

Abstract
The zebrafish (Danio rerio) is an established model organism for developmental and biomedical research. It is frequently used for high-throughput functional genomics experiments, such as genome-wide gene expression measurements, to systematically analyze molecular mechanisms. However, the use of whole embryos or larvae in such experiments leads to a loss of the spatial information. To address this problem, we have developed a tool called Zebrafish Expression Ontology of Gene Sets (ZEOGS) to assess the enrichment of anatomical terms in large gene sets. ZEOGS uses gene expression pattern data from several sources: first, in situ hybridization experiments from the Zebrafish Model Organism Database (ZFIN); second, it uses the Zebrafish Anatomical Ontology, a controlled vocabulary that describes connected anatomical structures; and third, the available connections between expression patterns and anatomical terms contained in ZFIN. Upon input of a gene set, ZEOGS determines which anatomical structures are overrepresented in the input gene set. ZEOGS allows one for the first time to look at groups of genes and to describe them in terms of shared anatomical structures. To establish ZEOGS, we first tested it on random gene selections and on two public microarray datasets with known tissue-specific gene expression changes. These tests showed that ZEOGS could reliably identify the tissues affected, whereas only very few enriched terms to none were found in the random gene sets. Next we applied ZEOGS to microarray datasets of 24 and 72 h postfertilization zebrafish embryos treated with beclomethasone, a potent glucocorticoid. This analysis resulted in the identification of several anatomical terms related to glucocorticoid-responsive tissues, some of which were stage-specific. Our studies highlight the ability of ZEOGS to extract spatial information from datasets derived from whole embryos, indicating that ZEOGS could be a useful tool to automatically analyze gene expression pattern features of any large zebrafish gene set.
Introduction
Zebrafish have become a well-established model organism to study development, fundamental biological mechanisms, and a variety of biomedically relevant processes. This is partly because functional genomics approaches allow researchers to perform high-throughput analyses of the zebrafish genome, transcriptome, and proteome under many different conditions, thus leading to a more systematic understanding and novel mechanistic insights.1,2 There are two alternative basic experimental designs possible for high-throughput experiments in zebrafish: isolation of a certain cell type or organ from embryos/larvae or adult animals, or performing such experiments on whole animals. The second alternative may be preferable if there is no information about which cells could be affected by the experimental manipulation, when a non-cell-autonomous effect is expected or when isolation of a particular tissue is problematic. Regardless of the experimental design chosen, functional genomics experiments generate information on hundreds to tens of thousands of genes, RNA species, proteins, or other molecular entities. The interpretation of such large datasets is challenging and their analysis requires statistical methods to extract or filter the useful and significant information. For example, approaches focusing on functional properties of genes include Gene Ontology (GO) annotation/enrichment,3 Kyoto Encyclopedia of Genes and Geomes (KEGG) pathway analysis,4 and Ingenuity pathway analyses (www.ingenuity.com). These methods search for functional properties that are significantly enriched in a certain gene set, therefore providing global insights into the most likely functions of these genes.
In addition to the identification of shared functional properties of genes in a dataset, datasets from whole animals can be used to identify overrepresented anatomical terms, thus shedding light on where in the animal these genes may be expressed. Zebrafish is highly suitable for this kind of analysis because a large amount of gene expression information, mostly from in situ hybridization experiments, is available in the Zebrafish Model Organism Database (ZFIN).5,6 The expression information is organized based on the Anatomical Ontology (AO), a controlled hierarchical vocabulary of terms describing anatomical structures of zebrafish and their relationships (see http://zfin.org/zf_info/anatomy/dict/sum.html). The zebrafish AO facilitates effective data dissemination, serves as a reference for zebrafish anatomy descriptions, and ensures the possibility of mapping homologous structures between anatomical ontologies of different species. The zebrafish AO has been further developed and modified in Developmental Anatomy Ontology of zebrafish for three-dimensional models of zebrafish anatomy7 and is used as the basis for a more general Teleost Anatomy Ontology.8
Current zebrafish-related resources are unable to provide summaries and enrichment of expression patterns for large sets of genes. Instead, they only allow extracting this information for single genes. For example, ZFIN supports single-gene identifier queries as well as queries based on anatomical terms combined with developmental stage limits. The first type of query results in a table of published gene expression patterns, whereas the second type returns a list of genes known to contain user-specified anatomical terms within the specified stage limits. Similar queries are offered by 4DXpress for zebrafish genes.9 In addition, 4DXpress provides the possibility to enter gene lists that will be linked to the corresponding gene pages, and it allows searching for gene homologs in zebrafish, medaka, mouse, Drosophila, and Platynereis to check for similarities in the expression patterns. However, ZFIN and other resources do not provide expression pattern summaries for sets of zebrafish genes, and generating a manual summary is tedious for large gene sets. This highlights the need for a tool capable to provide an overall picture of the most frequent or enriched anatomical terms in a large input gene set. To achieve this aim, we have developed a program called Zebrafish Expression Ontology of Gene Sets (ZEOGS). Here, the term “Expression Ontology of Gene Sets” is defined as the assignment of genes in a set to distinct enriched anatomical terms. Importantly, ZEOGS does not develop an expression ontology but rather uses public gene expression datasets from ZFIN to generate a summary of gene expression patterns for an input gene set. Further, ZEOGS finds significant associations between the genes in the set and anatomical terms. Here, we tested and validated the results generated by the ZEOGS tool on simulated random gene sets and on two datasets with predicted tissue-specific targets. As a less tissue-specific case study, we applied the ZEOGS software to lists of differentially expressed genes in zebrafish embryos treated with glucocorticoid hormone, which activates the glucocorticoid receptor (GR), a ligand-activated transcription factor. Zebrafish starts playing an important role in GR research because of its strengths in genetics research, imaging, functional genomics, and ease of experimental manipulation.10,11 Although GR is known to be expressed ubiquitously, its actions are highly tissue specific, and mediated by transcriptional activation, repression, or nongenomic actions.12,13 Therefore, we reasoned that GR signaling would be a good system to study the ability of ZEOGS to identify the anatomical structures in which GR is actively using genes with differential expression upon glucocorticoid treatment of whole zebrafish embryos. Anatomical terms related to glucocorticoid-responsive tissues such as muscle, cartilage, and liver were indeed found enriched by ZEOGS analyses for these datasets. Together, our studies showed that ZEOGS can be used to extract meaningful spatial information from large datasets derived from whole animal studies.
Materials and Methods
Zebrafish husbandry
AB wild-type zebrafish were maintained in the zebrafish facility of the Max-Planck Institute for Molecular Genetics according to standard protocols at 28.5°C.14 Embryos were collected and grown in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl, 0.33 mM MgSO4) at 28.5°C and staged by percentages of epiboly and somite numbers.15 All zebrafish experiments were approved by the Office for Health and Social Affairs Berlin.
