

Abstract
Purpose
Characterization of an approach to identify leukemia neoantigens arising in the context of drug resistance.
Experimental Design
We assessed whether leukemia neoantigens could be generated from drug-resistant mutations in BCR-ABL following imatinib relapse in chronic myelogenous leukemia (CML) patients.
Results
We computationally predicted that ~70 peptides derived from 26 BCR-ABL mutations would bind 8 common alleles of MHC class I (IC50<1000 nM). Seven of nine imatinib-resistant CML patients were predicted to generate at least one peptide that binds autologous HLA alleles. We predicted and confirmed that an E255K mutation-derived peptide would bind HLA-A3 with high affinity (IC50=28 nM), and demonstrated that this peptide is endogenously processed and presented. Polyfunctional E255K-specific CD8+ T cells were detected in two imatinib-resistant HLA-A3+ CML patients concurrent with an effective anti-CML response to further therapy.
Conclusions
Our in vitro studies support the hypothesis that leukemia-driven genetic alterations are targeted by the immune system in association with a clinical response, and suggest the possibility of immunizing relapsed CML patients against newly acquired tumor neoantigens.
Keywords: Mutated BCR-ABL, leukemia antigen, chronic myeloid leukemia, T cell
INTRODUCTION
Chronic myelogenous leukemia (CML) is a myeloproliferative disorder whose pathognomonic feature is translocation between chromosomes 9 and 22 to form the fusion protein BCR-ABL. This gene fusion results in constitutive activation of the ABL kinase whose oncogenic properties have been well-described (1,2). For decades, enduring curative therapy for young patients with CML has been achieved with allotransplantation (allo-HSCT) (although at significant risk of toxicities) and these results have provided clear demonstration of the potency of donor immunity in eradicating leukemia through graft-versus-leukemia (GvL) effects (3). The well-tolerated BCR-ABL targeting tyrosine kinase inhibitor (TKI) imatinib, however, has emerged as front-line therapy, since its FDA approval in 2001 (4). More potent second-generation TKIs, dasatinib and nilotinib, have also been developed and approved for treatment of CML with prior imatinib exposure (5-6). All three TKIs efficiently inhibit the catalytic activity of BCR-ABL by binding to the ATP-binding pocket of the ABL kinase domain (7).
While imatinib effectively controls CML with relatively minor toxicity, relapse and drug resistance remain as important problems, with annual progression rates of 3.3-7.5% during the first three years of treatment (8,9). The most common mechanism of drug resistance is the emergence of various BCR-ABL kinase domain mutations (10), with detection of BCR-ABL mutations varying from 35 to 89% of patients (11-13). Most mutations localize to the p-loop (or ATP binding site) of the fusion protein, followed by the imatinib binding site, catalytic domain, or activation loop, and have been reported to range from 25 to over 70 distinct mutations (10). Nearly two-thirds of all reported mutations localize to seven amino acids: G250 (10%), Y253 (11%), E255 (10.5%), T315 (13.5%), M351 (10%), F359 (6.5%) and H396 (5%) (10, 14, 15). Resistance to second-generation TKIs has been likewise observed, with each drug generating its own spectrum of BCR-ABL mutations (16-18).
Therapeutic alternatives that complement the potent cytolytic effects of TKIs and address drug resistance would substantively advance the treatment of patients with CML. In particular, an immunotherapeutic approach is promising for CML, due to its exquisite sensitivity to immune modulation (19). BCR-ABL has been suggested as an ideal immunologic target since, it is essential for leukemogenesis and its expression is specific to CML. However, chronic expression of this cancer-driving leukemia protein may result in selective host deletion of high avidity T cells directed against BCR-ABL expressing cells, as has been described for another CML-associated antigen PR-1 (20), or in reduced anti-tumor functionality. Consistent with this idea, T cells with specificity for BCR-ABL have been detected in CML patients, but they demonstrate poor cytolytic activity (21). Conversely, as tumors evolve, such as with drug resistance, newly-acquired genetic alterations may generate novel peptides that can stimulate new CTL clones, although immunosuppression exerted by leukemia cells may prevent these from controlling expansion of leukemia cells in vivo (22, 23). Fortunately, however, new potent adjuvants and checkpoint blockade inhibitors are now available that have the potential to circumvent these obstacles and will likely be incorporated in future immunotherapies (24-27).
Herein, we present a strategy to evaluate the immunogenicity of peptides generated from mutated BCR-ABL. This analysis is facilitated by the increasing accuracy of algorithms that predict binding capacity of peptides to a wide range of HLA alleles (28). We detected that T cell responses against processed mutated BCR-ABL peptides develop in association with tumor regression following allo-HSCT or second line TKIs. While the concept of tumor neoantigens as a target for immunotherapeutics has been long studied (29), our studies are the first to demonstrate that mutations that develop in the setting of drug resistance can be an abundant source of tumor-specific antigens. Our results provide a proof-of-concept for a systematic approach to discover immunogenic tumor antigens arising from evolving genetic alterations, with potential for developing novel immunotherapies.
METHODS
Patient samples
Heparinized blood was obtained from patients and normal donors enrolled on clinical research protocols at the Dana-Farber Harvard Cancer Center (DFHCC) approved by the DFHCC Human Subjects Protection Committee. Peripheral blood (PBMC) from normal donors and patients was isolated by Ficoll/Hypaque density-gradient centrifugation, cryopreserved with 10% DMSO, and stored in vapor-phase liquid nitrogen until the time of analysis. HLA typing was performed by either molecular studies or serotyping (Tissue Typing Laboratory, Brigham and Women’s Hospital, Boston).
Detection of BCR-ABL mutations
Mutations in BCR-ABL were detected by polony assay, described previously (30), or by routine molecular diagnostics at the Brigham and Women’s Hospital, Boston. In the latter process, total RNA was isolated from unfractionated peripheral blood or bone marrow cells, and reverse-transcribed with the ABL primer downstream of the kinase domain and PCR-amplified with primers across the BCR-ABL junction. The resulting PCR product was purified and analyzed by Sanger sequencing.
Prediction of peptide binding to HLA alleles
To discover peptide targets, MHC-binding affinity was predicted across all possible 9mer and 10mer peptides encoded by ABL mutations by using MHC-peptide binding prediction algorithms which have been independently verified as highly accurate (28, 31). The NetMHC algorithm met this criterion for 9mer prediction of HLA-A*02:01, *03:01, *11:01, *24:02, B*07:02, *08:01, and *15:01; and for 10mer prediction of HLA-A*01:01, *02:01, and B*07:02 (32). Alternatively, we used the IEDB SMM (IEDB) algorithm, which met the criterion for high accuracy for HLA-A*02:01, *03:01, *11:01, B*07:02, *08:01, and *15:01 (28). An IC50 < 50 nM was considered a predicted strong binder, an IC50 >50 and <500 nM was considered an intermediate binder, and IC50 between 500 and 1000 nM was considered a weak binder.
