

Abstract
The development of episomally maintained DNA vectors to genetically modify dividing cells efficiently and stably, without the risk of integration-mediated genotoxicity, should prove to be a valuable tool in genetic research. In this study, we demonstrate the utility of Scaffold/Matrix Attachment Region (S/MAR) DNA vectors to model the restoration of a functional wild-type copy of the gene folliculin (FLCN) implicated in the renal cancer Birt-Hogg-Dubé (BHD). Inactivation of FLCN has been shown to be involved in the development of sporadic renal neoplasia in BHD. S/MAR-modified BHD tumor cells (named UOK257-FS) show restored stable FLCN expression and have normalized downstream TGFβ signals. We demonstrate that UOK257-FS cells show a reduced growth rate in vitro and suppression of xenograft tumor development in vivo, compared with the original FLCN-null UOK257 cell line. In addition, we demonstrate that mTOR signaling in serum-starved FLCN-restored cells is differentially regulated compared with the FLCN-deficient cell. The novel UOK257-FS cell line will be useful for studying the signaling pathways affected in BHD pathogenesis. Significantly, this study demonstrates the suitability of S/MAR vectors to successfully model the functional expression of a therapeutic gene in a cancer cell line and will aid the identification of novel cancer markers for diagnosis and therapy.
Introduction
A variety of methods are currently used to genetically modify cells. Currently, the most popular method is the use of integrating viruses. While there are benefits to using these vectors, the inherent risk of genotoxicity by insertional mutagenesis due to random and even site-specific integration, always remains. Additionally, the effects of integration cannot be accurately predicted since the expression levels of surrounding and even more distant genes can be directly affected by the integrated construct.1 Furthermore, it has been noted that these integration events can even affect nuclear architecture and influence the nuclear location and state of chromatin condensation of the modified genome.2 The result of genetic integration therefore is the development of stable cell lines that potentially have very different gene expression profiles and significantly different cellular phenotypes from the originating lines thus reducing the reliability and comparability of these models. The development of alternative vector technologies, which can avoid the limitations of genetic integration, is therefore desirable.
We have previously shown the utility of a nonviral episomal DNA vector harboring an Scaffold/Matrix Attachment Region (S/MAR) element and a reporter gene for the generation of tumor cell lines, which produce sustained levels of detectable reporter gene expression over the lifetime of the cell and through countless cell divisions.3,4 The establishment of cancer cell lines stably expressing a reporter gene allows the development of xenograft models that can be monitored by noninvasive methods, such as by in vivo bioluminescent imaging. A further advantage is that xenografts can be tracked with high sensitivity in the same animals over a significant period of time thereby decreasing the number of animals required for any one experiment while still producing more reliable results. In this previous work, we showed that such S/MAR-luciferase-labeled tumor cells develop into tumors in vivo and that quantification of luciferase expression from the tumors over the experimental period provides a reliable indication of the increase in tumor mass. The histological appearance of the tumors isolated at the end of the experiment was identical with that of the original tumor from which the cell lines were derived and immunohistochemistry showed that each cell retained expression of the transfected luciferase transgene. Crucially, we show that the S/MAR vector does not integrate into the tumor cell genome but is retained episomally with 1–2 vector copies per cell.3
The generation of an S/MAR DNA vector expressing a gene of interest is simple and only requires active transcription upstream of the S/MAR sequence for its function. Mechanistically, the presence of the S/MAR module in the vector tethers the plasmid to the nuclear scaffold matrix by binding to nuclear matrix proteins such as scaffold attachment factor A and p300. This ‘piggy-back' machinery enables the S/MAR vector to be retained mitotically over apparently limitless cell divisions. The manner itself in which S/MAR attaches to the nuclear scaffold, in looped domains, also facilitates the maintenance of gene expression by preventing the spread of heterochromatin and therefore inhibiting epigenetic silencing which frequently occurs when using nonviral plasmid vectors. In addition, the S/MAR sequence itself is highly destabilized allowing greater access to transcription factors and thus providing high levels of gene expression from the DNA vector.4,5
In this current study, we show further development of the S/MAR DNA vector to model genes of interest, which will be significant in research investigating aberrant signaling feedback in cancer cell lines. Here, we demonstrate the utility of the S/MAR DNA vector to model the supplementation of a therapeutic gene in an inherited cancer model. Birt-Hogg-Dubé (BHD) syndrome is a rare autosomal dominant disorder that predisposes patients to developing fibrofolliculomas, lung cysts, and renal neoplasia.6 Previous studies have shown that the disorder is caused by a mutation of the BHD gene, which encodes a protein called folliculin (FLCN). In more than 50% of BHD cases, a cytosine insertion or deletion occurs in the mononucleotide tract of C8 in exon 11.7 A BHD cell line derived from a patient's renal rumor has been established and called UOK257. It has a cytosine insertion in the frequently mutated hotspot of exon 11.8 BHD tumors exhibit loss of heterozygosity consistent with the hypothesis that the FLCN encoding gene is a tumor suppressor.6,9 However, it is currently uncertain how FLCN functions to repress tumor progression. FLCN has no known functional domains and its contribution to the development of BHD-associated renal neoplasia is still uncertain. However, recent studies have implicated its role in the TGF-β signaling pathway, which is often deregulated in tumorigenesis.10,11 Other studies have shown an involvement of FLCN in the energy and nutrient-sensing mammalian target of rapamycin (mTOR) pathway via the FLCN-interacting proteins 1 and 2 (FNIP1/2) and 5′-AMP-activated protein kinase.12 BHD shares phenotypic similarities with other conditions, such as Von-Hippel-Lindau Syndrome, for which the deregulation of mTOR has also been implicated. The role of FLCN in the mTOR pathway is still being elucidated with so far contrasting reports showing up or downregulation of downstream mTOR substrates in different BHD animal models,13,14,15 which suggests that a variation of FLCN expression may have differential effects and may also be condition dependent.
Although nearly all germline BHD mutations result in the truncation of the FLCN protein, it is still unclear if the truncated FLCN protein has an oncogenic role in the development of the disorder. However, a previous study has shown that the transfer of a functional copy of FLCN-encoding gene into BHD cells had a therapeutic effect by normalizing the TGF-β pathways and preventing the development of tumors ex vivo.11 In this paper, Hong et al. developed a stable BHD cell line expressing FLCN (UOK257-2) using integrating lentiviruses. They showed an up to ~6.8-fold increase in FLCN mRNA levels in various stable clones over that of the original FLCN-deficient UOK257 cells and demonstrate growth suppression of the cells over a year-long xenograft study.
Here, we describe the generation of UOK257 cells, which stably express transgenic FLCN from episomally maintained S/MAR DNA vectors. The new UOK257 cell line (named UOK257-FS) is shown to produce sustained levels of FLCN over limitless cell divisions and to present a normalized expression of the downstream TGF-β regulators, SMAD3 and TGFβ2. Under normal conditions, UOK257-FS and UOK257 cells show comparable mTOR activity but when deprived of serum, we show the UOK257-FS to have a nearly complete inhibition of mTOR activity (indicated by loss of 4E-BP1 signal), which is hyperphosphorylated in BHD-/- embryonic stem cells16 in contrast to parental UOK257 cells. UOK257-FS cells display a reduction in proliferation in vitro and accordingly, show a complete suppression of tumor development in xenograft models. In conclusion, this study demonstrates for the first time a method for utilizing a S/MAR plasmid DNA vector for provision of a therapeutic gene in a cancer cell model. It also presents an investigation into the corresponding restoration of normal cellular biochemistry and morphological behavior of the genetically modified cells. The generation of UOK257-FS cells provides a novel BHD cell model in which transcriptional networks and signaling pathways involved in FLCN deregulation can be further analyzed.
Results
Generation of stable FLCN-expressing UOK257-FS cells
Based on a previously published S/MAR plasmid, pUbC-Luc-SMAR, which we have used to stably label cancer cells with a luciferase reporter gene,3 we constructed a novel S/MAR plasmid called pUbC-FLCN-SMAR. It contains the FLCN cDNA driven by the mammalian UbC promoter and harboring the S/MAR module downstream of the expression cassette (Figure 1a, left).
