

Synopsis
Kidney function declines with advancing age and mitochondria have been implicated. We have examined the integrated function of mitochondria isolated from kidneys of 6 and 24 month Fischer 344 rats. Oxidative phosphorylation (OXPHOS) of intact mitochondria and cytochrome c oxidase activity in permeabilized mitochondria were determined with polarographic assays. The activities of the electron transport chain (ETC) complexes and the cytochrome content in solubilized mitochondria were measured using spectrophotometric methods. The respiratory complexes were evaluated with blue-native gel electrophoresis. Mitochondrial preparations were evaluated by immunoblotting for cytochrome c, Smac/Diablo, and the voltage dependant ion channel (VDAC). Mitochondrial morphology was examined by electron microscopy. OXPHOS of mitochondria isolated from 24 month animals was decreased 15–25% with complex I, II, III, IV and fatty acid substrates. The electron microscopic appearance of mitochondria, the activity of the ETC complexes and the protein abundance of individual complexes and supercomplexes were unchanged. The content of cytochrome c was decreased by 37% in aged mitochondria as determined by spectrophotometric methods and confirmed with immunoblotting. Polarographic determination of cytochrome c oxidase activity with endogenous cytochrome c demonstrated a 23% reduction in aged mitochondria, which was corrected with the addition of exogenous cytochrome c. Renal mitochondrial OXPHOS decreased with aging in the Fischer 344 rat. Decreased mitochondrial cytochrome c content is a major factor contributing to the OXPHOS defect of mitochondria isolated from kidneys of elderly animals.
Keywords: Aging, mitochondria, oxidative phosphorylation, electron transport chain, cytochrome c, cytochrome c oxidase
Introduction
The age-related decline in kidney function in man has been recognized for many decades [1,2] and the prevalence of reduced kidney function in individuals >64 years of age is between 23.4 and 35.8% [3]. The incidence of acute kidney injury also is increased in older individuals for a variety of reasons [4]. The kidney has high energetic demands largely related to its active solute transport functions, accounting for 10% of whole body oxygen consumption, and as a whole the kidney utilizes mitochondrial oxidative phosphorylation to generate about 95% of its ATP [5]. Small amounts of reactive oxygen species (ROS) are produced in the mitochondria as a by-product of electron flux through the respiratory chain. The mitochondrial theory of aging states that mitochondrial ROS production leads to stochastic oxidative damage over time with the accumulation of mitochondrial DNA (mtDNA) mutations and decrements in mitochondrial function leading to the phenotypic changes of aging [6]. These features place the mitochondria in a central role for many of the mechanisms implicated in the progression of the aging process [6,7].
Rat models have provided valuable insights to the study of aging. Age-related decrements in mitochondrial oxidative phosphorylation (OXPHOS) and the electron transport chain (ETC) have been well described in several organs and tissues of the rat, but relatively little attention has been given to the kidney. One study used immunohistochemical methods to detect the age-related appearance of ETC-deficient renal tubular epithelial cells in the rat kidney [8]. The authors demonstrated that these effects were due to mtDNA deletions in the ETC-deficient cells, presumably generated via ROS mediated damage. There is evidence to support an age-related increase in ROS production within the rat kidney and a decline in the enzymes of the antioxidant systems [9,10]. However, to date there has been no detailed biochemical examination of the effects of aging on renal mitochondrial OXPHOS and ETC.
Therefore, we have undertaken these studies to examine isolated renal mitochondria for age-related decrements in function as have been described in other organs and tissues. Here we utilize spectrophotometric assays to evaluate the enzyme activities of the individual components of the electron transport chain and mitochondrial abundance of cytochromes. Blue-native gel electrophoresis is used to determine the relative abundance of the individual respiratory complexes as well as supercomplexes in mitochondria isolated from 6 and 24 month animals. Further, polarographic assays of oxygen consumption using freshly isolated renal mitochondria permits the evaluation of efficiency of ATP production in intact mitochondria. These assays depend upon the function of substrate transporters, dehydrogenases and respiratory complexes, while providing measures of the integrity of the isolated mitochondria via the respiratory control ratio (RCR) and coupling to the phosphorylation apparatus via ADP/O ratios. The use of a panel of metabolic substrates in conjunction with inhibitors and uncouplers of the respiratory chain permit the biochemical localization of the site of defects in mitochondrial oxidative phosphorylation.
Experimental Materials and Methods
Reagents
All chemicals were purchased from Sigma-Aldrich except where otherwise noted and were of the highest purity available.
Animals
Male Fischer 344 rats were obtained from National Institute of Aging colony (Harlan Laboratories Inc.) under an approved IACUC protocol (2006-0080). Rats were housed in the animal facility at the VA Medical Center and the investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The rats were fed ad libitum a 70717 NIH-31 Open Formula Rat diet (18% protein, 4% fat. 5% fiber, 8% ash) and had free access to water. Animals were obtained from the vendor at the age required for the study and were used after allowing a seven day adjustment period.