Experimental binding of peptides
Experimental confirmation of peptide-MHC prediction of a subset of peptides was performed using a competitive MHC allele binding assay to test binding to several common HLA alleles (A*02:01, A*03:01, A*11:01, B*07:02, B*15:01). This assay measures the ability of peptide ligands to inhibit the binding of a high affinity radiolabeled peptide to purified MHC molecules, and has been described in detail elsewhere (33). Briefly, purified MHC molecules, test peptides, and a radiolabeled probe peptide were incubated at room temperature in the presence of human β2-microglobulin and a cocktail of protease inhibitors. After a two-day incubation, binding of the radiolabeled peptide to the corresponding MHC class I molecule was determined by capturing MHC/peptide complexes on W6/32 antibody (anti-HLA A, B, and C) or B123.2 (anti-HLA B, C and some A) coated plates, and measuring bound cpm using a microscintillation counter. For competition assays, the concentration of peptide yielding 50% inhibition of radiolabeled peptide binding was calculated. Peptides were typically tested at 6 different concentrations covering a 100,000-fold dose range, and in 3 or more independent assays. Under the conditions utilized, where [label]<[MHC] and IC50 ≥ [MHC], the measured IC50 values are reasonable approximations of the true Kd values (34, 35).
Sources of antigen
Peptides were synthesized to >95% purity (confirmed by high performance liquid chromatography, New England Peptide; Gardner MA). Peptides were reconstituted in DMSO (10 mg/ml) and stored at −20°C until use. A minigene comprised of a sequence of 227bp encompassing the E255K mutation was PCR-cloned into the HindIII and EcoRI restriction enzyme sites of the expression vector pcDNA3.1 using the following primers: 5′-primer: AGCTTTAAGCTTCACATGTATGGTGTGTCCCCC; 3′primer: ATAGAATTCTGCAGTCAATGGTGATGGTGATGATGTCACTCTTCCACCTCCATGGT. To express the E255K mutation in target cells of T cell assays, 10 μg of the minigene plasmid was introduced into 2 million K562 cells (American Type Culture Collection, Manassus VA) together with 10 μg of a plasmid encoding HLA-A3 (in the vector pLNCX2, gift of Ellis Reinherz) by Amaxa nucleofection (Solution V, Program T16, Lonza Inc; Walkersville, MD). Expression of HLA-A3 was confirmed by flow cytometry using an anti-HLA-A3 mAb (One Lambda, Canoga Park, CA), followed by FITC-conjugated F(ab’)2 goat antimouse IgM (Zymed, South San Francisco, CA). Cells were incubated in DMEM (Cellgro; Manassas, VA), supplemented with 10% fetal bovine serum (Cellgro), 1% HEPES buffer (Cellgro), and 1% L-glutamine (Cellgro). The nucleofected cells were harvested 2 days later for immune assays.
Generation of antigen presenting cells (APCs)
Autologous dendritic cells (DCs) were generated from immunomagnetically-isolated CD14+ cells (Miltenyi Biotec, Auburn CA) that were cultured in RPMI (Cellgro) supplemented with 3% fetal bovine serum, 1% penicillin-streptomycin (Cellgro), 1% L-glutamine and 1% HEPES buffer in the presence of 120 ng/ml GM-CSF (Genzyme, Cambridge, MA) and 70 ng/ml IL-4 (R&D Systems, Minneapolis, MN). On days three and five, additional GM-CSF and IL-4 were added. On day six, cells were matured for 48 hours with 10 ng/ml TNF-α (Genzyme, Cambridge, MA) and 1 μg/ml prostaglandin E2 (Sigma-Aldrich, St. Louis MO). CD40-B cells were generated from PBMC by activation on CD40L-expressing irradiated feeder cells in the presence of IL-4 (R&D Systems) and cyclosporin A (Novartis, Basel, Switzerland) as described (36). The MHC class I-deficient immortalized B lymphoblastoid cell line 721.221 was retrovirally infected with a plasmid encoding the full-length A*03:01 (designated B721.221/A3 cells), and maintained in DMEM supplemented with 10% FCS, 2mM L-glutamine, antibiotics (Invitrogen), and 500 ng/mL of G418 (Cellgro).
Generation of antigen-specific T cells from normal volunteer and patient PBMC
To generate E255K-reactive T cells from a healthy adult volunteer, PBMC from an HLA-A3+ individual was cultured in 24-well plates (10×106 cells/well) in complete medium supplemented with 10% human AB serum and 10 ng/mL IL-7, together with autologous DCs (at 1:100 ratio) that were pulsed for 3 hours with 10 μM peptide. On day 6, CD8+ T cells were immunomagnetically isolated from PBMC (CD8+ Microbeads, Miltenyi, Auburn CA). CD8+ T cells were co-cultured with irradiated B721.221/A3 cells (3200 Gy) pulsed with 10μM peptide at a ratio of 10:1 in the presence of IL-7 (10 ng/mL) on days 7, 14, 21, and 28, and T cell specificity was tested one week after last stimulation. E255K-specific T cells were similarly expanded from cryopreserved patient PBMC, with ELISpottesting after two rounds of peptide stimulation.
Detection of antigen-specific T cells
ELISpot assay was performed using peptide-pulsed target cells (50,000 cells/well) coincubated with 50,000 immunomagnetically CD8+ T cells/well in duplicate on ELISpot plates (Millipore, Billerica, MA) for 24 hours. Interferon-γ secretion (IFNγ) was detected using capture and detection antibodies, as directed (Mabtech AB, Mariemont, OH), and imaged (ImmunoSpot Series Analyzer; Cellular Technology, Cleveland, OH). To test T cell reactivity dependence on class I, ELISpot plates were first coated with APCs co-incubated with class I blocking antibody (W6/32) for 2 hours at 37°C, prior to introduction of T cells into the wells. An HLA-A3 tetramer specific for the E255K-B peptide was generated by the NIH Tetramer Facility (Emory University, Atlanta GA). For tetramer staining, 5×105 cells were incubated 30 minutes at 4°C with 1 μg/mL tetramer, and then incubated with the addition of anti-CD8-FITC antibody (BD Biosciences, San Diego CA) for another 30 minutes at 4°C. A minimum of 5000 events were acquired per sample. Supernatants of cultured CD8+ T cells were also collected and analyzed for GM-CSF, TNF-α and IP-10 using a Luminex multiplex bead-based technology (Milliplex; Millipore, Billerica, MA), per the manufacturer’s recommendations. In brief, fluorescent labeled microspheres were coated with specific cytokine capture antibodies. After incubation with the culture supernatant sample, captured cytokines were detected by a biotinylated detection antibody, followed by a streptavidin-PE conjugate and median fluoresence intensity (MFI) was measured (Luminex 200 bead array instrument; Luminex Corporation, Austin TX). Based on a standard curve, cytokine levels were calculated in the Bead View Software program (Upstate, Millipore, Billerica).
