

Abstract
Astrocytes have many functions in the central nervous system (CNS). They support differentiation and homeostasis of neurons and influence synaptic activity. They are responsible for formation of the blood-brain barrier (BBB) and make up the glia limitans. Here, we review their contribution to neuroimmune interactions and in particular to those induced by the invasion of activated T cells. We discuss the mechanisms by which astrocytes regulate pro- and anti-inflammatory aspects of T-cell responses within the CNS. Depending on the microenvironment, they may become potent antigen-presenting cells for T cells and they may contribute to inflammatory processes. They are also able to abrogate or reprogram T-cell responses by inducing apoptosis or secreting inhibitory mediators. We consider apparently contradictory functions of astrocytes in health and disease, particularly in their interaction with lymphocytes, which may either aggravate or suppress neuroinflammation.
1. Introduction
Within the central nervous system (CNS), astrocytes are the most abundant cells. Their main task is to maintain the physiological homeostasis of neurons by providing a stable microenvironment and growth factors. Astrocytes form multicellular syncytia in vivo that ensure neuronal homeostasis by taking up excess neurotransmitters and buffering the ionic content of the extracellular medium in the brain. Astrocyte membranes contain numerous neurotransmitter receptors and transporters and can therefore sense and regulate formation, stability, and efficacy of synapses [1]. Recently, they have been shown to play a role in synaptic activity and regulating neuronal circuitry [2–4].
Astrocytes are dysfunctional in various neurological disorders such as epilepsy, amyotrophic lateral sclerosis, hepatic encephalopathy, stroke, and focal cerebral ischaemia (reviewed in [5]). Dysfunction is often accompanied by astrocytic hypertrophy and an increased number of astrocytic processes, termed astrogliosis [6]. Astrocytes also show these signs of activation in Alzheimer's disease [7, 8] and in Parkinson's disease [9] as well as in its rat model (Figure 1) [10]. Massive astrogliosis has been observed in postmortem tissue of Parkinsonian patients [9, 11–13]. These tissues demonstrated a lack of astrocyte-derived neurotrophins compared to control brains [14, 15]. Because astrocytes support and protect dopaminergic neurons in vitro [16], a functional failure of astrocytes may contribute to CNS pathology.
Figure 1.
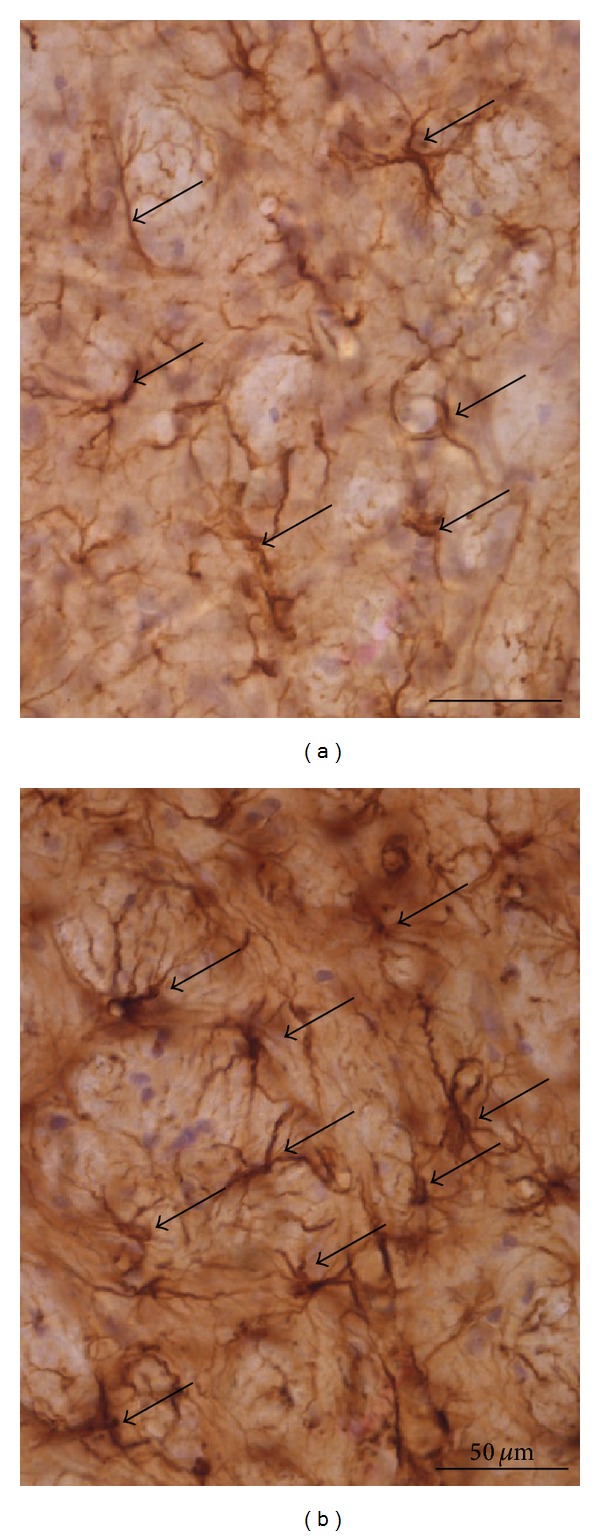
Astrocytes activated in a rat model of Parkinson's disease. Astrocytes (arrows) in the globus pallidus of rats after unilateral striatallesion of dopaminergic neurons by injection of 6-hydroxydopamine (6-OHDA). (a) Contralateral hemisphere; astrocytes have short cellular processes. (b) Ipsilateral hemisphere; astrocytes are in an activated state characterised by long cellular processes and enlarged cell bodies with an intense staining. Staining of glial fibrillary acidic protein (GFAP). For detailed information, see [10].
The potential for antigen presentation and production of proinflammatory cytokines by astrocytes has been studied in the neuroinflammatory disease multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE). They can protect against neuroinflammation by T cells invading the CNS. Thus, they contribute to the immune privilege of the CNS. The privilege is not simply the absence of immune reactions but rather a complicated network of passive and active barriers and of brain tissue. It can modify immune reactions in the CNS so as to minimize the danger of destructive side effects in a tissue with limited ability to regenerate [17]. In this review, we focus on astrocyte functions in health and disease, particularly on their interaction with lymphocytes.