Microarray-based expression profiling of beclomethasone-induced genes
Zebrafish AB embryos at 24 and 72 hours postfertilization (hpf) were treated with either 25 μM beclomethasone or 0.1% DMSO for 3 h, anesthetized with 0.02% of tricaine (Sigma; E10521), and frozen in liquid nitrogen. RNA was extracted using the RNeasy Mini Kit (Qiagen; 74106) and genomic DNA was then removed using the TurboDNA-free kit (Ambion; AM1907). RNA (3 μg) was sent to the Microarray Analysis Facility at the Max-Planck Institute for Cell Biology and Genetics and one-color Cy3 hybridizations were performed to Agilent V3: 026437 microarray slides in triplicate. Resulting images were quantified and text files containing raw values were analyzed. Data preprocessing, differential expression analysis, and gene annotation were done in R, using available Bioconductor packages (www.bioconductor.com). First, the signals were background corrected with the normexp method16 (Limma package17), and an offset of 1 was added to the intensities before normalization and log transformation to ensure that all intensity values are positive. After background correction, the data were normalized between arrays using the quantile method.18 Differentially expressed probes among different conditions were identified by means of the linear model implemented in the Limma package.1 In addition, the empirical Bayes method was used to construct moderated t-statistics and adjust for multiplicity of the tests. The Benjamini and Hochberg's method was used to control the false discovery rate.19 The biomaRt annotation package20 was used to assign the corresponding gene accessions (Ensembl IDs, Entrez Gene Ids, and ZFIN gene symbols) to each Agilent probe ID. Each gene was then assigned to the median expression value from all the corresponding probes. Genes with a false discovery rate less than 0.1 and a fold change higher than 2 were considered differentially expressed. The data are available at the Gene Expression Omnibus website (www.ncbi.nlm.nih/gov/geo) (accession number GSE41904).
GO and KEGG pathway enrichment
Database for Annotation, Visualization, and Integrated Discovery (DAVID) Bioinformatics Resources website21 was used to assess whether certain sets of up- or downregulated genes showed statistically significant enrichment of GO categories or KEGG pathways when comparing two different conditions.
Implementation
Data sources
All of the zebrafish gene expression data used in ZEOGS come from the ZFIN database, where in situ hybridization staining experiments from high-throughput expression screens and individual publications have been annotated in detail using the AO system. ZFIN provides two views of the anatomical information on gene expression: first, anatomical terms for individual genes on ZFIN are listed on the gene summary pages without stage information (e.g., http://zfin.org/action/marker/view/ZDB-GENE-000426-1) (“gene summary” option); second, gene expression patterns of the same genes are annotated with curated stages when their expression was observed (see http://zfin.org/data_transfer/Downloads/wildtype-expression.txt) (“wild-type expression” option). As genes can be expressed in different anatomical structures, depending on the developmental stage, it is crucial to extract the stage information for any kind of analysis. For both data source options, this information can be extracted either by a direct link between anatomical terms and stage of development(see http://zfin.org/data_transfer/Downloads/staged_anatomy.txt), or directly from the experimental data records, although data are not always available for each stage. Thus, when the “gene summary” option is chosen, anatomical terms are restricted based on their existence at the selected stage (Fig. 1A), while with the “gene summary” option, anatomical terms are selected if the user-defined stage matches one of their curated stages (Fig. 1B).
FIG. 1.

Data source options of ZEOGS. (A) Filtering by the existence of an anatomical term: When this option is selected, the program tests which anatomical terms are present at the input stage. This option is predictive of where genes are expressed based on the current knowledge but may not be empirically based at each particular stage. (B) Filtering by actual observations of gene expression in an anatomical term: The selection of this option forces the program to consider only the anatomical terms with direct empirical evidence for a certain gene to be expressed in these anatomical structures at the selected stage, making the results of the program fully empirically based.
The ZEOGS software
ZEOGS is a web-based program implementing an algorithm written in Perl/CGI, which summarizes the most over-represented anatomical terms in an input gene set and their statistical significance. Figure 2 illustrates the workflow of the program. As input, the user provides a list of gene identifiers (either gene symbols of ZFIN IDs) and selects the data source by means of the “Data source” button (Fig. 2); since in most experiments not all genes are tested, the user can directly enter the actual list of genes analyzed in a particular experiment (“Tested genes” option). In detail, the data source option “Anatomical terms on ZFIN gene summary pages+stage filter” (abbreviated as “gene summary”) (Fig. 2) uses the anatomical terms listed on ZFIN gene summary pages. Furthermore, the user also has to select a stage (“Stage of development”), which will be used for filtering of anatomical terms known for the selected stage (Fig. 1A). The data source option “Wild-type expression: manually annotated data for some stages” (abbreviated as “wild-type expression”) is stricter and will retrieve those anatomical terms that are experimentally validated by in situ hybridization experiments, as explained previously (Fig. 1B). For this option, the stage filter option has also been implemented. Finally, anatomical terms are tested for over-representation using the hypergeometric test with Benjamini–Hochberg multiple testing correction, and the user can choose how to sort them (“Sorting option”).
FIG. 2.

Scheme of the overall ZEOGS software algorithm. To run ZEOGS, the user chooses a data source, a gene identifier list (either gene symbol or ZFIN ID), stage of development for which the output will be generated, a list of tested genes, and a sorting option for the retrieved anatomical terms. Input items are surrounded by a gray area. Next, the input gene identifiers are converted to ZFIN IDs to query the gene expression datasets according to the user-selected data source and the stage of development. The output consists of a list of anatomical terms assigned to genes in the sample, followed by a list of genes and their associated counts for all retrieved anatomical terms. The above procedure can be performed either for all the zebrafish genes with available expression information or for the list of genes actually tested in an experiment. Enrichment of anatomical terms is assessed by means of the p-value from a hypergeometric test. Finally, anatomical terms are sorted according to the user-selected sorting option.
The input gene identifiers are converted to ZFIN IDs, which are used to query expression datasets stored on the server as flat files. Next, the program obtains anatomical terms for the input gene identifiers from either all known zebrafish genes or from the input genes provided with the “Tested genes” option. During this process, anatomical terms are filtered based on their observation at the selected stage of development. In the next step, the algorithm generates the gene lists and gene counts for each encountered anatomical term and determines which anatomical terms are significantly over-represented by means of a hypergeometric test and Benjamin–Hochberg correction for the p-values. The program provides two tables as output: one contains all the enriched anatomical terms, sorted according to a user-specified option, together with gene counts and p-values, and the second one contains tables of gene identifiers and descriptions for each anatomical term.
Statistical testing of the output results
The frequency distribution of genes among anatomical terms in the summary output is useful for gaining a first qualitative understanding of which genes are mostly expressed in specific anatomical structures, but does not contain a quantitative statistical assessment of enriched anatomical terms compared with the whole set of genes with known expression patterns. We assessed the enrichment of anatomical terms by means of a hypergeometric distribution. The p-value of the hypergeometric test represents the probability of randomly selecting a certain number x or more genes with an anatomical term, given the gene set size, the total number of genes described by anatomical terms, and the total number of genes annotated with this particular anatomical term. In detail,
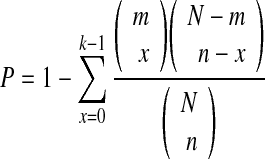 |
where N is the total number of genes that are annotated on ZFIN with specific anatomical terms (all terms are included except for “unspecified” and “whole organism”), n is the number of genes annotated with a specific anatomical term, m is the number of genes in the input gene set, k is the number of input genes annotated with a specific anatomical term, and x is the random variable of the hypergeometric distribution. The current value of N is 6427, and the list is available for download from a link on the ZEOGS websites (http://zeogs.x10.bz/The_list_total_genes_N.txt). Alternatively, N can be derived from the user-specified list of tested genes by querying this list against all of the available zebrafish expression pattern information. In addition, since ZEOGS is testing the hypothesis of enrichment for many anatomical terms, we applied Benjamini–Hochberg method for multiple testing correction.19 The anatomical terms with p-values smaller than 0.1 after the correction are then regarded as significantly enriched.