Statistical considerations
Comparisons between T cell responses to APCs pulsed with wild-type and mutated peptide on ELIspot; or between peptide-pulsed APCs or E255K minigene-transfected APCs were calculated as two sided p values using a two–sample t-test.
RESULTS
Common imatinib-induced BCR-ABL mutations frequently generate peptides predicted to bind MHC Class I alleles
Twenty-six BCR-ABL mutations have been reported to arise in association with CML relapse following imatinib exposure, with T315I, E255K and M351T as the most common (21%, 19% and 15%, respectively) (Figure 1C), and with the remaining mutations occurring at a frequency of 6.8% or less (37). To explore the potential of mutated BCR-ABL to generate novel immunogenic peptides, we applied the validated prediction algorithm NetMHC to the amino acid sequences encompassing these common BCR-ABL mutations. NetMHC has been demonstrated to accurately predict 9-and 10-mers binding to 8 common Class I HLA alleles: A*02:01, A*03:01, A*11:01, B*07:02, B*08:01, B*15:01, A*01:01, and A*24:02 (28).
Figure 1. Predicted Class I HLA binding peptides based on common BCR-ABL mutations using the NetMHC prediction server.
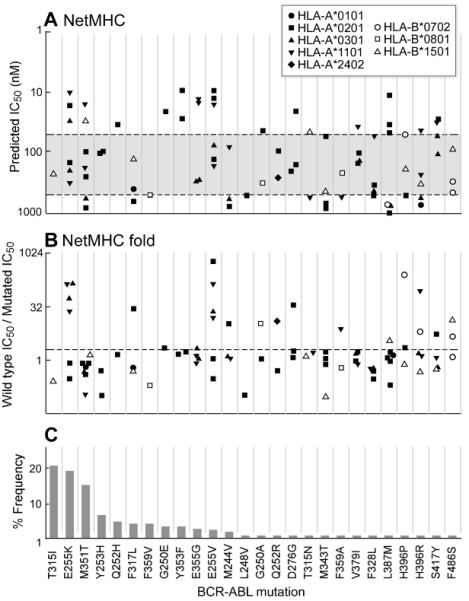
Twenty-six BCR-ABL mutations, ordered in decreasing frequency from left to right per Deininger et al (37) (C) were tested by NetMHC for peptides predicted to bind common class I HLA alleles. A. The binding affinities of the predicted high affinity peptides (IC50<1000 nM) per BCR-ABL mutation are displayed. Strong binders are defined as IC50<50 nM (region above the upper dotted line). Intermediate binders are defined as 50 nM≤IC50<500 nM (between the two dotted lines). Weak binders are defined as 500 nM≤IC50<1000 nM (in the region below the lower dotted line). B. Fold predicted binding of mutated peptide compared to wild-type. Points above the dotted line represent peptides predicted to bind with at least two-fold greater affinity compared to wild-type.
By tiling 9- and 10-mers around each mutation, we identified 71 distinct peptides predicted by NetMHC to bind one or more common class I HLA alleles with IC50<1000 nM for a total of 84 peptide-allele combinations. As shown in Figure 1A, NetMHC predicted 25 of 84 (30%) as high binders (IC50<50 nM), 41 (49%) as intermediate binders (IC50=50-500 nM), and 18 (21%) as weak binders (1000> IC50>500 nM). Predicted high binding peptides were evenly distributed across the 26 mutations. Of note, E255K produced 3 high and 3 intermediate affinity Class I HLA candidate binding peptides, and M351T produced 2 high and 4 intermediate affinity candidate binding peptides. In contrast, the highly aggressive T315I mutation yielded only one peptide of intermediate binding affinity, predicted to bind HLA-B*1501.
A priority in the field of immunotherapy is the identification of targetable tumor-specific epitopes that do not elicit cross-reactivity to normal tissue. We therefore examined whether the predicted mutated peptides had increased binding affinity compared to respective parental peptides. As shown in Figure 1B, 24 of 84 predicted mutated peptides (29%) demonstrated at least two-fold higher predicted binding affinity to HLA compared to parental peptides (median 1.2, range 0.02 to 622). Of the three most frequent mutations, 4 of 6 E255K peptides (but none of the T315I or M351T peptides) were predicted to bind with at least two-fold higher affinity compared to parental peptides.
Similar results were observed using an alternate established algorithm, IEDB, for prediction of peptides derived from mutated BCR-ABL peptides binding to HLA-A*02:01, HLA-A*03:01, HLA-A*11:01, HLA-B*07:02, HLA-B*08:01, and HLA-B*15:01. Forty-five distinct peptides were predicted to bind with IC50< 1000 nM, with 60 peptide-allele combinations (Supplemental Figure 1). Twenty-three (38%) had at least two-fold greater affinity for HLA binding than the wild-type parental peptide. Overall, 46 peptide-allele combinations were common to both the NetMHC and IEDB servers. These analyses demonstrate that mutated BCR-ABL has the potential to generate neopeptides that can bind to common HLA alleles.
Imatinib-resistant CML patients can potentially express immunogenic mutated BCR-ABL epitopes
To establish the frequency that immunogenic peptides arising from mutated BCR-ABL can potentially be generated from CML patients, we analyzed nine imatinib-resistant CML patients, whose HLA typing and BCR-ABL mutation status were known. All patients had been exposed to imatinib for a median of 6 months (range 1-33) before developing drug resistance (Table 1). All leukemia samples harbored a median of 1 BCR-ABL mutation (range 1-3). In total, 20 distinct HLA-A and -B alleles were represented among the 9 patients, and 8 distinct mutations were detected, 5 of which were unique to particular patients.. The three most common HLA alleles were A1, A2 and B7, represented in 4, 5 and 4 patients, respectively.
Table 1.