Mitochondrial Isolation
Rats were weighed and then killed at 6 or 24 months by decapitation according to the protocol. Mitochondrial isolation was adapted from previously reported methods [11] and is briefly summarized as follows. Kidneys were extirpated and rinsed in ice-cold MSM (220 mM D-mannitol, 70 mM sucrose, 5 mM MOPS, pH=7.4). Kidneys were decapsulated, rinsed in MSM and weighed. Kidneys were minced, rinsed twice in MSM and suspended in 10ml of MSM+ (MSM, 2mM EDTA, 0.2% defatted BSA) per gm wet-weight. The suspension was homogenized with three strokes of a loose fitting pestle in a Potter-Elvehjem homogenizer at ~1,000rpm. The homogenate was centrifuged at 1,600 rpm in a Sorvall SS-34 rotor (300 × g) at 4°C and supernatant was decanted through gauze. The low-speed pellet was resuspended in MSM+, homogenized and centrifuged, as above. The supernatant was decanted through gauze and combined with the first supernatant. Combined supernatants were centrifuged at 7,650 rpm using the SS-34 rotor (7,000 × g) at 4°C. Supernatant was decanted and the pellet was resuspended in MSM and centrifuged at 7,000 × g two more times. The final mitochondrial pellet was resuspended in 0.2 ml MSM/gm wet weight of starting material. Protein concentration was determined using biuret reagent with bovine serum albumin as a standard. Mitochondria were maintained on ice till used in subsequent assays on preparations done that day or stored at −80°C for future use.
OXPHOS assays
Measurements of OXPHOS were performed as previously described [12] on freshly isolated mitochondria in a Clark-type oxygen electrode (Strathkelvin, North Lanarkshire, Scotland). Assays were performed by adding mitochondria to 0.5 ml of air-saturated respiration buffer (100 mM KCl, 5 mM KH2PO4, 1 mM EGTA, 1 mg/ml defatted BSA, 50 mM MOPS, pH=7.4) equilibrated to 30° C. Rotenone solubilized in DMSO was added in a volume of 5 μl to the respiration buffer prior to assays using the substrates, succinate, duroquinol (DHQ) and N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD), at a final concentration of 7.5 μM. Endogenous substrates were depleted by the addition of ADP to a concentration of 75 μM. Substrate or substrate combinations were then added to the chamber in volumes ranging from 5 to 25 μl per component to achieve the final concentrations as follows: glutamate [20 mM], malate [5 mM], pyruvate [10 mM], malonate [10 mM], α-ketoglutarate [10 mM], succinate [20 mM], DHQ [1 mM], TMPD [1 mM], ascorbate [10 mM], acetylcarnitine [5 mM], palmitoylcarnitine [0.04 mM], carnitine [5 mM], palmitoyl-CoA [0.02 mM]. State 3 (ADP-stimulated) and state 4 (ADP-limited) respiratory rates were recorded after the addition of ADP to [150 μM] or [75 μM] for succinate and DHQ. State 3 rates were recorded in the presence of high ADP concentration [2 mM]. Rates of uncoupled respiration were recorded after the addition of dinitrophenol to [0.2 mM] (Fisher Scientific, Fairlawn, NJ, USA). Respiratory control ratios (RCR, State 3 divided by State 4) reflects the integrity of the mitochondrial preparation and control of oxygen consumption by phosphorylation (“coupling”). The ADP/O ratios (number of ADP molecules added for each oxygen atom consumed) is an index of the efficiency of OXPHOS[13,14].
Citrate Synthase
Citrate Synthase activity was determined as described [15] using a diode array spectrophotometer at a wavelength of 412 nm.
ETC assays
Samples of fresh rat kidney mitochondria (RKM) were treated with 10 mg cholate/mg mitochondrial protein, taken to a final volume of 1 mg/ml MSM-EDTA buffer supplemented with 1 μl/ml mammalian protease inhibitor cocktail. For the assay, the samples were diluted 1:10. For COX activity, 0.1 mg fresh intact mitochondria were suspended in 100 mM KCl, 50 mM MOPS, 0.5 mM EGTA, pH=7.4 and assayed with and without dodecyl β-D-maltoside. ETC enzyme activities were measured spectrophotometrically as specific donor-acceptor oxidoreductase activities in 0.1M phosphate buffer. Both donor and acceptors span specific regions of the ETC [16–18]. Rotenone-sensitive NADH-cytochrome c reductase assesses complexes I and III. NADH coenzyme Q reductase was measured as the rotenone-sensitive oxidation of NADH with decylubiquinone as acceptor, and assesses only complex I. NADH ferricyanide reductase measures NADH dehydrogenase and mitochondrial outer membrane cytochrome b5 (rotenone-insensitive NADH-reductase). Antimycin A-sensitive succinate-cytochrome c reductase assesses complexes II and III. Complex II activity was measured as the thenoyltrifluoroacetone (TTFA)-sensitive reduction of 2,6-dicholorophenolindophenol with succinate as substrate, whereas total complex II was measured in the same manner but with duroquinone added as a source of exogenous coenzyme Q. Succinate dehydrogenase measures the first two subunits of complex II [19]. Decylubiquinol-cytochrome c oxidoreductase was measured as the antimycin A-sensitive reductase to assess complex III [20]. Cytochrome c oxidase (COX) was measured as the oxidation of reduced cytochrome c and expressed as the first order rate constant.
Electron Microscopy
Electron Microscopic analysis was performed as described in [17] with changes as noted in the following. RKM were prepared from 6 and 24 month animals as described. An aliquot of RKM was added to an equal volume of phosphate-buffered, half-strength Karnovsky’s fixative, mixed, and immediately spun down for 30 s in a microcentrifuge. The fixation continued in fresh one-quarter-strength Karnovsky’s fixative for a total of 2 hours at room temperature. The pellets were thoroughly rinsed in distilled water, then postfixed for 2 hours in an unbuffered 1:1 mixture of 2% osmium tetroxide and 3% potassium ferricyanide. After rinsing with distilled water, the specimens were soaked overnight in an acidified solution of 0.25% uranyl acetate. After another rinse in distilled water, they were dehydrated in ascending concentrations of ethanol, passed through propylene oxide, and embedded in Poly/Bed 812 embedding media (Polysciences, Warrington, PA). Thin sections (70nm) cut on a RMC MT6000-XL ultramicrotome were mounted on Gilder square 300 mesh nickel grids (Electron Microscopy Sciences, PA) and then sequentially stained with acidified methanolic uranyl acetate followed by lead. These were coated in a Denton DV-401 carbon coater (Denton Vacuum LLC, NJ), and examined in a Zeiss CEM 902 electron microscope (Oberkochen, Germany).