Statistical methods
The data from the random sampling simulations were plotted using the GraphPad Prism 5.03 software. We used the same software to analyze the correlation between the total number of genes associated with an anatomical term and its frequency of enrichment or its enrichment p-value. Specifically, we used the Spearman correlation coefficient because gene numbers associated with anatomical terms may not be normally distributed.
ZEOGS interfaces
The ZEOGS website can be accessed at http://zeogs.molgen.mpg.de or http://zeogs.x10.bz. The ZEOGS website has two basic interfaces for interacting with the user: an input and an output interface, which are both linked to several additional web pages explaining the ZEOGS algorithm, as well as providing help, links to related websites, and contact information. Additionally, the output interface provides links to temporarily stored files with all the results from the current program execution, a table with all anatomical terms associated to the input gene set, links with detailed description of each term, gene counts and p-values associated with each anatomical term, and summary information on the overall results from the program run. The results from each program execution are stored in a temporary folder on the web server, which is accessible for at least 24 h after which these folders are deleted.
Results
Comparison of public data sources for zebrafish gene expression information
To choose the most complete data source as the basis of our software, we compared several databases containing gene expression pattern information for zebrafish genes. The most important of these databases are ZFIN, Ensembl BioMart, and 4DXpress. These databases differ in their query mechanisms: BioMart allows queries for gene lists outputting gene–anatomical term pairs, whereas the other two databases are designed mainly for single-gene queries. We compared the contents of these databases by counting the total number of genes with available gene expression pattern annotations and the total number of annotated gene–anatomical term pairs. This comparison is summarized in Table 1 and revealed that ZFIN gene pages and the ZFIN wild-type expression file were the most comprehensive sources of zebrafish gene expression patterns, containing 10,506 and 10,945 genes, as well as 64,125 and 68,437 gene–anatomical term pairs, respectively. By contrast, Ensembl BioMart contained fewer gene expression information for zebrafish, with a total number of genes equal to 6384 and 31,188 gene–anatomical term pairs. Finally, 4DXpress had the smallest amount of information with 9319 genes and 25,783 gene–anatomical term pairs. These results convincingly show that ZFIN is the most complete database, and therefore we used it as the data source for ZEOGS software.
Table 1.
Comparison of Different Data Sources of Expression Information for Zebrafish
| ZFIN gene pages | ZFIN wild-type expression file | Ensembl BioMart | 4DXpress | |
|---|---|---|---|---|
| Total genes with expression | 10,506 | 10,945 | 6384 | 9319 |
| Total pairs of genes and anatomical terms | 64,125 | 68,437 | 31,188 | 25,783 |
| Average number of anatomical terms/gene | 6.1 | 6.3 | 4.9 | 2.8 |
The results in this table correspond to the data obtained in April 2012 and will change regularly as the databases evolve and are updated.
Comparison of data source options in ZEOGS
ZEOGS allows the user to use data source options “gene summary” or “wild-type expression,” which represent two different views on the curated zebrafish gene expression data (see Implementation section). This raises the question whether the choice of the data source option influences the results. The “gene summary” option contains more annotation about anatomical terms, but it is partially based on predictions, while the “wild-type expression” option focuses only on developmental stages for which the actual data are available; therefore, this option is more reliable, but it contains less information. We compared both options by applying them systematically to the whole set of zebrafish genes at all known zebrafish developmental stages. For each option we evaluated, first, the number of genes that could be associated to an anatomical term and, second, the number of anatomical terms encountered for all of the genes. Running ZEOGS with the “gene summary” option revealed that both gene counts and anatomical term counts follow a sigmoid curve pattern with a plateau at the final developmental stages (Fig. 3A, B). This can be explained by growing anatomical diversity of zebrafish as development proceeds. The plateau for the “gene summary” curves in Figure 3A and B after stage 32 (Pharyngula:High-pec) is caused by the strong similarities between known anatomical terms during larval stages of zebrafish development, which become more diverse and numerous at the adult stage (Fig. 3B). In contrast, the results from the “wild-type expression” option, which is based on empirical evidence, showed a peaked distribution for both gene and anatomical term counts at intermediate developmental stages (Fig. 3A, B). This can be explained by the fact that the majority of the data in the “wild-type expression” option is available from stage 17 (Gastrula: 50%-epiboly) to stage 33 (Hatching:Long-pec). Interestingly, with the same option, the decrease in the number of encountered anatomical terms with developmental time was not as steep as the number of genes associated with anatomical terms because of multiple anatomical terms associated with each gene. Thus, the stage at which a gene set was generated determines which data source option should be selected. The “gene summary” option is generally applicable and better suited for samples obtained after 72 h of development, whereas the “wild-type expression” option is most suitable for earlier developmental stages.
FIG. 3.

The numbers of genes and anatomical terms in the output by ZEOGS with all zebrafish genes as input with “gene summary” and “wild-type expression” data source options. The total number of genes with known specific anatomical terms (A) and the number of encountered anatomical terms (B) for these genes were quantified across all of the zebrafish developmental stages for “gene summary” and “wild-type expression” data source options. The anatomical terms “unspecified” and “whole organism” were not included in this analysis as they do not convey specific information.
Assessment of the false-positive rate of ZEOGS using random gene sets
Before applying ZEOGS to real biological datasets, we assessed its expected false-positive rate defined as the fraction of enriched anatomical terms when a random selection of genes is used. Therefore, we randomly sampled 500 genes from all known zebrafish genes and ran the ZEOGS algorithm using these genes as input. Random sampling simulations generally provide a good reference for determining the significance for the observed results of real biological datasets. The aim of these simulations was to assess the false-positive rate of ZEOGS and to assess whether there is an enrichment bias toward anatomical terms with higher associated gene counts. Stages 17–33 (5–60 hpf) were chosen for the analysis because they cover most of the zebrafish embryonic development and because, at these stages, both data source options have comparable contents. We repeated these random sampling procedures 200 times at each stage, and each time we determined the proportion of enriched anatomical terms and their enrichment p-values. The results of the simulations indicated, first, that setting the enrichment cutoff p-value at 0.1 led to comparable rates of enrichment for both “gene summary” (mean=0.1232; standard deviation [SD]=0.0138) and “wild-type expression” data source options (mean=0.1667; SD=0.0208), confirming the random nature of these simulation experiments (Fig. 4A). In addition, the application of the Benjamini–Hochberg correction method resulted in dramatically reduced rates of enrichment for both “gene summary” (mean=0.0039; SD=0.0047) and “wild-type expression” (mean=0.0068; SD=0.0055) options at all stages examined (Fig. 4A).
FIG. 4.

Random sampling experiment using ZEOGS to assess the false-discovery rate and the possible bias toward high gene counts. A basic ZEOGS algorithm was run 200 times at stages 17–33 (5–60 hpf) for both “gene summary” and “wild-type expression” data source options using a random selection of 500 genes. The proportion of anatomical terms for which the enrichment p-value was below 0.1 (enrichment threshold) was recorded as well as the number of associated genes. Data points represent fractions of enriched anatomical terms at individual stages for both data source options and a 0.1 p-value cutoff are presented when standard hypergeometric p-value calculations or the Benjamini–Hochberg-corrected p-values (BH) were done (A). Plots of the number of times each anatomical term was found enriched (enrichment frequency) relative to the log2 of the anatomical term's total gene count for all stages combined show a modest correlation between these two variables (Spearman coefficient r) for both “gene summary” (B) and “wild-type expression” (C) data source options. The extent of enrichment measured by the negative log10 of p-values did not correlate with log2 of the total gene counts of anatomical terms for either of “gene summary” (D) or “wild-type expression” (E) data source options.