2. Functions of Astrocytes at the Blood-Brain Barrier (BBB)
The BBB limits exchange of solutes between capillaries and the brain parenchyma. Brain capillaries are about 50 to 100 times tighter than peripheral capillaries. This is achieved by complex tight junctions. Astrocytes influence tightness of the BBB by soluble factors that affect endothelial cells [18]. The perivascular space is separated from the brain parenchyma by the basement membrane and the glia limitans, made up of astrocytic end-feet, reviewed in [19]. Notably, it is not the direct contact of astrocytic end-feet with endothelial cells that induces the tightness but soluble factors secreted by them. The presence of numerous astrocytic end-feet close to the BBB allows for a rapid regulation of BBB permeability. Humoural agents that are able to increase BBB permeability and may be secreted by astrocytes include endothelin-1, glutamate, interleukin- (IL-) 1β, IL-6, tumour necrosis factor (TNF), macrophage inflammatory protein- (MIP-) 2, and nitric oxide [20]. Soluble astrocytic factors that induce tight junction formation at the BBB are less well characterized. A recent study has shown that sonic hedgehog, a member of the hedgehog signalling pathway family, is produced by astrocytes. Sonic hedgehog promotes BBB formation and integrity, and hedgehog-mediated signals induce immune quiescence in the CNS [21]. Thus, inhibition of hedgehog signalling exacerbates EAE by increasing demyelination, accumulation of leukocytes in the CNS, and production of interferon- (IFN-) γ and IL-17 by infiltrating T cells [21].
3. Pro- and Anti-Inflammatory Mediators Produced by Astrocytes
Astrocytes are capable of producing a range of proinflammatory cytokines that have been found in the brain of Alzheimer's disease patients such as IL-1α, IL-1β, IL-6, and TNF [22]. It has been shown that amyloid-β 25–35 in combination with bacterial cell wall lipopolysaccharide (LPS) induced a strong astrocytic production of IL-6 and TNF while neither of the substances alone did [23]. Others found that LPS induced the production of TNF, IL-6, and IL-1 in microglia but not in astrocytes while astrocytes responded neither to LPS nor TNF but to IL-1β by producing TNF and IL-6 [24]. This indicates that astrocytes may be regulated by microglial IL-1β. Microglial cells produce free radicals and proinflammatory cytokines such as TNF-α when exposed to amyloid-β 1–42 [25, 26]. TNF and superoxide anion production by macrophages cocultured with amyloid-β 1–42 was strongly reduced in the presence of primary human astrocytes or astrocytoma cells. Interestingly, astrocytes bound amyloid-β 1–42 and showed activation of the transcription factor NFκB in that study, but unlike in macrophages this activation did not result in TNF production. This indicates that distinct signal transduction pathways are activated in macrophages and astrocytes by inflammation [27]. Indeed, astrocytes can also downregulate microglial activation by the secretion of anti-inflammatory substances such as transforming growth factor- (TGF-) β and prostaglandin E2 (PGE2) [28, 29] and may thereby limit inflammation-induced neurodegeneration. However, activated microglia can also reduce amyloid-β accumulation by phagocytosing and degrading it [30]. Thus, the clinical relevance of both astrocytic and microglial activation has not yet been fully elucidated.