Figure 1.

Characterization of UOK257-FS cells. (a) Schematic representation of plasmids pUbC-FLCN-SMAR (left) and pUbC-Luc-SMAR (right) used for transfection. The S/MAR module is located downstream of the expression cassette. Digestion sites used for restriction analysis of plasmid rescue are shown. The arrows depict the positions of PCR primers used for sequencing. (b) Western analysis of FLCN expression in stable FLCN-restored UOK257-FS cells (1), UOK257-Luc cells (2) and UOK257 cells (3). (c) FLCN mRNA levels as measured by quantitative RT-PCR. Columns indicate mean values ± SEM (n = 3). (d) Morphology of UOK257-FS (left panel) and UOK257 cells (right panel) on adherent plates (top panel) and in 3D culture (bottom panel). Magnification 4x. Scale bar = 500 μm.
UOK257 cells were transfected with plasmid pUbC-FLCN-SMAR and cultured for 4 weeks in the presence of G418 (400 μg/ml). Colonies that formed after this period were isolated and expanded in normal medium. A stable colony named UOK257-FS was selected for further investigations. We confirmed FLCN expression by Western analysis (Figure 1b) and detected ~15.9-fold higher levels of FLCN mRNA in UOK257-FS compared with endogenous RNA levels of FLCN in the parental UOK257 cells (Figure 1c).
It was immediately evident following stable colony formation that the morphology of FLCN-expressing UOK257-FS cell line differed from the original UOK257 cells. On adherent plates, UOK257-FS cells display loss of cell-cell contact (Figure 1d, top left) in contrast UOK257 cells grew in tight islets with defined borders exhibiting the loss of contact inhibition (Figure 1d, top right). Following these observations, we went on to investigate the effect of FLCN in a 3D culture on ultralow attachment plates. The difficulty in proper spatial orientation, which is required for cell coordination in a 3D environment, is revealed by an even more contrasting phenotype difference between the two cell lines. UOK257-FS cells form tightly-bound round spheres (Figure 1d, lower left) while only amorphous cell clusters are seen with FLCN-deficient UOK257 cells (Figure 1d, lower right). These results are in accordance with a previous study that showed that downregulation of FLCN disrupts its interaction with a junction protein, p0071, resulting in vastly affected junction formation and cell polarity.17
To generate a stable control cell line expressing a mock gene (luciferase) as a control, plasmid pUbC-Luc-SMAR (Figure 1a, right) was transfected into UOK257 cells and the cells were placed in selective medium (400 µg/ml) for 4 weeks. A stable UOK257 cell line expressing luciferase was obtained, named UOK257-Luc. The method for obtaining stable UOK257-Luc cells using pUbC-Luc-SMAR is represented schematically in see Supplementary Figure S1a online. No significant differences in the FLCN mRNA levels (Figure 1c) or in cell morphology (see Supplementary Figure S1b online) were detected when comparing UOK257-Luc with the parental UOK257 cells. It should be noted that when a non-S/MAR control plasmid pUbC-Luc, was used for transfection, luciferase expression was lost within a week post-transfection, in accordance to previous observations that the presence of the S/MAR is required to reveal stable transgenic clones.4,18
Upregulation of TGF-β downstream components SMAD3, TGFβ2, and SMAD7 in UOK257-FS cells
To investigate whether FLCN supplementation in UOK257-FS cells had any effect on the regulation of TGF-β signaling, we performed Western analysis against TGF-β mediator SMAD3. We observed a clear upregulation of phosphorylated SMAD3 (pSMAD3) and SMAD3 expression in UOK257-FS cells in comparison with UOK257 cells. Higher levels of pSMAD3 and SMAD3 signals were observed in the stable UOK257-FS cells (Figure 2a, 2) compared with cells transiently transfected with pUbC-FLCN-SMAR in UOK257 cells (Figure 2a, 4). These separate Western analyses of FLCN from the same cells indicate that stably maintained levels of FLCN are necessary for normalized molecular TGF-β signals in BHD.
Figure 2.
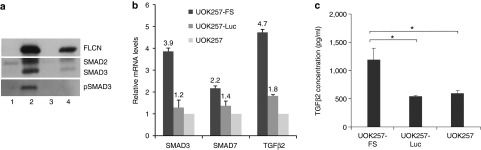
Restoration of TGF-β signaling in UOK257-FS cells. The compilation of protein bands from separate Western blots show the (a) upregulation of SMAD3 and pSMAD3 expression in stable UOK257-FS cells (2) compared with stable UOK257-Luc cells (1), untreated UOK257 cells (3) and UOK257 cells following transient 24 hours transfection with pUbC-FLCN-SMAR (4). (b) Upregulation of SMAD3, SMAD7, and TGFβ2 mRNA levels in UOK257-FS cells relative to endogenous UOK257 mRNA levels. No differences in SMAD3, SMAD7, and TGFβ2 levels were detected in UOK257-Luc cells over UOK257 mRNA levels. Columns represent mean ± SEM (n = 3). (c) Significantly higher levels of secreted TGFβ2 protein in measured the medium of UOK257-FS cells compared with UOK257 cells (*P < 0.05), as measured by ELISA.
Figure 4.

Evaluation of mTOR activity in UOK257-FS and UOK257 cells under serum-starved conditions. Protein levels of mTOR, Raptor, p70-S6 (Thr389), pS6R (Ser235/236) and p4E-BP1 of both cell lines were determined by Western analysis under normal serum (S+) and serum-deprived conditions (S-) as shown by the compilation of Western blots. A portion of UOK257-FS cells was stimulated with serum again following serum starvation (S++). Under normal serum conditions (S+), no differences in mTOR activity is detected between UOK257-FS (1) and UOK257 cells (2). Following serum-deprivation for 48 hours (S-), loss of mTOR signaling is detected in the FLCN-restored UOK257-FS cells (3), indicated by loss of p4E-BP1 signal. Re-establishment of 4E-BP1 phosphorylation is seen following restoration of serum for 48 hours (S++) in UOK257-FS cells (5). Under serum-starved conditions (S-) no suppression of mTOR activity is detected in UOK257 cells (4) with an increase in the α-isoform of p4E-BP1.
Accordingly, we see a higher induction of SMAD3 mRNA, and other downstream TGF-β proteins SMAD7 and TGFβ2 mRNA, in UOK257-FS (~3.9-fold, 2.2-fold and 4.7-fold increase, respectively) relative to endogenous UOK257 levels (Figure 2b). SMAD7 is upregulated by TGF-β activation and under normal oxygenated conditions, expression of SMAD7 has been shown to inhibit cancer proliferation.19 In addition, to confirm that the increase in TGFβ2 mRNA levels correlates with secreted protein levels, we measured TGFβ2 in the media of cells and show a twofold increase in TGFβ2 protein secretion over parental UOK257 cells (P < 0.05) (Figure 2c). No differences in SMAD3, SMAD7, and TGFβ2 levels were detected between UOK257-Luc and UOK257 cells, indicating that expression of a reporter gene had no effect on regulation of TGF-β (Figure 2b). These results here show that UOK257-FS cells have restored TGF-β levels.
Reduced proliferation rate of UOK257-FS cells
As the general loss of normal TGF-β signaling leads to abnormal apoptotic regulation and increased cell growth,20 we went on to compare cell proliferation rates of both UOK257-FS and UOK257 cell lines. Cells were plated onto 96-well plates and cell numbers were counted over a 20-day period. We showed that UOK257-FS cells grew ~20% slower than the original UOK257 cell line, with a doubling rate of once every 63.3 (± 9 hours), compared with UOK257 cells, which doubled once every 50.4 (± 5 hours). No comparable differences in growth rates were observed between UOK257 and UOK257-Luc cells confirming that the expression of the reporter gene had no effect on the cell propagation (Figure 3a).
Figure 3.

Proliferation rates of cells in vitro. (a) Growth curve of UOK257-FS, UOK257-Luc, and UOK257 cells on plates, showing a reduced growth proliferation of UOK257-FS cells on adherent assays. (b) Increased numbers of colonies formed in soft agar assays of UOK257-Luc and UOK257 cells after a four week incubation, compared with UOK257-FS (*P < 0.05), as quantified using ImageQuant TL Software (GE Healthcare, UK). Columns represent mean ± SD (n = 3).