We next tested whether the frequency of enriched terms (the number of times the p-value was below 0.1 for a certain term) and the significance of the enrichment (negative logarithm of the p-value) were correlated with the number of genes associated to a certain term.
Both of these analyses were done for all of the stages 5–33, but since there were no significant differences between stages (data not shown), all of the data were pooled. The values of the correlations between anatomical term enrichment frequencies and gene numbers associated with a term showed that there might be a bias of enriched terms toward high gene counts (Spearman r=0.4607 and r=0.4185 for “gene summary” and “wild-type expression” options, respectively) (Fig. 4B, C). However, when looking at correlations between negative logarithms of p-values and number of genes associated with a certain term, we found no evidence of positive correlation between the two variables (Spearman r=−0.038 and r=−0.3295 for both data source options; Fig. 4D, E). In conclusion, our random sampling experiments show that running ZEOGS on randomly selected genes returns few false positives and that there was no bias toward anatomical terms with larger numbers of associated genes when these are ranked by their p-values.
ZEOGS settings for gene set analyses
For the biological datasets we defined as ”enriched” those anatomical terms that had hypergeometric p-values less than 0.1, and “highly enriched” those terms that had corrected p-values less than 0.1. The choice of 0.1 as the p-value for the current ZEOGS version is motivated by the fact that we are exploring a variety of more or less noisy biological datasets, and we think that a p-value threshold, less stringent than 0.05, better captures those instances of enriched anatomical terms, which are statistically, as well as biologically significant. It is worth pointing out that all of the anatomical terms predicted by ZEOGS analyses may be biologically relevant. Nevertheless, focusing on enriched anatomical terms allows one to predict the main sites of expression changes.
Testing the performance of ZEOGS on tissue-specific zebrafish microarray datasets
We next tested if ZEOGS is able to detect enrichment of anatomical terms in datasets where differentially expressed genes are expected to be tissue specific. The first dataset we selected was a comparison of zebrafish retinas with or without the retinal pigment epitheliums (RPE) at 52 hpf22 (GSE5048). We applied ZEOGS with the “gene summary” option using as input genes with higher expression in the retina with RPE (Supplementary Table 1; Supplementary Data available online at www.liebertonline.com/zeb) to determine the enriched anatomical terms. Consistent with expectations, the two most highly enriched anatomical terms were “pigment cell” and “retinal pigment epithelium” (Supplementary Table 1), thus validating ZEOGS output for this dataset. We then tested ZEOGS on a set of genes downregulated by cyclopamine, a hedgehog signaling inhibitor, and upregulated by overexpression of the dominant-active protein kinase A, a potent activator of the hedgehog signaling, both at 24 hpf.23 The differentially expressed genes in this gene set are thus likely target genes of transcription factors downstream of the hedgehog signaling pathway. Indeed, application of ZEOGS to this dataset revealed that the most enriched terms were several anatomical terms related to the floor plate of the neural tube and brain and forebrain (Supplementary Table 2), fully consistent with the expression patterns of hedgehog ligands in zebrafish.24 In conclusion, using two very different examples we confirmed the usefulness of ZEOGS in cases where gene expression changes are tissue specific.
GO and KEGG pathway analysis of beclomethasone-regulated gene sets reveals their functions
As a validation example of the ZEOGS application, we used it in our own research project aimed at understanding tissue-specific actions of the GR. GR belongs to the steroid hormone receptor family and mediates cell responses to endogenous corticosteroids and a variety of synthetic glucocorticoids. GR is conserved among vertebrates, including zebrafish, at sequence, structural, and functional levels.10 GR can stimulate gluconeogenesis; regulate protein, amino acid, and lipid metabolism; induce stress responses; inhibit immune responses and inflammation; affect neural activity and behavior; and is involved in the regulation of cell proliferation and survival. We performed microarray-based expression profiling of whole zebrafish embryos at 24 and 72 hpf after 3 h treatment with 25 μM of beclomethasone, a potent glucocorticoid with known activity in zebrafish11 or with 0.1% DMSO as control. The regulated gene sets at both stages (Supplementary Tables 3 and 4) were subsequently used as test cases for ZEOGS to predict anatomical structures in the embryo where GR is active.
To gain insight into the functions of GR-regulated genes in zebrafish, we first performed GO and KEGG pathway analyses on both datasets to identify enriched functional terms. The majority of GO- and KEGG-enriched terms among upregulated genes at both 24 and 72 hpf are related to carbohydrate, purine and steroid metabolism, oxidative processes, ion transport, steroid receptor signaling, cell death regulation, and, interestingly, heart development (Supplementary Tables 5 and 6) consistent with a crucial role of GR in metabolism25 and apoptosis.26 Moreover, several downregulated genes (mcm2, mcm3, mcm5, mcm6, gins2) at 72 hpf were associated with DNA replication, suggesting that repression of these genes by GR may lead to inhibition of proliferation.
Application of ZEOGS to the set of beclomethasone-regulated genes at 24 hpf
Using the “gene summary” option, we found that several anatomical terms related to the developing musculature (“myotome,” “adaxial cell,” “trunk musculature,” “musculature system,” and “somite”) (Table 2; Supplementary Table 7), three of which (“myotome,” “musculature system,” and “somite”) were also identified using the “wild-type expression” option (Table 3; Supplementary Table 7). Since pharmacological use of glucocorticoids results in muscle wasting,27 these results reinforce the idea that usage of ZEOGS can identify the sites of action of GR signaling. In addition, we found terms related to heart and vasculature (“pericardium,” “primitive heart tube,” “vein,” “artery,” “posterior caudal vein”) with the “gene summary” option, but only heart-related anatomical terms were common to both data source options (Tables 2 and 3). Heart tissue and vasculature are indeed transcriptionally and physiologically responsive to both mineralocorticoids28 and glucocorticoids,29,30 with demonstrably different regulated sets of genes.31,32 Finally, anatomical terms related to zebrafish ear development (“immature anterior and posterior maculas”) were common to both data source options (Tables 2 and 3). Although, ear is known to respond to glucocorticoids,33 the significance of the result is unclear because of very different anatomies of mammalian and zebrafish ears and the fact that the enrichment is only based on two associated genes. Other enriched anatomical terms were found only with a single data source option. The analysis with the “gene summary” found “YSL” (“yolk syncitial layer”) associated with 18 genes. Furthermore, there were several notochord and nervous system-related anatomical terms enriched in the same analysis (“axial mesoderm,” “chordo neural hinge,” “roof plate,” “peripheral nervous system”) (Table 2). Several anatomical terms found only with the “wild-type expression” option were related to glia (“immature Schwann cell,” “glial cell,” “perineuronal satellite cell”) (Table 3). Such observations of predicted GR-regulated gene expression in neurons and glia are consistent with the expression of GR and mineralocorticoid receptor (MR) in rat neurons34,35 and glia,36 although further studies will be necessary to confirm their sites of expression and regulation. Identification of pronephros (“pronephric proximal convoluted tubule”) and “interrenal primordium” terms in this analysis may be significant since in the mammalian system kidney is a primary target of mineralocorticoids, but not of glucocorticoids because of glucocorticoid inactivation by the HSD11B2 enzyme.37 However, the situation in embryonic zebrafish may not be the same since MR is apparently not expressed in pronephros of embryonic zebrafish (ZFIN Gene Expression database).
Table 2.
