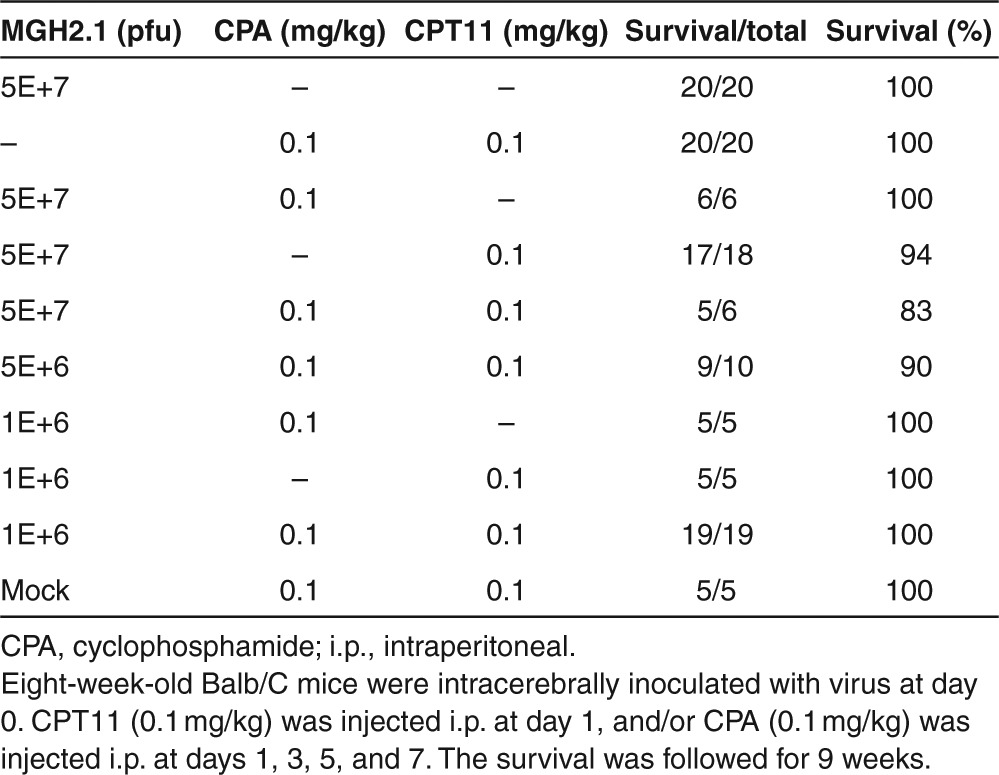
Next, we evaluate the lethality of MGH2.1 in combination with prodrugs in young (aged 8 weeks) Balb/C mice (Table 3). Virus was injected intracranially at day 0, CPT11 (0.1 mg/kg) was injected i.p. at day 1, and/or CPA (0.1 mg/kg) was injected i.p. at days 1, 3, 5, and 7. The combination of prodrugs in the absence of MGH2.1 was not lethal and did not cause significant body weight reduction other than the first week (Supplementary Figure S2b). While CPA in combination with 5 × 107 pfus of MGH2.1 was not lethal, CPT11 with MGH2.1 led to the death of one out of 18 mice. The combination of virus at 5 × 107 pfus and both prodrugs also caused death in one out of six mice. MGH2.1 was still lethal at the dose of 5 × 106 pfus when combined with both prodrugs. However, all of 19 mice injected with MGH2.1 at a dose of 1 × 106 pfus in combination with CPA and CPT11 survived through the course of the experiment. The maximum tolerated dose for injection in a brain without tumor of MGH2.1 + CPA was not achieved (i.e., above 5 × 107 pfus) and for MGH2.1 + CPT11, it was 106 pfus (Table 3). The LD50 of MGH2.1 in combination with CPA was thus not calculable, that in combination with CPT11 was 1.24 × 1041 pfus, while that in combination with both prodrugs was 3.53 × 108 pfus.
Table 3. Lethality of MGH2.1 after intracerebral injection in combination with systemic prodrugs in Balb/C mice.
Discussion
We have previously shown that oncolytic virus-mediated activation of the prodrugs, CPA and/or CPT11, produced more in vitro cytotoxicity against glioma cells and led to significantly increased survivorship of mice harboring brain glioma xenografts, when compared with treatment with prodrugs alone.17 In this report, we show that the combination of MGH2.1, CPA, and CPT11 was cytotoxic to human glioma cells, including glioma “stem-like” cells and did not significantly affect the viability of a panel of normal human and mouse cells. Importantly, the intracerebral administration of MGH2.1, expressing the two prodrug-activating transgenes, also did not alter the endogenous metabolism of CPA and CPT11. MGH2.1 DNA and transcripts were largely confined to the brain after intracranial injection and did not end up in organs outside the central nervous system even in the presence of CPA and/or CPT11. MGH2.1 intracerebral injection alone was not associated with lethality at doses up to 108 pfus, while the combination of MGH2.1 (intracerebral) and systemic CPA + CPT11 was not associated with lethality at a dose of 106 pfus. Therefore, these studies establish an animal toxicology and biodistribution set of findings that can be useful for further design of a clinical trial of MGH2.1 with and without CPA and/or CPT11 in humans with malignant glioma.
