

Abstract
Mesenchymal stem cell (MSC) transplantation after ischemia/reperfusion (I/R) injury reduces infarct size and improves cardiac function. We used mouse ventricular myocytes (VMs) in an in vitro model of I/R to determine the mechanism by which MSCs prevent reperfusion injury by paracrine signaling. Exposure of mouse VMs to an ischemic challenge depolarized their mitochondrial membrane potential (Ψmito), increased their diastolic Ca2+, and significantly attenuated cell shortening. Reperfusion of VMs with Ctrl tyrode or MSC-conditioned tyrode (ConT) resulted in a transient increase of the Ca2+ transient amplitudes in all cells. ConT-reperfused cells exhibited a decreased number early after depolarization (EADs) (ConT: 6.3% vs. Ctrl: 28.4%) and prolonged survival (ConT: 58% vs. Ctrl: 33%). Ψmito rapidly recovered in Ctrl as well as ConT-treated VMs on reperfusion; however, in Ctrl solution, an exaggerated hyperpolarization of Ψmito was determined that preceded the collapse of Ψmito. The ability of ConT to attenuate the hyperpolarization of Ψmito was suppressed on inhibition of the PI3K/Akt signaling pathway or IK,ATP. However, protection of Ψmito was best mimicked by the reactive oxygen species (ROS) scavenger mitoTEMPO. Analysis of ConT revealed a significant antioxidant capacity that was linked to the presence of extracellular superoxide dismutase (SOD3) in ConT. In conclusion, MSC ConT protects VMs from simulated I/R injury by its SOD3-mediated antioxidant capacity and by delaying the recovery of Ψmito through Akt-mediated opening of IK,ATP. These changes attenuate reperfusion-induced ROS production and prevent the opening of the permeability transition pore and arrhythmic Ca2+ release.
Introduction
The benefit of transplantation of bone marrow-derived mesenchymal stem cells (MSCs) after cardiac infarction has been evaluated in many preclinical and clinical studies [1,2]. Although most studies indicate an improved left ventricular ejection, a comparison between studies is difficult due to variations in (1) cell selection, (2) number of cells transplanted, (3) time of treatment, (4) method of cell delivery, and (5) time of follow up. The differences in cell application may affect the mechanism by which cardioprotection is achieved. Potential mechanisms of MSC-mediated protection from ischemic injury are the differentiation of MSCs into cardiomyocytes, the stimulation of cardiac stem cell proliferation, and/or cells that enhance vascularization [3]. However, the low retention rate of MSCs after transplantation and their low propensity to differentiate into a cardiac phenotype [4] make it unlikely that cell replacement is the primary mechanism of benefit. Since MSCs secrete an array of cytokines and growth factors, recent research has focused on the relevance of paracrine signaling in MSC-mediated cardioprotection [5]. It was demonstrated that MSC-conditioned medium can promote vascularization, enhance the activation of body-owned stem cells [3,5], increase cell survival [6], and enhance contractility or cellular remodeling. For vascular endothelial growth factor [7], transforming growth factor [8], and tumor necrosis factor [9], it could be demonstrated that changes in their secretion level correlate with modifications in the MSC-induced signaling.
Ischemia in the cardiac muscle results in the loss of energy production and changes in the intracellular ion homeostasis [10]. While rapid re-oxygenation is the treatment of choice, reperfusion itself causes further damage to the myocardium. pH recovery through the sodium hydrogen exchanger increases [Na]i and promotes Ca2+ entry through reverse mode sodium calcium exchanger (NCX), which can lead to Ca2+ overload-induced arrhythmia [11]. The mitochondrial membrane potential (Ψmito) depolarizes during ischemia followed by a loss of ATP production [12]. While mitochondria can repolarize during reperfusion, the reestablished oxygen supply promotes a surge of reactive oxygen species (ROS) production that can lead to oxidation of proteins of the electron transport chain or opening of the permeability transition pore (PTP) and collapse of Ψmito [12–14]. Prevention of Ca2+ overload as well as delay of Ψmito recovery were shown to be cardioprotective [13,15], and PI3K/Akt signaling has been postulated as a mediator of postconditioning [10,16]. MSC-mediated cardioprotection from ischemia/reperfusion (I/R) injury has been demonstrated through preconditioning of the cardiac tissue [17]. In these cases, postinfarct recovery was enhanced when MSCs expressed increased levels of interleukin-18, and depended on the secretion of the vascular endothelial growth factor [8,18–20]. However, a postconditioning benefit of MSCs was also demonstrated when cells were injected 2 h after ischemic injury [21].
We have previously demonstrated [22] that conditioned tyrode (ConT) obtained from MSCs enhances cardiac excitation–contraction coupling (ECC) by Akt-mediated activation of endothelial nitric oxide synthase (eNOS) and a subsequent increase in L-type Ca2+ current (ICa,L) and enhances Ca2+ uptake through the sarcoplasmic reticulum Ca2+ ATPase (SERCA). While we could also describe an enhanced survival of isolated mouse ventricular myocytes (VMs) in ConT, the context of this anti-apoptotic mechanism during I/R injury remained to be determined. To test the hypothesis that ConT confers a postconditioning benefit, we used an in vitro model of simulated I/R and determined the physiological properties of isolated VMs by monitoring field stimulation-induced changes in [Ca2+]i and Ψmito.
Methods
Ethical approval
All animal procedures were performed with the approval of IACUC at the University of Illinois at Chicago and in accordance with the National Institute of Health's Guide for the Care and Use of Laboratory Animals.
Cardiomyocyte isolation and MSC-ConT
VMs were isolated from 3–5 month-old C57BL/6 mice by Langendorff perfusion. The mice were anesthetized by inhalation of Isoflurane (3%). Anesthesia was confirmed by lack of response to toe pinch before the aorta was transected and the heart was excised [22,23]. Cells were plated on laminin-coated (Sigma Aldrich; 1 mg/mL) 25 mm coverslips in standard tyrode solution (in mmol/L: NaCl 130, KCl 5.4, CaCl2 1, MgCl2 1.5, NaHCO3 10, Glucose 10, HEPES 25, l-glutamine 4, nonessential amino acids 0.1; pH 7.4). Mouse bone marrow was isolated from tibia and femur bones [4,24], and MSCs were enriched for CD5, CD45R, CD11b, Gr1, and TER119 (Murine Progenitor Enrichment Cocktail, Stemcell Technologies) and were found to be Sca-1+, CD34+, ckit+, CD105+, and CD90.1+ positive and negative for CD45−. Cells were cultured in Mesencult medium (Stemcell Technologies) supplemented with penicillin and streptomycin. ConT was obtained by overnight incubation (15 h) of 80% confluent MSC cultures with tyrode (in mmol/L: NaCl 130, KCl 5.4, CaCl2 1, MgCl2 1.5, NaHCO3 10, glucose 10, HEPES 25, l-glutamine 4, nonessential amino acids 0.1; pH 7.4) at 37°C as previously described [22]. All experiments were performed at room temperature.