Next, we investigated the potential for neoplastic transformation of UOK257-FS compared with UOK257 cells in a colony forming soft agar assay. Here, cells (2 × 103) were suspended in soft agar and incubated over a 4-week period. The number of colonies obtained was quantified at the end of the experiment and we show a significantly increased number of colonies obtained in the UOK257 plates compared with UOK257-FS cells (P < 0.05) (Figure 3b). It was also noted that colonies formed with UOK257 cells were overall larger in size compared with the colonies obtained with the FLCN-restored UOK257-FS cell line. This indicates that growth of UOK257-FS cells are suppressed in an anchorage-independent assay.
Inhibition of mTOR signaling in serum-starved UOK257-FS cells
Previous studies have shown a contradictory role for mTOR signals in BHD-related neoplasia. It was shown that FLCN is a downstream signaling component of both mTOR and 5′-AMP-activated protein kinase and its expression has both positively and negatively regulated mTOR depending on the variation in the levels of FLCN as well as the differences in experimental conditions.12,16 To examine mTOR regulation in our UOK257-FS cell line under serum-starved conditions, UOK257-FS and UOK257 cells were grown to confluence and then starved of serum. A portion of UOK257-FS cells were then stimulated with serum again and then lysed for analysis. We investigated the phosphorylation of eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), a well-known substrate of mTOR. The 4E-BP1 protein resolves as three bands on SDS-PAGE, where the top band represents the hyperphosphorylated ‘γ-isoform', which binds weakly to eIF4E, and the lowest band is the least phosphorylated ‘α-isoform', which binds the strongest to eIF4E. Under normal serum conditions, 4E-BP1 phosphorylation remains unchanged regardless of FLCN levels. However, when serum-deprived, we show a complete inhibition of mTOR activation (indicated by the absence of any detectable p4E-BP1 band) in UOK257-FS cells (Figure 4, 3). This would indicate loss of eIF4E binding and subsequent suppression of protein synthesis. Upon serum restimulation, we show upregulation of p4E-BP1-indicating restoration of mTOR activation Figure 4, 5. Interestingly, under serum-starved conditions, the majority of 4E-BP1 is hypophosphorylated resolving mainly as an ‘α-isoform' in UOK257 cells (Figure 4, 4) that strongly interacts with eIF4E. Overexpressed eIF4E is strongly associated with tumor progression.21 UOK257 cells have also been shown to favor aerobic glycolysis over lipid oxidation and therefore may have proliferative advantage over UOK257-FS cells in serum-starved conditions.22 We also looked at phosphorylation levels of pS6R, a known readout for mTOR/S6K activity. Unlike 4E-BP1, serum-starved UOK257 cells with or without FLCN levels of p70-S6/pS6 were not altered. This result points to the modulation of 4E-BP1/eIF4E interaction as major contributor to FLCN-mediated cell cycle arrest in BHD and further implicate the role of mTOR signaling in BHD.
Figure 5.
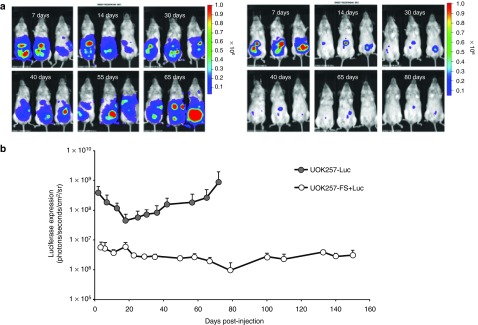
Longitudinal analysis of luciferase in UOK257-FSLuc and UOK257-Luc xenografts. Two groups of mice (n = 5) were innoculated intraperitoneally with 3 × 106 cells of either UOK257-FSLuc or UOK257-Luc. (a) Representative photos from three animals from each group showing the level of luciferase expression are shown over time. The intensity of luciferase is represented from high to low as red to blue, as indicated by the colored bar. (b) Luciferase expression is shown up to day 72 for both groups (n = 5) and up to day 150 for animals treated with UOK257-FSLuc. Columns represent mean ± SEM (n = 3).
Suppression of tumor formation in UOK257-FS xenograft models
Next, we investigated the tumorigenic potential of UOK257-FS by generating UOK257-FS xenograft models. To enable noninvasive monitoring of xenograft formation in vivo, we generated a new S/MAR FLCN construct coexpressing luciferase as a reporter gene. The new DNA vector named pUbC-FLCN-Luc-S/MAR (see Supplementary Figure S2a online) was used to generate a new UOK257 cell line stably expressing FLCN and luciferase called UOK257-FSLuc. Expression of FLCN was confirmed by Western analysis and qRT-PCR (see Supplementary Figure S2b and Figure S2d online). Luciferase expression was established by bioluminescent imaging (see Supplementary Figure S2d online) and we measured luciferase expression on increasing numbers of cells to show limits of signal detection, which were found to be between 250 and 150 cells (see Supplementary Figure S2c online).
The protein levels of FLCN derived from UOK257-FSLuc were shown to be significantly lower than FLCN expression from UOK257-FS cells when analyzed by Western blot (see Supplementary Figure S2b online). This correlated with the corresponding levels of transgenic mRNA levels in both cell lines where we detected about half the level of FLCN mRNA in UOK257-FSLuc cell lines compared with UOK257-FS (7.8-fold versus 15.9-fold, over the knockout UOK257 cell line) (see Supplementary Figure S3 online). This is most likely due to the clonal variation; although S/MARs function by reducing the occurrence of epigenetic silencing within a clonal population, S/MARs do not prevent clonal variation between different populations.18 Nevertheless, we see a restoration and upregulation of the TGF-β signaling mediators SMAD3, SMAD7, and TGFβ2 in UOK257-FSLuc cells (2.3-, 2.2- and 5.5-fold increase, respectively, relative to the parental UOK257 cells) comparable with the levels detected in UOK257-FS cells (see Supplementary Figure S3 online) and a reduced growth proliferation rate in vitro (23% reduced growth rate compared with UOK257 cells, data not shown) suggesting that in both constructs, folliculin levels were sufficient to restore the cells' normal biochemistry.
We also show that luciferase expression from UOK257-FSLuc was approximately one order of magnitude lower than luciferase expression from the UOK257-Luc cell line (see Supplementary Figure S2d online), which is not unexpected due to position of the luciferase cDNA as the second gene in the bicistronic expression cassette.
To investigate the tumorigenic potential in vivo, UOK257-FSLuc and UOK257-Luc cells were separately administered to two groups of immunodeficient mice (n = 5) at doses of 3 × 106 cells. The animals were monitored for tumor development using bioluminescent imaging (Figure 5a), where the increase in luciferase expression correlates to the increase in tumor growth. Luciferase expression was detected in both groups 24 hours following inoculation of cells and tracked over a 150-day experimental period (Figure 5b). In the animals treated with UOK257-Luc, we observed an initial decrease in the levels of luciferase in the first 2 weeks following injection, which is most likely due to the loss of cells prior to establishment of vascularization required for growth. However, this reduction in luciferase levels was not observed in the cohort treated with UOK257-FSLuc although the reason for this is unclear. From day 18 onward, we detected a sharp rise in luciferase expression in the UOK257-Luc cohort within the same abdominal region in all animals. Between days 18 and 72, luciferase expression increased 18-fold which indicated rapid proliferation of cells and possible development of tumor masses. In one animal, a palpable tumor was detectable at day 72 postinjection and due to terms of our procedural license, the UOK257-Luc-treated animals were culled at this time point. In all animals, tumors were evident in the peritoneal region and ranged from 0.5 to 1.5 cm in diameter. No metastasis was observed outside the peritoneal cavity and invasion of viscera was not detected. A representative photo of the tumor is shown in Figure 6c (top panel). Each of the tumors dissected contained numerous solid tumor nodules with slightly lobulated surfaces. In some of the tumors, calcification was detected which is most likely due to necrosis or pseudo cysts due to formation by neoplastic cysts.