MGH2.1 provides multimodal therapeutics in the context of a single bioagent for a cancer, such as glioblastoma, where multiple genes are mutated and multiple signaling pathways are deregulated. Therefore, the multiple modes of tumor killing may potentially circumvent the tumor cell's ability to escape from anyone single treatment. In fact, tumor cell toxicity arises from: (i) the direct cytotoxic action of MGH2.1 viral infection and lysis of tumors; (ii) the alkylation of tumor cell DNA by CPA's activated metabolites that can also diffuse out and kill other tumor cells, even if MGH2.1 did not infect them;44 (iii) the inhibition of tumor cell topoisomerase I activity by CPT11's activated metabolite that can also kill other noninfected tumor cells, by diffusion of the metabolite or of the prodrug-activating enzyme22; and (iv) the enhancement of MGH2.1 replication by the immunomodulatory effects of CPA.35 Results from the in vitro cytotoxicity studies show that MGH2.1, in combination with prodrugs, was more toxic to human glioma cell lines and primary glioma cells than MGH2.1 alone, providing evidence for the effectiveness of multiple mechanisms of tumor cell kill. It should be noted that the experiments shown in Figure 1d–f measure cytotoxicity mediated by only a 4-hour infection and replicative cycle of MGH2.1 due to the 37 to 39.8 °C temperature shift38 combined with virus-encoded transgene–mediated conversion of CPA and/or CPT11, whereas the experiments shown in Figure 1g measure cytotoxicity mediated by the fully replicative oncolytic virus with and without prodrugs. In this context, when multiple mechanisms of tumor cell toxicity are operative, one concern is that they can also lead to increased toxicity against normal cells. At least in vitro, this was not the case: even combined at their highest tested dose (MOI = 10 for MGH2.1, 1000 µmol/l for CPA, and 0.2 µmol/l for CPT11), there was no significant effect on human astrocyte viability or on the viability of a panel of normal cells. Therefore, in vitro the combination of MGH2.1 and its two prodrugs increases glioma cell cytotoxicity without affecting the viability of normal cells.
Both CPA and CPT11 are metabolized by endogenous enzymes. CPA is primarily metabolized by the hepatic cytochrome P450 system with rat CYP2B1 being the most active catalytic isozyme for this reaction.25 CPA is first metabolized into the unstable intermediate 4-hydroxy CPA which spontaneously degrades into PM and acrolein.39 PM is the active anticancer agent that alkylates DNA and, thus, leads to DNA damage and cell death.40 CPT11 is metabolized by serum CEs into its active anticancer metabolite, SN-38, that inhibits topoisomerase I, thus leading to DNA damage.36 In addition, a number of intracellular CEs can also catalyze this reaction and shiCE is such an isozyme.22,23,43 We had previously shown that the active metabolites of CPA (4-hydroxy CPA and its metabolites)45 and that of CPT11 (SN-38) were cytotoxic to glioma cells.17 In addition, expression of CE and CYP2B1 in the context of the oHSV1, MGH2 (i.e., MGH2.1 with a GFP transgene) was highly effective against intracranial gliomas in mice when combined with systemic CPA and CPT11.17 The rationale for this is that, in spite of endogenous bioconversion of the two prodrugs, there was still sufficient prodrug not being converted that could be activated locally in tumor by MGH2. Evidence for this mechanism was shown previously where local conversion of CPA by a replicating HSV expressing CYP2B1 was shown.45 However, one concern may be that the local intratumoral bioconversion of the two prodrugs could alter or change systemic endogenous pharmacokinetics. The data in this paper show that intracranial inoculation of MGH2.1 or mock followed by i.p. injection of CPA and CPT11 into immunocompetent Balb/C mice did not alter the pharmacokinetics of prodrugs and their metabolites in brain, serum, and liver. This suggests that MGH2.1 replication/infection is minimal in naive brains and the observed expression of shiCE and CYP2B1 (Figure 4) would not be meaningful enough to alter the endogenous metabolism of the two prodrugs. Although the same amount of CPA and CPT11 were injected, the concentrations of CPA and its metabolite PM were higher than those of CPT11 and SN-38 by an order of magnitude in brain, which likely reflect the differences in regard to half-life of the prodrugs and metabolites, ability of prodrugs to cross brain–blood barrier, and enzyme activity. Therefore, the endogenous kinetics of prodrug metabolism were not altered by the injection of virus in brains.