In vitro model of simulated I/R
To mimic ischemic conditions in vitro, VMs were superfused with a solution that mimics hypoxia, hyperkalemia, acidosis, and lactate accumulation [25]. The solution contained (mmol/L): NaCl 113, NaHCO3 6, NAH2PO4 0.9, KCl 8, MgCl2 1.5, CaCl2 1, Na/lactate 20, (pH 6.8). The solution was gassed with 95% N2, 5% CO2. Ischemia was maintained for 15 min before reperfusion with either control (Ctrl) or ConT solution. Using a trypan blue dye exclusion assay, the effect of ConT on cell survival was evaluated at the end of reperfusion (15 min). Trypan blue was added for a final concentration of 0.4%, and slides were evaluated for the percentage of surviving cells [26].
Detection of protein carbonylation
VMs were plated on laminin-coated culture dishes at 70% density and exposed to the simulated I/R protocol. During the procedure, the cells were field stimulated (0.5 Hz; multi-channel stimulator; IonOptix, LLC). After 15 min of reperfusion, the myocytes were recovered and processed for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting as previously described (8). Ninety micrograms of cell lysate was loaded per well. For the detection of ROS-mediated oxidative modification (carbonylation) of proteins, introduced carbonyl groups were derivatized to 2,4-dinitrophenyl (DNP)-hydrazone in a reaction with DNP-hydrazine solution for 5 min by directly treating the blotted nitrocellulose membrane. Western blotting was completed using an anti-DNP moiety antibody (OxyBlot Kit No. S7150; Millipore). To evaluate equivalent sample loading, the blot was stripped and re-probed with primary antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH- No. 5174; Cell Signaling Technology). To determine the induction of apoptosis, cell lysates were probed for cleaved caspase-9 (Asp330; No. 9501; Cell Signaling Technology).
Measurements of [Ca2+]i, mitochondrial membrane potential and ROS production
Measurements of [Ca2+]i were performed as previously described [22]. For recordings of Ψmito, VMs were loaded with tetramethylrhodamine, methyl, ester, and perchlorate (TMRM: 100 nmol/L) for 40 min; after that, 40 nmol/L TMRM was maintained in all extracellular solutions [27]. Dynamic changes in Ψmito were determined by superfusing VMs with (1) oligomycin (OM: 10 μmol/L), a blocker of proton entry via ATP-synthase, which causes a hyperpolarization of Ψmito or (2) fluoromethoxy-phenylhydrazone (FCCP), which induces a collapse of Ψmito [28] (not shown). No significant changes in Ψmito were determined when VMs were superfused with either 15 mmol/L [K]o or pHo 6.7, ruling out changes in TMRM due to experimental conditions alone (not shown). Dynamic changes in mitochondrial ROS production were identified by loading VMs with the mitochondrial dye MitoSox Red (20 min, 0.5 μmol/L). MitoSox fluorescence was measured by taking 2D confocal images (0.5 Hz; excitation: 400 nm; emission: 561 nm). ROS production was quantified as the change in MitoSox fluorescence over time (Δfluorescence a.u./5 min)[29].
Nitro blue tetrazolium in gel assay for superoxide dismutase activity
Superoxide dismutase (SOD; Sigma-S5395) in units of activity and ConT proteins were directly compared using an in-gel assay for enzymatic activity. For this assay, in situ visualization of anti-oxidant/SOD protein activity occurs as an achromatic gel band resulting from the inhibition of nitro blue tetrazolium (NBT) chromogenic reduction within the gel [30,31]. Briefly, protein samples were mixed with 5X-loading buffer (1.5 mol/L Tris, pH 8.3) without 2-mercaptoethanol, SDS, or heating and loaded directly onto precast 4%–20% Novex tris-glycine gels (Invitrogen) for nondenaturing PAGE. After the dye front reached the bottom of the gel, it was transferred to a solution containing 28 μmol/L riboflavin and 28 mmol/L TEMED in potassium phosphate buffer (pH 7.8). A 20 min incubation in the above solution was followed by incubation in 2.45 mmol/L NBT until the visualization of SOD activity was optimal for evaluation and image acquisition.
Chemicals
Fluo-4/AM and TMRM were purchased from Invitrogen, LY294002 from Cayman Chemicals, 5-hydroxydecanoate (5-HD), diazoxide (DZX), oligomycin (OM) and carbonyl cyanide-p-tri-FCCP from Sigma, the Akt inhibitor V (triciribine) from EMD/Calbiochem, and mitoTEMPO from Enzo Lifesciences.
Statistics
Data sets other than for cell survival were statistically evaluated using paired and un-paired t test. Cell survival was shown as a Kaplan–Meier curve, and differences in viability to control were evaluated with a log-rank test [26] (GraphPad Prism software). All averaged data are presented as means±SEM.
Results
ConT enhances cell survival and attenuates arrhythmic activity after simulated I/R
We have previously demonstrated that MSC-derived ConT significantly enhances cell survival of isolated VMs in culture [22]. To determine whether this translates into the protection of VMs during simulated I/R injury, we monitored intracellular Ca2+ transients from isolated, field-stimulated VMs that were loaded with Fluo-4/AM. Figure 1A and B shows representative F/F0 plots in preischemic (pI) and ischemic conditions (15 min), as well as 1 min, and 5 min after reperfusion. While all VMs were treated identically during pI and ischemic conditions, reperfusion was performed with either Ctrl solution (Fig. 1A) or ConT (Fig. 1B). Cell survival after reperfusion (Fig. 1D) and the occurrence of EAD (Fig. 1C: EADs: NoEAD×100/Notwitch during 10 s) were quantified [32]. Analysis of VMs 15 min after reperfusion by a trypan blue exclusion assay showed that ConT promoted cell survival after simulated I/R injury (ConT: 56.9%±2.2%, n=9 slides, 2,123 VMs vs. Ctrl: 35.8%±1.3%, n=9 slides, 2,485 cells; P<0.05; Fig. 1D) and suppressed EADs at 5 min and 10 min of reperfusion (Ctrl: 19.8%±5.5% and 28.4%±5%, n=21 vs. ConT: 3.7%±1.4% and 6.3%±2%, n=29). An examination of oxidative protein modifications (carbonylation) in whole cell lysates recovered 15 min after reperfusion showed overall increased levels of detectable carbonylation in different protein bands. Carbonylation levels were comparable to untreated VMs when ConT was used for reperfusion (n=3, 2 shown; Fig. 1E). In addition, ConT reperfusion prevented the increase in cleaved caspase-9 levels following the simulated I/R protocol when Ctrl solution was used for reperfusion (Fig. 1F, n=3). The results indicate that ConT mediates a postconditioning benefit during reperfusion after an ischemic challenge.
FIG. 1.