Glia maturation factor (GMF) is produced by astrocytes. It is not only necessary for the growth and maturation of neurons and glia cells, but can also induce the production of proinflammatory cytokines. Overexpression of GMF in astrocytes induces the production and secretion of granulocyte-macrophage-colony stimulating factor (GM-CSF), an activation of microglia and the expression of proinflammatory genes including major histocompatibility complex- (MHC-) II, IL-1β, and MIP-1β [31]. Knockdown of GMF reduces the production of the proinflammatory cytokines and chemokines responsible for EAE [32, 33]. Interestingly, it also inhibits growth of glioblastoma cells by inducing G0/G1 cell cycle arrest in vitro [34, 35]. In the brain of Alzheimer's disease patients, GMF is upregulated [36, 37]. However, what drives astrocytes to upregulate GMF to a level where it contributes to tissue damage is unknown.
Astrocytes produce or take up, store, and reexocytose a range of neurotrophins neuroprotective in EAE [38–41], dementia of the Alzheimer type [42], and Parkinson's disease [43, 44]. Astrocytes are the major source of nerve growth factor (NGF) and glial cell line-derived neurotrophic factor (GDNF) in the CNS [45–47]. In brain tissue of Parkinson's disease patients, GDNF, NGF, and brain-derived neurotrophic factor (BDNF) are deficient [14, 15], hence the clinical trials of therapeutic GDNF injection into the brain of Parkinson's patients. While intraputaminal infusion of GDNF was safe and improved motor functions in a small group of patients over one [48] and two years [49], a randomized placebo-controlled trial found that motor function has not improved [50]. Notably, from all the 32 genes associated with astrocyte function described in this review, only GDNF was found to be associated with a disease: “major depressive disorder.” For this, see the NCBI catalog of genomewide association studies (GWAS) (http://www.genome.gov/gwastudies/).
On the other hand, as mentioned above, astrocytes are a major source of the proinflammatory cytokines IL-1β and IL-6 in the brain [51, 52]. Transgenic mice that lack IL-6 production are resistant to EAE induction [53, 54]. This is due to a blockade of activation and differentiation of autoreactive T cells in the periphery with both T helper (Th) 1 and Th2 cells differentiation being affected [53]. Very recently, dendritic cells have been identified as a sufficient and probably the main source for EAE induction [55]. Whether astrocytic IL-6 plays a decisive role in the etiogenesis of EAE has been ruled out in animal models. Transgenic mice that overexpress IL-6 in astrocytes but are otherwise deficient in IL-6 develop a mild form of ataxia, but no symptoms of lymphocyte-driven EAE. These mice had indeed cellular infiltrates in the cerebellum independent of MOG immunisation [56]. Thus, the observed ataxia may be a result of a general inflammatory process in the brain.
It is known that IL-1β plays an important role in MS and EAE. Families with a high IL-1β over IL-1 receptor antagonist (IL-1Ra) production ratio have a higher risk to have a patient relative with MS than families with a low ratio [57]. Mice deficient in IL-1 receptor type I (IL-1RI−/−) are resistant to EAE induction [58, 59]. Apparently, IL-1β is necessary for the induction of IL-17-producing T cells (Th17) [59]. IL-17 has been shown to be crucial for the development of EAE [60, 61]. However, both IL-6 and IL-1β do not necessarily have only detrimental effects. Recently, IL-6 has been demonstrated to induce IL-10 in T cells and thus inhibit proinflammatory responses of Th1 cells [62]. The production of IL-1β and IL-6 does not necessarily lead to neuronal damage because these cytokines also induce upregulation of Fas ligand (FasL) in astrocytes, which may induce T-cell apoptosis [63] (see below). In addition, IL-1β and IL-6 are messengers between the brain, particularly the hypothalamic-pituitary-adrenal axis, and the immune system. Thus, IL-1β produced during EAE upregulates glucocorticoid production which has a downregulatory effect on inflammation [64].