Measurement of cytochrome spectra
The mitochondrial content of cytochromes aa3, b, c1 and c was quantitatively determined using the difference spectra according to the method of Williams [21]. Briefly, mitochondria stored at −80 °C were thawed on ice and 0.25 mg of mitochondrial protein was solubilized in reaction buffer (2.1% sodium deoxycholate, 5 mM sodium L-ascorbate, 25 mM NaH2PO4, pH=7.0). The absorption spectra between 500 and 660 λ was recorded using an HP 8453 UV-visible ChemStation spectrophotometer with ChemStation software for each sample when oxidized and reduced. The difference spectra (reduced-oxidized) was used for the solution of simultaneous equations to quantitatively determine the content of these cytochromes in the mitochondrial samples as described by Williams [21].
Complex IV Oxidase Assay
Mitochondria stored at −80°C were thawed on ice and 0.25 mg of mitochondrial protein was diluted to a final concentration of [1 mg/ml] in KPi/EDTA buffer (20 mM KH2PO4, 0.1 mM EDTA, pH=7.4). KPi buffer without or with exogenous cytochrome c, final concentration of [32 μM], (Calbiochem, San Diego, CA, USA) was added to the chamber of a clark-type oxygen electrode (Strathkelvin, North Lanarkshire, Scotland). After equilibration of 500 μl of KPi buffer in the reaction chamber to 30°C, 25 μl of KPi treated mitochondria were added and baseline rates recorded. Substrate was added [1 mM TMPD + 10 mM ascorbate] and the substrate-stimulated rate was recorded. Sodium azide [2 mM] was added and the azide-insensitive rate was recorded. The complex IV azide-sensitive rates were determined for each assay by determining the difference between the substrate-stimulated rates and the azide-insensitive rates.
Blue Native gel electrophoresis
Blue native gel electrophoresis was performed according to previously reported methods [22] and are briefly summarized as follows. An aliquot of freshly isolated mitochondria containing 300 μg of mitochondrial protein was resuspended in buffer and solubilized with digitonin (Calbiochem, LaJolla, CA) or dodecyl maltoside by incubation for 30 minutes on ice. Unsolubilized membranes were pelleted by centrifugation for 10 min at 100,000 rpm in airfuge. Supernatant was removed and 20 μl of sample buffer was added as described [22]. Samples were divided into 20 μl aliquots and stored at −80°C for future use. Samples were loaded (75 μgms in a volume of 15 μl) onto a 3–12% precast NativePAGE Novex Bis-Tris gradient gel (Invitrogen) for analysis of digitonin solubilized samples or a 4–16% precast NativePAGE Novex Bis-Tris gradient gel (Invitrogen) for analysis of dodecyl maltoside solubilized samples. Samples were separated with electrophoresis at a voltage of 75 V for a total of 1300 volt-hours. Gels were destained and imaged with GeneGenius Bioimaging System (Syngene, Fredrick, MD, USA). Gel images were processed with image J software for background subtraction using a rolling ball algorithm based upon previous methods [23] with a rolling ball radius of 50 pixels. Background subtracted images were imported into MutliGauge v2.0 software (Fujifilm, Tokyo, Japan) for calculation of band density by calculation of the area under the curve for each of the respective respiratory complex bands.
Immunoblotting
Mitochondrial samples, previously stored at −80 °C, were thawed on ice and solubilized in SDS -PAGE loading buffer and boiled for 5 minutes. Samples were loaded onto a 12.5 % acrylamide gel (50 μgm of mitochondrial protein/lane) and separated with SDS-PAGE. Separated proteins were transferred to PVDF membrane and allowed to air dry before blocking for 1 hr at 21 °C in TBS, pH=7.4 with 5% non-fat dry milk. Membranes were then incubated for 2 hr at 21 °C in blotting buffer (1% non-fat dry milk, 0.1% Tween-20, TBS, pH=7.4) with primary antibodies against cytochrome c at 1:500 (Epitomics Inc., Burlingame, CA, USA), Smac/Diablo at 1:1000 (Epitomics Inc., Burlingame, CA, USA), and VDAC at 1:1000 (Mitosciences, Eugene, OR, USA). Membranes were washed three times for 10 min. at 21 °C in blotting buffer, incubated with HRP-conjugated secondary antibodies at 1:5,000 in blotting buffer for 1 hr at 21 °C, followed by three washes in blotting buffer and one wash in TBS. A chemiluminescent detection kit (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) was used for HRP detection and membranes were exposed to film for times appropriate to obtain non-saturating exposures and developed. Film was scanned as TIFF file and band density was determined using image J software available from the National Institutes of Health.
Statistics
Results are reported as mean ± standard deviation. Comparisons between groups are made by two-tailed t-test and reported as two-tailed p-value. Statistical significance is considered to be p-value of <0.05.