Patient characteristics of 9 patients with known mutations in BCR-ABL following the development of imatinib resistance.
| Patient | Age/Sex | Months on Imatinib until resistance |
HLA-A and -B alleles |
BCR-ABL Mutations |
|---|---|---|---|---|
| 1 | 64/M | 32 | A3, A11, B7, B52 | L150M, E255K, T315I, |
| 2 | 44/M | 6 | A2, A3, B7, B49 | E255K |
| 3 | 59/M | 2 | A2, A3, B7, B44 | E255K |
| 4 | 53/M | 6 | A2, A33, B37, B14 | E255K, T315I |
| 5 | 70/M | 1 | A1, A2, B8, B38 | M244V, E281K, T315A |
| 6 | 61/M | 14 | A2, A2, B40, B51 | G250E |
| 7 | 48/F | 2 | A1, A30, B18, B57 | V379I |
| 8 | 58/F | 7 | A1, A31, B7, B57 | T315I |
| 9 | 64/F | 33 | A1, A24, B8, B57 | E281K, G254R |
As shown in Figure 2, 7 of 9 imatinib-resistant CML patients could potentially generate at least one mutated BCR-ABL derived peptide with predicted binding affinity to autologous HLA alleles of IC50<1000 nM (median number of predicted binders/patient was 2, range 0-5). None of the 3 patients with the T315I mutation (Patients 1, 4, 8) expressed HLA-B15 (the only allele predicted to strongly bind a T315I-derived peptide). On the other hand, CML cells from 3 of 4 patients harboring an E255K mutation also expressed HLA-A3 (Patients 1-3), and this mutation was predicted to generate 2 promising candidate HLA-A3 binding peptides: E255K-A247-255 (KLGGGQYGK, IEDB IC50=113 nM; NetMHC IC50=192 nM) and E255K-B255-263 (KVYEGVWKK, IEDB IC50=29 nM; NetMHC IC50=28 nM). Moreover, both peptides were predicted to bind HLA-A*03:01 with at least ten-fold greater affinity than parental peptides. Additionally, three of four patients harboring an E255K mutation expressed HLA-A2 (Patients 2-4); E225K was predicted to generate two other promising HLA-A2 binding candidate peptides by NetMHC: E255-C247-256 (KLGGGQYGKV; IC50=145 nM) and E255K-D251-260 (GQYGKVYEGV; IC50=16 nM), but not by IEDB.
Figure 2. Individual class I HLA binding of peptides derived from mutated BCR-ABL in CML patients.

Eight distinct BCR-ABL mutations were detected among 9 patients with imatinib resistant CML. Displayed are the 5 of 8 mutations for which HLA binding peptides could be predicted from the individual patients, and are ordered by decreasing frequency based on larger patient series (37). For each mutation, class I HLA binding peptides using only the HLA alleles represented within the set of 9 patients that are predicted by the NetMHC algorithm are provided. Not shown are the three mutations that were present in patient leukemia cells, but based on the patients’ HLA typing, were not predicted to generate any HLA-binding peptides (T315I [for Pts 1, 4, 8); E281K [for Pts 5,9], and T315A [Pt 5]). Black boxes - patients expressing both the HLA allele and the BCR-ABL mutation that would generate a binding peptide with affinity of IC50<1000 nM. White boxes - patients without expression of a particular BCR-ABL mutation. White box with “m”-patients harboring a particular BCR-ABL mutation, but not an HLA allele that would be predicted to bind the mutated peptide.
Experimental validation of HLA-binding of predicted BCR-ABL peptides
To validate the HLA binding predictions, we synthesized fifty-five 9- and 10-mers derived from mutated and wild-type BCR-ABL and determined their binding capacity (IC50 nM) using a well-established competitive MHC binding assay (33). We focused on peptides predicted to have high binding affinity (IC50 < than 100 nM) by NetMHC, for which IEDB binding predictions were also available.
We observed that experimental binding was highly related to the extent of concordance between NetMHC and IEDB prediction results. As shown in Figure 3, 14 of 19 (74%) peptides with predicted IC50<100 nM by both NetMHC and IEDB, were experimentally confirmed to have IC50<100 nM. Of the 35 peptides with high binding per NetMHC but intermediate binding per IEDB (IC50 between 100 and 1000 nM), 11 (34%) were verified to have IC50<100 nM, and 21 (66%) with IC50 between 100 and 1000 nM. The only peptide with high binding per NetMHC, but weak predicted binding per IEDB (IC50>1000 nM) demonstrated experimental IC50>1000 nM.
Figure 3. Experimental validation of Class I HLA-peptide binding predictions.

The binding affinities of a subset of mutated BCR-ABL peptides with predicted IC50 <100 nM (by NetMHC) were tested experimentally. Experimental results are grouped based on the predicted affinities per IEDB: filled black circles - predicted high binders by IEDB (IC50<100 nM); grey circles – peptides with predicted IC50 between 100 nM and 1000 nM by IEDB; open circles - peptides with predicted IC50 > 1000 nM by IEDB
We subsequently focused our attention on peptides E255K-A and E255K-B. Each was predicted to strongly bind to HLA-A*03:01 with at least ten-fold greater affinity than their parental peptides by both NetMHC and IEDB. Both were experimentally confirmed to bind HLA-A3 with high affinity (IC50 scores of 208 nM and 17 nM, respectively, Table 2). In addition, E255K-B also bound other HLA-A3 superfamily members HLA-A*11:01, HLA-A*30:01, HLA-A*31:01 and HLA-A*68:01 with high to moderate affinity (IC50: 39-603 nM).
Table 2.
Results of experimental binding studies to HLA-A3 and related supertype family HLA alleles for mutated peptides E255K-A and E255K-B. Dashed lines indicate IC50>70,000 nM.
| HLA-A03 supertype binding capacity (IC50 nM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Peptide | Sequence | A*0301 | A*1101 | A*3001 | A*3101 | A*3301 | A*6801 | |
| E225K-A | WT | KLGGGGQYGE | 2283 | 2281 | 10112 | 9871 | 63631 | 4205 |
| MUT | KLGGGQYGK | 208 | 1074 | 27784 | 31762 | -- | 57106 | |
| E225K-B | WT | EVYEGVWKK | 1074 | 63 | -- | -- | 60 | 10 |
| MUT | KVYEGVWKK | 17 | 39 | 603 | 202 | 42860 | 45 | |
Mutated BCR-ABL peptide E255K-B is immunogenic
T cell lines were generated against the E255K-B peptide but not E255K–A from PBMC of a normal HLA-A3+ volunteer, following four rounds of weekly stimulation of T cells against mutated peptide-pulsed autologous APCs. To assess the specificity of T cell response, we generated HLA-A3 expressing APCs by transfecting a plasmid encoding HLA-A3 into K562 cells (Figure 4A-right panel). As shown in Figure 4B, the expanded T cell lines against E255K-B demonstrated greater IFNγ-ELISpot reactivity against the mutated peptide (2330±325 SFC/million cells, p=0.045) than the parental peptide (1270±42 SFC/million cells). Moreover, T cell reactivity against HLA-A3 expressing APCs that were transfected with a minigene (Figure 4A-left panel) encompassing 227 base pairs surrounding the E255K mutation (1900±85 SFC/million cells) was increased compared to unpulsed APCs (710±70 SFC/million cells) or to APCs pulsed with wild-type peptide (p=0.011). Further confirming that E255K-B is a processed and surface-displayed peptide, we detected an expanded and discrete population of E255K-B reactive CD8+ T cells within E255K-B stimulated T cell lines (at a frequency of 0.64%) compared to control PBMC populations from normal adult volunteers (frequency <0.01%), using an E255K/A3-specific tetramer (Figure 4C). These data together support E255K-B as an endogenously processed and presented peptide epitope in the context of HLA-A3.