4. Interactions of Astrocytes and T Lymphocytes
4.1. Induction of Apoptosis in Activated T Cells
Activated T cells can cross the BBB not only in neuroinflammatory diseases but also in the healthy brain [65, 66]. Later, it has been shown that in macrophage-depleted mice, activated T cells which extravasate are not able to enter the brain parenchyma via the basement membrane but accumulate in the perivascular spaces [67]. Matrix metalloproteinases (MMP-) 2 and -9 are necessary to cross the basement membrane after local digestion [68]. These enzymes could be produced by perivascular macrophages.
These infiltrating T cells may combat infection, but damage to tissue needs to be avoided, and in particular that mediated by Th1 and cytotoxic T cells and accompanied by inflammation. Inflammatory cytokines such as TNF-α are neurotoxic. Given that neurons have a very limited capacity to regenerate in the mature brain, side effects could be detrimental. One mechanism preventing damage is elimination of T cells: astrocytes induce apoptosis in these cells [69–71]. This effect is mediated by the expression of FasL (CD95L) by astrocytes [63, 72, 73]. In EAE, FasL expressing astrocytes exist in close vicinity to apoptotic T cells [74, 75]. The same mechanism of enforcing immune-privilege has been observed in placenta [76–79], testes [80], and anterior chamber of the eye [81]. A downside of this mechanism is that astrocytoma express FasL and thus escape immune attack [82, 83].
4.2. Astrocytes as Antigen-Presenting Cells in Neuroinflammation
In neuroinflammation, astrocytes can act as antigen-presenting cells (APCs) [84, 85]. While microglia express MHC-II readily upon activation in vivo and in vitro, astrocyte MHC-II expression occurs only during prolonged inflammation in vivo [86] or in vitro under stimulation by interferon- (IFN-) γ [87]. This MHC-II induction may be suppressed by neurons via a mechanism that has not fully been elucidated. One study claims that cell-cell contact is required [88] while another one found that secreted glutamate and norepinephrine could inhibit IFN-γ induced MHC-II expression in astrocytes [89]. In keeping with this, neuronal loss induces MHC-II expression in astrocytes [88, 90], supporting the view that astrocytes can present antigen only during severe neuroinflammation. The expression of costimulatory B7 molecules by astrocytes both in vivo and in vitro has been controversially discussed. While some authors found B7 expression on astrocytes [91–94], others did not [95, 96]. Functioning as APCs in vitro, astrocytes have been found to stimulate differentiated T cells; and interestingly, they stimulate Th2 cells more efficiently than Th1 cells [87, 97]. Th2 cells may be less damaging than the cellular immune responses, and hence the preferred agents of protection against infection in the CNS. Thus, astrocytes from transgenic mice expressing MS-associated MHC-II human haplotypes HLA-DR2 and HLA-DR4 induced a mixed Th1/Th2 cytokine response in MOG-specific T cells, whereas dendritic cells induced a Th1 response [98]. One can only speculate about the biological relevance of an astrocyte-mediated Th2 bias. In EAE, T cells typically enter the CNS as activated, differentiated Th1 cells. However, the T-cell population may not consist exclusively of Th1 cells. If astrocytes preferentially restimulate Th2 cells [87, 97], the proportion of these cells could increase, thus favouring an anti-inflammatory microenvironment. Also, memory T cells are recruited to the CNS during EAE [99]. Memory cells are heterogeneous and part of the population is not biased for a certain Th subpopulation, yet. Thus, it is tempting to speculate that astrocytes may prevent induction of a Th1 cytokine profile of memory cells in the CNS [100]. The astrocyte-mediated bias towards Th2 responses cannot be explained by their cytokine secretion as astrocytes do not produce IL-4, which is the main inductor of Th2 responses, but might rather reflect the signal strength of the MHC-II-T-cell receptor (TCR) interaction. Lowering the signal strength has been found to favour Th2 differentiation [101]. For instance, the surface density of MHC-II expression determines the cytokine profile of T cells with low MHC-II expression levels favouring Th2 responses [102]. Astrocytes do not readily express MHC-II molecules and are thus likely to deliver a weaker TCR signal than “professional” APCs with higher density of MHC-II molecules on their surface.