Reperfusion with conditioned tyrode (ConT) protects ventricular myocytes (VMs) early after depolarization (EADs) and cell death. F/F0 plots show changes in [Ca2+]i in field-stimulated VMs during preischemic (pI) and ischemic conditions and after 1 and 5 min of reperfusion with Ctrl (A) solution or ConT (B). Bar graphs show the percentage of EADs (C: *P<0.05 compared with Ctrl) after 5 min and 10 min of reperfusion in Ctrl (black) and ConT (gray). (D) Trypan blue exclusion assay after 15 min of reperfusion (Ctrl: nine slides, 2,485 VMs; ConT: nine slides, 2,123 VMs; *P<0.05); (E) Total VM lysates resolved on a 4%–20% gradient gel following the simulated ischemia/reperfusion (I/R) protocol. Anti-2,4-dinitrophenyl antibody (Ab) western blotting for protein carbonylation is shown for untreated cells (Ctrl) and cells after 15 min of reperfusion with either Ctrl (CtrlR) or ConT. GAPDH immunoblotting is presented as a loading control. (F) Representative western blotting panel (n=3) showing cleaved caspase-9 levels in cardiomyocytes following a simulated I/R protocol. GAPDH is the loading control.
Changes in stimulation-induced Ca2+ transients during simulated I/R
To determine whether ConT-induced changes in ECC [22] contribute to changes in VM survival [Ca2+]i, cell shortening and SR load were recorded during the simulated I/R protocol. During ischemia, no significant change in Ca2+ transient amplitude (Fig. 2B) or diastolic cell length (Fig. 2D) was determined. However, diastolic Ca2+ (Fig. 2A) and the Ca2+ transient decay constant (τCa, Fig. 2C) increased significantly, and cell shortening almost ceased (Fig. 2E). On reperfusion, the Ca2+ transient amplitudes significantly increased (ΔF/F0, Ctrl: pI: 2.98±0.2; R1′: 6.26±0.44; n=12, P<0.01) before returning to pI levels. Concomitant with this increase, cell shortening was enhanced [cell length (μm): Ctrl: pI: 1.83±0.27; R1′: 7.2±1.99; n=8, P<0.05), but incomplete relaxation left the cell in a state of contracture (Fig. 2D). ConT reperfusion did not prevent the increase in [Ca2+]i (ΔF/F0, ConT: 2.05±0.14; R1′: 4.83±0.37; n=12, P<0.01) or decrease in τCa (Fig. 2B, C), but the diastolic cell length remained comparable to pI conditions (Fig. 2D, E) although cell shortening was enhanced during reperfusion [(μm), ConT: preischemia: 1.13±0.29; R1′: 6.12±1.43; n=6, P<0.01; Fig. 2E).
FIG. 2.

ConT does not change cellular Ca2+ handling during reperfusion. The change in diastolic Ca2+ (A), Ca2+ transient amplitude (B), Ca2+ transient decay constant (τCa, C), and diastolic cell length (D; n=24) normalized to pI conditions. Changes are shown for ischemic (I) conditions and reperfusion (R) for Ctrl (n=12) and ConT (n=12)-treated cells after 1 min (1′R) and 15 min (15′R). (E) Representative stimulation induced changes in cell length from a Ctrl (top) and a ConT (bottom)-reperfused cell. (F) Caffeine transient amplitude during pI and reperfusion (R) in Ctrl (F) and ConT (G)-reperfused VMs, (*P<0.05 compared with pI; &P<0.05 compared with ConT).
ConT is known to enhance SERCA-mediated Ca2+ uptake, resulting in an increased SR load [22]. Caffeine (10 mmol/L) was applied to identify differences in SR load between Ctrl and ConT reperfused VMs under pI and ischemic conditions, and during reperfusion. The caffeine transient amplitude was different in ConT and Ctrl-reperfused VMs compared with preischemia. In Ctrl-reperfused cells (Fig. 2F), it decreased over the time of reperfusion (ΔF/F0: Ctrl pI: 6.62±0.33; R10′: 5.61±0.3; n=4, P<0.05); whereas in ConT-reperfused VMs (Fig. 2G), it was transiently increased at R3′ (ConT: pI: 5.4±0.36; R3′: 6.0±0.4; n=8, P<0.05). Due to increased cell death in Ctrl-reperfused cells, the second caffeine transient had to be induced at 10 min in Ctrl rather than at 20 min for the ConT-reperfused cells. No difference in the caffeine transient amplitude was determined between the two treatment groups. The results indicate that there was no ConT induced change in ECC during reperfusion; however, it preserves SR load likely by the suppression of EADs.
ConT preserves the mitochondrial membrane potential (Ψmito) during simulated I/R
The recovery of VMs from I/R injury depends on the recovery of the cells energy metabolism [14,33]. To determine whether differences in cell survival in ConT and Ctrl-reperfused cells are based on I/R damage of cardiac mitochondria, we measured Ψmito with the voltage-sensitive dye TMRM during the simulated I/R protocol. During ischemia, Ψmito depolarized but rapidly recovered on reperfusion with Ctrl solution or ConT (Fig. 3A). In Ctrl-reperfused VMs, an exaggerated hyperpolarization of Ψmito could be observed that exceeded pI fluorescence by 111.7%±5.9% (Fig. 3D; n=9; at R5′ P<0.05 to pI). During reperfusion with ConT, Ψmito recovered to 95.5%±2.3% (Fig. 3D; n=5; at R5′ P<0.05 to Ctrl) of pI fluorescence. In Ctrl VMs, this transient hyperpolarization was followed by a continuous depolarization of Ψmito, which reached significantly lower levels compared with ConT-treated cells at 15 min of reperfusion (Ctrl: 81.3%±3.1%; n=9; ConT: 101.1%±2.6%; n=5; at R15′ P<0.05). The difference was independent of the cells' contractile properties (not shown). The results indicate that ConT treatment on reperfusion can better preserve Ψmito.
FIG. 3.

Changes in Ψmito of VMs during simulated I/R injury. (A) Time-dependent change of TMRM fluorescence during the different phases of simulated I/R for VMs reperfused with Ctrl tyrode (black, n=9) or ConT (gray, n=5). [*P<0.05 compared with preischemia; &P<0.05 compared with ConT at 5 min of reperfusion (R5′)]. (B) Representative images of TMRM loaded VMs at time points indicated. (C) Change in TMRM fluorescence during simulated I/R for VMs reperfused with LY294002 (LY: 10 μmol/L) supplemented ConT (black circles, n=3). Mean fluorescence of ConT-treated cells is shown as a reference (−). (D) Ψmito at 5 min of reperfusion normalized to pI values for Ctrl (black), ConT (gray), and ConT supplemented with AktiV (AktiV: 20 μmol/L; white, n=4) or +LY (hatched, n=3) (*P<0.05 compared with pI; &P<0.05 compared with Ctrl at R5′).
Mechanism of ConT-mediated protection of Ψmito
ConT was previously determined to be an activator of the PI3K/Akt signal transduction pathway [22]. To determine the role of PI3K/Akt in the protection of Ψmito, we supplemented ConT during reperfusion with inhibitors of PI3K (LY294002, LY: 10 μmol/L) or Akt [Akt inhibitor V (Akt iV): 20 μmol/L] [22]. In the presence of LY or Akt iV, ConT no longer prevented the exaggerated hyperpolarization of Ψmito on reperfusion (Fig. 3C, D: LY: 139.1%±14.9%; n=3; Akt iV: 114.7%±4.7%; n=4, at 5 min of reperfusion: P<0.05). The results indicate that ConT attenuates the hyperpolarization of Ψmito through PI3K/Akt-dependent signaling. In the following, we used the degree of hyperpolarization on reperfusion as a measure of ConT action.