Results
Comparisons were made between the 6 and 24 month animals for body weight, kidney weight, mitochondrial yield, and the activity of the mitochondrial marker enzymes, citrate synthase and succinate dehydrogenase in isolated mitochondria (Table 1). There was no significant difference in body weight between the two groups. The kidney size, as expressed in grams wet weight, was significantly greater in the 24 month animals. The mitochondrial yield, as expressed in mg of mitochondrial protein per gram wet weight of kidney tissue, was significantly lower in the 24 month animals. The efficiency of mitochondrial recovery was 22% ± 4 vs. 23% ± 8 in the 6 and 24 month groups respectively. Recovery was determined by the percentage of citrate synthase activity recovered in the final mitochondrial isolate obtained per gram of kidney weight to the total citrate synthase activity in the initial homogenate per gram of kidney weight. The decreased mitochondrial yield per gram of kidney in the context of equivalent recovery efficiency suggest that the lower yield is a reflection of decreased mitochondrial protein content per gram of kidney tissue in the 24 month animals. The relative equivalence of the mitochondrial preparations, with respect to mitochondrial purity, was assessed by the activities of the mitochondrial marker enzymes, citrate synthase and succinate dehydrogenase. There were no significant differences in the mitochondrial preparations obtained from the 6 and 24 month animals for either marker enzyme (Table 1). Citrate synthase activity per mg wet weight of kidney and succinate dehydrogenase activity per mg wet weight of kidney were lower in the older animals, but this trend did not reach statistical significance.
Table 1.
Body and kidney weight, mitochondrial yield and mitochondrial marker enzymes in 6 and 24 month animals
| 6 Month (n=8) | 24 Month (n=8) | p-value | |
|---|---|---|---|
| Body Weight (grams) | 381.3±6.4 | 386.3±28.8 | 0.6386 |
| Kidney weight (grams) | 2.2±0.2 | 2.7±0.6 | 0.0340 |
| Citrate Synthase (nmol/min/mg wet wt) | 23.8±3.2 | 20.2±5.6 | 0.1895 |
| Succinate Dehydrogenase (nmol/min/mg wet wt) | 8.5±1.2 | 6.8±2.3 | 0.1317 |
| Mitochondrial yield (mg/gm wet wt) | 16.8±1.0 | 12.3±2.3 | 0.0002 |
| Citrate Synthase (nmol/min/mg mito protein) | 301.4±51.0 | 354.7±78.8 | 0.1306 |
| Succinate Dehydrogenase (nmol/min/mg mito protein) | 139.8±38.1 | 120.8±26.6 | 0.2668 |
OXPHOS was evaluated in freshly isolated, intact mitochondria. The state 3 and state 4 respiratory rates, the respiratory control ratio (RCR), the rate of respiration obtained in the presence of high ADP [2 mM], the uncoupled rate of respiration obtained in the presence of dinitrophenol (DNP) and the ADP/O ratio were recorded, as shown for the substrate combination of glutamate and malate (Table 2). Using the substrate combination of glutamate and malate, the state 3, state 4, and high [ADP] respiratory rates were decreased in the 24 month animals by 13%, 21% and 13%, respectively (Table 2). The uncoupled respiratory rate, the RCR, a metric of mitochondrial integrity and coupling, and the ADP/O ratio, a reflection the control and efficiency of the phosphorylation apparatus, were not significantly different between the two groups (Table 2).
Table 2.
Polarographic assay of 6 and 24 month rat kidney mitochondria using the substrate combination glutamate and malate
| Substrate: Glutamate + Malate | 6 Month N=8 | 24 Month N=8 | P-value |
|---|---|---|---|
| State 3 (nmol O/min/mg) | 159.9±14.5 | 139.6±15.3 | 0.0165 |
| State 4 (nmol O/min/mg) | 28.2±2.8 | 22.4±3.1 | 0.0015 |
| RCR | 5.7±0.6 | 6.3±0.7 | 0.0870 |
| High ADP (2 mM) (nmol O/min/mg) | 215.0±17.9 | 187.5±24.2 | 0.0216 |
| Uncoupled Rate (nmol O/min/mg) | 213.5±27.3 | 190.8±24.2 | 0.1448 |
| ADP/O Ratio | 2.84±0.23 | 2.76±0.18 | 0.4514 |
A panel of substrates, including complex I, II, III, IV, and fatty acid substrates, were used to evaluate the rates of OXPHOS in intact mitochondrial preparations isolated from the kidneys of 6 and 24 month animals. The rates of state 3 respiration were slower in 24 month animals for the substrates, glutamate plus malate, α-ketoglutarate plus malonate, DHQ, and the fatty acid substrates (Figure 1). The rate of state 3 respiration in the presence of high ADP [2 mM] was significantly slower in mitochondria obtained from 24 month animals for all substrates tested except for two of the complex I substrates, glutamate and the combination of pyruvate and malate (Figure 1).
Figure 1. Oxidative phosphorylation rates in 6 and 24 month kidney mitochondria.

Substrates for polarographic assays of OXPHOS through complex I (glutamate), complex II (succinate), complex III (DHQ), and complex IV (TMPD) are shown (A). ADP-stimulated (2 mM) rates of state 3 respiration were determined in freshly isolated mitochondria from 6 month (light bars, n=8) and 24 month (dark bars, n=8). Uncoupled rates determined after the addition of DNP are shown for substrates: glutamate, succinate, and DHQ. ADP-stimulated (2 mM) rates of state 3 respiration are shown for the substrate combinations: pyruvate and malate (P+M), α-ketoglutarate (αKG) and malonate, acetyl-carnitine and malate (AC+M), palmitoyl-carnitine and malate (PC+M), and palmitoyl-CoA with malate (PCoA+M) and carnitine (B). Bars represent the mean value of each experimental group and the error bars indicate the standard deviation from the mean. P-values <0.05 are denoted by an asterisk (*) and p-values <0.01 are denoted by a cross (†) above the indicated pair and were calculated from non-paired two-tailed student t-test.