Anatomical Terms Enriched for Genes Upregulated by Beclomethasone Treatment at 24 hpf: ZEOGS Analysis with the “Gene Summary” Option
| Anatomical term | Sample gene count | Total gene count | p-value | Corrected p-value | Below threshold? | Enriched for “wt_expression” |
|---|---|---|---|---|---|---|
| Myotome | 32 | 703 | 5,29E-01 | 0.00065 | Yes | ✓ |
| Trunk musculature | 8 | 82 | 0.00022 | 0.01360 | Yes | |
| Adaxial cell | 18 | 355 | 0.00024 | 0.00967 | Yes | |
| Musculature system | 12 | 246 | 0.00382 | 0.11648 | No | ✓ |
| Pericardium | 2 | 5 | 0.00388 | 0.09468 | Yes | |
| Immature posterior macula | 2 | 5 | 0.00388 | 0.07890 | Yes | ✓ |
| Ysl | 20 | 531 | 0.00440 | 0.07664 | Yes | |
| Somite | 35 | 1158 | 0.00641 | 0.09770 | Yes | ✓ |
| Immature anterior macula | 2 | 7 | 0.00793 | 0.10754 | No | ✓ |
| Vein | 8 | 155 | 0.01271 | 0.15504 | No | |
| Artery | 7 | 130 | 0.01565 | 0.17362 | No | |
| Primitive heart tube | 7 | 146 | 0.02765 | 0.28112 | No | ✓ |
| Vent | 1 | 2 | 0.03994 | 0.37480 | No | |
| Axial mesoderm | 4 | 67 | 0.04566 | 0.39794 | No | |
| Chordo neural hinge | 3 | 40 | 0.04607 | 0.37471 | No | |
| Pharyngeal pouch 1 | 1 | 3 | 0.05930 | 0.45217 | No | |
| Peripheral nervous system | 2 | 20 | 0.06052 | 0.43433 | No | |
| Roof plate | 1 | 4 | 0.07827 | 0.53052 | No |
Significantly enriched anatomical terms were identified for the input gene list using ZEOGS with the “gene summary” option at the 24–30 hpf developmental stage and presented in the table together with the sample and total gene counts, hypergeometric p-values (with 0.1 being the enrichment threshold), Benjamini–Hochberg-corrected p-values, indications whether corrected p-values are <0.1 (gray rows), and whether the anatomical terms were also found enriched when the analysis was run with the “wild-type expression” option (✓). The number of genes with anatomical terms for all genes and the sample were 5842 and 118, respectively. ZEOGS encountered 346 anatomical terms for all genes and 122 for the sample genes.
Table 3.
Anatomical Terms Enriched for Genes Upregulated by Beclomethasone Treatment at 24 hpf: ZEOGS Analysis with the “Wild-Type Expression” Option
| Anatomical term | Sample gene count | Total gene count | p-value | Corrected p-value | Below threshold? | Enriched for “gene_summary” |
|---|---|---|---|---|---|---|
| Myotome | 31 | 598 | 1,45E-02 | 0.00001 | Yes | ✓ |
| Heart rudiment | 9 | 168 | 0.00536 | 0.25172 | No | |
| Interrenal primordium | 2 | 6 | 0.00543 | 0.17020 | No | |
| Primitive heart tube | 8 | 149 | 0.00846 | 0.19876 | No | ✓ |
| Somite | 18 | 548 | 0.01858 | 0.34937 | No | ✓ |
| Pronephric proximal convoluted tubule | 2 | 11 | 0.01868 | 0.29261 | No | |
| Perineuronal satellite cell | 1 | 1 | 0.01962 | 0.26353 | No | |
| Glial cell | 1 | 1 | 0.01962 | 0.23058 | No | |
| Immature Schwann cell | 1 | 1 | 0.01962 | 0.20496 | No | |
| Pronephric proximal straight tubule | 2 | 12 | 0.02213 | 0.20801 | No | |
| Skeletal muscle cell | 2 | 13 | 0.02582 | 0.22065 | No | |
| Immature posterior macula | 1 | 2 | 0.03886 | 0.30442 | No | ✓ |
| Immature anterior macula | 1 | 2 | 0.03886 | 0.28100 | No | ✓ |
| Gut | 5 | 106 | 0.05631 | 0.37808 | No | |
| Musculature system | 1 | 3 | 0.05772 | 0.36172 | No | ✓ |
| Common myeloid progenitor | 1 | 4 | 0.07621 | 0.44774 | No |
Significantly enriched anatomical terms were identified for the input gene list using ZEOGS with the “wild-type expression” option at the 24–30 hpf developmental stage and are presented in the table together with the sample and total gene counts, hypergeometric p-values (with 0.1 being the enrichment threshold), Benjamini–Hochberg-corrected p-values, indications whether corrected p-values are <0.1 (gray rows), and whether the anatomical terms were also found enriched when the analysis was run with the “gene summary” option (✓). The numbers of genes with anatomical terms for all genes and the sample were 4784 and 94, respectively. ZEOGS encountered 500 anatomical terms for all genes and 94 for the sample genes.
Application of ZEOGS to analyze the set of beclomethasone-regulated genes at 72 hpf
In contrast to the results at 24 hpf, the analysis at 72 hpf did not show an overlap for the two data source options and showed very different numbers of genes associated with anatomical terms (90 for the “gene summary” and 19 for the “wild-type expression”). Moreover, enrichments of anatomical terms for the “wild-type expression” option were calculated on the basis of only one or two genes. These results indicate that the “wild-type expression” option of ZEOGS should not be used beyond the stage 33 (60 hpf), which is also consistent with the data in Fig. 3A. Therefore, we focused only on results obtained with the “gene summary” option (Table 4; Supplementary Table 8). The top and highly significant anatomical term identified was “liver,” a well-characterized site of GR action,38 where it induces gluconeogenesis. Anatomical terms related to cartilage and presumptive skeletal structures (“chondrocranium,” “palatoquadrate cartilage,” “vertebra,” “notochord,” and “teeth”) were also enriched, which is in line with pharmacological effects of glucocorticoids in regulation of gene expression in chondrocytes,39,40 inhibition of their proliferation, survival, and differentiation.41,42 Neural-related anatomical terms “epiphysis” and “tract of the post-optic commisure” were likewise strongly enriched, epiphysis being represented by 14 genes. This result is of much interest, because several recent studies indicate that glucocorticoids are implicated into control of melatonin production during the light-dark cycle43–45 and gene regulation in the pineal gland.46 Further, the cardiovascular system was represented by “heart,” “posterior cardinal vein,” and “posterior caudal vein” terms. This overlaps with our findings at 24 hpf and is consistent with the mammalian studies on glucocorticoid and MR function in the heart.29,31,32 Similarly to the results at 24 hpf, ear-related anatomical terms “semi-circular canal” and “otolith organ” were identified as enriched terms. “Somite,” a strongly enriched term at 24 hpf, was also enriched at 72 hpf, suggesting that the genes expressed in the muscle are regulated by GR at both stages. Finally, “pronephric duct” was another anatomical term shared between the results of 24 and 72 hpf experiments, further reinforcing the significance of GR-mediated gene regulation in this anatomical structure.
Table 4.