The recovery of Ψmito depends on the functional recovery of proton pumps, which allow for the simultaneous e− flow across the electron transport chain [13]. Potassium influx through the mitochondrial ATP-sensitive potassium channel (IK,ATP) can counteract the recovery of Ψmito. Since IK,ATP is proposed to be activated downstream of the PI3K/Akt pathway [34], we hypothesized that ConT activates IK,ATP. To test this hypothesis, ConT was supplemented with the IK,ATP inhibitor 5-HD (500 μmol/L) [35]. During ConT5-HD reperfusion, the action of ConT was attenuated and Ψmito now exhibited an increased hyperpolarization (Fig. 4A, B; ConT5-HD: 124.4±3; n=6 at 5 min of reperfusion). In addition, five out of seven cells died within the first 3 min of reperfusion. Next, we aimed at determining whether the opening of IK,ATP can mimic the attenuated recovery of Ψmito during ConT treatment. For this purpose, Ctrl solution was supplemented with the IK,ATP opener DZX (200 μmol/L; CtrlDZX) [36]. On reperfusion with CtrlDZX, no increase of Ψmito above pI levels was determined (CtrlDZX: 99.3±4.8; n=7 at 5 min of reperfusion) and now, seven out of seven VMs survived for approximately 20 min of reperfusion. No significant difference of Ψmito between ConT5-HD and CtrlDZX could be determined at 15 min of reperfusion. The experiments support the hypothesis that ConT promotes cell survival by Akt-dependent activation of the uncoupling protein IK,ATP attenuating the rapid recovery of Ψmito.
FIG. 4.

Reperfusion-induced Ψmito hyperpolarization depends on IK,ATP open probability. (A) Changes in TMRM fluorescence during the simulated I/R protocol normalized to the fluorescence during preischemia is shown for VMs reperfused with Ctrl solution supplemented with 200 μmol/L diazoxide (DZX) (−: n=7) or ConT supplemented with 500 μmol/L 5-hydroxydecanoate (5-HD) (black circles: n=6). (B) Bar graph presents normalized TMRM fluorescence at 5 min of reperfusion (R5′). (C) Change in TMRM fluorescence during superfusion with 5-HD (top) or DZX (bottom). (D) Normalized TMRM fluorescence during the I/R protocol. VMs were reperfused with Ctrl solution (gray circles: n=5) or ConT (black circles: n=5) supplemented with the permeability transition pore (PTP) inhibitor cyclosporin A (1 μmol/L). Data are summarized in (E) [*P<0.05 compared with pI; &P<0.05 compared with Ctrl+DZX at 5 min of reperfusion (R5′)].
Inhibition of the mitochondrial PTP has been described to protect VMs from I/R injury; however, early and transient opening of the channel could also attenuate Ψmito hyperpolarization [37]. To determine the role of PTP in Ψmito recovery after ischemia, we supplemented Ctrl as well as ConT with the PTP inhibitor cyclosporin A (CsA: 1 μmol/L). CtrlCsA reperfusion could not prevent the exaggerated hyperpolarization at 5 min of reperfusion; however, in contrast to Ctrl-reperfused cells, Ψmito remained constant for the duration of reperfusion (Fig. 4D, E), indicating that the PTP opening might be responsible for the decay of Ψmito after reperfusion. In ConTCsA-reperfused VMs, Ψmito recovery was not significantly different from ConT-reperfused cells. The data support the hypothesis that the PTP opening does not play a role in the delayed hyperpolarization of Ψmito, but the opening contributes to the collapse of Ψmito in the later phase of reperfusion.
ConT prevents ROS production and concomitant arrhythmic Ca2+ release
ROS production is significantly enhanced during reperfusion when oxygen supply is re-established and Ψmito as well as the intracellular pH return to physiological levels [12,14]. To determine whether ROS production influences the recovery of Ψmito, in subsequent EADs and cell survival, we supplemented Ctrl reperfusion solution with the mitochondrial ROS scavenger mitoTEMPO (5 μmol/L) [12]. As shown in Fig. 5C and D, mitoTEMPO significantly enhanced cell survival and attenuated the occurrence of EADs, thereby reproducing the benefit of ConT. In addition, mitoTEMPO even reduced the exaggerated hyperpolarization of Ψmito (Fig. 5A, B) (significantly different at 15 min compared with Ctrl), suggesting that reperfusion-induced mitochondrial ROS production further enhances Ψmito hyperpolarization. To identify whether ConT changes ROS production during the reperfusion period, we exposed mitoSOX-loaded VMs to the I/R protocol. Constant stimulation led to a steady increase of fluorescence with an attenuated slope during ischemic conditions. Reperfusion in Ctrl, ConT, and Ctrl+mitoTEMPO induced a rapid increase in ROS production; however, the total amount of fluorescent change was significantly reduced in ConT and Ctrl+mitoTEMPO-treated VMs (Fig. 6A), indicating that ROS production and Ψmito hyperpolarization are closely linked events and have an adverse effect on cell survival.
FIG. 5.

Scavenging of intra-mitochondrial ROS enhances cell survival on I/R. (A) Changes in TMRM fluorescence during the simulated I/R protocol normalized to preischemia for VMs reperfused with Ctrl+mitoTempo (circles: mitoT: 5 μmol/L; n=6; &P<0.05 compared with Ctrl at 5 min of reperfusion). Mean fluorescence of Ctrl-treated cells is shown as reference (−). MitoTEMPO prevents the reperfusion-induced increase in TMRM fluorescence (B). Time-dependent cell survival on reperfusion with Ctrl, ConT, or Ctrl+mitoTempo presented as a Kaplan–Meier curve (C). Bar graph reflecting the number of EADs at 5 and 10 min of reperfusion in the conditions indicated (D). (*,&,#P<0.05 compared with Ctrl, ConT, and mitoT at R1′, respectively).
FIG. 6.
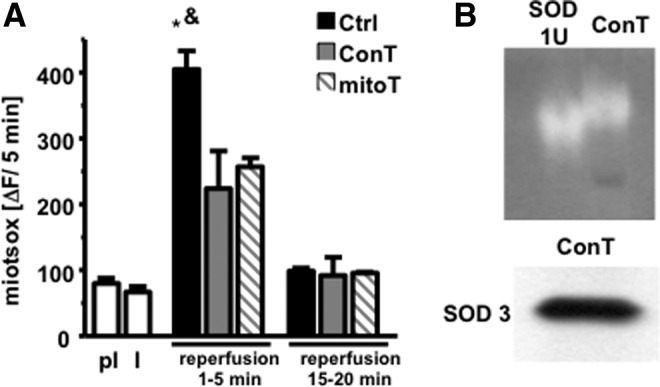
ConT and mitoTEMPO attenuate reperfusion-induced ROS production. (A) The slope of mitoSOX fluorescence during the I/R protocol under pI and ischemic (I) conditions (n=10) and during reperfusion with either Ctrl (black; n=5), ConT (gray; n=4), or Ctrl+mitoT (hatched; n=4) solution (*P<0.05 compared with pI; &P<0.05 compared with ConT at 1–5 min of reperfusion). (B) Lack of NBT oxidation in a gel in which superoxide dismutase (SOD) and ConT were blotted (top); the lower panel shows positive staining for SOD3 in ConT.