Figure 6.
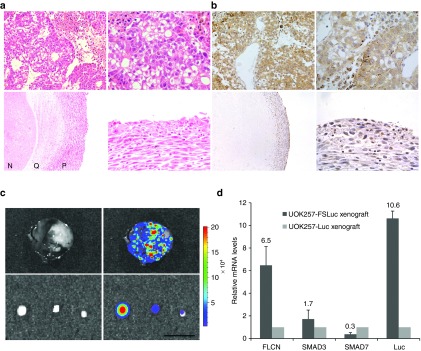
Analysis of xenografts following tumor isolation. (a) H&E staining and (b) antiluciferase immunohistochemistry of tumor sections from UOK257-Luc xenografts at day 72 postinjection (top row) and from UOK257-FSLuc xenografts at day 150 postinjection (bottom row) at 10x (left panel) and 40x (right panel) magnification. The UOK257-Luc xenograft display high-grade clear cell histologies with large abnormal vascular systems whereas the xenograft from UOK257-FSLuc-treated animals form small compacted cell bodies displaying necrotic (N), quiescent (Q) and proliferating (P) spheroid microregions typical of a cell spheroid. Immunohistochemistry show positive staining indicating retention of the encoded luciferase transgene. (b) Representative photo of a tumor isolated from an animal treated with UOK257-Luc at day 72 postinjection (top panel) and of cell bodies isolated from an animal treated with UOK257-FSLuc at day 150 postinjection (bottom panel). Luciferase expression is shown from high to low as red to blue, as indicated by the colored bar. (c) mRNA levels of FLCN, SMAD3, SMAD7 and luciferase (Luc) as measured by quantitative RT-PCR from total RNA isolated from xenografts. Columns indicate mean ± SEM (n = 3).
Conversely, in the animals treated with UOK257-FSLuc, we detected no increase in luciferase levels throughout the experimental period. Indeed, a small decline in luciferase levels (approximately half a log) was seen between days 18 and 72 (Figure 5b). At day 72, two animals were culled for analysis. Patches of cells (between 0.2 and 0.5 cm2 in size) strongly expressing luciferase were isolated from the peritoneal region of the animals. In both animals, the cells were found attached to the peritoneum near the site of injection. To ensure no potential further development of tumors in this cohort, monitoring of luciferase expression was continued up to day 150 postinjection in the remaining three animals (Figure 5). We observed a small increase in luciferase expression of approximately one-third of a log from day 72 to 150; however, no exponential rise in luciferase expression was detected that would indicate the formation of tumors. At day 150, the remaining three animals were sacrificed and round compacted cell clumps ranging from 0.1 to 0.4 cm2 were isolated from one of the animals. A representative photo is shown in Figure 6b (bottom panel). In the remaining two animals, the cells expressing luciferase were not evident once dissected and no cell clumps expressing luciferase could be detected for isolation. The lack of detectable luciferase expression following dissection of the animals is very likely due to the reduced level of oxygen in the cadaver affecting light emission in the time required to look for the cells. Nevertheless, the tissue obtained from one of the animals was used for further analysis.
Hemotoxylin and eosin (H&E) staining of tumor tissue in the UOK257-Luc-treated group show high-grade tumors presenting mainly clear cell histologies with pronounced cell membranes (Figure 6a). In contrast, H&E staining of xenograft isolated from the UOK257-FSLuc-treated animal shows viable cells (Figure 6a, bottom panel) surrounding necrotic centers, often seen in tumor spheroids above 500 μm in diameter.23 Importantly, antiluciferase immunohistochemistry of both xenografts show isolated positive staining isolated indicating maintenance of the encoded luciferase transgene (Figure 6a and 6b).
Retention of FLCN expression in the UOK257-FSLuc cells
In order to show retention of FLCN expression in UOK257-FSLuc cells isolated from the animals, we performed quantitative PCR on mRNA isolated from the xenografts at the end of the experiment. We show approximately sevenfold increase in FLCN mRNA levels in cells isolated from the UOK257-FSLuc cohort compared with the UOK257-Luc tumors, similar to the levels obtained in vitro, indicating the UOK257-FSLuc cells are able to retain FLCN over at least 50 doublings ex vivo.
In order to determine whether the downstream TGF-β genes were differentially regulated by FLCN in UOK257-FSLuc and UOK257-Luc xenografts ex vivo, we measured the expression of the downstream TGF-β signaling proteins SMAD3 and SMAD7 at the mRNA level (Figure 6c). We detected a reduced level of SMAD7 expression in UOK257-FSLuc cells ex vivo compared with UOK257-Luc. It has been shown that under hypoxic conditions, increased SMAD7 has been linked to malignant transformation and increased tumorigenesis19 and it is likely that the decreased level of SMAD7 seen here may play a role in the prevention of UOK257-FSLuc cell growth. Similar to observations in vitro, the SMAD3 mRNA levels in UOK257-FSLuc cells ex vivo remained higher than the SMAD3 mRNA levels in UOK257-Luc tumors.
Although luciferase expression from UOK257-FSLuc on in vitro plates was approximately one order of magnitude lower than that from the UOK257-Luc cell line, as measured by bioluminescent imaging, the 10-fold higher luciferase mRNA levels seen in UOK257-FSLuc xenografts compared with UOK257-Luc tumors is not unexpected and most likely due to the additional cells in the differentiated UOK257-Luc tumor, for example, the recruitment of vascular and stromal cells, resulting in proportionately less luciferase-expressing cells (Figure 6).
Episomal retention of S/MAR plasmid in UOK257-Luc xenografts
To provide physical evidence for the molecular retention of the S/MAR plasmid in xenografts, we performed plasmid rescue experiments on UOK257-Luc xenografts obtained at the end of the study. DNA isolated from the tumors was transformed into bacterial cells and all 14 colonies obtained were analyzed by restriction digest. A representative photo of two colonies digested separately with HpaI and PvuII is shown in see Supplementary Figure S4a. The expected restriction patterns that were obtained are similar to the original plasmid, indicating intact extrachromosomal maintenance of the pUbC-Luc-SMAR in UOK257 xenografts. Due to the small size of the xenografts isolated from the animals treated with UOK257-FS, we did not have sufficient material to isolate the high concentration of DNA required for efficient bacterial transformation. However, due to the retention of episomal expression of pUbC-Luc-SMAR in the UOK257-Luc xenograft and increased mRNA levels of FLCN and luciferase in UOK257-FS compared with UOK257 xenografts as well as based on our previous data showing episomal retention of S/MAR vectors in vitro,4,24 in vivo,25,26 and ex vivo,3 we expect plasmid pUbC-FLCN-Luc-SMAR to be similarly retained.
To verify the stability of the plasmid at the end of the experiment, two clones (out of the 14 obtained) were selected for sequencing. No differences in DNA sequences were detectable between the two clones and the original pUbC-Luc-SMAR indicating maintenance of plasmid integrity over the 72-day period in vivo (data not shown).
Discussion
Signaling pathways controlling cell growth and differentiation are almost invariably altered in cancer. The elucidation of key cellular pathways disrupted in tumorigenesis provides valuable insight into the cause of the disease. This enables the identification of mutated genes, which can cause cancer thus providing potential gene targets for diagnosis and therapy. The rapid and simple generation of genetically modified cell lines facilitates the analysis and understanding of the regulation of the different genes affected in various pathways.
Modeling the expression of a gene of interest in a cell line without the risk of random integration is important for studying signaling pathways, where changes in the regulation of a protein would provide mechanistic insights into the genetic defects that occur in tumorigenesis. Significantly, the modeling of genes suspected to have therapeutic benefit in cancer cell lines will enable the development of novel markers cancer diagnosis and potentially for treatment as well.
The use of the S/MAR system for genetic modification of cells has several benefits over standard protocols using integrating viral vectors. One is simply, the ease by which S/MAR plasmids are able to stably transfect cell lines allowing the generation of a stable cell line within a month after transfection. Another is the easy and relatively cheap production of S/MAR plasmid DNA at high concentration. Furthermore, the S/MAR vector has a virtually unlimited genetic capacity allowing delivery of a complete genomic locus27 and thereby enabling expression of a transgene at normal physiological levels.