In order to determine if the defects observed in OXPHOS were attributable to intrinsic deficits of the respiratory complexes, the enzyme activities of the ETC were assayed in freshly isolated mitochondria. We did not detect a significant difference in the activities of any of the components of the ETC (Supplementary Figure 1). Consistent with this observation, the protein abundance of the individual ETC complexes was similar between the two groups (Supplementary Figure 2A, C, E, G). In order to determine if the abundance of the ETC supercomplexes differed between the two groups, mitochondria were solubilized with digitonin and separated using blue native gel electrophoresis (Supplementary Figure 2B). There were no significant differences in the protein abundance of supercomplexes: S0, S1 or S2, between the two groups (Supplementary Figure 2D, F, H).
The morphology of mitochondria isolated from a 6 month and 24 month animal was evaluated by electron microscopy (Figure 2). Isolated mitochondria are predominantly of a proximal and distal tubule origin. There were no obvious morphologic differences between the 6 and 24 month mitochondrial preparations.
Figure 2. Transmission electron micrographs of mitochondria isolated from kidneys of 6 and 24 month Fischer 344 rats.

Mitochondria were isolated from 6 month (A) and 24 month (B) animals using differential centrifugation and fixed for transmission electron microscopy, shown at 18,600 x magnification. Scale bars represent 1.0 μm.
The content of cytochromes aa3, b, c1 and c were assayed using a spectrophotometric method to determine if differences in the cytochromes could be contributing to the decrement in OXPHOS capacity seen in the mitochondria of 24 month animals. We found a 23% and 37% decrease in the abundance of cytochrome b and cytochrome c, respectively, in mitochondria isolated from the kidneys of 24 month animals (Figure 3A). The abundance of cytochrome aa3 and cytochrome c1 was not significantly different between the two groups (Figure 3A). Immunoblotting confirmed a decrease in cytochrome c in the mitochondria isolated from 24 month animals relative to 6 month animals (Figure 3C). The abundance of cytochrome c was decreased by 31% by band densitometry in mitochondria isolated from the 24 month animals compared to 6 month controls (Figure 3B). The decrease in cytochrome c was not related to differences in mitochondrial loading or loss of integrity of the outer mitochondrial membrane as the abundance of VDAC, a marker of the outer mitochondrial membrane, and Smac/Diablo, a soluble protein in the intermembranous space, was unchanged between the two groups (Supplementary Figure 3).
Figure 3. Determination of cytochrome content in 6 and 24 month RKM.

RKM from 6 and 24 month animals (n=7 for each group) were solubilized with deoxycholate and the cytochrome content was determined spectrophotometrically using the difference spectra (A). Bars represent the mean value of each experimental group and the error bars indicate the standard deviation. Cytochrome c was detected using a monoclonal antibody on immunoblots of RKM isolated from 6 and 24 month animals (n=4 for each group) separated with SDS-PAGE and transferred to PVDF membranes (C). Band densitometry confirmed a significant decrease in cytochrome c content of RKM isolated from 24 month animals (B) Significant p-values (<0.05) are denoted by an asterisk (*) above the indicated pair and were calculated from non-paired, two-tailed student t-test.
In order to confirm the functional relevance of the decrease in the cytochrome c abundance observed in the mitochondria of 24 month animals, we performed a polarographic assay of cytochrome oxidase on permeabilized mitochondria without and with the addition of 16 nanomoles of exogenous cytochrome c. Without the addition of exogenous cytochrome c, the rate of oxygen consumption through cytochrome oxidase in mitochondria isolated from 24 month animals was 645 nmol O/min/mg of mitochondrial protein, 23% slower than the rate in 6 month animals, 836 nmol O/min/mg of mitochondrial protein, and was highly significant (p=0.002) (Figure 4). This difference was corrected by the addition of exogenous cytochrome c.
Figure 4. Polarographic cytochrome oxidase assay.
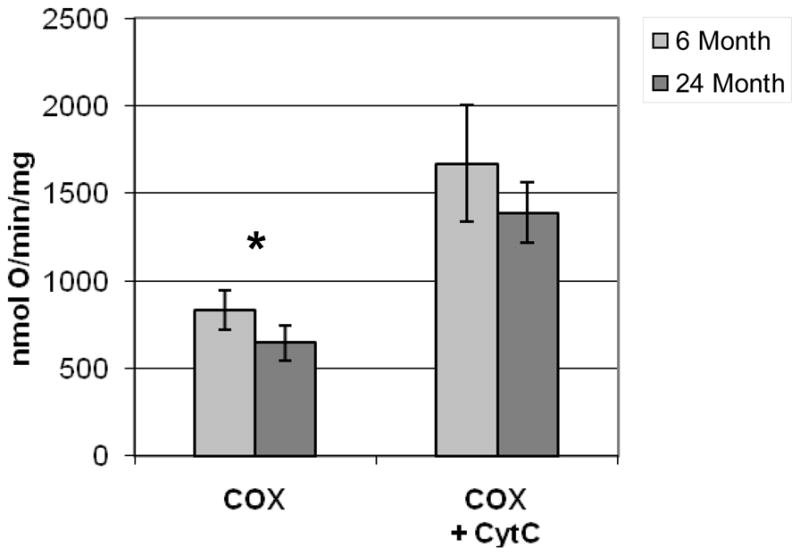
RKM from 6 month (n=7) and 24 month (n=7) animals were permeabilized and cytochrome oxidase activity was evaluated by the rate of oxygen consumption after the addition of TMPD and ascorbate with endogenous cytochrome c (COX) and with the addition of exogenous cytochrome c (COX + CytC). Bars represent the mean value of each experimental group and the error bars indicate the standard deviation. Significant p-values (<0.005) are denoted by an asterisk (*) above the indicated pair and were calculated from non-paired, two-tailed student t-test.