To evaluate the possibility that ConT can have a direct function as an antioxidant, we separated ConT proteins with PAGE and soaked the gel in NBT (2.5 mmol/L) and riboflavin (28 mmol/L). NBT serves as a substrate for the superoxide-inducing riboflavin so that proteins with antioxidant properties in the gel can be identified as areas in which NBT oxidation was suppressed. Since the presence of SOD in MSC conditioned media had been suggested [38], we ran the antioxidant SOD (1 U) as a control in the lane adjacent to ConT. The gel shown in Fig. 6B displayed areas of oxidized NBT in both lanes at a comparable molecular weight. In addition, western blotting of ConT proteins identified the extracellular copper/zinc-SOD (SOD3) at a molecular weight of 30 kDa, supporting the capacity of ConT as an antioxidant.
Discussion
We demonstrate that ConT from mouse bone marrow-derived MSCs exerts a postconditioning benefit on VMs when applied during reperfusion. ConT prevents an exaggerated reperfusion-induced hyperpolarization of Ψmito, subsequent PTP opening, and arrhythmic Ca2+ release (EADs). Mechanisms of cardioprotection are (1) the SOD3-mediated antioxidant capacity of ConT that reduces ROS and (2) the Akt/PI3K-mediated opening of IK,ATP. Both mechanisms prevent the exaggerated hyperpolarization of Ψmito that further enhances reperfusion-induced ROS and, ultimately, cell death (Fig. 7).
FIG. 7.

Schematic summary of the experimental results illustrates the proposed signal transduction pathway.
In vitro model of simulated I/R
We used an in vitro model of simulated I/R that was previously used to analyze ischemia-induced changes in protein expression as well as cardiac stunning [25]. Ischemia induced an increase in basal [Ca2+]i, cessation of contractile activity and depolarization of Ψmito. While we observed a 30% decrease in TMRM fluorescence during 15 min of ischemia, more extensive depolarizations of Ψmito are reported in other models [39]; in these cases, a recovery of Ψmito was rarely determined on reperfusion. In our experiments, the rapid recovery of Ψmito on reperfusion indicates that no permanent damage to the mitochondria had been induced. While one could argue that the time for ischemia compared with effective in vivo models is relatively short, one has to remember that ischemic conditions do not develop gradually in the model used. Changes in oxygen, pH, and ionic balance are immediate, and the increased cell death observed indicates that the duration of ischemia applied in this in vitro model is effective. Overall, the described changes in [Ca2+]i [25], contractility, and Ψmito [40] are in good agreement with other models of I/R. The amount of [Ca2+]i increase or Ψmito depolarization during ischemia was previously described to correlate with the cells' propensity for death on reperfusion [39]. In our model, all VMs were treated identically until the onset of reperfusion; ischemia-induced changes in Ca2+ and Ψmito, therefore, should not contribute to the differences in survival, EADs, and Ψmito observed between ConT and Ctrl-reperfused cells.
Reperfusion-induced changes in cardiac ECC
Our experiments demonstrate that the addition of ConT on reperfusion maintains contractility, suppresses EADs, and prevents an exaggerated hyperpolarization of Ψmito. While changes in Ψmito and [Ca2+]i signaling can enhance cell death individually, both parameters are highly interdependent [33,40]. The ischemia-induced change in the ionic balance, particularly the increase in [Na]i, results in enhanced Ca2+ entry on reperfusion. In our experiments, this rise in [Ca2+]i is reflected in increased Ca2+ transient amplitudes (Figs. 1 and 2) [11, 25]. Limiting Ca2+ entry and enhancing Ca2+ extrusion during this period through the block of the Na/H exchanger [13,15], inhibition of reverse mode NCX [11] or enhanced SERCA activity [41], can decrease infarct size and limit the number and duration of arrhythmic events. There is experimental evidence that reperfusion-induced spontaneous Ca2+ oscillations are the initial trigger for the increase in mitochondrial Ca2+ and, consequently, PTP opening [42]; however, other experimental approaches showed that reperfusion-induced ROS production remained elevated even when spontaneous Ca2+ release events were suppressed [43]. We have previously reported that ConT changes ECC in VMs by increasing ICa,L and enhancing SERCA activity [22]; however, on reperfusion, no significant differences in Ca2+ transient amplitude or τCa were determined between Ctrl and ConT-reperfused cells (Fig. 2). These observations could be due to the fact that (1) ConT-mediated changes in ICa,L and SERCA are delayed; (2) the ConT-induced increase is masked by the reperfusion-induced Ca2+ entry through reverse mode NCX [11]; and (3) the ConT-induced nitrosylation of SERCA is masked by Ca2+, CaMKII-induced phospholamban phosphorylation [44]. The lack of difference in [Ca2+]i between Ctrl and ConT-treated groups implies that the ConT-mediated survival benefit does not occur through a change of the Ca2+ handling properties on reperfusion. The difference in contractility was observed at 1 min of reperfusion between Ctrl and ConT-treated cells. Since no change in [Ca2+]i occurs, it is likely based on an increased myofilament Ca2+ sensitivity in ConT-treated cells. At this point, it is difficult to evaluate the mechanism, as significance between the two treatment groups is only transient.
Reperfusion-induced ROS production and its consequences
Reperfusion restores the availability of nutrients as well as the supply of oxygen to the myocytes. The concomitant reactivation of the electron transport chain promotes the recovery of Ψmito (Fig. 3A), which can lead to a hyperpolarization that exceeds pI values in VMs [45]. This enhanced hyperpolarization of Ψmito can be induced experimentally by H2O2 [46], high glucose, and increased cytosolic Ca2+ [47] and is followed by mitochondrial depolarization and cell death [33]. This is in agreement with our results, where the prevention of Ψmito hyperpolarization enhances cell survival during reperfusion. The role of Ψmito hyperpolarization in reperfusion-induced cell damage is further supported by the fact that a delay in Ψmito recovery through reduction in the extracellular pH, inhibition of Na/H-exchanger [13], or opening of mitochondrial ion channels such as IK,ATP (Fig. 4A, B) [36,48] or Cx43 [49] reduces ROS production and mitochondrial Ca2+ loading.