Another significant benefit of using S/MAR vectors is their ability to maintain transgene expression episomally.28,29 Episomal maintenance systems offer many advantages over integrating vectors as they avoid unpredictable integration into the host genome and the associated potential risk of cellular transformation. We, and others, have shown that the S/MAR DNA is persistently maintained without integration over countless cell divisions.30 In addition, we have shown that the S/MAR plasmid replicates episomally within mammalian cells, losing its bacterial methylation pattern and gaining a mammalian pattern of methylation, by undergoing at least two rounds of cell divisions in mammalian cells.3,4 In the present study, we show plasmid rescue of whole intact pUbC-Luc-SMAR DNA from tumor cells, which indicates extrachromosomal retention of the plasmid as an entity within the cells.
Here, we use a model of the renal cancer BHD to demonstrate the suitability for the S/MAR vector to stably restore functional expression of a tumor suppressor gene FLCN in the BHD UOK257 cell line. The levels of transgenic folliculin expression detected in these genetically modified cells are at least equivalent to those described in normal human cells.11 These cells, which were cultured from a biopsy of a BHD tumor, have lost their wild-type FLCN expression. Restoration of FLCN expression in UOK257-FS cells conferred by the S/MAR vector is shown to restore normal levels of the TGF-β signaling pathway by upregulation of SMAD3, SMAD7, and TGFβ2 levels. The TGF-β superfamily is involved in diverse array of differentiation, adhesion, and migration programs. TGF-β has been shown to function as a tumor suppressor in early stages of cancer, but can also promote metastasis in the later stages.20 Restoration of FLCN expression in stable UOK257-FS cells results in restored levels of the TGF-β-mediated growth modulators pSMAD3 and SMAD3 in comparison with the parental UOK257 cells where SMAD3 expression is absent or at very low levels. Similarly, low levels of SMAD3 and of SMAD3/SMAD2 ratios have been reported in BHD patient tumors in comparison with normal kidneys.11 The increased rate of proliferation observed in UOK257 cells is possibly due to the low levels of SMAD3 and a corresponding reduction in its suppressive effects. Accordingly, low levels of SMAD3 in gastric tumors and cancer cells expressing SMAD3, show a decrease tumorigenicity in vivo31 and restoration of SMAD3 expression has also been reported to suppress tumor development in a gastric cancer cell model.32
SMAD3 has been implicated in the TGF-β mediation of epithelial-to-mesenchymal transition that is hypothesized to promote the dissemination of cancer cells in the intraperitoneal cavity or metastasis into other organs. Cancer cells that undergo epithelial-to-mesenchymal transition lose their cell-cell contact and cell polarity allowing increased motility.33 Downregulation of SMAD3 in ovarian cancer cells has been shown to inhibit the loss of cell-cell adhesion and the transition to mesenchymal morphology.34 Accordingly, following the upregulation of SMAD3 levels in UOK257-FS cells, we observe a loss of cell-cell adhesion on plates and normalized cell polarity in 3D cultures (Figure 1). Nutrient limitation in UOK257 cells as previously reported22 may play a role in the loss of spatial orientation seen as impaired spheroid growth in the 3D culture. In a recent study, Medvetz et al. reported the interaction of FLCN with p0071, a junctional protein, and that downregulation of FLCN expression increases cell-cell adhesion with defective cell polarity.17 These observations are consistent with the results of our study even though it is surprising given the conventional view that loss of cell-cell adhesion leads to tumorigenesis. However, Medvetz et al. suggest that the overenhanced cell-cell adhesion resulting from deficient FLCN-p0071 complex may contribute to the tumorigenesis. It is likely that FLCN is involved in the Wnt signaling pathway known for establishing cellular orientation and that the disrupted cell polarity observed here may be due to deregulation of Wnt activity. Further investigations looking at the interaction of FLCN with β-catenins will be of interest. Nevertheless, the results here confirm the structural role of FLCN in cell junction organization which has recently been shown to play an increasingly important role in tumorigenesis.
We also observed a small upregulation of SMAD7 mRNA levels in UOK257-FS cells in accordance with previous work carried out using (lentiviral-mediated) FLCN-expressing UOK257-2 cells.11 SMAD7 has been reported to have contradictory roles in cancer progression. Under normal (well-oxygenated) conditions, SMAD7 expression is found to suppress cell motility and invasion. Increased SMAD7 expression has been reported to impair the invasive capacity of melanoma cancer cell's invasive capacity and to reduce anchorage-independent cell growth in vitro by inhibiting matrix metalloproteinases.35 However, under the hypoxic conditions as seen in solid tumors,23 increased SMAD7 expression is linked to malignant transformation and tumor growth. Indeed, increased SMAD7 expression has been shown across a number of cancers including renal carcinoma.36 In our study, we show an approximate twofold increase in SMAD7 RNA levels in UOK257-FS cells in vitro suggesting a possible explanation in the suppression of cell proliferation on plates and lack of colony formation in adherence-free assays. Conversely, SMAD7 RNA levels are downregulated in UOK257-FS xenografts suggesting that under hypoxic conditions, FLCN restoration is able to suppress tumor progression by inhibiting SMAD7/TGF-β-mediated growth.19 Interestingly, our UOK257-FSLuc xenografts formed compact spheroids mimicking the 3D formation in culture. This further confirms the restoration of proper spatial orientation by FLCN supplementation in UOK257 cells increasing polarity, which reduces the likelihood for epithelial-to-mesenchymal transition.
The restoration of FLCN expression in UOK257 cells is attributed to the ability of functional FLCN expression to delay progression through the late S and G2/M phase of the cell cycle.37 Accordingly, we show an approximate 20% reduction in cell proliferation of UOK257-FS cells compared with the FLCN-deficient UOK257 cells. In a separate study comparing proliferation of UOK257 cells stably expressing FLCN from a lentiviral vector (UOK257-2) and original UOK257 cells, no difference in cell proliferation between both cell lines was detected on adherent plates.11 While the reason for the differences between the observations of this study and ours are unclear, the level of FLCN expression conferred may be important. Hong et al. 11 showed up to a 6.8-fold increase in FLCN RNA levels in UOK257-2 cells over that of the parental UOK257 cell line whereas UOK257-FS cell line in this study had an approximate 15-fold increase in FLCN levels. The increased levels in FLCN may be attributed to the presence of the S/MAR element itself which is highly destabilized permitting greater access to transcription factors as well as the advantage of the mammalian UbC promoter-driving expression in the S/MAR vectors over the CMV promoter used in the viral vector. Another possible reason for the difference between the two studies could be the method by which the stable FLCN-expressing UOK257 cells are obtained. Lentiviral vectors are liable to alter the behavior of infected cells unpredictably through integration-mediated modification of the cells genome and the resulting cell lines are therefore not likely to represent reliable and reproducible cellular models. Nevertheless, in both, our study using UOK257-FS cells and in Hong et al.'s investigations with UOK257-2 cells, a reduced rate of proliferation was detected in anchorage-independent assays, which suggest a reduced potential for neoplastic transformation of the UOK257 cells following FLCN restoration. Further investigations comparing both the lentiviral-transduced and the S/MAR-transfected FLCN UOK257 cell lines may be beneficial.