Anatomical Terms Enriched for Genes Upregulated by Beclomethasone Treatment at 72 hpf: ZEOGS Analysis with the “Gene Summary” Option
| Anatomical term | Sample gene count | Total gene count | p-value | Corrected p-value | Below threshold? |
|---|---|---|---|---|---|
| Liver | 32 | 1066 | 0.00044 | 0.04805 | Yes |
| Epiphysis | 15 | 458 | 0.00907 | 0.49416 | No |
| Otolith organ | 1 | 2 | 0.03330 | 1.00 | No |
| Tract of the postoptic commissure | 1 | 3 | 0.04954 | 1.00 | No |
| Chondrocranium | 2 | 24 | 0.06065 | 1.00 | No |
| Palatoquadrate cartilage | 2 | 24 | 0.06065 | 1.00 | No |
| Somite | 26 | 1158 | 0.06621 | 1.00 | No |
| Semicircular canal | 2 | 26 | 0.06991 | 0.95247 | No |
| Pharynx | 7 | 220 | 0.07604 | 0.92092 | No |
| Posterior cardinal vein | 3 | 59 | 0.07616 | 0.83017 | No |
| Heart | 16 | 653 | 0.07750 | 0.76800 | No |
| Pronephric duct | 15 | 605 | 0.07962 | 0.72321 | No |
| Paraxial mesoderm | 5 | 138 | 0.08191 | 0.68675 | No |
| Intestinal bulb | 7 | 229 | 0.08965 | 0.69796 | No |
| Posterior caudal vein | 1 | 6 | 0.09661 | 0.70206 | No |
Significantly enriched anatomical terms were identified for the input gene list using ZEOGS with the “gene summary” option at the 72–96 hpf developmental stage and presented in the table together with sample and total gene counts, hypergeometric p-values (with 0.1 being the enrichment threshold), Benjamini–Hochberg-corrected p-values, and indications whether corrected p-values are <0.1 (gray rows). The numbers of genes with anatomical terms for all genes and the sample were 6066 and 102, respectively. ZEOGS encountered 377 anatomical terms for all genes and 109 for the sample genes.
Discussion
The advent of high-throughput gene expression profiling technologies in the zebrafish model system has enabled systematic analysis of transcriptomes, but often at the expense of spatial resolution when whole embryos or larvae are used for the analysis. The question of where in the embryo certain expression changes occur can thus become impossible to answer if one only has gene lists from such high-throughput experiments. However, the availability of large amounts of detailed gene expression pattern information in the zebrafish makes it possible to address this question using prediction approaches without direct experimental validation. In this article, we describe ZEOGS, a web-based computational method to predict the anatomical region(s) in which changes in expression patterns take place for a given set of input genes. The prediction is based on the expression information available in the ZFIN database and the AO of zebrafish. The latter provides a unified vocabulary for describing zebrafish anatomical structures and a hierarchical structure for the relationships among the anatomical terms. The principle of our method is to obtain a list of relevant anatomical structures for each gene in an input set and for all known or tested genes, and perform a hypergeometric test to identify anatomical structures that have a statistically significant enrichment. Previously, Packham and colleagues47 studied transcriptional changes caused by blocked blood flow in zebrafish embryos and tabulated all of the regulated genes with the corresponding anatomical terms from ZFIN, conceptually similar to what ZEOGS does. What is new about ZEOGS is that it is automatic and incorporates statistical methods, and, to our knowledge, this method has not been applied in this form to zebrafish or other animal model systems. The inspiration and the basic statistical procedure in ZEOGS were adopted from GO term enrichment tools, which are used for interpreting high-throughput datasets.3,48,49 The statistical procedure implemented in ZEOGS is based on the calculation of a one-tailed hypergeometric p-value given its focus on enriched anatomical terms, which may be different for statistical testing in some GO tools.48 Another difference is that the GO terms are derived from different types of experimental and computational annotations,49 whereas ZEOGS uses almost exclusively the data from RNA in situ hybridization stainings available on ZFIN.
During the development of ZEOGS, we also compared different sources of zebrafish gene expression pattern information and found that unsurprisingly ZFIN was more complete than Ensembl BioMart50 and 4DXpress.9 The smaller amount of gene expression data in both Ensembl BioMart and 4DXpress suggests that their contents are not frequently synchronized with ZFIN and indeed 4DXpress has not been updated since its launch in 2008, which explains its current state. Ensembl, on the other hand, may be more focused on providing genomic coordinates, sequences, and variation information than on serving as a hub for gene expression pattern information.
There are two main ways to obtain gene expression pattern information on ZFIN, the first being from gene summary pages (“gene summary”), where all annotated anatomical terms for a specific gene are listed, and the second being derived from manually curated published results of in situ hybridization experiments (“wild-type expression”). When applying the “gene summary” option in ZEOGS, anatomical terms are filtered based on their presence at specified stages. We found that this option could be applied successfully to both 24 and 72 hpf datasets, but it is less empirically based than the “wild-type expression” option, for which the data are mostly available from ∼6–60 hpf. Haudry and colleagues9 previously reached a very similar conclusion concerning the developmental stage distribution of gene expression data on ZFIN. These stage distribution quantifications can be explained partially by the biological focus of the investigators but mostly by limitations of the RNA in situ hybridization techniques that can successfully stain zebrafish embryos only until about 3–4 days of development. Thus, the zebrafish community clearly needs stage-independent gene expression visualization technologies. Once technical challenges are overcome, our vision is that, for each gene at each stage of zebrafish development, we will have a detailed and highly specific anatomical term annotation, which would increase the prediction power of ZEOGS-like tools and obviate the need for two different data source options.
As negative control, we applied ZEOGS to randomly chosen gene sets and showed that the proportion of enriched terms was comparable with the p-value threshold, whereas when we applied the Benjamini–Hochberg correction for multiple testing, there were almost no enriched terms. The results with random gene sets mean that those anatomical terms passing the significance threshold after the Benjamini–Hochberg correction for real datasets are likely significant. Importantly, the terms with larger p-values may also be biologically significant since gene list inputs to ZEOGS are already coming from experiments where false-positive rates have been controlled. We also addressed the issue of a possible bias in the results toward heavily studied anatomical terms. Indeed, there was a modest correlation between the enrichment likelihood of an anatomical term and the number of genes associated with it. However, no correlation was found between enrichment p-values and extents of anatomical term annotations. Thus, enrichment p-values are of greater use for judging enrichment of anatomical terms. The opposite situation occurs when anatomical terms with 1 or very few associated genes are identified as enriched. Here we recommend the user to pay attention to anatomical term annotations and judge whether such terms may indeed be enriched and biologically relevant.
We also applied ZEOGS to biological datasets we generated obtained by treating zebrafish embryos at 24 and 72 hpf stages with 25 μM beclomethasone and performing gene expression profiling. We found that most of the identified enriched anatomical terms showed overlap with mammalian tissues known to be responsive to glucocorticoids. The extent of enrichment was different between stages and there were common as well as stage-specific enriched anatomical terms, indicating that tissues show stage-specific sensitivities to glucocorticoids. The choice of the data source option was also very important, since the “gene summary” option was applicable to any developmental stage, whereas the “wild-type expression” option was applicable to stages before 72 hpf. Our studies with GR were a good test case for ZEOGS on a ubiquitously expressed transcription factor where we expected effects on several tissues. We also tested ZEOGS on datasets where much more tissue-specific effects were expected. Indeed, we found that the term “retinal pigment epithelium” was highly enriched in the expression profiling datasets of retinas with or without the RPE. Similarly, a gene set containing putative hedgehog signaling target genes allowed us to identify enrichment of anatomical terms, which are well-established sites of hedgehog signaling. The larger numbers of highly enriched anatomical terms in these two latter datasets compared with the former dataset also suggest that the extent of anatomical term enrichment may be greater when expression changes are more tissue specific due to similar expression patterns of large subsets of these genes. Together, these example applications of ZEOGS argue for its ability to identify anatomical features of the gene expression patterns of genes in large datasets irrespective of how tissue specific these gene expression changes are.