In terms of immune reactions, intracranial injection of MGH2.1 in immunocompetent Balb/C did not elicit induction of serum cytokines (TNF-α, IFN-α, IFN-β, or IFN-γ), and only a marginal increase of IL-6 was detected at the various time points. We do anticipate that MGH2.1 administration may induce anticancer immune response through viral replication followed by cell lysis in tumor-bearing animals. Modulating host immunity in viral cancer therapy is tricky. Antiviral immune response should be suppressed until infection and replication is established in target tumor cells. At this juncture, antitumor immune response should be induced by viral molecules produced upon successful viral replication, such as dsRNA, and/or processed antigens specific to virally infected cells. CPA, an alkylating agent widely used in various cancer treatments, has also immunosuppressive effect and has been shown to improve replication of oHSV in tumor cells both in vitro and in vivo.9,27,28,29,31,33,34,35,46,47 We observed that there was an expected CPA effect on enhancement of in vitro cytotoxicity of MGH2.1 in the present study, as well.
Seven days after intracranial virus injection, viral genomic DNA was observed in all brain samples regardless of prodrug administration. In some of prodrug-treated animals, TG also exhibited viral DNA (Figure 3a). This shows that prodrugs (likely CPA) may enhance the ability of virus to travel to TG, where the virus likes to harbor in a latent state. By 60 days after virus injection without prodrug treatment, brains, but not TG, still harbored viral genomic DNA (Figure 3b). HSV1 is thought to establish latency in TG in both human and mouse. Intracerebrally injected HSV was observed to spread via retrograde transport to neurons.48,49,50 RNA and DNA of LAT and viral genes were detected in TG of mice latently infected HSV1 at 60 days after infection, although there was no conclusive data regarding their replication.51 Systemic dissemination of intracranially administered MGH2.1 was assayed in naive immunocompetent Balb/C mice by Q-PCR. Various organs were harvested from animals killed 7 days after virus injection. All brain samples expressed transgenes and LAT genes regardless of prodrug treatment. Other than brain tissue, only one TG harvested from CPA-treated animal exhibited a LAT transcript. Significantly, there was no evidence of viral gene expression in other organs, even in CPA-treated animals. The lack of transcript expression in organs other than the brain (and TG) and the lack of the CYP2B1 transcript that is under control of the HSV IE4/5 promoter in the brain at 60 days (Figure 3c) argues that MGH2.1 transcripts would be unlikely to be found in organs such as liver at 60 days. The lack of oHSV dissemination in the presence of CPA is consistent with a previous result that pretreatment of CPA did not cause systemic distribution of oHSV1.32,33,46,52 In these studies, we did not observe viral gene expression even in CPA-treated TG. However, samples were harvested only 12 hours after virus injection into the intracranial tumor mass, suggesting that time may have been too short for the virus to travel to TG. Taken together, our study along with previously cited reports confirms that oHSV injected in brains of mice remains localized to the central nervous system even in the presence of prodrugs that are activated by the oHSV.
The primary objective of this study was to show the safety profile of MGH2.1 with/without its prodrugs upon intracerebral injection. Previously, we have shown its efficacy in animal models of glioma.17 The LD50 for MGH2.1 in combination with CPA was not reached, and the LD50 for the virus in combination with CPT11 was calculated as 1.24 × 1041 pfus, which is not achievable. MGH2.1 intracerebral administration by itself was not lethal to mice at the highest tested dose (108 pfus), but we did establish an LD50 when it was combined with systemic administration with prodrugs at a dose of 3.53 × 108 pfus. However, we were able to find a dose (106 pfus) that was not associated with animal death. In our previously published study,17 this dose was effective in an animal model of glioma and, thus 106 pfus is the No Observed Adverse Event Level dose in mice. This, thus, provides a starting dose in an eventual Phase I clinical trial for human subjects with malignant gliomas taking into account differences in brain weight. There was some dose-dependent weight loss in mice intracranially injected with MGH2.1 at the highest dose and systemic prodrugs exacerbated this adverse effect, but this was a transient effect with animal weight gain returning to the original level by a few weeks after surgery. Since the PBS-injected control group also showed some weight loss, albeit to a lesser degree, a portion of this adverse effect can be attributed to the anesthesia and surgery.
In summary, these studies provide feasibility data related to the toxicology and biodistribution of this novel oHSV in combination with its prodrugs in preparation for an eventual clinical trial.