Other studies have implicated the mTOR pathway in BHD, which regulates cell growth and size through stimulation of protein synthesis. This is dependent on several inputs, such as the availability of amino acids, energy and growth factors, by regulating the phosphorylation of p70-S6 kinase and 4E-BP1, and by the classical mTORC1 (mTOR/Raptor) function. FLCN has been shown to bind to FNIP1/2 and to complex with 5′-AMP-activated protein kinase in the mTOR pathway12 and FLCN has been implicated as a downstream signaling component of mTOR and 5′-AMP-activated protein kinase.38 Our results corroborate with these studies as no differences were detected for mTOR activation, regardless of FLCN expression in both normal and serum-deprived conditions. However, serum-deprived UOK257-FS cells show a significant reduction of mTOR substrate 4E-BP1 signal indicating a loss of mTOR-mediated protein synthesis. In contrast, FLCN-null UOK257 cells show a hypophosphorylation of 4E-BP1 signal indicating strong expression of eIF4E, a proto-oncogene, which is highly overexpressed in many human tumors. Since UOK257 cells has been shown to favor glycolytic rather than oxidative lipid metabolism, the so-called ‘Warburg effect',22 the results indicate that 4E-BP1/eIF4E may be central for driving deregulated protein synthesis and cell transformation in BHD. In a separate study, Baba et al. showed a similar inhibition of mTOR signaling as a result of serum starvation in FLCN-restored UOK257-2 cells shown by general loss of 4E-BP1 signal.12 However, amino acid deprivation had the opposite effect inhibiting mTOR signaling more effectively in FLCN-null UOK257 cells. This may be attributed to the higher dependency of UOK257 cells on glycolysis.22 Unlike phosphorylation of 4E-BP1, we showed no change in activated levels of p70-S6 or its target S6 following serum starvation of UOK257-FS. This is in contrast to the loss of pS6R signal following serum deprivation of FLCN-restored UOK257-2 cells observed by Baba et al. The reason for the different observations is unclear, but in our current study, it appears that serum depletion modulates the dynamics of mTOR/Raptor to inhibit 4E-BP1 but not S6K phosphorylation. Further investigations will be necessary to elucidate the complex feedback mechanisms involved in BHD mTOR signaling.
In conclusion, we have shown for the first time the therapeutic application of a tumor suppressor gene expressed from a nonviral S/MAR DNA vector in a cancer model. The novel UOK257-FS cell line expressing FLCN conferred by the episomal S/MAR vector is able to sustain ~15-fold higher levels of FLCN over endogenous UOK257 FLCN levels. The new cell line shows clear phenotypic differences compared with the original cell line with regards to restoration of the normal TGFβ pathways, which lead to suppression of proliferation, migration, and transformation in in vitro and in vivo assays. We expect that further investigations using the UOK257-FS cell line will provide a deeper insight into the role of FLCN in kidney cancer and may lead to the development of possible therapeutic interventions. Importantly, we show proof of principle for the ability of a S/MAR vector to mediate the therapeutic effects of FLCN in BHD as well as proof of a novel method to genetically correct cancer cells using an episomally maintained nonviral vector. The S/MAR system is able to mediate similar results to viral systems with the added advantage of being set up readily with significant impact on signaling pathways. Such high levels of FLCN restoration seen here may not be necessary to restore normal biochemistry in BHD but the ability of the S/MAR system to restore such levels may be advantageous in other syndromes. Other work will include the generation of a stable UOK257 cell line expressing the full genomic locus of FLCN conferred by a S/MAR vector and controlled by native promoters of this gene, enabling its expression at normal physiological levels with correct alternative splicing and promoter usage mechanisms. This will provide an ideal cell line for further BHD investigations. Further development of the S/MAR vector for therapeutic use in BHD will involve applying newly generated S/MAR vectors to animal models of BHD in order to investigate the efficacy of the S/MAR vector to rescue the affected phenotype in vivo.
Materials and methods
DNA vectors. The FLCN cDNA (kindly provided by Dr Laura Schmidt, National Institute of Health, Bethesda, MD) was PCR amplified with FLCN forward (5′ATGAATGCCATCGTGGCTCTCTGCC 3′) and reverse (5′ TCAGTTCCGAGACTCCGAGGCTGTG 3′) primers. The FLCN PCR product was inserted into the SmaI site in the multiple cloning site of pIRES2-GFP (Clontech, CA) by blunt-end ligation to generate a vector named pFLCN-GFP. To construct pUbC-FLCN-SMAR, the FLCN cDNA was excised from pFLCN-GFP with NheI/BamHI restriction digest, blunt-ended and inserted into the SmaI site into pUbC-MCS-SMAR. To construct pUbC-FLCN-LUC-SMAR, the FLCN-IRES sequence was excised from pFLCN-GFP with NheI/NcoI digestion and inserted blunt into pUbC-Luc-SMAR (kindly provided by Dr Carsten Rudolph, University of Munich, Germany) which had been previously linearized with AflII between the promoter and the luciferase gene. The newly generated plasmids were verified by sequencing with UbC Fwd (5′ ATGCCATCGTGGCTCT 3′) primers. Plasmids used in this study were amplified in Escherichia coli DH5α using Purelink HiPure Plasmid Filter Maxiprep Kit (Invitrogen, UK).
Establishment of stable cell lines. UOK257 cells (kindly provided by Dr Laura Schmidt, National Institute of Health) were cultured at 37°C/5% CO2 in DMEM (Invitrogen) supplemented with 1 mmol/l sodium pyruvate, 10% fetal calf serum, and 1% penicillin/streptomycin. For generation of stably transfected cells, UOK257 cells were transfected with X-tremeGENE HP DNA Transfection Reagent (Roche Diagnostics, UK) at a 4:1 ratio of μl reagent: μg DNA according to manufacturer's instructions. Transfected cells were grown under selection with 400 μg/ml of G418 (Sigma, Poole, UK) for 3–4 weeks. Single colonies were isolated and expanded in normal medium.
Western analysis. UOK257 cells and tumor tissue was lysed in Tris-HCL buffer (10 mmol/l Tris pH 7.4, 2% SDS) containing protease inhibitors (Roche Diagnostics, UK). For SDS page electrophoresis, 3–5 μg of protein was denatured and separated on Mini Protean TGX 4–20% gels (Bio-Rad, UK) before blotting onto PVDF membranes (Milipore, UK). The membranes were blocked in 5% nonfat milk in PBS followed by overnight incubations with primary antibodies at 4°C. The following antibodies were used in this study: anti-FLCN, anti-phospho-mTOR, anti-Raptor, anti-mTOR, anti-phospho-p70 S6 Kinase (Thr389), anti-phospho-4E-BP1 (Thr37/46), anti-phospho-S6 Ribosomal Protein (Ser235/236), anti-SMAD3, anti-GAPDH (all from Cell Signaling Technology, UK) and anti-phospho-SMAD2/3 (Santa Cruz Biotechnology, Dallas, TX). The blots were then washed and incubated with HRP-conjugated secondary antibodies before visualization with ECL (GE Healthcare, UK).
Growth proliferation assay. To measure cell growth, 100 cells were seeded onto each well on 96-well black-walled tissue culture plates (Corning) with medium refreshed every three days. Cell numbers were assayed in triplicate using CyQuant Direct Cell Proliferation Assay NF (Invitrogen) at days 0, 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, and 20. Quantification of cell numbers was performed using ImageQuant TL software (GE Healthcare).
Colony formation assay. Cells (2 × 103) were suspended in 1 ml of 0.3% agar in DMEM containing 10 mmol/l sodium pyruvate, 10% fetal calf serum, and 1% penicillin/streptomycin. The cells were overlaid on 2 ml of 0.6% agar in the same medium on 30 mm plates and incubated for 4 weeks at 37°C/5% CO2. Colonies were analyzed using CyQuant Direct Cell Proliferation Assay (Invitrogen) and counted using ImageQuant TL software (GE Healthcare) with the following settings: Parameter sensitivity 7500/Operator size 99/Noise factor 3/Background 1.
3D culture. Cells (5 × 104) were suspended in 96-well Lipidure-coated plates overnight in triplicate. The resulting cell formation was visualized using Colourview Soft Imaging System on an Olympus CKX41 microscope.
Histology and immunohistochemistry. Tumor tissue was fixed in 4% paraformaldehyde and paraffin-embedded before sectioning (4 μm in thickness). For observing tissue morphology, sections were rehydrated through a series of decreasing concentrations of ethanol prior to staining with hemotoxylin and eosin. For immunohistochemical staining of tissue sections, endogenous peroxidase activity was blocked by incubation of sections in 3% hydrogen peroxide and rehydrated through decreasing concentrations of ethanol. Sections were then heated in 10 mmol/l sodium citrate buffer and treated with avidin and biotin (Vector Laboratories, CA). The sections were incubated with antiluciferase antibody (Santa Cruz Biotechnology) overnight. Following this, the sections were incubated with biotin-conjugated secondary antibodies followed by signal amplification with Vectastain ABC Complex (Vector Laboratories) according to the manufacturer's instructions. Color development was performed using DAB substrate (Vector Labs). Sections were counterstained with hemotoxylin and dehydrated with increasing concentrations of ethanol before being mounted onto coverslips. The resulting slides were visualized using Leica DM4000 B microscope with a Leica DFC420 camera on an inverted microscope. Image acquisition was performed using Leica LAS software (Lite version).