In this study, we have developed a novel tool called ZEOGS for calculating enrichment of zebrafish gene expression pattern features using straightforward but powerful algorithms and publicly available zebrafish gene expression data. To ensure the reliability and usefulness of the software, we carefully studied the properties of data sources it is based upon and tested its applications to random and biological datasets. Its performance on biological datasets was highly consistent with our expectations, but also led to some novel predictions. Thus, we expect the software to be suitable for finding novel anatomical term associations with large gene sets. The input for ZEOGS are lists of genes obtained from high-throughput experiments, whereas the output may inform new experiments to identify expression patterns of novel genes without associated expression information, as well as to study more in detail the expression patterns of genes with known function (Fig. 5). These experiments, if submitted, could further enrich the ZFIN database and, consequently, the data used for ZEOGS algorithm, thus enabling a productive cycle of database and tool improvement.
FIG. 5.

Information flow of ZEOGS. ZEOGS directly uses expression pattern information available in ZFIN databases and typically generated in high-throughput experiments whose results are large gene sets. The output of ZEOGS (anatomical terms, list of genes for anatomical terms, and a list of genes without annotations) may guide focused experiments to determine gene expression patterns further expanding the information available on ZFIN and through an update mechanism improving the output of ZEOGS.
Authors' Contributions
S.V.P. conceived the idea of the method, programmed the software, performed computational and biological experiments, and wrote the article. A.M. analyzed the microarray data, drafted the microarray analysis section of the article, and edited the article. S.H.M. was involved in the design and analysis of experiments and writing of the article.
Supplementary Material
Acknowledgments
We would like to thank Julia Jarrells for carrying out microarray hybridizations, Edda Einfeldt and Katja Borzym for technical assistance in the lab, Dmitry Shaposhnikov for creative website banner design and recommendations on optimal color schemes for the website, and Irene Galkina for corrections of the article.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Teh C. Parinov S. Korzh V. New ways to admire zebrafish: progress in functional genomics research methodology. Biotechniques. 2005;38:897–906. doi: 10.2144/05386RV01. [DOI] [PubMed] [Google Scholar]
- 2.Alestrom P. Holter JL. Nourizadeh-Lillabadi R. Zebrafish in functional genomics and aquatic biomedicine. Trends Biotechnol. 2006;24:15–21. doi: 10.1016/j.tibtech.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Ashburner M. Ball CA. Blake JA. Botstein D. Butler H. Cherry JM, et al. Gene ontology: tool for the unification of biology. Gene Ontol Consort Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanehisa M. Goto S. Hattori M. Aoki-Kinoshita KF. Itoh M. Kawashima S, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sprague J. Bayraktaroglu L. Bradford Y. Conlin T. Dunn N. Fashena D, et al. The Zebrafish Information Network: the zebrafish model organism database provides expanded support for genotypes and phenotypes. Nucleic Acids Res. 2008;36:D768–D772. doi: 10.1093/nar/gkm956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sprague J. Bayraktaroglu L. Clements D. Conlin T. Fashena D. Frazer K, et al. The Zebrafish Information Network: the zebrafish model organism database. Nucleic Acids Res. 2006;34:D581–D585. doi: 10.1093/nar/gkj086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belmamoune M. Verbeek FJ. Developmental anatomy ontology of zebrafish: an integrative semantic framework. J Integr Bioinform. 2007;4:65. [Google Scholar]
- 8.Dahdul WM. Lundberg JG. Midford PE. Balhoff JP. Lapp H. Vision TJ, et al. The teleost anatomy ontology: anatomical representation for the genomics age. Syst Biol. 2010;59:369–383. doi: 10.1093/sysbio/syq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haudry Y. Berube H. Letunic I. Weeber PD. Gagneur J. Girardot C, et al. 4DXpress: a database for cross-species expression pattern comparisons. Nucleic Acids Res. 2008;36:D847–D853. doi: 10.1093/nar/gkm797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schaaf MJ. Chatzopoulou A. Spaink HP. The zebrafish as a model system for glucocorticoid receptor research. Comp Biochem Physiol A Mol Integr Physiol. 2009;153:75–82. doi: 10.1016/j.cbpa.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Schoonheim PJ. Chatzopoulou A. Schaaf MJ. The zebrafish as an in vivo model system for glucocorticoid resistance. Steroids. 2010;75:918–925. doi: 10.1016/j.steroids.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Heitzer MD. Wolf IM. Sanchez ER. Witchel SF. DeFranco DB. Glucocorticoid receptor physiology. Rev Endocr Metab Disord. 2007;8:321–330. doi: 10.1007/s11154-007-9059-8. [DOI] [PubMed] [Google Scholar]
- 13.Gross KL. Cidlowski JA. Tissue-specific glucocorticoid action: a family affair. Trends Endocrinol Metab. 2008;19:331–339. doi: 10.1016/j.tem.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westerfield M The Zebrafish Book. 4th. Eugene, OR: University of Oregon Press; 2000. A Guide for the Laboratory Use of the Zebrafish (Danio rerio) [Google Scholar]
- 15.Kimmel CB. Ballard WW. Kimmel SR. Ullmann B. Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 16.Ritchie ME. Silver J. Oshlack A. Holmes M. Diyagama D. Holloway A. Smyth GK. A comparison of background correction methods for two-colour microarrays. Bioinformatics. 2007;23:2700–2707. doi: 10.1093/bioinformatics/btm412. [DOI] [PubMed] [Google Scholar]
- 17.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 18.Smyth GK. Speed T. Normalization of cDNA microarray data. Methods. 2003;31:265–273. doi: 10.1016/s1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- 19.Benjamini Y. Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol) 1995;57:289–300. [Google Scholar]
- 20.Durinck S. Moreau Y. Kasprzyk A. Davis S. De Moor B. Brazma A. Huber W. BioMart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 21.Huang da W. Sherman BT. Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 22.Leung YF. Ma P. Dowling JE. Gene expression profiling of zebrafish embryonic retinal pigment epithelium in vivo. Invest Ophthalmol Vis Sci. 2007;48:881–890. doi: 10.1167/iovs.06-0723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu J. Srinivas BP. Tay SY. Mak A. Yu X. Lee SG, et al. Genomewide expression profiling in the zebrafish embryo identifies target genes regulated by Hedgehog signaling during vertebrate development. Genetics. 2006;174:735–752. doi: 10.1534/genetics.106.061523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ekker SC. Ungar AR. Greenstein P. von Kessler DP. Porter JA. Moon RT. Beachy PA. Patterning activities of vertebrate hedgehog proteins in the developing eye and brain. Curr Biol. 1995;5:944–955. doi: 10.1016/s0960-9822(95)00185-0. [DOI] [PubMed] [Google Scholar]