Discussion
Here we show that RKM develop a defect in OXPHOS with age (Figure 1). Assays of OXPHOS capacity in intact mitochondria test not only the activities of the ETC complexes, but also substrate transporters and dehydrogenases, membrane integrity and elements which transfer electrons between ETC complexes (ie.. coenzyme Q and cytochrome c). Our use of a comprehensive panel of substrates to test the integrated mitochondrial function in OXPHOS assays has permitted the biochemical dissection of the pathway and allowed the site of the defect to be localized.
Our data demonstrate an age-related OXPHOS defect with the use of substrates, which enter the ETC at complexes I, II, III, and cytochrome c, and is not relieved by the addition of an uncoupler demonstrating the site of the defect to occur at cytochrome c or complex IV. Variance in the degree of decline in state 3 rates between different metabolic substrates suggests that substrate transporters and dehydrogenases also may have a contributory role in the age-related decline of substrate utilization for OXPHOS within the kidney. The first order rate constant of complex IV was unchanged in spectrophotometric assays of solubilized mitochondria from aged kidney in the presence of exogenous, reduced cytochrome c (Supplementary Figure 1), arguing that an intrinsic defect in complex IV is not the principal cause of the diminished OXPHOS capacity in mitochondria isolated from 24 month animals.
mtDNA deletions and concomitant loss of ETC activity has been convincingly shown to occur in an age-dependant fashion in the rat kidney [8]. The principle defect in OXPHOS related to mtDNA deletions should be a decrease in activity of the respiratory complexes containing subunits encoded on the mtDNA (i.e. complexes I, III, IV). We found no significant changes in the respiratory complexes with spectrophotometric measurement of activity or BN-gel assessment of protein abundance (Supplementary Figure 1 and 2). Therefore, our results do not support a defect in the respiratory complexes as the principal cause of the reduction in OXPHOS observed and suggest that mtDNA deletions are not responsible for the OXPHOS defect in these studies. This may not be surprising given the observation that the ETC-deficient renal tubular cells only comprised ~0.2% of the cortical volume at 24 months [8], and mitochondria derived from these tubules would therefore be expected to make a relatively minor contribution to the total mitochondrial population.
Therefore, we sought alternative mechanisms, including a decrease in mitochondrial cytochrome c content, to explain the age-dependant defects in OXPHOS that we have observed. The cytochrome b and cytochrome c content in mitochondria isolated from kidneys of 24 month animals was 23% and 37% lower, respectively (Figure 3). The total cytochrome b content is distributed between complexes II and III within the mitochondria [24]. We judged the decline in cytochrome b to be non-limiting, as the activities of complex II (CoQ-reductase) and complex III (ubiquinol-cytochrome c oxidoreductatase) were no different between the two groups (Supplementary Figure 1). The fact that the cytochrome b content is divided between the two complexes may attenuate the effect a reduction in the total cytochrome b content has on either complex individually and may contribute to the lack of functional differences in these complexes between the two groups. This suggested that the observed decline in OXPHOS capacity of the 24 month RKM was due to the decreased cytochrome c content. To test this hypothesis we performed polarographic assays of cytochrome oxidase, complex IV, without and with exogenous cytochrome c. The addition of exogenous cytochrome c corrected the defect in the polarographic activity of cytochrome c oxidase. This demonstrates that the depletion of cytochrome c is the dominant mechanism for the decrease in OXPHOS capacity observed in mitochondria obtained from the kidneys of 24 month animals (Figure 4).
The observed decrease of cytochrome c content in mitochondria isolated from kidneys of 24 month animals is not explained by leak of cytochrome c across the outer mitochondrial membrane (OMM), as we demonstrated that the OMM was impermeable to cytochrome c prior to addition of the detergent, dodecyl maltoside, in the spectrophotometric assay of cytochrome oxidase activity (Supplementary Figure 1). It is known that the transcription of cytochrome c declines with age in human and murine kidneys [25,26]. This leads to the hypothesis that an age-related decline in cytochrome c transcription underlies the reduction in mitochondrial cytochrome c abundance, which we have observed in aged animals. However, transcriptional microarray experiment of rat kidney failed to identify age-related changes in cytochrome c transcription [27] and alternative explanations including increased rates of cytochrome c turnover or decreased capacity for cytochrome c binding to the inner mitochondrial membrane also must be considered as possibilities. The age-related transcriptional decline of cytochrome c has not been described for extra-renal organs and the reasons underlying this tissue-specific finding have yet to be examined.
Modulation of flux through the ETC in direct relation to mitochondrial cytochrome c content, independent of the activities and protein abundance of the respiratory complexes, is not completely novel. Serum stimulated murine fibroblasts demonstrated an increase in oxygen consumption related to an increase in cytochrome c content [28]. Further, fibroblasts isolated from individuals with early onset Parkinson’s disease and mutations in PINK1 were demonstrated to have decreased respiratory capacity related to a decrease in cytochrome c content [29]. Cultured neurons derived from 24 month rat brains appear to have reduced cytochrome c content and complex IV activity when compared with 9 month controls [30].
The decline in OXPHOS efficiency is demonstrated by a decrease in State 3 and state 4 rates. Thus, there is not only a slower rate of ADP consumption, implying a slower rate of ATP production, in mitochondria isolated from 24 month animals, there also is a slower rate of proton leak across the inner mitochondrial membrane or a decline in ATPase activity unassociated with the F1F0 ATPase. Interestingly, the integrity of the mitochondria, as reflected by the RCR, and coupling to the phosphorylation apparatus, as reflected by the ADP/O, in renal mitochondria appear to be preserved with aging in the kidney. This result is contrary to a previous report in which both the RCR and ADP/O were decreased in mitochondria isolated from the kidneys of 22 month Wistar rats compared to 4 month controls [9], but agrees with two others [31,32] demonstrating preservation of renal mitochondrial RCR and ADP/O ratios with aging.