ELISA. Cells (1 × 105) were cultured on six well plates for 3 days and culture medium was collected for analysis. Serum TGF-β2 levels in the media was quantified by Human TGF-β2 DuoSet (R&D systems, Minneapolis, MN), following the manufacturer's protocol. Data were analyzed using the Student's t-test.
Quantitative real-time reverse transcription-PCR. Total RNA was isolated from UOK257 cells and tumor tissue using the RNAeasy mini kit with additional DNase treatment (Qiagen, Crawley, UK) following manufacturer's instructions. Levels of mRNA expression was calculated by real-time PCR using DyNAmo SYBR Green 2-Step qRT-PCR (Fisher Scientific, UK) on an Applied Biosystems 7500 Fast Real-Time PCR System, with 40 cycles per sample. Identical reactions were performed without reverse transcription as negative controls. Cycling temperatures are as follows: denaturing 95°C, annealing and extension 60°C. Primers specific for GAPDH gene were used to enable normalization between the samples through calculation of the number of cells used as input. The following primers were used: Luciferase Fwd 5′ GGCGCGTTATTTATCGGAGTT 3′ Rev 5′-CCATACTGTTGAGCAATTCACGTT-3′ GAPDH Fwd 5′-ACCACAGTCCATGCCATCAC-3′ Rev: 5′-TCCACCCTGTTGCTGTA 3′, human FLCN, SMAD7, SMAD3, and TGFβ2 primers used were as previously published.11 All samples were tested in triplicate.
Relative amounts of plasmid DNA in UOK257-FS and UOK257-FSLuc were calculated by real-time PCR as described above, with FLCN cDNA used to determine amounts of S/MAR plasmid. Serial dilutions of plasmids were used to produce a standard amplification curve for quantification and all samples were tested in triplicate.
Xenograft models. Animal studies were carried out in accordance with Home Office guidelines under a UK Home Office License (PPL 70/7305). NOD-SCID (Non-Obese Diabetic/Severe Combined Immuno-Deficient) mice (Harlan, UK) were injected intraperitoneally with 3 × 106 cells suspended in 150 μl of PBS under sterile conditions. The mice were imaged twice weekly for the first 2 months and once weekly thereafter for bioluminescence using the IVIS Image Spectrum as described below.
Bioluminescent imaging. For bioimaging of cells, D-luciferin (150 μg/ml) (Gold Biotechnology, St Louis, MO) was diluted in DMEM and added to cells for 10 minutes prior to imaging using IVIS Spectrum Imager (Xenogen, Alameda, CA). The background level of bioluminescence on untreated cells is 5 × 104 photons/seconds/cm2/seradian (sr). For bioimaging of xenografts in vivo, mice were injected intraperitoneally with 300 μl D-luciferin (15 mg/ml) 10 minutes before imaging under anesthesia in a light-tight chamber. The background level of bioluminescence in PBS-treated animals is 5 × 105 photons/seconds/cm2/sr. Analysis was performed using the Living Image 2.50 software.
Plasmid rescue experiments. Stbl3 E. coli cells (Invitrogen) were transformed by heat-shock using 20 μg DNA prepared by Genomic DNA Isolation Kit and Genomic DNA Clean and Concentrator kit (Zymo Research, CA) according to the manufacturer's instructions. Transformed colonies were selected on agar plates containing 30 μg/ml kanamycin. Plasmid DNA was isolated from individual colonies and analyzed with HpaI and PvuII restriction digestion (New England Biolabs, UK) and by sequencing with with UbC Fwd (5′ ATGCCATCGTGGCTCT 3′); Luciferase Fwd (5′-GGCGCGTTATTTATCGGAGTT- 3′); and Backbone Fwd (5′AGCGCACGAGGGAGCT3′) primers (Eurofins MWG Operon, UK).
SUPPLEMENTARY MATERIAL Figure S1. Generation of UOK257-Luc cells using pUbC-Luc-SMAR. Figure S2. Analysis of UOK257-FSLuc. Figure S3. Quantitative RT-PCR of FLCN, SMAD3, SMAD7 and TGFβ2 expression levels in UOK257-FSLuc, UOK257-FS and UOK257 cells. Figure S4. Episomal maintenance of S/MAR plasmids in xenografts.
Acknowledgments
We thank Dr Laura Schmidt (National Institute of Health, Besthesda, USA) for kindly providing the FLCN cDNA and UOK257 cells, and Dr Carsten Rudolph (University of Munich, Germany) for kindly providing the pUbC-Luc-SMAR vector. We also thank Lorraine Lawrence for tissue sectioning, Dhani Tracey-White for her assistance with animal care, John-Poul Ng-Blichfelt for constructing the cloning vector pFLCN-GFP and Dr Stephen Land of Dundee University for his insightful contribution to the preparation of this manuscript. This work was supported by the Myrovlitis Trust.
Supplementary Material
Generation of UOK257-Luc cells using pUbC-Luc-SMAR.
Analysis of UOK257-FSLuc.
Quantitative RT-PCR of FLCN, SMAD3, SMAD7 and TGFβ2 expression levels in UOK257-FSLuc, UOK257-FS and UOK257 cells.
Episomal maintenance of S/MAR plasmids in xenografts.
References
- Pannell D, Ellis J. Silencing of gene expression: implications for design of retrovirus vectors. Rev Med Virol. 2001;11:205–217. doi: 10.1002/rmv.316. [DOI] [PubMed] [Google Scholar]
- Nagel J, Gross B, Meggendorfer M, Preiss C, Grez M, Brack-Werner R, et al. Stably integrated and expressed retroviral sequences can influence nuclear location and chromatin condensation of the integration locus. Chromosoma. 2012;121:353–367. doi: 10.1007/s00412-012-0366-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyros O, Wong SP, Gowers K, Harbottle RP. Genetic modification of cancer cells using non-viral, episomal S/MAR vectors for in vivo tumour modelling. PLoS ONE. 2012;7:e47920. doi: 10.1371/journal.pone.0047920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyros O, Wong SP, Fedonidis C, Tolmachov O, Waddington SN, Howe SJ, et al. Development of S/MAR minicircles for enhanced and persistent transgene expression in the mouse liver. J Mol Med. 2011;89:515–529. doi: 10.1007/s00109-010-0713-3. [DOI] [PubMed] [Google Scholar]
- Jenke AC, Stehle IM, Herrmann F, Eisenberger T, Baiker A, Bode J, et al. Nuclear scaffold/matrix attached region modules linked to a transcription unit are sufficient for replication and maintenance of a mammalian episome. Proc Natl Acad Sci USA. 2004;101:11322–11327. doi: 10.1073/pnas.0401355101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt LS, Nickerson ML, Warren MB, Glenn GM, Toro JR, Merino MJ, et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet. 2005;76:1023–1033. doi: 10.1086/430842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157–164. doi: 10.1016/s1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]
- Yang Y, Padilla-Nash HM, Vira MA, Abu-Asab MS, Val D, Worrell R, et al. The UOK 257 cell line: a novel model for studies of the human Birt-Hogg-Dubé gene pathway. Cancer Genet Cytogenet. 2008;180:100–109. doi: 10.1016/j.cancergencyto.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson F, Gatalica Z, Grossmann P, Perez Montiel MD, Alvarado Cabrero I, Bulimbasic S, et al. Sporadic hybrid oncocytic/chromophobe tumor of the kidney: a clinicopathologic, histomorphologic, immunohistochemical, ultrastructural, and molecular cytogenetic study of 14 cases. Virchows Arch. 2010;456:355–365. doi: 10.1007/s00428-010-0898-4. [DOI] [PubMed] [Google Scholar]