Figure 4. E255K-B is immunogenic in a normal HLA-A3+ volunteer.

A. Western Blot confirming E255K minigene expression (left panel) and flow cytometry confirmation of transient HLA-A3 expression in K562 cells (right panel). B. ELISpot assay results (left panel) demonstrating increased IFNγ secretion following exposure to the mutated peptide E255K-B and the minigene encoding the E255K mutation compared to the parental peptide by a T cell line that was generated following repeated peptide stimulations of PBMC derived from a normal adult volunteer. Shown are duplicate wells for each test condition. APCs used for this assay were HLA-A3+ expressing K562 cells. Right panel - Graphical representation of ELISpot results. SFC-spot forming cells; PHA-phytohemagluttinin. C. Flow cytometric detection of a discrete population of E255K-B-reactive CD8+ T cells within the bulk T cell line by immunostaining with a PE-conjugated E255K-B-HLA-A3+ specific tetramer together with anti-CD8-FITC (top panel), compared to a control population of PBMC (bottom panel).
Mutated BCR-ABL peptide can elicit memory responses from imatinib-resistant patients
We next examined the potential of E255K-B for stimulating T cell responses in CML patients. We focused on Patients 2 and 3, from whom cryopreserved PBMC were available. PBMC from patient samples were stimulated twice with E255K-B pulsed APCs, and the expanded CD8+ T cells were subsequently tested for evidence of memory T cell responses by ELISpot, tetramer and cytokine secretion assays. Although Patients 2 and 3 underwent different therapy subsequent to imatinib resistance, both patients demonstrated that T cell immunity to E255K-B can develop in vivo in the setting of effective CML cytoreduction.
The results of the T cell studies from Patients 2 and 3 are shown in Figures 5 and 6, respectively. Patient 2 developed rising BCR-ABL PCR levels while on imatinib and dasatinib, and subsequently initiated treatment with nilotinib, to which a major cytogenetic response was achieved. PBMC collected at the time of nilotinib exposure were stimulated against E255K-B peptide-pulsed A3-expressing APCs, and tested for reactivity by ELISpot assay. Compared to wild-type E255K-B peptide (390±14 SFC/million cells), increased IFNγ secretion was detected against both the mutated E255K-B peptide (1050±71 SFC/million cells; p=0.006) and APCs expressing the E255K minigene (590±14 SFC/million cells; p=0.005) (Figures 5A,B). T cell responses were abrogated by the presence of class I blocking antibody w6/32, down to 360±28 SFC/million cells against E255K-B peptide, and 180±28 SFC/million cells against APCs expressing the E255K minigene. T cells reactive to the mutated peptide were polyfunctional, expressing GM-CSF, IP-10 and TNF-α following exposure to mutated peptide, but not to wild-type peptide (Figure 5C).
Figure 5. The peptide encoded by mutated E255K elicits a memory response in association with disease control following nilotinib therapy.
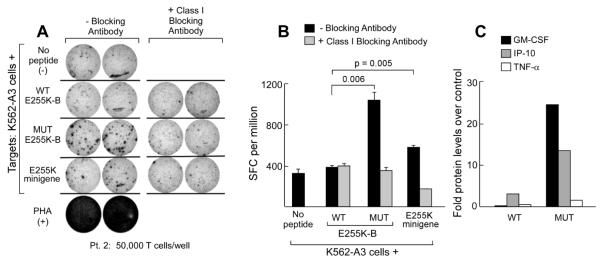
A. ELIspot results from PBMC of Patient 2, collected at the time of complete response to nilotinib, demonstrating IFNγ secretion following exposure to both the E255K-B peptide and the E255K minigene. Results from duplicate wells are displayed. T cell responses were abrogated by blocking with the Class I MHC blocking antibody w6/32, confirming the class I restriction of these T cell responses. B. Graphical representation of ELISpot results. C. T cells reactive to the mutated peptide are polyfunctional, with secretion of IP-10, GM-CSF and TNF-α, measured by Luminex assay. Cytokine secretion results were depicted as fold over control cells (background cytokine secretion by T cells lines exposed to the antigen presenting cells (K562-A3) alone).
Figure 6. The peptide encoded by mutated E255K elicits a memory response in association with disease control following allogeneic HSCT.

A. Graphical representation of ELISpot results showing IFNγ secretion in response to E255K-B and the E255K minigene by T cells expanded from Patient 3 following allo-HSCT, at molecular remission (left panel). Increased reactivity against E255K-B and the E255K minigene was not observed in T cells expanded from control Patients A-C (HLA-A3+, but lacking the mutation E255K) after allo-HSCT (right panel). B. Flow cytometric detection of cytotoxic CD8+ T cells specific for E255K-B in the context of HLA-A3 by tetramer staining in expanded post-allo-HSCT T cells from Patient 3. C. Post-HSCT E255K-B reactive T cells are polyfunctional, secreting increased GM-CSF, IP-10, and TNF-α following exposure to E255K-B peptide or the E255K minigene, compared to pre-HSCT T cells. Cytokine secretion was detected by luminex assay. Cytokine secretion results were depicted as fold over control cells (background cytokine secretion by T cell lines exposed to the antigen presenting cells (K562-A3) alone).