Our observations demonstrate an age-related mitochondrial defect in the pool of mitochondria obtained from whole kidney. Future efforts will be needed to determine if the decline in OXPHOS shown here is a generalized phenomenon in renal mitochondria or if it localizes to mitochondria isolated from a defined nephron segment. The nephron travels a circuitous route through the cortex and medulla and is composed of multiple segments often containing multiple cell types. Renal mitochondria are not distributed evenly in all cell types with the highest density of mitochondria found in the medullary and cortical thick ascending limb of Henle’s loop followed by the proximal tubule [33]. In addition, the kidney is able to metabolize a broad array of substrates for energy production and substrate preferences have been shown to vary along the length of the nephron [34]. Finally, the renal microenvironment varies dramatically in a radial fashion from the cortex to the medulla with increasing tonicity and decreasing oxygen tension.
From these studies we can conclude that the mitochondrial content per gram of kidney weight is lower in 24 month animals compared to 6 month animals. The mitochondria isolated from the elderly animals demonstrate a decrease in OXPHOS capacity mediated by decreases in state 3 and state 4 respiration, which could not be explained by decrements in the biochemical activities of the individual components of the ETC or the protein abundance of the ETC protein complexes. The mitochondrial content of cytochrome c is decreased in mitochondria isolated from the kidneys of 24 month animals. The decrease in cytochrome c significantly contributes to the decreased rate of oxygen consumption by cytochrome c oxidase observed in the mitochondria of 24 month animals. The decline in cytochrome c and cytochrome oxidase activity results in a global decrease in OXPHOS capacity, as all of the OXPHOS substrates tested require electron flux through cytochrome c and cytochrome oxidase.
Supplementary Material
Acknowledgments
The authors would like to gratefully acknowledge the support and technical advice provided by David Kehres, William Parland, Edwin Vazquez and Mariana Rosca.
Funding
JFO is supported by grants from the NIH (DK071108) and the Kidney Foundation of Ohio. CLH is supported by a grant from the NIH (PO1AG015885).
Abbreviations
- OXPHOS
oxidative phosphorylation
- ETC
electron transport chain
- ROS
reactive oxygenspecies
- mtDNA
mitochondrial DNA
- RCR
respiratory control ratio
- RKM
rat kidney mitochondria
- DHQ
duroquinol
- TMPD
N,N,N′,N′-tetramethyl-p-phenylenediamine
- COX
cytochrome c oxidase
- DNP
dinitrophenol
- OMM
outer mitochondrial membrane
Footnotes
The authors have no conflicts to declare.
Reference List
- 1.Davies DF, Shock NW. Age changes in glomerular filtration rate, effective renal plasma flow, and tubular excretory capacity in adult males. J Clin Invest. 1950;29:496–507. doi: 10.1172/JCI102286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindeman RD, Tobin J, Shock NW. Longitudinal studies on the rate of decline in renal function with age. J Am Geriatr Soc. 1985;33:278–285. doi: 10.1111/j.1532-5415.1985.tb07117.x. [DOI] [PubMed] [Google Scholar]
- 3.Zhang QL, Rothenbacher D. Prevalence of chronic kidney disease in population-based studies: systematic review. BMC Public Health. 2008;8:117. doi: 10.1186/1471-2458-8-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lameire N, Van BW, Vanholder R. The changing epidemiology of acute renal failure. Nat Clin Pract Nephrol. 2006;2:364–377. doi: 10.1038/ncpneph0218. [DOI] [PubMed] [Google Scholar]
- 5.Gullans SR. In: The Kidney. Brenner BM, editor. W.B. Saunders Company; Philadelphia: 2000. pp. 215–246. [Google Scholar]
- 6.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guarente L. Mitochondria--a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKiernan SH, Tuen VC, Baldwin K, Wanagat J, Djamali A, Aiken JM. Adult-onset calorie restriction delays the accumulation of mitochondrial enzyme abnormalities in aging rat kidney tubular epithelial cells. Am J Physiol Renal Physiol. 2007;292:F1751–F1760. doi: 10.1152/ajprenal.00307.2006. [DOI] [PubMed] [Google Scholar]
- 9.de Cavanagh EM, Piotrkowski B, Basso N, Stella I, Inserra F, Ferder L, Fraga CG. Enalapril and losartan attenuate mitochondrial dysfunction in aged rats. FASEB J. 2003;17:1096–1098. doi: 10.1096/fj.02-0063fje. [DOI] [PubMed] [Google Scholar]
- 10.Meng Q, Wong YT, Chen J, Ruan R. Age-related changes in mitochondrial function and antioxidative enzyme activity in fischer 344 rats. Mech Ageing Dev. 2007;128:286–292. doi: 10.1016/j.mad.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Sachan DS, Hoppel CL. Carnitine biosynthesis. Hydroxylation of N6-trimethyl-lysine to 3-hydroxy-N6-trimethyl-lysine. Biochem J. 1980;188:529–534. doi: 10.1042/bj1880529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiMarco JP, Hoppel C. Hepatic mitochondrial function in ketogenic states. Diabetes, starvation, and after growth hormone administration. J Clin Invest. 1975;55:1237–1244. doi: 10.1172/JCI108042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem. 1955;217:409–427. [PubMed] [Google Scholar]