- Hong SB, Oh H, Valera VA, Baba M, Schmidt LS, Linehan WM. Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLoS ONE. 2010;5:e15793. doi: 10.1371/journal.pone.0015793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SB, Oh H, Valera VA, Stull J, Ngo DT, Baba M, et al. Tumor suppressor FLCN inhibits tumorigenesis of a FLCN-null renal cancer cell line and regulates expression of key molecules in TGF-β signaling. Mol Cancer. 2010;9:160. doi: 10.1186/1476-4598-9-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci USA. 2006;103:15552–15557. doi: 10.1073/pnas.0603781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasumi H, Baba M, Hong SB, Hasumi Y, Huang Y, Yao M, et al. Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene. 2008;415:60–67. doi: 10.1016/j.gene.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman TR, Nicolas E, Klein-Szanto A, Al-Saleem T, Cash TP, Simon MC, et al. The role of the Birt-Hogg-Dubé protein in mTOR activation and renal tumorigenesis. Oncogene. 2009;28:1594–1604. doi: 10.1038/onc.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudon V, Sabourin S, Dydensborg AB, Kottis V, Ghazi A, Paquet M, et al. Renal tumour suppressor function of the Birt-Hogg-Dubé syndrome gene product folliculin. J Med Genet. 2010;47:182–189. doi: 10.1136/jmg.2009.072009. [DOI] [PubMed] [Google Scholar]
- Cash TP, Gruber JJ, Hartman TR, Henske EP, Simon MC. Loss of the Birt-Hogg-Dubé tumor suppressor results in apoptotic resistance due to aberrant TGFß-mediated transcription. Oncogene. 2011;30:2534–2546. doi: 10.1038/onc.2010.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvetz DA, Khabibullin D, Hariharan V, Ongusaha PP, Goncharova EA, Schlechter T, et al. Folliculin, the product of the Birt-Hogg-Dube tumor suppressor gene, interacts with the adherens junction protein p0071 to regulate cell-cell adhesion. PLoS ONE. 2012;7:e47842. doi: 10.1371/journal.pone.0047842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbete JL, Buceta M, Mermod N. MAR elements regulate the probability of epigenetic switching between active and inactive gene expression. Mol Biosyst. 2009;5:143–150. doi: 10.1039/b813657b. [DOI] [PubMed] [Google Scholar]
- Heikkinen PT, Nummela M, Jokilehto T, Grenman R, Kähäri VM, Jaakkola PM. Hypoxic conversion of SMAD7 function from an inhibitor into a promoter of cell invasion. Cancer Res. 2010;70:5984–5993. doi: 10.1158/0008-5472.CAN-09-3777. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- Nathan CO, Amirghahari N, Abreo F, Rong X, Caldito G, Jones ML, et al. Overexpressed eIF4E is functionally active in surgical margins of head and neck cancer patients via activation of the Akt/mammalian target of rapamycin pathway (2004). Clin Cancer Res. 10:5820–5827. doi: 10.1158/1078-0432.CCR-03-0483. [DOI] [PubMed] [Google Scholar]
- Preston RS, Philp A, Claessens T, Gijezen L, Dydensborg AB, Dunlop EA, et al. Absence of the Birt-Hogg-Dubé gene product is associated with increased hypoxia-inducible factor transcriptional activity and a loss of metabolic flexibility. Oncogene. 2011;30:1159–1173. doi: 10.1038/onc.2010.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinci M, Gowan S, Boxall F, Patterson L, Zimmermann M, Court W, et al. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012;10:29. doi: 10.1186/1741-7007-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenke BH, Fetzer CP, Stehle IM, Jönsson F, Fackelmayer FO, Conradt H, et al. An episomally replicating vector binds to the nuclear matrix protein SAF-A in vivo. EMBO Rep. 2002;3:349–354. doi: 10.1093/embo-reports/kvf070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyros O, Wong SP, Niceta M, Waddington SN, Howe SJ, Coutelle C, et al. Persistent episomal transgene expression in liver following delivery of a scaffold/matrix attachment region containing non-viral vector. Gene Ther. 2008;15:1593–1605. doi: 10.1038/gt.2008.113. [DOI] [PubMed] [Google Scholar]
- Wong SP, Argyros O, Coutelle C, Harbottle RP. Non-viral S/MAR vectors replicate episomally in vivo when provided with a selective advantage. Gene Ther. 2011;18:82–87. doi: 10.1038/gt.2010.116. [DOI] [PubMed] [Google Scholar]
- Lufino MM, Manservigi R, Wade-Martins R. An S/MAR-based infectious episomal genomic DNA expression vector provides long-term regulated functional complementation of LDLR deficiency. Nucleic Acids Res. 2007;35:e98. doi: 10.1093/nar/gkm570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenke AC, Eisenberger T, Baiker A, Stehle IM, Wirth S, Lipps HJ. The nonviral episomal replicating vector pEPI-1 allows long-term inhibition of bcr-abl expression by shRNA. Hum Gene Ther. 2005;16:533–539. doi: 10.1089/hum.2005.16.533. [DOI] [PubMed] [Google Scholar]
- Jenke AC, Scinteie MF, Stehle IM, Lipps HJ. Expression of a transgene encoded on a non-viral episomal vector is not subject to epigenetic silencing by cytosine methylation. Mol Biol Rep. 2004;31:85–90. doi: 10.1023/b:mole.0000031363.35839.46. [DOI] [PubMed] [Google Scholar]
- Argyros O, Wong SP, Harbottle RP. Non-viral episomal modification of cells using S/MAR elements. Expert Opin Biol Ther. 2011;11:1177–1191. doi: 10.1517/14712598.2011.582035. [DOI] [PubMed] [Google Scholar]
- Han SU, Kim HT, Seong DH, Kim YS, Park YS, Bang YJ, et al. Loss of the Smad3 expression increases susceptibility to tumorigenicity in human gastric cancer. Oncogene. 2004;23:1333–1341. doi: 10.1038/sj.onc.1207259. [DOI] [PubMed] [Google Scholar]
- Takagi Y, Kohmura H, Futamura M, Kida H, Tanemura H, Shimokawa K, et al. Somatic alterations of the DPC4 gene in human colorectal cancers in vivo. Gastroenterology. 1996;111:1369–1372. doi: 10.1053/gast.1996.v111.pm8898652. [DOI] [PubMed] [Google Scholar]
- Grünert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4:657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- Do TV, Kubba LA, Du H, Sturgis CD, Woodruff TK. Transforming growth factor-β1, transforming growth factor-β2, and transforming growth factor-β3 enhance ovarian cancer metastatic potential by inducing a Smad3-dependent epithelial-to-mesenchymal transition (2008). Mol Cancer Res. 6:695–705. doi: 10.1158/1541-7786.MCR-07-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javelaud D, Mohammad KS, McKenna CR, Fournier P, Luciani F, Niewolna M, et al. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 2007;67:2317–2324. doi: 10.1158/0008-5472.CAN-06-3950. [DOI] [PubMed] [Google Scholar]
- Higgins JP, Shinghal R, Gill H, Reese JH, Terris M, Cohen RJ, et al. Gene expression patterns in renal cell carcinoma assessed by complementary DNA microarray. Am J Pathol. 2003;162:925–932. doi: 10.1016/S0002-9440(10)63887-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviolette L, Wilson J, Koller J, Zimmer M, Iliopoulos O. Abstract 2171: Human FLCN delays cell cycle progression through late S and G2/M-phases: Effect of phosphorylation and tumor-associated mutations. Can Res. 2012;72 doi: 10.1371/journal.pone.0066775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Keller JR, Sun HW, Resch W, Kuchen S, Suh HC, et al. The folliculin-FNIP1 pathway deleted in human Birt-Hogg-Dubé syndrome is required for murine B-cell development. Blood. 2012;120:1254–1261. doi: 10.1182/blood-2012-02-410407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Generation of UOK257-Luc cells using pUbC-Luc-SMAR.
Analysis of UOK257-FSLuc.
Quantitative RT-PCR of FLCN, SMAD3, SMAD7 and TGFβ2 expression levels in UOK257-FSLuc, UOK257-FS and UOK257 cells.
Episomal maintenance of S/MAR plasmids in xenografts.