In another example, Patient 3 developed a partial response to dasatinib for treatment of imatinib-resistant disease, and subsequently underwent myeloablative allogeneic HSCT. PBMC collected before and after allo-HSCT were tested for reactivity to E255K-B. As shown in Figure 6, reactivity was expanded in the setting of donor-derived engraftment following HSCT, concurrent with attainment of molecular remission. Increased reactivity by ELISpot (Figure 6A) was seen in post-HSCT CD8+ T cells tested against APCs pulsed with mutated E255K-B peptide (2120±340 SFC/million) but not to wild-type E255K peptide (1140±28 SFC/million, p=0.067). Increased IFNγ secretion was also observed against APCs expressing the E255K minigene (1810±325 SFC/million). Again, reactivity was abrogated in the presence of blocking antibody with 810±156 SFC/million cells detected against E255K-B, and 1260±113 SFC/million cells to APCs expressing the E255K minigene. From the post-HSCT T cells, an expanded population of E255K-B reactive T cells was detected by tetramer staining (0.83%) (Figure 6B). Finally, post-but not pre-transplant T cells from Patient 3 were detected to be polyfunctional on the basis of secretion of GM-CSF, IP-10, and TNF-α following stimulation. In contrast, we were unable to elicit E255K-B specific T cell reactivity from 3 HLA-A3+ CML patients with imatinib resistance, but who did not harbor the E255K mutation, and had undergone allogeneic HSCT (Figure 6A). While polyclonal alloimmune responses are certainly generated following allo-HSCT, our results demonstrate the potential for tumor-specific T cell immunity to also develop in vivo, which may contribute to the GvL responses present following effective HSCT
DISCUSSION
Gene alterations in malignant cells can drive cancers, lead to disease progression, or generate resistance to drug therapy. These same events, on the other hand, have the potential to create novel immunological targets. As they are unique to cancer cells, mutations that generate novel tumor-specific peptides can be targeted immunologically; this may lead to tumor elimination with minimal toxicity. Our current study provides proof-of-concept of the feasibility of an approach to systematically define potential immunogenic epitopes arising from a mutated tumor protein. While the immunogenicity of individual examples of tumor neoantigens has been established (29), our studies focusing on mutated BCR-ABL in CML are the first to demonstrate that tumor-specific gene alterations, arising commonly as a result of drug resistance during the treatment of CML, can generate immunogenic epitopes. Beginning with a set of known common BCR-ABL mutations, we identified peptides encoded by mutations that were predicted to bind to high frequency HLA class I alleles using well-validated prediction algorithms. The majority could be reliably validated through experimental binding studies. We used the example of a novel predicted HLA-A3+ restricted epitope derived from the common E255K mutation to demonstrate that mutated BCR-ABL peptides can stimulate CD8+ T cell responses that arise in association with effective therapeutic response. Our findings not only provide a rationale for developing tumor-specific immunotherapy for CML, but also support the feasibility of a broadly applicable approach for identifying neoplasm-specific neoantigens.
Our study predicted an unexpectedly large number of peptides encoded by common resistance mutations with potential to tightly bind multiple common HLA class I alleles. Our findings are consistent with recent studies that demonstrate that a bioinformatic approach can systematically identify a larger spectrum of immunogenic epitopes than by conventional T cell cloning-based tumor antigen discovery approaches (38). We used both NetMHC and IEDB, as several comparative analyses have identified them as highly sensitive and specific across many common HLA class I alleles (28). In agreement with Trost et al., we found that the intersecting results from muliple prediction tools more accurately predicted MHC class I peptide binding (39). Of the three candidate peptides that we characterized in cellular assays, only one (E255K-B) was predicted as a high binder by both NetMHC and IEDB, and was determined to be a processed and presented epitope. Together, these findings suggest that a screening strategy using binding prediction criteria can efficiently generate a list of promising tumor neoepitopes. Furthermore, they highlight the increasing reliability of class I prediction servers for peptide binding against an expanded repertoire of HLA class I alleles beyond an examination restricted to HLA-A2 (conventionally, the focus of peptide prediction studies).
While BCR-ABL has long been considered a promising immunogen for tumor-specific immunotherapy, clinical studies overall have not been encouraging. This may be related in part to the previously unappreciated restricted spectrum of HLA haplotypes that have been confirmed to present BCR-ABL epitopes and thus have the potential to stimulate T cell responses (HLA-A3, -A11, and -B8 alleles) (40, 41). Vaccine trials using wild-type BCR-ABL junctional peptides with or without heteroclitic peptides have not definitively demonstrated anti-tumor responses (42-44). Furthermore, chronic persistence of BCR-ABL+ tumor cells appears to actively delete high-avidity CML-specific T cells over time (20), and thus many barriers exist to mounting an effective cytolytic T cell response against an existing tumor antigen.
In contrast, our findings support the notion that peptides generated from a newly arising tumor mutation are not tolerogenic. We identified the development of CD8+ T cell responses in two examples of therapeutic response. In the first, BCR-ABL mutation-specific T cell responses developed following institution of a second generation TKI that effectively cytolysed tumor. Imatinib and the other TKIs have been variably reported to impair or enhance anti-tumor immunity (45, 46). Independent of the potential immunosuppressive properties of TKIs, recent studies have revealed multiple mechanisms by which tumor cells can directly exert profound local immunosuppressive effects on T cell function (22, 23). For example, Mumprecht et al. detected increased expression of PD-1 in CD8+ T cells in CML patients that was associated with an exhausted phenotype and with disease progression (22). Thus, for Patient 2, effective elimination of tumor cells by nilotinib presumably contributed to restoration of T cell immunity. In the case of Patient 3, E255K-B specific T cell responses were detected following allo-transplant, a setting that enables the development of robust T cell immunity against tumor-associated antigens (47). In both the cases of Patient 2 and 3, we could show the limited potential for cross-reactivity to the associated wild-type peptide.
Our toolkit for promoting effective tumor immunity has expanded rapidly in recent years, and now include potent adjuvants, a variety of potentially efficacious delivery systems, and agents that can overcome checkpoint blockade (48). Thus, a priority is to identify antigens with truly tumor-specific expression so that a focused immune response can be generated without inciting potentially detrimental immune responses against normal tissue. While the specific application of our studies are limited to a small subset of TKI-treated CML patients who are both HLA-A3 positive and harbor the E255K mutation, our studies provide a rational basis for developing immunotherapy for any TKI-treated CML patient. Larger-scale studies to test immunogenicity of other imatinib-associated BCR-ABL mutation-related peptides would be anticipated to reveal a wider spectrum of immunogenic peptides that could elicit reactivity from a broader selection of HLA alleles. Hence, one could envision an immunotherapeutic approach that could be personalized based on the individual’s HLA typing and BCR-ABL mutation genotype (49, 50). Our approach is further generally applicable to other mutations related to resistance to other TKIs, that have been described to induce an alternate spectrum of BCR-ABL mutations (see Supplemental Figure 2 for analysis of potential peptide binders for common mutations related to dasatinib and nilotinib). BCR-ABL mutations “drive” CML relapse, and would be expected to be expressed in all CML cells—even the malignant progenitor cell populations that have been demonstrated to be generally resistant to TKIs. Thus, an attractive future strategy that could potentially provide durable responses with minimal toxicity would be to combine effective cytoreductive molecularly targeting agents together with polyclonal leukemia-targeting immune therapy.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
Tyrosine kinase inhibitors are currently front-line therapy for chronic myelogenous leukemia (CML), but relapse from drug resistance remains an important problem. Because BCR-ABL mutations arise in the setting of drug resistance, we sought to identify potential neoantigens derived from mutated BCR-ABL that could be targeted for the treatment of this immune-responsive malignancy. We identified polyfunctional E255K-specific CD8+ T cells from two imatinib-resistant HLA-A3+ CML patients arising in the setting of effective anti-CML response to further therapy. Our studies demonstrate that leukemia-driven genetic alterations can be targeted by the immune system in association with a clinical response. Our studies support the approach of immunizing relapsed CML patients against newly acquired tumor neoantigens as a promising anti-leukemia treatment strategy.