Materials and methods
Engineering of MGH2.1. MGH1 is a double-mutant oHSV1 derived from wild-type F strain containing deletions of both copies of ICP34.5 and an insertion in UL39.53 To enhance its anticancer therapeutic efficacy, two transgene cassettes encoding the prodrug-activating enzymes, rat CYP2B1 and shiCE, were integrated into MGH1 and designated as MGH2.17 CYP2B1 and shiCE convert CPA and irinotecan (CPT11) into their active metabolites, PM and SN-38, respectively. For potential clinical trials, the GFP transgene cassette was removed from the MGH2 sequence due to its possible immunogenicity (Supplementary Figure S1a). Details of the engineering are described in Supplementary Materials and Methods. This new oHSV was designated as MGH2.1 and three different isolates (1-1-5, s1-2-6, and 1-2-8) were initially plaque-purified. Of the three MGH2.1 isolates, 1-2-6 was selected because it showed the highest CE activity, the retention of the transgene cassettes, and the deletion of the GFP expression cassette. To confirm the structural stability of transgenes in its genome, MGH2.1 isolate, 1-2-6, was plaque-purified on Master Cell Bank cells B7 and N23 (obtained from William B Goins, University of Pittsburgh, Pittsburgh, PA) for 20 passages, viral DNA was extracted every five passages, and gross genomic structure was verified by Southern blot analysis by following published procedures17,54 (Supplementary Figure S1b). CYP2B1 and shiCE sequences were excised from the transfer plasmid pT-oriS-IE4/5-CMV-hiCEC and used as probes. A parental strain rHSVQ1 (rH) which lacks the transgenes CYP2B1 and shiCE was used as a negative control. A single band of 4.8 kb was detected in each viral DNA sample of MGH2.1 digested with HindIII by using the shiCE fragment as a probe. The CYP2B1 probe detected 1.4 and 2.2 kb fragments in the samples digested with HindIII and with SacII and BglII, respectively. The lack of detection of hybridizing fragments for DNA isolated after five passages on N23 was likely due to technical issues since later passages showed hybridizations.
In vitro bioequivalence studies were conducted to show that MGH2.1 retained secreted intestinal CE activity and tumor cell oncolysis to the same magnitude as MGH2 (Supplementary Figure S3). CE activity and production rate of its metabolite SN-38 was measured in cultured media and cell lysate prepared from Vero cells infected with the virus stocks.17 CE activity was higher in cell extracts than medium in all samples, which was consistent with production rate of SN-38 (Supplementary Figure S3a,b). CYP2B1 function was confirmed in terms of cytotoxicity of MGH2.1 in the presence of CPA (Supplementary Figure S3c). Taken together, the newly generated MGH2.1 was confirmed to sustain oncolytic activity through prodrug conversion activity comparative to its parent strain, MGH2.
Cell culture. Vero cells and human glioma cell lines (U251, Gli36, and U87) were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 2 or 10% heat-inactivated fetal bovine serum (Invitrogen), 100 U/ml of penicillin (Invitrogen), and 10 mg/ml of streptomycin (Invitrogen). Primary human glioma cells (G35, G68, G97, OG02, and X12) were cultured as spheres under conditions to enrich for the glioma “stem-like” cell subpopulation in brain tumor stem cell medium consisting of Neurobasal medium (Invitrogen) supplemented with GlutaMAX (Invitrogen), B27 supplements (Invitrogen), 20 ng/ml of human recombinant basic FGF (Peprotech, Rocky Hill, NJ), and 20 ng/ml of human recombinant EGF (Peprotech). The day before infection, primary glioma sphere cells were trypsinized and seeded onto plates coated with poly-d-lysine (Invitrogen) containing brain tumor stem cell medium with reduced concentration of fibroblast growth factor and epidermal growth factor (5 ng/ml for each) to form monolayers. Primary human cells (astrocyte, pulmonary fibroblast, hepatocyte, renal epithelial cells, skeletal muscle cells, smooth muscle cells, and human umbilical venous endothelial cell: umbilical vein smooth muscle cells) and mouse neural cells were purchased from ScienCell Research Laboratories (Carlsbad, CA), and cultured in the corresponding medium following the manufacturer's instructions. The primary normal cells used in this study were passaged no more than five times, and the aliquots were stored in Bambanker serum-free cell freezing medium (Wako Chemicals USA, Richmond, VA) at – 80 °C until use. All cells were grown at 37 °C in an atmosphere containing 5% CO2.
In vitro virus cytotoxicity assay. Cytotoxicity was evaluated and reported as the fraction of cells surviving after oncolytic virus infection compared with those treated with vehicle (fractional cell ratio). The day before infection, 5 × 103 cells were plated onto 96-well plates in medium prepared following manufacturer's instructions for normal primary cells, brain tumor stem cell medium for primary glioma cells or Dulbecco's modified Eagle's medium supplemented with 2% fetal bovine serum for glioma cell lines and allowed to adhere. The medium for normal cells was changed to basal medium a few hours after cell preparation. The following day, viruses were added at various MOIs, as indicated in figure legends. MGH2.1, inactivated with ultraviolet radiation, was used as a mock control. One hour after infection, cells were washed with glycine saline solution (10 mmol/l glycine, 137 mmol/l NaCl, 24.1 mmol/l KCl, 0.49 mmol/l MgCl2, and 0.68 mmol/l CaCl2, pH 3) followed by PBS to remove unattached viruses and fresh medium was then added. Cells were incubated at 37 °C in an atmosphere containing 5% CO2. Five days after infection, virus cytotoxicity was assayed as the amount of lactase dehydrogenase released upon cell lysis with CytoTox96 Non-Radioactive Cytotoxicity Assay kit (Promega, Madison, WI). For prodrug functional assays, cells were prepared in the same manner as described above. After 1-hour incubation in viral solution at 37 °C, fresh medium containing CPT11 (APP Pharmaceuticals, Schaumburg, IL) and/or CPA (Baxter, Deerfield, IL) at indicated concentrations was added. Viral replication was then stopped 3 hours later by transferring the plates to a 39.8 °C incubator.38 Cytotoxicity was measured 4 days later with CytoTox96 Non-Radioactive Cytotoxicity Assay kit. For some cell lines, cytotoxicity was measured by enumeration through a Coulter counter (Beckman Coulter, Indianapolis, IN). The day before infection, 5 × 104 cells were seeded onto 12-well plates. Infection, drug treatment and incubation were performed in the same manner as described above. Cells were incubated for 5 days for dose–response assays and 4 days for prodrug functional assays and surviving cells were counted with a Coulter counter.