The mechanism by which this exaggerated hyperpolarization occurs is not entirely understood. The delayed recovery of Ψmito in the presence of mitoTEMPO indicates a major role for ROS in this process (Fig. 5A). In brain tissue, ROS was shown to enhance cytochrome C oxidase (COX) activity directly [46,50] or decrease its phosphorylation [51]. Here, we have identified SOD3 in ConT, indicating secretion from MSCs, which is consistent with previous reports by Kemp et al. [38]. The anti-oxidative effect of ConT determined in a SOD activity assay was equivalent to 1U of purified and activity-assayed SOD1 (U/mg protein), which would be compared with approximately 200 pg SOD in ConT that is again comparable to the previously reported release of 0.046 pg/cell [38]. SOD3 is actively secreted by cells through the trans-golgi network and while it only constitutes approximately 1% to 5% of the total cellular SOD, its loss has been shown to result in vascular dysfunction and during trans-aortic constriction, to enhance cellular remodeling and fibrosis [52]. SOD3 deletion was further shown to impair tissue recovery after renal ischemia, which is consistent with the proposed benefit of SOD3 during reperfusion.
Our experimental results further support the fact that ConT induces postconditioning by activation of the PI3K/Akt pathway. Akt-induced postconditioning has been described to lead to the opening of IK,ATP through eNOS/NO-mediated activation of guanylate cyclase and protein kinase G [34,53]. The sensitivity of the ConT benefit to blockers of IK,ATP and the ability of IK,ATP openers to mimic its effect underline the involvement of this mitochondrial ion channel in the regulation of Ψmito on reperfusion. However, in addition, an NO-dependent inhibition of COX [51] could contribute to an attenuated hyperpolarization of Ψmito. There are multiple factors described in ConT that could induce the activation of the PI3K/Akt pathway; however, it is interesting to point out that aSOD3-dependent increase in Akt signaling was described in neuronal cells [38].
Our experimental results show that addition of the membrane-permeable ROS scavenger mitoTEMPO attenuates EADs and cell death in VMs (Fig. 5). mitoTEMPO allows ROS scavenging in the extracellular solution, the cytoplasm, and the mitochondria; while superfusion of an anti-oxidant such as SOD3 would only target extracellular ROS. In this case, the location of ROS scavenging does not indicate the site of ROS production. For example, superoxide has limited membrane permeability, but can be released from mitochondria by PTP opening, or diffuse through the plasma membrane after intracellular SOD-dependent conversion to H2O2. The increase in mitochondrial ROS (Fig. 6A) that we observed on reperfusion, however, indicates mitochondria as one source of ROS in our model. The membrane-bound NADPH oxidase 2 (NOX2) could be another potential source, but experiments on NOX2-deficient mice indicate that lack of NOX2 does not confer a benefit for I/R injury [54]. However, recent data obtained from an in vitro model demonstrate NOX2-dependent ROS production on reperfusion. Nevertheless, our data indicate that independent of the source of ROS, extracellular anti-oxidant activity supplements the overburdened cell mechanisms of ROS removal. Therefore, ConT-dependent extracellular ROS scavenging at the time of reperfusion can attenuate the downward spiral of ROS-induced mitochondrial ROS production, mitochondrial ROS release, and ROS-dependent oxygenation of cardiac proteins, thereby preventing arrhythmic Ca2+ release events such as EADs [55]. This is further supported by the reduced oxidative protein modifications determined in ConT-reperfused myocytes. As indicated by the activation of Akt, ConT contains an array of signaling components. At the current point, we cannot rule out the fact that other intracellular anti-oxidative mechanisms are activated or up-regulated by ConT as previously described for the conditioned media isolated from endothelial progenitor cells [56]. Nevertheless, the immediate effect of ConT on ROS production limits the role of ConT-induced changes in protein expression.
Conclusion
We demonstrate that tyrode conditioned by bone marrow-derived MSCs mediates a postconditioning benefit which protects cardiomyocytes from simulated I/R injury. Primary mechanisms involve the SOD3-dependent extracellular scavenging of ROS, the PI3K/Akt-mediated activation of IK,ATP, and, subsequently, the attenuation of Ψmito hyperpolarization. The changes in Ψmito prevent excessive ROS production and subsequent damage through PTP opening and ROS release. The mechanism of MSC-mediated cardioprotection indicates that the use of MSC-ConT can enhance cell survival in the acute phase of I/R. The experiments presented, therefore, bridge a gap in knowledge between the benefits of MSC transplantation seen in vivo and the understanding of the cellular mechanisms by which these benefits are induced.
Acknowledgments
The work was supported by grants from the National Institutes of Health (HL089617 and HL089617-03S1 to K.B.).
Author Disclosure Statement
No competing financial interests exist
References
- 1.van der Spoel TI. Jansen of Lorkeers SJ. Agostoni P. van Belle E. Gyongyosi M. Sluijter JP. Cramer MJ. Doevendans PA. Chamuleau SA. Human relevance of pre-clinical studies in stem cell therapy: systematic review and meta-analysis of large animal models of ischaemic heart disease. Cardiovasc Res. 2011;91:649–658. doi: 10.1093/cvr/cvr113. [DOI] [PubMed] [Google Scholar]
- 2.Williams AR. Trachtenberg B. Velazquez DL. McNiece I. Altman P. Rouy D. Mendizabal AM. Pattany PM. Lopera GA, et al. Intramyocardial stem cell injection in patients with ischemic cardiomyopathy: functional recovery and reverse remodeling. Circ Res. 2011;108:792–796. doi: 10.1161/CIRCRESAHA.111.242610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatzistergos KE. Quevedo H. Oskouei BN. Hu Q. Feigenbaum GS. Margitich IS. Mazhari R. Boyle AJ, et al. Bone marrow mesenchymal stem cells stimulate cardiac stem cell proliferation and differentiation. Circ Res. 2010;107:913–922. doi: 10.1161/CIRCRESAHA.110.222703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grajales L. Garcia J. Banach K. Geenen DL. Delayed enrichment of mesenchymal cells promotes cardiac lineage and calcium transient development. J Mol Cell Cardiol. 2010;48:735–745. doi: 10.1016/j.yjmcc.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirotsou M. Jayawardena TM. Schmeckpeper J. Gnecchi M. Dzau VJ. Paracrine mechanisms of stem cell reparative and regenerative actions in the heart. J Mol Cell Cardiol. 2011;50:280–289. doi: 10.1016/j.yjmcc.