- 25.Rose AJ. Vegiopoulos A. Herzig S. Role of glucocorticoids and the glucocorticoid receptor in metabolism: insights from genetic manipulations. J Steroid Biochem Mol Biol. 2010;122:10–20. doi: 10.1016/j.jsbmb.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 26.Schlossmacher G. Stevens A. White A. Glucocorticoid receptor-mediated apoptosis: mechanisms of resistance in cancer cells. J Endocrinol. 2011;211:17–25. doi: 10.1530/JOE-11-0135. [DOI] [PubMed] [Google Scholar]
- 27.Hasselgren PO. Alamdari N. Aversa Z. Gonnella P. Smith IJ. Tizio S. Corticosteroids and muscle wasting: role of transcription factors, nuclear cofactors, and hyperacetylation. Curr Opin Clin Nutr Metab Care. 2010;13:423–428. doi: 10.1097/MCO.0b013e32833a5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viengchareun S. Le Menuet D. Martinerie L. Munier M. Pascual-Le Tallec L. Lombes M. The mineralocorticoid receptor: insights into its molecular and (patho)physiological biology. Nucl Recept Signal. 2007;5:e012. doi: 10.1621/nrs.05012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu B. Strom J. Chen QM. Dexamethasone induces transcriptional activation of Bcl-xL gene and inhibits cardiac injury by myocardial ischemia. Eur J Pharmacol. 2011;668:194–200. doi: 10.1016/j.ejphar.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodwin JE. Zhang J. Gonzalez D. Albinsson S. Geller DS. Knockout of the vascular endothelial glucocorticoid receptor abrogates dexamethasone-induced hypertension. J Hypertens. 2011;29:1347–1356. doi: 10.1097/HJH.0b013e328347da54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Latouche C. Sainte-Marie Y. Steenman M. Castro Chaves P. Naray-Fejes-Toth A. Fejes-Toth G, et al. Molecular signature of mineralocorticoid receptor signaling in cardiomyocytes: from cultured cells to mouse heart. Endocrinology. 2010;151:4467–4476. doi: 10.1210/en.2010-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshikawa N. Nagasaki M. Sano M. Tokudome S. Ueno K. Shimizu N, et al. Ligand-based gene expression profiling reveals novel roles of glucocorticoid receptor in cardiac metabolism. Am J Physiol Endocrinol Metab. 2009;296:E1363–E1373. doi: 10.1152/ajpendo.90767.2008. [DOI] [PubMed] [Google Scholar]
- 33.Tahera Y. Meltser I. Johansson P. Bian Z. Stierna P. Hansson AC. Canlon B. NF-kappaB mediated glucocorticoid response in the inner ear after acoustic trauma. J Neurosci Res. 2006;83:1066–1076. doi: 10.1002/jnr.20795. [DOI] [PubMed] [Google Scholar]
- 34.Rosenfeld P. van Eekelen JA. Levine S. de Kloet ER. Ontogeny of corticosteroid receptors in the brain. Cell Mol Neurobiol. 1993;13:295–319. doi: 10.1007/BF00711575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cintra A. Bhatnagar M. Chadi G. Tinner B. Lindberg J. Gustafsson JA, et al. Glial and neuronal glucocorticoid receptor immunoreactive cell populations in developing, adult, and aging brain. Ann N Y Acad Sci. 1994;746:42–61. doi: 10.1111/j.1749-6632.1994.tb39210.x. discussion 61–43. [DOI] [PubMed] [Google Scholar]
- 36.Bohn MC. Howard E. Vielkind U. Krozowski Z. Glial cells express both mineralocorticoid and glucocorticoid receptors. J Steroid Biochem Mol Biol. 1991;40:105–111. doi: 10.1016/0960-0760(91)90173-3. [DOI] [PubMed] [Google Scholar]
- 37.Mune T. White PC. Apparent mineralocorticoid excess: genotype is correlated with biochemical phenotype. Hypertension. 1996;27:1193–1199. doi: 10.1161/01.hyp.27.6.1193. [DOI] [PubMed] [Google Scholar]
- 38.Feigelson P. Beato M. Colman P. Kalimi M. Killewich LA. Schutz G. Studies on the hepatic glucocorticoid receptor and on the hormonal modulation of specific mRNA levels during enzyme induction. Recent Prog Horm Res. 1975;31:213–242. doi: 10.1016/b978-0-12-571131-9.50010-4. [DOI] [PubMed] [Google Scholar]
- 39.James CG. Ulici V. Tuckermann J. Underhill TM. Beier F. Expression profiling of dexamethasone-treated primary chondrocytes identifies targets of glucocorticoid signalling in endochondral bone development. BMC Genomics. 2007;8:205. doi: 10.1186/1471-2164-8-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jux C. Leiber K. Hugel U. Blum W. Ohlsson C. Klaus G. Mehls O. Dexamethasone impairs growth hormone (GH)-stimulated growth by suppression of local insulin-like growth factor (IGF)-I production and expression of GH- and IGF-I-receptor in cultured rat chondrocytes. Endocrinology. 1998;139:3296–3305. doi: 10.1210/endo.139.7.6099. [DOI] [PubMed] [Google Scholar]
- 41.van der Eerden BC. Karperien M. Wit JM. Systemic and local regulation of the growth plate. Endocr Rev. 2003;24:782–801. doi: 10.1210/er.2002-0033. [DOI] [PubMed] [Google Scholar]
- 42.Olney RC. Mechanisms of impaired growth: effect of steroids on bone and cartilage. Horm Res. 2009;72(Suppl 1):30–35. doi: 10.1159/000229761. [DOI] [PubMed] [Google Scholar]
- 43.Fernandes PA. Bothorel B. Clesse D. Monteiro AW. Calgari C. Raison S, et al. Local corticosterone infusion enhances nocturnal pineal melatonin production in vivo. J Neuroendocrinol. 2009;21:90–97. doi: 10.1111/j.1365-2826.2008.01817.x. [DOI] [PubMed] [Google Scholar]
- 44.Ferreira ZS. Fernandes PA. Duma D. Assreuy J. Avellar MC. Markus RP. Corticosterone modulates noradrenaline-induced melatonin synthesis through inhibition of nuclear factor kappa B. J Pineal Res. 2005;38:182–188. doi: 10.1111/j.1600-079X.2004.00191.x. [DOI] [PubMed] [Google Scholar]
- 45.Couto-Moraes R. Palermo-Neto J. Markus RP. The immune-pineal axis: stress as a modulator of pineal gland function. Ann N Y Acad Sci. 2009;1153:193–202. doi: 10.1111/j.1749-6632.2008.03978.x. [DOI] [PubMed] [Google Scholar]
- 46.Dagnino-Subiabre A. Zepeda-Carreno R. Diaz-Veliz G. Mora S. Aboitiz F. Chronic stress induces upregulation of brain-derived neurotrophic factor (BDNF) mRNA and integrin alpha5 expression in the rat pineal gland. Brain Res. 2006;1086:27–34. doi: 10.1016/j.brainres.2006.02.118. [DOI] [PubMed] [Google Scholar]
- 47.Packham IM. Gray C. Heath PR. Hellewell PG. Ingham PW. Crossman DC, et al. Microarray profiling reveals CXCR4a is downregulated by blood flow in vivo and mediates collateral formation in zebrafish embryos. Physiol Genomics. 2009;38:319–327. doi: 10.1152/physiolgenomics.00049.2009. [DOI] [PubMed] [Google Scholar]
- 48.Rivals I. Personnaz L. Taing L. Potier MC. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics. 2007;23:401–407. doi: 10.1093/bioinformatics/btl633. [DOI] [PubMed] [Google Scholar]
- 49.du Plessis L. Skunca N. Dessimoz C. The what, where, how and why of gene ontology—a primer for bioinformaticians. Brief Bioinform. 2011;12:723–735. doi: 10.1093/bib/bbr002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kinsella RJ. Kahari A. Haider S. Zamora J. Proctor G. Spudich G, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011 doi: 10.1093/database/bar030. bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.