Animal studies. All animal studies were performed in accordance with guidelines issued by The Ohio State University Institutional Animal Care and Use Committee, utilizing an approved animal protocol. Viral inoculation and care of animals harboring virus were conducted in approved BL2 laboratory rooms. BALB/c mice were obtained from Charles River Laboratories (Wilmington, MA) or the National Cancer Institute (Frederick, MD). For neurotoxicity experiments, 8-week-old mice were anesthetized by i.p. administration of ketamine (100 mg/kg) and xylazine (20 mg/kg). Oncolytic virus was then stereotactically injected into the right frontal lobe of brain (2 mm lateral and 1 mm anterior to the bregma at a depth of 3 mm). PBS and ultraviolet-radiated MGH2.1 were used as negative and mock control, respectively. The survival time for each group was monitored for 60 days after virus injection and body weights were measured weekly. CPA (2 mg in 100 µl PBS) was administered i.p. at 1, 3, 5, and 7 days after virus injection, and CPT11 (2 mg in 100 µl PBS) was injected i.p. 1 day after virus injection.
Metabolite assays. CPA, CPT11, and their active metabolites, PM and SN-38, were measured with the API-3000 triple quadrupole mass spectrometer (ABSciex, Framingham, MA) in mouse plasma, brain, and liver tissue. Thirty-six Balb/C mice were divided into two groups which were injected intracranially with either mock or 106 pfus of MGH2.1. The next day, mice were treated with i.p. injections of CPA (2 mg) and/or CPT11 (2 mg). Blood, brain, and liver tissues were collected 10 minutes and 30 minutes, 1, 2, 4 and 6 hours after drug administration. Blood was collected into heparin tubes placed on ice, followed by 10 minutes centrifugation (6,000 rpm) at 4 °C. A 200-µl plasma aliquot was transferred into an Eppendorf tube with 20 µl of 2 mol/l semicarbazide in 50 mmol/l potassium phosphate buffer (pH 7.4) and frozen in dry ice. Two-hundred milligrams of mouse brain or liver was transferred to an Eppendorf tube with 500 µl PBS and 50 µl of 2 mol/l semicarbazide solution, then homogenized on ice (15 seconds with a 10-second stop, repeat three times). After 10 minutes centrifugation (6,000 rpm) at 4 °C, the supernatant was transferred to a new Eppendorf tube and frozen in dry ice. All samples were stored in −80 °C before analysis. Refer to Supplementary Materials and Methods for detailed descriptions of measurement conditions.
PCR and RT-PCR. Brain and organs were harvested from animals euthanized at 7 or 60 days after virus inoculation at the dose of 5 × 107 pfus (7 days) and 107 or 108 pfus (60 days). CPA or CPT11 was injected i.p. at 1, 3, 5, and 7 days (CPA) or 1 day (CPT11) after virus inoculation. A small piece of tissue excised from each organ was immediately placed into RNA later tissue storage reagent (QIAGEN, Germantown, MD) and stored at 4 °C until use, for a period up to 4 weeks. Viral genomic DNA was extracted from the tissues using QIAamp DNA Mini Kit (QIAGEN) following the manufacturer's protocol. Total mRNA extraction and first-strand cDNA synthesis was conducted using OneStep RT-PCR system (QIAGEN) following the manufacturer's protocol. Primer sequences and annealing temperatures are listed in Supplementary Table S1. The cDNA products of the reverse transcription reaction were denatured at 95 °C for 15 minutes followed by a 35-cycle PCR reaction (94 °C for 30 seconds, 56 °C or 63 °C for 30 seconds, and 72 °C for 2 minutes). PCR products were separated by agarose gel electrophoresis to confirm the size of products.
ELISA assay. Blood was collected from three Balb/C mice per each time point before and 3, 7, 14 and 60 days after MGH2.1 (5 × 107 pfus) inoculation into Balb/C mouse brain. Serum was separated by brief centrifugation and used for ELISA assays to quantify the concentration of TNF-α (Hycult Biotechnology, Uden, Netherlands), IL-6 (Cell Science, Canton, MA), IFN-α (Cell Science), IFN-β (Cell Science), and IFN-γ (Cell Science), following manufacturer's instructions.