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohnishi S. Yanagawa B. Tanaka K. Miyahara Y. Obata H. Kataoka M. Kodama M. Ishibashi-Ueda H. Kangawa K. Kitamura S. Nagaya N. Transplantation of mesenchymal stem cells attenuates myocardial injury and dysfunction in a rat model of acute myocarditis. J Mol Cell Cardiol. 2007;42:88–97. doi: 10.1016/j.yjmcc.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 7.Markel TA. Wang Y. Herrmann JL. Crisostomo PR. Wang M. Novotny NM. Herring CM. Tan J. Lahm T. Meldrum DR. VEGF is critical for stem cell-mediated cardioprotection and a crucial paracrine factor for defining the age threshold in adult and neonatal stem cell function. Am J Physiol Heart Circ Physiol. 2008;295:H2308–H2314. doi: 10.1152/ajpheart.00565.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herrmann JL. Abarbanell AM. Wang Y. Weil BR. Poynter JA. Manukyan MC. Meldrum DR. Transforming growth factor-alpha enhances stem cell-mediated postischemic myocardial protection. Ann Thorac Surg. 2011;92:1719–1725. doi: 10.1016/j.athoracsur.2011.06.057. [DOI] [PubMed] [Google Scholar]
- 9.Kelly ML. Wang M. Crisostomo PR. Abarbanell AM. Herrmann JL. Weil BR. Meldrum DR. TNF receptor 2, not TNF receptor 1, enhances mesenchymal stem cell-mediated cardiac protection following acute ischemia. Shock. 2010;33:602–607. doi: 10.1097/SHK.0b013e3181cc0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perrelli MG. Pagliaro P. Penna C. Ischemia/reperfusion injury and cardioprotective mechanisms: role of mitochondria and reactive oxygen species. World J Cardiol. 2011;3:186–200. doi: 10.4330/wjc.v3.i6.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei GZ. Zhou JJ. Wang B. Wu F. Bi H. Wang YM. Yi DH. Yu SQ. Pei JM. Diastolic Ca2+ overload caused by Na+/Ca2+ exchanger during the first minutes of reperfusion results in continued myocardial stunning. Eur J Pharmacol. 2007;572:1–11. doi: 10.1016/j.ejphar.2007.05.065. [DOI] [PubMed] [Google Scholar]
- 12.Liang HL. Sedlic F. Bosnjak Z. Nilakantan V. SOD1 and MitoTEMPO partially prevent mitochondrial permeability transition pore opening, necrosis, and mitochondrial apoptosis after ATP depletion recovery. Free Radic Biol Med. 2010;49:1550–1560. doi: 10.1016/j.freeradbiomed.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toda T. Kadono T. Hoshiai M. Eguchi Y. Nakazawa S. Nakazawa H. Higashijima N. Ishida H. Na+/H+ exchanger inhibitor cariporide attenuates the mitochondrial Ca2+ overload and PTP opening. Am J Physiol Heart Circ Physiol. 2007;293:H3517–H3523. doi: 10.1152/ajpheart.00483.2006. [DOI] [PubMed] [Google Scholar]
- 14.Loor G. Kondapalli J. Iwase H. Chandel NS. Waypa GB. Guzy RD. Vanden Hoek TL. Schumacker PT. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim Biophys Acta. 2011;1813:1382–1394. doi: 10.1016/j.bbamcr.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villa-Abrille MC. Cingolani E. Cingolani HE. Alvarez BV. Silencing of cardiac mitochondrial NHE1 prevents mitochondrial permeability transition pore opening. Am J Physiol Heart Circ Physiol. 2011;300:H1237–H1251. doi: 10.1152/ajpheart.00840.2010. [DOI] [PubMed] [Google Scholar]
- 16.Hausenloy DJ. Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–253. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Wairiuko GM. Crisostomo PR. Wang M. Morrell ED. Meldrum KK. Lillemoe KD. Meldrum DR. Stem cells improve right ventricular functional recovery after acute pressure overload and ischemia reperfusion injury. J Surg Res. 2007;141:241–246. doi: 10.1016/j.jss.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Wang M. Tan J. Wang Y. Meldrum KK. Dinarello CA. Meldrum DR. IL-18 binding protein-expressing mesenchymal stem cells improve myocardial protection after ischemia or infarction. Proc Natl Acad Sci U S A. 2009;106:17499–17504. doi: 10.1073/pnas.0908924106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abarbanell AM. Wang Y. Herrmann JL. Weil BR. Poynter JA. Manukyan MC. Meldrum DR. Toll-like receptor 2 mediates mesenchymal stem cell-associated myocardial recovery and VEGF production following acute ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2010;298:H1529–H1536. doi: 10.1152/ajpheart.01087.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo Y. Wang Y. Poynter JA. Manukyan MC. Herrmann JL. Abarbanell AM. Weil BR. Meldrum DR. Pretreating mesenchymal stem cells with interleukin-1beta and transforming growth factor-beta synergistically increases vascular endothelial growth factor production and improves mesenchymal stem cell-mediated myocardial protection after acute ischemia. Surgery. 2012;151:353–363. doi: 10.1016/j.surg.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 21.Fang J. Chen L. Fan L. Wu L. Chen X. Li W. Lin Y. Wang W. Enhanced therapeutic effects of mesenchymal stem cells on myocardial infarction by ischemic postconditioning through paracrine mechanisms in rats. J Mol Cell Cardiol. 2011;51:839–847. doi: 10.1016/j.yjmcc.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 22.Desantiago J. Bare DJ. Semenov I. Minshall RD. Geenen DL. Wolska BM. Banach K. Excitation-contraction coupling in ventricular myocytes is enhanced by paracrine signaling from mesenchymal stem cells. J Mol Cell Cardiol. 2012;52:1249–1256. doi: 10.1016/j.yjmcc.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolska BM. Solaro RJ. Method for isolation of adult mouse cardiac myocytes for studies of contraction and microfluorimetry. Am J Physiol Heart Circ Physiol. 1996;271:H1250–H1255. doi: 10.1152/ajpheart.1996.271.3.H1250. [DOI] [PubMed] [Google Scholar]
- 24.Schuleri KH. Amado LC. Boyle AJ. Centola M. Saliaris AP. Gutman MR. Hatzistergos KE. Oskouei BN. Zimmet JM, et al. Early improvement in cardiac tissue perfusion due to mesenchymal stem cells. Am J Physiol Heart Circ Physiol. 2008;294:H2002–H2011. doi: 10.1152/ajpheart.00762.2007. [DOI] [PubMed] [Google Scholar]
- 25.Louch WE. Ferrier GR. Howlett SE. Changes in excitation-contraction coupling in an isolated ventricular myocyte model of cardiac stunning. Am J Physiol Heart Circ Physiol. 2002;283:H800–H810. doi: 10.1152/ajpheart.00020.2002. [DOI] [PubMed] [Google Scholar]
- 26.O'Brien JD. Ferguson JH. Howlett SE. Effects of ischemia and reperfusion on isolated ventricular myocytes from young adult and aged Fischer 344 rat hearts. Am J Physiol Heart Circ Physiol. 2008;294:H2174–H2183. doi: 10.1152/ajpheart.00058.2008. [DOI] [PubMed] [Google Scholar]