ACKNOWLEDGEMENTS
We would like to acknowledge the generous support from the DFCI Pasquarello Tissue Bank, and from the clinical leukemia and transplant teams at the DFCI, Boston. We would especially like to thank excellent technical help from Maris Handley and invaluable advice and assistance from Drs. Pedro Reche, Nir Hacohen, Ellis Reinherz, and Jerome Ritz. A.C. acknowledges support from the Howard Hughes Medical Institute. C.J.W acknowledges support from the Department of Defense (W81XWH-07-1-0080), the Claudia Adams Barr Program Award, the Miles and Eleanor Shore Award, NCI (5R21CA115043-2), the Early Career Physician-Scientist Award of the Howard Hughes Medical Institute, and is a Damon-Runyon Clinical Investigator supported in part by the Damon-Runyon Cancer Research Foundation (CI-38-07).
Footnotes
AUTHORSHIP CONTRIBUTIONS AC, DK and CJW designed the study. AC and CJW wrote the manuscript. AC performed experiments and analyzed data. AA, DD, WZ, NH, JS, GZ and VB performed critical experiments and analyzed data. VN identified BCR-ABL mutations in our patients. RMS provided patient samples. All authors edited the paper.
CONFLICT OF INTEREST DISCLOSURES None of the authors have conflicts of interest to disclose.
REFERENCES
- 1.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2(5):561–66. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 2.Tracy GL, Ann-Marie P, Alexander JM, Owen NW. Tyrosine Kinase Activity and Transformation Potency of bcr-abl Oncogene Products. Science. 1990;247(4):1079–82. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 3.Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75(3):555–62. [PubMed] [Google Scholar]
- 4.Cortes J, Hochhaus A, Hughes T, Kantarjian H. Front-Line and Salvage Therapies With Tyrosine Kinase Inhibitors and Other Treatments in Chronic Myeloid Leukemia. J Clin Oncol. 2011;29(5):524–31. doi: 10.1200/JCO.2010.31.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, et al. Dasatinib versus Imatinib in Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. New England Journal of Medicine. 2010;362(24):2260–70. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- 6.Saglio G, Kim D-W, Issaragrisil S, le Coutre P, Etienne G, Lobo C. Nilotinib versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia. New England Journal of Medicine. 2010;362(24):2251–59. doi: 10.1056/NEJMoa0912614. et. [DOI] [PubMed] [Google Scholar]
- 7.Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7(2):129–41. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 8.O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 9.Druker BJ, Guilhot Fo, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-Year Follow-up of Patients Receiving Imatinib for Chronic Myeloid Leukemia. New England Journal of Medicine. 2006;355(23):2408–17. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 10.Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8(11):1018–29. doi: 10.1016/S1470-2045(07)70342-X. [DOI] [PubMed] [Google Scholar]
- 11.Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A, et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006;12(24):7374–79. doi: 10.1158/1078-0432.CCR-06-1516. [DOI] [PubMed] [Google Scholar]
- 12.Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102(1):276–83. doi: 10.1182/blood-2002-09-2896. [DOI] [PubMed] [Google Scholar]
- 13.Lahaye T, Riehm B, Berger U, Paschka P, Muller MC, Kreil S, et al. Response and resistance in 300 patients with BCR-ABL-positive leukemias treated with imatinib in a single center: a 4.5-year follow-up. Cancer. 2005;103(8):1659–69. doi: 10.1002/cncr.20922. [DOI] [PubMed] [Google Scholar]
- 14.Skaggs BJ, Gorre ME, Ryvkin A, Burgess MR, Xie Y, Han Y, et al. Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutants. Proceedings of the National Academy of Sciences. 2006;103(51):19466–71. doi: 10.1073/pnas.0609239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griswold IJ, MacPartlin M, Bumm T, Goss VL, O’Hare T, Lee KA, et al. Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib. Mol Cell Biol. 2006;26(16):6082–93. doi: 10.1128/MCB.02202-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.le Coutre P, Ottmann OG, Giles F, Kim D-W, Cortes J, Gattermann N, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood. 2008;111(4):1834–39. doi: 10.1182/blood-2007-04-083196. [DOI] [PubMed] [Google Scholar]
- 17.Apperley JF, Cortes JE, Kim D-W, Roy L, Roboz GJ, Rosti G, et al. Dasatinib in the Treatment of Chronic Myeloid Leukemia in Accelerated Phase After Imatinib Failure: The START A Trial. Journal of Clinical Oncology. 2009;27(21):3472–79. doi: 10.1200/JCO.2007.14.3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cortes J, Kim DW, Raffoux E, Martinelli G, Ritchie E, Roy L, et al. Efficacy and safety of dasatinib in imatinib-resistant or -intolerant patients with chronic myeloid leukemia in blast phase. Leukemia. 2008;22(12):2176–83. doi: 10.1038/leu.2008.221. [DOI] [PubMed] [Google Scholar]
- 19.Wu CJ, Ritz J. Induction of tumor immunity following allogeneic stem cell transplantation. Adv Immunol. 2006;90:133–73. doi: 10.1016/S0065-2776(06)90004-2. [DOI] [PubMed] [Google Scholar]
- 20.Molldrem JJ, Lee PP, Kant S, Wieder E, Jiang W, Lu S, et al. Chronic myelogenous leukemia shapes host immunity by selective deletion of high-avidity leukemia-specific T cells. J Clin Invest. 2003;111(5):639–47. doi: 10.1172/JCI16398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gannage M, Abel M, Michallet AS, Delluc S, Lambert M, Giraudier S, et al. Ex vivo characterization of multiepitopic tumor-specific CD8 T cells in patients with chronic myeloid leukemia: implications for vaccine development and adoptive cellular immunotherapy. J Immunol. 2005;174(12):8210–18. doi: 10.4049/jimmunol.174.12.8210. [DOI] [PubMed] [Google Scholar]
- 22.Mumprecht S, Schurch C, Schwaller J, Solenthaler M, Ochsenbein AF. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood. 2009;114(8):1528–36. doi: 10.1182/blood-2008-09-179697. [DOI] [PubMed] [Google Scholar]
- 23.Mumprecht S, Claus C, Schurch C, Pavelic V, Matter MS, Ochsenbein AF. Defective homing and impaired induction of cytotoxic T cells by BCR/ABL-expressing dendritic cells. Blood. 2009;113(19):4681–89. doi: 10.1182/blood-2008-05-156471. [DOI] [PubMed] [Google Scholar]