4.3. Suppression of T-Cell Functions
In EAE, infiltrating T cells do not proliferate in the target organ [103]; this has been ascribed to the influence of astrocytes [104]. In vitro, astrocytes can either suppress [105–107] or stimulate [87, 97, 108] T-cell functions. In coculture studies, astrocytes induce hyporesponsiveness in T cells. This was interpreted as a result of downregulation of the TCR [105] and insufficient stimulation by low levels of ICAM-1 on astrocytes [106]; this would limit adhesion of T cells to astrocytes, so that the two cells ignore each other. As this would not silence invading T cells in the CNS, other mechanisms may have been involved which are not fully understood, yet.
T-cell activation is tightly regulated by surface molecules, providing scope for immunotherapy [109–111]. While the primary costimulatory molecule CD28 and its homologue CTLA-4 (cytotoxic T-lymphocyte-associated antigen-4, CD152) on T cells engage the same ligands B7-1 (CD80) and B7-2 (CD86) on APCs, CTLA-4 binds with 10–100-fold higher affinity than CD28 [110, 112]. CD28 signaling initiates, sustains, and enhances T-cell activation while CTLA-4 signaling inhibits T-cell activation and attenuates ongoing responses [110, 113, 114]. The relevance of this has been demonstrated by genetic inactivation of CTLA-4 in mice, which leads to lymphoproliferative disease and early death [110, 112]. T cells of this mouse strain proliferate spontaneously ex vivo and show an activated phenotype stressing the central role of CTLA-4 in attenuating unwanted T-cell responses. In contrast to CD28, which is constitutively expressed on the surface of T cells, CTLA-4 is not detectable on resting T cells [114]. Expression of CTLA-4 mRNA and CTLA-4 protein on the T-cell surface is induced upon activation. CTLA-4 is stored intracellularly, and its surface expression is strictly controlled with a peak after 48 h–72 h after T-cell stimulation [114, 115]. Blockade of CTLA-4 in mouse models of autoimmune diseases increases the incidence of EAE [111, 116]. Short blockade of CTLA-4 during priming of the immune response has lasting effects, suggesting that failure in the regulation of CTLA-4 would have long-lasting impact on immune responses including autoimmunity [117]. Thus, giving agonistic CTLA-4 signals might be a promising strategy for controlling inflammatory responses in the CNS, particularly as CTLA-4 is highly expressed on the T cells which accumulate there [118].
Our own study showed that astrocytes inhibit T-cell proliferation, production of IL-2 and IL-10, and expression of the IL2R α-chain (CD25) [107]. Functionally, astrocytes mediated these effects by upregulating CTLA-4 on Th1 and Th2 cells. Although inhibition did not require astrocyte contact with T cells, the mechanism was independent of the major inhibitory cytokine TGF-β. The study provided optimal stimulation for T cells by having professional APCs and antigen in the cultures when astrocytes were added. Thus, astrocytic inhibitory or stimulatory effects could be discerned from baseline effects occurring during T cell-APC interaction. In this way, we also avoided differences in the stimulatory capacity of astrocytes towards Th1 versus Th2 cells [87, 97]. The interpretation is supported by a recent study showing that astrocytes inhibited proliferation and IFN-γ, interleukin- (IL-) 4, IL-17, and TGF-β secretion levels of encephalitic T cells in vitro unless they were pretreated with IFN-γ. They even promoted T-cell proliferation, presumably by additional antigen presentation [119]. The inhibitory effect of astrocytes could be ameliorated by IL-27 neutralisation [119]. IL-27 has been shown to suppress Th17 cells and thereby EAE [120, 121]. Also, it negatively regulates Th17 cells during chronic inflammation of the CNS resulting from chronic infection with Toxoplasma gondii [122]. Coculture of astrocytoma cell lines with CD3/CD28-activated T cells revealed suppression of T-cell proliferation. The effect was more pronounced when direct contact was allowed between astrocytes and T cells but remained strong when astrocytes and T cells were separated by cell culture inserts [123]. The finding that T-cell proliferation was still inhibited by astrocytes when astrocytes and T cells were separated by a cell culture insert or a transwell-membrane showed that a soluble factor produced by astrocytes is responsible for this inhibition [107, 123, 124]. However, astrocytes might conceivably have protruded cellular nanotubes through the cell culture inserts so as to contact the T cells. The separating membranes had pore sizes of 200 nm [123] or 400 nm [107, 124]. An electron-microscopical study of astrocytes growing on engineered surfaces showed that astrocytes extend nanotubes with a diameter below 100 nm to make contact with other cells and may even exchange substances via these nanotubes [125]. This may be a mechanism which allowed astrocytes to contact the T cells physically. Cell-cell contact did not bear sole responsibility for the control of T-cell proliferation, since astrocyte-conditioned supernatant also inhibited T-cell proliferation [124]. Despite being of interest for immunotherapy, the nature of this soluble inhibitory factor remains unclear. Blockade of TGF-β had no [124] or only a minor effect [107] on the inhibition of T-cell proliferation. Inhibition of nitric oxide production also did not reverse the inhibitory effect [123, 124]. Furthermore, inhibition of indoleamine-2,3 dioxygenase (IDO) by methyltryptophan did not affect astrocyte-mediated inhibition of T-cell proliferation [123].