Statistical analyses. For MGH2.1 dose effect analyses, a square root transformation was used to stabilize the variances. Dunnet's method was used to adjust for multiple comparisons. For dose effect of CPA or CPT11, linear models (analysis of variance) were used to compare cytotoxicity among groups and days. Bonferroni was used to control for type I error. For primary glioma cells and normal cells, t test (one-tailed, unequal variance) was performed.
SUPPLEMENTARY MATERIAL Figure S1. Engineering of MGH2.1. Figure S2. Effect of intracerebrally injected MGH2.1 ± CPA and ±CPT11 on body weights of Balb/C mice. Figure S3. In vitro functional assays of transgenes (shiCE and CYP2B1) in MGH2.1. Table S1. List of primers utilized in study.
Acknowledgments
These studies were funded by the Alliance for Cancer Gene Therapy (Greenwich, CT) to E.A.C. and NCI 1P01CA163205-01A1 to E.A.C. and W.B.G. Work in the Potter lab is supported in part by NIH grant CA108775, a Cancer Center Core grant CA21765, and by the American Lebanese Syrian Associated Charities (ALSAC) and St. Jude Children's Research Hospital (SJCRH). The authors declared no conflict of interest.
Supplementary Material
Engineering of MGH2.1.
Effect of intracerebrally injected MGH2.1 ± CPA and ±CPT11 on body weights of Balb/C mice.
In vitro functional assays of transgenes (shiCE and CYP2B1) in MGH2.1.
List of primers utilized in study.
References
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Kohler BA, Ward E, McCarthy BJ, Schymura MJ, Ries LA, Eheman C, et al. Annual report to the nation on the status of cancer, 1975-2007, featuring tumors of the brain and other nervous system. J Natl Cancer Inst. 2011;103:714–736. doi: 10.1093/jnci/djr077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick A, Patel D, Hadziahmetovic M, Chakravarti A, Mehta M. Current therapeutic paradigms in glioblastoma. Rev Recent Clin Trials. 2010;5:14–27. doi: 10.2174/157488710790820544. [DOI] [PubMed] [Google Scholar]
- Ballman KV, Buckner JC, Brown PD, Giannini C, Flynn PJ, LaPlant BR, et al. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro-oncology. 2007;9:29–38. doi: 10.1215/15228517-2006-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- Westphal M, Lamszus K. The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat Rev Neurosci. 2011;12:495–508. doi: 10.1038/nrn3060. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiocca EA. Oncolytic viruses. Nat Rev Cancer. 2002;2:938–950. doi: 10.1038/nrc948. [DOI] [PubMed] [Google Scholar]
- Mohyeldin A, Chiocca EA. Gene and viral therapy for glioblastoma: a review of clinical trials and future directions. Cancer J. 2012;18:82–88. doi: 10.1097/PPO.0b013e3182458b13. [DOI] [PubMed] [Google Scholar]
- Shen Y, Nemunaitis J. Herpes simplex virus 1 (HSV-1) for cancer treatment. Cancer Gene Ther. 2006;13:975–992. doi: 10.1038/sj.cgt.7700946. [DOI] [PubMed] [Google Scholar]
- Cassady KA, Parker JN. Herpesvirus vectors for therapy of brain tumors. Open Virol J. 2010;4:103–108. doi: 10.2174/1874357901004010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi P, Peruzzi P, Reinhart B, Cohen JB, Chiocca EA, Glorioso JC. Design and application of oncolytic HSV vectors for glioblastoma therapy. Expert Rev Neurother. 2009;9:505–517. doi: 10.1586/ern.09.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todo T. Oncolytic virus therapy using genetically engineered herpes simplex viruses. Front Biosci. 2008;13:2060–2064. doi: 10.2741/2823. [DOI] [PubMed] [Google Scholar]
- Manservigi R, Argnani R, Marconi P. HSV Recombinant Vectors for Gene Therapy. Open Virol J. 2010;4:123–156. doi: 10.2174/1874357901004010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulkkanen KJ, Yla-Herttuala S. Gene therapy for malignant glioma: current clinical status. Mol Ther. 2005;12:585–598. doi: 10.1016/j.ymthe.2005.07.357. [DOI] [PubMed] [Google Scholar]
- Tyminski E, Leroy S, Terada K, Finkelstein DM, Hyatt JL, Danks MK, et al. Brain tumor oncolysis with replication-conditional herpes simplex virus type 1 expressing the prodrug-activating genes, CYP2B1 and secreted human intestinal carboxylesterase, in combination with cyclophosphamide and irinotecan. Cancer Res. 2005;65:6850–6857. doi: 10.1158/0008-5472.CAN-05-0154. [DOI] [PubMed] [Google Scholar]