- 24.Caskey M, Lefebvre F, Filali-Mouhim A, Cameron MJ, Goulet JP, Haddad EK, et al. Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J Exp Med. 2011;208:2357–2366. doi: 10.1084/jem.20111171. Br. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and Activity of Anti-PD-L1 Antibody in Patients with Advanced Cancer. N Eng J Med. 2012 Jun 2; doi: 10.1056/NEJMoa1200694. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N Engl J Med. 2012 Jun 2; doi: 10.1056/NEJMoa1200690. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010 Aug 19;363(8):711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin HH, Ray S, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC class I peptide binding prediction servers: applications for vaccine research. BMC Immunol. 2008;9(1):8–8. doi: 10.1186/1471-2172-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sensi M, Anichini A. Unique tumor antigens: evidence for immune control of genome integrity and immunogenic targets for T cell-mediated patient-specific immunotherapy. Clin Cancer Res. 2006;12(17):5023–32. doi: 10.1158/1078-0432.CCR-05-2682. [DOI] [PubMed] [Google Scholar]
- 30.Nardi V, Raz T, Cao X, Wu CJ, Stone RM, Cortes J, et al. Quantitative monitoring by polymerase colony assay of known mutations resistant to ABL kinase inhibitors. Oncogene. 2007;27(6):775–82. doi: 10.1038/sj.onc.1210698. [DOI] [PubMed] [Google Scholar]
- 31.Zhang GL, Ansari HR, Bradley P, Cawley GC, Hertz T, Hu X, et al. Machine learning competition in immunology-Prediction of HLA class I molecules. Journal of Immunological Methods. 2009 doi: 10.1016/j.jim.2011.09.010. (0) [DOI] [PubMed] [Google Scholar]
- 32.Nielsen M, Lundegaard C, Worning P, Hvid CS, Lamberth K, Buus S, et al. Improved prediction of MHC class I and class II epitopes using a novel Gibbs sampling approach. Bioinformatics. 2004;20(9):1388–97. doi: 10.1093/bioinformatics/bth100. [DOI] [PubMed] [Google Scholar]
- 33.Sidney J, Southwood S, Oseroff C, del Guercio MF, Sette A, Grey HM. Measurement of MHC/peptide interactions by gel filtration. Curr Protoc Immunol. 2001 doi: 10.1002/0471142735.im1803s31. Chapter 18:Unit-Uni3. [DOI] [PubMed] [Google Scholar]
- 34.Gulukota K, DeLisi C. Neural network method for predicting peptides that bind major histocompatibility complex molecules. Methods Mol Biol. 2001;156:201–09. doi: 10.1385/1-59259-062-4:201. [DOI] [PubMed] [Google Scholar]
- 35.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 36.Schultze JL, Michalak S, Seamon MJ, Dranoff G, Jung K, Daley J, et al. CD40-activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J Clin Invest. 1997;100(11):2757–65. doi: 10.1172/JCI119822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105(7):2640–53. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 38.Ofran Y, Kim HT, Brusic V, Blake L, Mandrell M, Wu CJ, et al. Diverse Patterns of T-Cell Response against Multiple Newly Identified Human Y Chromosome-Encoded Minor Histocompatibility Epitopes. Clin Cancer Res. 2010;16(5):1642–51. doi: 10.1158/1078-0432.CCR-09-2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trost B, Bickis M, Kusalik A. Strength in numbers: achieving greater accuracy in MHC-I binding prediction by combining the results from multiple prediction tools. Immunome Res. 2007;3(1):5–5. doi: 10.1186/1745-7580-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark RE, Dodi IA, Hill SC, Lill JR, Aubert G, Macintyre AR, et al. Direct evidence that leukemic cells present HLA-associated immunogenic peptides derived from the BCR-ABL b3a2 fusion protein. Blood. 2001;98(10):2887–93. doi: 10.1182/blood.v98.10.2887. [DOI] [PubMed] [Google Scholar]
- 41.Bocchia M, Korontsvit T, Xu Q, Mackinnon S, Yang SY, Sette A, et al. Specific human cellular immunity to bcr-abl oncogene-derived peptides. Blood. 1996;87(9):3587–92. [PubMed] [Google Scholar]
- 42.Pinilla-Ibarz J, Cathcart K, Korontsvit T, Soignet S, Bocchia M, Caggiano J, et al. Vaccination of patients with chronic myelogenous leukemia with bcr-abl oncogene breakpoint fusion peptides generates specific immune responses. Blood. 2000;95(5):1781–87. [PubMed] [Google Scholar]
- 43.Bocchia M, Gentili S, Abruzzese E, Fanelli A, Iuliano F, Tabilio A, et al. Effect of a p210 multipeptide vaccine associated with imatinib or interferon in patients with chronic myeloid leukaemia and persistent residual disease: a multicentre observational trial. The Lancet. 2005;365(9460):657–62. doi: 10.1016/S0140-6736(05)17945-8. [DOI] [PubMed] [Google Scholar]
- 44.Rojas JM, Knight K, Wang L, Clark RE. Clinical evaluation of BCR-ABL peptide immunisation in chronic myeloid leukaemia: results of the EPIC study. Leukemia. 2007;21(11):2287–95. doi: 10.1038/sj.leu.2404858. [DOI] [PubMed] [Google Scholar]
- 45.Wang H, Cheng F, Cuenca A, Horna P, Zheng Z, Bhalla K, et al. Imatinib mesylate (STI-571) enhances antigen-presenting cell function and overcomes tumor-induced CD4+ T-cell tolerance. Blood. 2005;105(3):1135–43. doi: 10.1182/blood-2004-01-0027. [DOI] [PubMed] [Google Scholar]
- 46.Dietz AB, Souan L, Knutson GJ, Bulur PA, Litzow MR, Vuk-Pavlovic S. Imatinib mesylate inhibits T-cell proliferation in vitro and delayed-type hypersensitivity in vivo. Blood. 2004;104(4):1094–99. doi: 10.1182/blood-2003-12-4266. [DOI] [PubMed] [Google Scholar]
- 47.Zhang W, Choi J, Zeng W, Rogers SA, Alyea EP, Rheinwald JG, et al. Graft-versus-leukemia antigen CML66 elicits coordinated B-cell and T-cell immunity after donor lymphocyte infusion. Clin Cancer Res. 2010;16(10):2729–39. doi: 10.1158/1078-0432.CCR-10-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brusic A, Wu CJ. Enhancing graft-versus-leukemia after transplant: the rise of anti-cancer vaccines. Front Biosci. 2012;17:635–55. doi: 10.2741/3949. [DOI] [PubMed] [Google Scholar]
- 49.Chu S, Xu H, Shah NP, Snyder DS, Forman SJ, Sawyers CL, et al. Detection of BCR-ABL kinase mutations in CD34+ cells from chronic myelogenous leukemia patients in complete cytogenetic remission on imatinib mesylate treatment. Blood. 2005;105(5):2093–98. doi: 10.1182/blood-2004-03-1114. [DOI] [PubMed] [Google Scholar]
- 50.Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–25. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.