IDO is a tryptophan-degrading enzyme and as such inhibits T-cell proliferation. It has been proposed as a major player in the immune privilege of the placenta [126]. Astrocytes and microglia are capable of expressing IDO in vitro and in vivo upon activation with IFN-γ [127]. IDO blockade in EAE mediates disease exacerbation, suggesting that IDO induction by Th1-derived IFN-γ may play a role in self-limiting autoimmune inflammation during EAE and MS [128]. IDO can also induce tolerance of tumours in the CNS [129]. PGE2 induces IDO in dendritic cells [130, 131]. Systemic administration of cytosine-phosphate-guanine dinucleotide (CpG), a frequent dinucleotide in bacterial DNA and therefore detected by pattern recognition receptor Toll-like receptor-9 (TLR-9), upregulates IDO in plasmacytoid dendritic cells, where it is required for activation of regulatory T cells (Tregs), and blocks their conversion into Th17 cells [132]. Although likely, whether IDO induction in astrocytes by PGE2 or CpG plays a role in the CNS and whether astrocytes can induce Treg activation is one of the open questions concerning astrocytes so far. IDO-deficient mice develop exacerbated EAE with enhanced Th1 and Th17 responses [133]. In this model, not only tryptophan depletion was responsible for the effect on T cells but also a downstream tryptophan metabolite from the kynurenine pathway, 3-hydroxyanthranilic acid (3-HAA), was. The kynurenine pathway starts with tryptophan degradation by IDO or tryptophan-2,3 dioxygenase (TDO) leading to 3-HAA. 3-HAA was shown to increase the percentage of Tregs and inhibited Th1 and Th17 cells leading to EAE amelioration [133]. 3-HAA has been shown to be neuroprotective in cytokine-mediated inflammation in vitro [134] while other metabolites of the kynurenine pathway such as 3-hydroxykynurenine and quinolinic acid (QUIN) appear to be neurotoxic [135]. Another metabolite of the IDO-kynurenine pathway is kynurenic acid (KYNA) which has been shown to be neuroprotective [136]. Interestingly, activated human astrocytes have been shown to produce large amounts of KYNA but almost no QUIN [137]. Thus, astrocytic IDO activation may lead to various effects which are mostly beneficial.
Astrocytes in a rat EAE model could induce development of Tregs, as has been shown in a study where T cells that had been cocultured with astrocytes not only lost ability to proliferate and inhibit proliferation of antigen-stimulated T cells but also markedly alleviated the disease [138]. Also in this study a heat-sensitive soluble factor was implicated, other than IL-10 or TGF-β [138].
Another surface molecule, B7-H1 (PD-L1), might downregulate T-cell responses in the CNS; it is a member of the B7-family known to downmodulate T-cell activity [139]. In a model of fiber tract injury in the hippocampus of adult mice, it is strongly upregulated on astrocytes while T-cell recruitment to the site of injury was not accompanied by autoimmune demyelination [140].
4.4. Astrocyte-Released Signals That May Influence T-Cell Influx
Astrocytes are efficiently activated by the IFN-γ produced by Th1 cells (see above). Under the influence of IFN-γ, astrocytoma cells upregulate expression of chemokines including CCL3, CCL5, CXCL8, and CXCL10, as well as proinflammatory cytokines such as IL-6 and IL-1β (but also an anti-inflammatory IL-1 receptor antagonist) [123]. Most of these chemokines attract Th1 cells more than Th2 cells, thus aggravating neuroinflammation. Thus, astrocytes may inhibit and delay neuroinflammation, but in case of sustained inflammation accompanied by high IFN-γ levels, they may switch to become potent APCs and even promotors of inflammation [119].