- Aghi M, Visted T, Depinho RA, Chiocca EA. Oncolytic herpes virus with defective ICP6 specifically replicates in quiescent cells with homozygous genetic mutations in p16. Oncogene. 2008;27:4249–4254. doi: 10.1038/onc.2008.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1:938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- Wei MX, Tamiya T, Chase M, Boviatsis EJ, Chang TK, Kowall NW, et al. Experimental tumor therapy in mice using the cyclophosphamide-activating cytochrome P450 2B1 gene. Hum Gene Ther. 1994;5:969–978. doi: 10.1089/hum.1994.5.8-969. [DOI] [PubMed] [Google Scholar]
- Hicks LD, Hyatt JL, Stoddard S, Tsurkan L, Edwards CC, Wadkins RM, et al. Improved, selective, human intestinal carboxylesterase inhibitors designed to modulate 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (Irinotecan; CPT-11) toxicity. J Med Chem. 2009;52:3742–3752. doi: 10.1021/jm9001296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danks MK, Morton CL, Pawlik CA, Potter PM. Overexpression of a rabbit liver carboxylesterase sensitizes human tumor cells to CPT-11. Cancer Res. 1998;58:20–22. [PubMed] [Google Scholar]
- Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol. 2011;33:369–383. doi: 10.1007/s00281-011-0245-0. [DOI] [PubMed] [Google Scholar]
- Clarke L, Waxman DJ. Oxidative metabolism of cyclophosphamide: identification of the hepatic monooxygenase catalysts of drug activation. Cancer Res. 1989;49:2344–2350. [PubMed] [Google Scholar]
- Sensenbrenner LL, Marini JJ, Colvin M. Comparative effects of cyclophosphamide, isophosphamide, 4-methylcyclophosphamide, and phosphoramide mustard on murine hematopoietic and immunocompetent cells. J Natl Cancer Inst. 1979;62:975–981. [PubMed] [Google Scholar]
- Alvarez-Breckenridge CA, Yu J, Kaur B, Caligiuri MA, Chiocca EA. Deciphering the Multifaceted Relationship between Oncolytic Viruses and Natural Killer Cells. Adv Virol. 2012;2012:702839. doi: 10.1155/2012/702839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurozumi K, Hardcastle J, Thakur R, Yang M, Christoforidis G, Fulci G, et al. Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. J Natl Cancer Inst. 2007;99:1768–1781. doi: 10.1093/jnci/djm229. [DOI] [PubMed] [Google Scholar]
- Fulci G, Dmitrieva N, Gianni D, Fontana EJ, Pan X, Lu Y, et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007;67:9398–9406. doi: 10.1158/0008-5472.CAN-07-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamfers ML, Fulci G, Gianni D, Tang Y, Kurozumi K, Kaur B, et al. Cyclophosphamide increases transgene expression mediated by an oncolytic adenovirus in glioma-bearing mice monitored by bioluminescence imaging. Mol Ther. 2006;14:779–788. doi: 10.1016/j.ymthe.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambara H, Saeki Y, Chiocca EA. Cyclophosphamide allows for in vivo dose reduction of a potent oncolytic virus. Cancer Res. 2005;65:11255–11258. doi: 10.1158/0008-5472.CAN-05-2278. [DOI] [PubMed] [Google Scholar]
- Wakimoto H, Ikeda K, Abe T, Ichikawa T, Hochberg FH, Ezekowitz RA, et al. The complement response against an oncolytic virus is species-specific in its activation pathways. Mol Ther. 2002;5:275–282. doi: 10.1006/mthe.2002.0547. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Ichikawa T, Wakimoto H, Silver JS, Deisboeck TS, Finkelstein D, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med. 1999;5:881–887. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- Alvarez-Breckenridge CA, Yu J, Price R, Wojton J, Pradarelli J, Mao H, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, Yu J, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci USA. 2006;103:12873–12878. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Hosokawa M, Atsumi R, Suzuki W, Hakusui H, Nagai E. Metabolic activation of CPT-11, 7-ethyl-10-[4-(1-piperidino)-1- piperidino]carbonyloxycamptothecin, a novel antitumor agent, by carboxylesterase. Biol Pharm Bull. 1994;17:662–664. doi: 10.1248/bpb.17.662. [DOI] [PubMed] [Google Scholar]
- Vredenburgh JJ, Desjardins A, Reardon DA, Friedman HS. Experience with irinotecan for the treatment of malignant glioma. Neuro-oncology. 2009;11:80–91. doi: 10.1215/15228517-2008-075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghi M, Chou TC, Suling K, Breakefield XO, Chiocca EA. Multimodal cancer treatment mediated by a replicating oncolytic virus that delivers the oxazaphosphorine/rat cytochrome P450 2B1 and ganciclovir/herpes simplex virus thymidine kinase gene therapies. Cancer Res. 1999;59:3861–3865. [PubMed] [Google Scholar]