- 14.Estabrook RW. Mitochondrial respiratory control and polarographic measurement of ADP/O ratios. Methods Enzymol. 1967;10:41–47. [Google Scholar]
- 15.Matsuoka Y, Srere PA. Kinetic studies of citrate synthase from rat kidney and rat brain. J Biol Chem. 1973;248:8022–8030. [PubMed] [Google Scholar]
- 16.Hoppel CL, Kerr DS, Dahms B, Roessmann U. Deficiency of the reduced nicotinamide adenine dinucleotide dehydrogenase component of complex I of mitochondrial electron transport. Fatal infantile lactic acidosis and hypermetabolism with skeletal-cardiac myopathy and encephalopathy. J Clin Invest. 1987;80:71–77. doi: 10.1172/JCI113066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–8739. [PubMed] [Google Scholar]
- 18.Sordahl LA, McCollum WB, Wood WG, Schwartz A. Mitochondria and sarcoplasmic reticulum function in cardiac hypertrophy and failure. Am J Physiol. 1973;224:497–502. doi: 10.1152/ajplegacy.1973.224.3.497. [DOI] [PubMed] [Google Scholar]
- 19.Hoppel C, Cooper C. The action of digitonin on rat liver mitochondria. The effects on enzyme content. Biochem J. 1968;107:367–375. doi: 10.1042/bj1070367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krahenbuhl S, Talos C, Wiesmann U, Hoppel CL. Development and evaluation of a spectrophotometric assay for complex III in isolated mitochondria, tissues and fibroblasts from rats and humans. Clin Chim Acta. 1994;230:177–187. doi: 10.1016/0009-8981(94)90270-4. [DOI] [PubMed] [Google Scholar]
- 21.Williams JN., Jr A method for the simultaneous quantitative estimation of cytochromes a, b, c1, and cin mitochondria. Arch Biochem Biophys. 1964;107:537–543. doi: 10.1016/0003-9861(64)90313-3. [DOI] [PubMed] [Google Scholar]
- 22.Wittig I, Braun HP, Schagger H. Blue native PAGE. Nat Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 23.Sternberg SR. Biomedical image processing. IEEE Computer. 1983;16 (1):22–34. [Google Scholar]
- 24.Benard G, Faustin B, Passerieux E, Galinier A, Rocher C, Bellance N, Delage JP, Casteilla L, Letellier T, Rossignol R. Physiological diversity of mitochondrial oxidative phosphorylation. Am J Physiol Cell Physiol. 2006;291:C1172–C1182. doi: 10.1152/ajpcell.00195.2006. [DOI] [PubMed] [Google Scholar]
- 25.Rodwell GE, Sonu R, Zahn JM, Lund J, Wilhelmy J, Wang L, Xiao W, Mindrinos M, Crane E, Segal E, Myers BD, Brooks JD, Davis RW, Higgins J, Owen AB, Kim SK. A transcriptional profile of aging in the human kidney. PLoS Biol. 2004;2:e427. doi: 10.1371/journal.pbio.0020427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zahn JM, Sonu R, Vogel H, Crane E, Mazan-Mamczarz K, Rabkin R, Davis RW, Becker KG, Owen AB, Kim SK. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006;2:e115. doi: 10.1371/journal.pgen.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen G, Bridenbaugh EA, Akintola AD, Catania JM, Vaidya VS, Bonventre JV, Dearman AC, Sampson HW, Zawieja DC, Burghardt RC, Parrish AR. Increased susceptibility of aging kidney to ischemic injury: identification of candidate genes changed during aging, but corrected by caloric restriction. Am J Physiol Renal Physiol. 2007;293:F1272–F1281. doi: 10.1152/ajprenal.00138.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herzig RP, Scacco S, Scarpulla RC. Sequential serum-dependent activation of CREB and NRF-1 leads to enhanced mitochondrial respiration through the induction of cytochrome c. J Biol Chem. 2000;275:13134–13141. doi: 10.1074/jbc.275.17.13134. [DOI] [PubMed] [Google Scholar]
- 29.Piccoli C, Sardanelli A, Scrima R, Ripoli M, Quarato G, D’Aprile A, Bellomo F, Scacco S, De MG, Filla A, Iuso A, Boffoli D, Capitanio N, Papa S. Mitochondrial respiratory dysfunction in familiar parkinsonism associated with PINK1 mutation. Neurochem Res. 2008;33:2565–2574. doi: 10.1007/s11064-008-9729-2. [DOI] [PubMed] [Google Scholar]
- 30.Jones TT, Brewer GJ. Critical age-related loss of cofactors of neuron cytochrome C oxidase reversed by estrogen. Exp Neurol. 2009;215:212–219. doi: 10.1016/j.expneurol.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinbach EC, Garbus J. Coenzyme A content and fatty acid oxidation in liver and kidney mitochondria from aged rats. Gerontologia. 1959;3:253–260. doi: 10.1159/000210904. [DOI] [PubMed] [Google Scholar]
- 32.Gold PH, Gee MV, Strehler BL. Effect of age on oxidative phosphorylation in the rat. J Gerontol. 1968;23:509–512. doi: 10.1093/geronj/23.4.509. [DOI] [PubMed] [Google Scholar]
- 33.Pfaller W, Rittinger M. Quantitative morphology of the rat kidney. Int J Biochem. 1980;12:17–22. doi: 10.1016/0020-711x(80)90035-x. [DOI] [PubMed] [Google Scholar]
- 34.Wirthensohn G, Guder WG. Renal substrate metabolism. Physiol Rev. 1986;66:469–497. doi: 10.1152/physrev.1986.66.2.469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.