- 27.Dedkova EN. Wang YG. Ji X. Blatter LA. Samarel AM. Lipsius SL. Signalling mechanisms in contraction-mediated stimulation of intracellular NO production in cat ventricular myocytes. J Physiol. 2007;580:327–345. doi: 10.1113/jphysiol.2006.126805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perry SW. Norman JP. Barbieri J. Brown EB. Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques. 2011;50:98–115. doi: 10.2144/000113610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dedkova EN. Blatter LA. Measuring mitochondrial function in intact cardiac myocytes. J Mol Cell Cardiol. 2012;52:48–61. doi: 10.1016/j.yjmcc.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beauchamp C. Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 31.Chen J. Liao C. Mao SJ. Chen T. Weng C. A simple technique for the simultaneous determination of molecular weight and activity of superoxide dismutase using SDS-PAGE. J Biochem Biophys Methods. 2001;47:233–237. doi: 10.1016/s0165-022x(00)00162-7. [DOI] [PubMed] [Google Scholar]
- 32.Nakayama H. Bodi I. Maillet M. DeSantiago J. Domeier TL. Mikoshiba K. Lorenz JN. Blatter LA. Bers DM. Molkentin JD. The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ Res. 2010;107:659–666. doi: 10.1161/CIRCRESAHA.110.220038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kadenbach B. Ramzan R. Moosdorf R. Vogt S. The role of mitochondrial membrane potential in ischemic heart failure. Mitochondrion. 2011;11:700–706. doi: 10.1016/j.mito.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Egom EE. Mohamed TM. Mamas MA. Shi Y. Liu W. Chirico D. Stringer SE. Ke Y. Shaheen M, et al. Activation of Pak1/Akt/eNOS signaling following sphingosine-1-phosphate release as part of a mechanism protecting cardiomyocytes against ischemic cell injury. Am J Physiol Heart Circ Physiol. 2011;301:H1487–H1495. doi: 10.1152/ajpheart.01003.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Penna C. Rastaldo R. Mancardi D. Raimondo S. Cappello S. Gattullo D. Losano G. Pagliaro P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res Cardiol. 2006;101:180–189. doi: 10.1007/s00395-006-0584-5. [DOI] [PubMed] [Google Scholar]
- 36.Murata M. Akao M. O'Rourke B. Marban E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: possible mechanism of cardioprotection. Circ Res. 2001;89:891. doi: 10.1161/hh2201.100205. [DOI] [PubMed] [Google Scholar]
- 37.Korge P. Yang L. Yang JH. Wang Y. Qu Z. Weiss JN. Protective role of transient pore openings in calcium handling by cardiac mitochondria. J Biol Chem. 2011;286:34851–34857. doi: 10.1074/jbc.M111.239921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kemp K. Hares K. Mallam E. Heesom KJ. Scolding N. Wilkins A. Mesenchymal stem cell-secreted superoxide dismutase promotes cerebellar neuronal survival. J Neurochem. 2010;114:1569–1580. doi: 10.1111/j.1471-4159.2009.06553.x. [DOI] [PubMed] [Google Scholar]
- 39.Levraut J. Iwase H. Shao ZH. Vanden Hoek TL. Schumacker PT. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol. 2003;284:H549–H558. doi: 10.1152/ajpheart.00708.2002. [DOI] [PubMed] [Google Scholar]
- 40.Kim JS. Jin Y. Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 41.del Monte F. Lebeche D. Guerrero JL. Tsuji T. Doye AA. Gwathmey JK. Hajjar RJ. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci U S A. 2004;101:5622–5627. doi: 10.1073/pnas.0305778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruiz-Meana M. Abellan A. Miro-Casas E. Agullo E. Garcia-Dorado D. Role of sarcoplasmic reticulum in mitochondrial permeability transition and cardiomyocyte death during reperfusion. Am J Physiol Heart Circ Physiol. 2009;297:H1281–H1289. doi: 10.1152/ajpheart.00435.2009. [DOI] [PubMed] [Google Scholar]
- 43.Talukder MA. Kalyanasundaram A. Zhao X. Zuo L. Bhupathy P. Babu GJ. Cardounel AJ. Periasamy M. Zweier JL. Expression of SERCA isoform with faster Ca2+ transport properties improves postischemic cardiac function and Ca2+ handling and decreases myocardial infarction. Am J Physiol Heart Circ Physiol. 2007;293:H2418–H2428. doi: 10.1152/ajpheart.00663.2007. [DOI] [PubMed] [Google Scholar]
- 44.Vila-Petroff M. Salas MA. Said M. Valverde CA. Sapia L. Portiansky E. Hajjar RJ. Kranias EG. Mundina-Weilenmann C. Mattiazzi A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc Res. 2007;73:689–698. doi: 10.1016/j.cardiores.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 45.Abdallah Y. Iraqi W. Said M. Kasseckert SA. Shahzad T. Erdogan A. Neuhof C. Gunduz D. Schluter KD, et al. Interplay between Ca(2+) cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes. J Cell Mol Med. 2010;15:2478–2485. doi: 10.1111/j.1582-4934.2010.01249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi K. Kim J. Kim GW. Choi C. Oxidative stress-induced necrotic cell death via mitochondira-dependent burst of reactive oxygen species. Curr Neurovasc Res. 2009;6:213–222. doi: 10.2174/156720209789630375. [DOI] [PubMed] [Google Scholar]
- 47.Mansfield K. Pucci B. Adams CS. Shapiro IM. Induction of apoptosis in skeletal tissues: phosphate-mediated chick chondrocyte apoptosis is calcium dependent. Calcified Tissue Int. 2003;73:161–172. doi: 10.1007/s00223-002-1056-z. [DOI] [PubMed] [Google Scholar]
- 48.Murphy E. Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boengler K. Dodoni G. Rodriguez-Sinovas A. Cabestrero A. Ruiz-Meana M. Gres P. Konietzka I. Lopez-Iglesias C. Garcia-Dorado D, et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc Res. 2005;67:234–244. doi: 10.1016/j.cardiores.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 50.Sedlic F. Sepac A. Pravdic D. Camara AK. Bienengraeber M. Brzezinska AK. Wakatsuki T. Bosnjak ZJ. Mitochondrial depolarization underlies delay in permeability transition by preconditioning with isoflurane: roles of ROS and Ca2+ Am J Physiol Cell Physiol. 2010;299:C506–C515. doi: 10.1152/ajpcell.00006.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huttemann M. Helling S. Sanderson TH. Sinkler C. Samavati L. Mahapatra G. Varughese A. Lu G. Liu J, et al. Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta. 2011;1817:598–609. doi: 10.1016/j.bbabio.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gongora MC. Harrison DG. Sad heart from no SOD. Hypertension. 2008;51:28–30. doi: 10.1161/HYPERTENSIONAHA.107.101162. [DOI] [PubMed] [Google Scholar]
- 53.Das S. Wong R. Rajapakse N. Murphy E. Steenbergen C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ Res. 2008;103:983–991. doi: 10.1161/CIRCRESAHA.108.178970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cave AC. Brewer AC. Narayanapanicker A. Ray R. Grieve DJ. Walker S. Shah AM. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 55.Terentyev D. Gyorke I. Belevych AE. Terentyeva R. Sridhar A. Nishijima Y. de Blanco EC. Khanna S. Sen CK, et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Z. von Ballmoos MW. Faessler D. Voelzmann J. Ortmann J. Diehm N. Kalka-Moll W. Baumgartner I. Di Santo S. Kalka C. Paracrine factors secreted by endothelial progenitor cells prevent oxidative stress-induced apoptosis of mature endothelial cells. Atherosclerosis. 2010;211:103–109. doi: 10.1016/j.atherosclerosis.2010.02.022. [DOI] [PubMed] [Google Scholar]