- Struck RF, Kirk MC, Witt MH, Laster WR Jr. Isolation and mass spectral identification of blood metabolites of cyclophosphamide: evidence for phosphoramide mustard as the biologically active metabolite. Biomed Mass Spectrom. 1975;2:46–52. doi: 10.1002/bms.1200020109. [DOI] [PubMed] [Google Scholar]
- Colvin M, Hilton J. Pharmacology of cyclophosphamide and metabolites. Cancer Treat Rep. 1981;65 suppl. 3:89–95. [PubMed] [Google Scholar]
- Boddy AV, Yule SM. Metabolism and pharmacokinetics of oxazaphosphorines. Clin Pharmacokinet. 2000;38:291–304. doi: 10.2165/00003088-200038040-00001. [DOI] [PubMed] [Google Scholar]
- Roy P, Yu LJ, Crespi CL, Waxman DJ. Development of a substrate-activity based approach to identify the major human liver P-450 catalysts of cyclophosphamide and ifosfamide activation based on cDNA-expressed activities and liver microsomal P-450 profiles. Drug Metab Dispos. 1999;27:655–666. [PubMed] [Google Scholar]
- Potter PM, Wolverton JS, Morton CL, Wierdl M, Danks MK. Cellular localization domains of a rabbit and a human carboxylesterase: influence on irinotecan (CPT-11) metabolism by the rabbit enzyme. Cancer Res. 1998;58:3627–3632. [PubMed] [Google Scholar]
- Wei MX, Tamiya T, Rhee RJ, Breakefield XO, Chiocca EA. Diffusible cytotoxic metabolites contribute to the in vitro bystander effect associated with the cyclophosphamide/cytochrome P450 2B1 cancer gene therapy paradigm. Clin Cancer Res. 1995;1:1171–1177. [PubMed] [Google Scholar]
- Ichikawa T, Petros WP, Ludeman SM, Fangmeier J, Hochberg FH, Colvin OM, et al. Intraneoplastic polymer-based delivery of cyclophosphamide for intratumoral bioconversion by a replicating oncolytic viral vector. Cancer Res. 2001;61:864–868. [PubMed] [Google Scholar]
- Wakimoto H, Fulci G, Tyminski E, Chiocca EA. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide's enhancement of viral oncolysis. Gene Ther. 2004;11:214–223. doi: 10.1038/sj.gt.3302143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase M, Chung RY, Chiocca EA. An oncolytic viral mutant that delivers the CYP2B1 transgene and augments cyclophosphamide chemotherapy. Nat Biotechnol. 1998;16:444–448. doi: 10.1038/nbt0598-444. [DOI] [PubMed] [Google Scholar]
- Markovitz NS, Baunoch D, Roizman B. The range and distribution of murine central nervous system cells infected with the γ(1)34.5- mutant of herpes simplex virus 1. J Virol. 1997;71:5560–5569. doi: 10.1128/jvi.71.7.5560-5569.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMenamin MM, Byrnes AP, Pike FG, Charlton HM, Coffin RS, Latchman DS, et al. Potential and limitations of a gamma 34.5 mutant of herpes simplex 1 as a gene therapy vector in the CNS. Gene Ther. 1998;5:594–604. doi: 10.1038/sj.gt.3300639. [DOI] [PubMed] [Google Scholar]
- Chiocca EA, Choi BB, Cai WZ, DeLuca NA, Schaffer PA, DiFiglia M, et al. Transfer and expression of the lacZ gene in rat brain neurons mediated by herpes simplex virus mutants. New Biol. 1990;2:739–746. [PubMed] [Google Scholar]
- Kramer MF, Coen DM. Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J Virol. 1995;69:1389–1399. doi: 10.1128/jvi.69.3.1389-1399.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Wakimoto H, Ichikawa T, Jhung S, Hochberg FH, Louis DN, et al. Complement depletion facilitates the infection of multiple brain tumors by an intravascular, replication-conditional herpes simplex virus mutant. J Virol. 2000;74:4765–4775. doi: 10.1128/jvi.74.10.4765-4775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramm CM, Chase M, Herrlinger U, Jacobs A, Pechan PA, Rainov NG, et al. Therapeutic efficiency and safety of a second-generation replication-conditional HSV1 vector for brain tumor gene therapy. Hum Gene Ther. 1997;8:2057–2068. doi: 10.1089/hum.1997.8.17-2057. [DOI] [PubMed] [Google Scholar]
- Terada K, Wakimoto H, Tyminski E, Chiocca EA, Saeki Y. Development of a rapid method to generate multiple oncolytic HSV vectors and their in vivo evaluation using syngeneic mouse tumor models. Gene Ther. 2006;13:705–714. doi: 10.1038/sj.gt.3302717. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Engineering of MGH2.1.
Effect of intracerebrally injected MGH2.1 ± CPA and ±CPT11 on body weights of Balb/C mice.
In vitro functional assays of transgenes (shiCE and CYP2B1) in MGH2.1.
List of primers utilized in study.