4.5. T-Cell-Mediated Induction of Nerve Growth Factor
Nerve growth factor (NGF) is a member of the neurotrophin family. Growth, differentiation, survival, and maintenance of peripheral and central neurons are facilitated by NGF [143]. NGF administered intracerebroventricularly into marmosets delays the onset of EAE and reduces lesion formation [41]. Subsequent to induction of EAE, mice treated with NGF by intraperitoneal injection exhibited a delayed onset of disease in combination with lower clinical disease scores [144]. Moreover, myelin basic protein- (MBP-) specific T cells retrovirally transduced to secrete high levels of NGF are unable to mediate clinical EAE and suppress induction of EAE by nontransduced MBP-specific T cells in rats [40]. Astrocyte-T-cell interaction results in increased NGF production by astrocytes. This upregulation was found to be dependent on antigen recognition as blockade of MHC-II abrogated the effect, and resting astrocytes which were not able to present antigens did not show an upregulation of NGF production. Neutralisation of the cytokines IFN-γ, IL-4, and IL-10 produced in the cocultures did not affect NGF production [142]. This finding suggests a neuroprotective role of astrocytes during T-cell-mediated inflammation in the CNS. Conversely, cells of the immune system carry NGF receptors, and NGF signalling modulates immune function. Perivascular infiltrates of NGF-treated marmosets decrease IFN-γ and increase IL-10 expression [145]. NGF inhibits the MHC-II inducibility of microglia, thereby limiting antigen-presentation in the CNS [146].
Mechanisms by which astrocytes maintain immune privilege or limit inflammation-induced damage are summarised in Figure 2.
Figure 2.
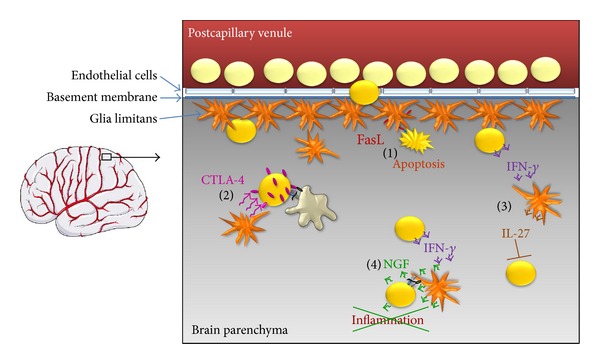
Astrocytes enforce the immune privilege of the CNS (left) at multiple checkpoints employing various mechanisms (right). Astrocytes in the glia limitans are responsible for the exceptional tightness of endothelial tight junctions by producing soluble factors [18]. Despite the BBB, activated T cells (yellow) are able to enter the brain parenchyma (grey) [65]. (1) At the same time, astrocytes in the glia limitans and in the parenchyma may express FasL while activated T cells may express Fas [63, 72, 73]. The ligation of Fas and FasL induces apoptosis of T cells [71]. (2) As this does not fully eradicate infiltrating T cells, the surviving T cells may be restimulated by activated microglia presenting CNS-specific antigens on MHC-II. In the presence of astrocytes, T cells upregulate CTLA-4 [107] which upon ligation of B7 molecules induces a stop of proliferation and anergy of the T cells. (3) IFN-γ produced by invading T cells stimulates astrocytic IL-27 production which suppresses Th17 cells [120, 121, 141]. (4) During sustained T-cell-mediated inflammation, IFN-γ secreted by T cells activates astrocytes to gain the ability to present antigen on MHC-II and costimulate T cells. While this cognate interaction may exacerbate neuroinflammation, it simultaneously leads to an upregulation of NGF production that counteracts neuroinflammation [142]. Also, astrocytes acting as APCs appear to promote Th2 responses and the formation of regulatory T cells [138]. Astrocytes: orange cells; pink: effects leading to CTLA-4 upregulation; green: effects of NGF; dark red: blood; grey: brain parenchyma.
5. Conclusions
For a long time, the CNS has been considered immune-privileged. However, the initial explanation of a strictly sealed BBB weakened when activated T cells were found to cross the BBB in the healthy brain. Clearly, various cells contribute to the phenomenon, including astrocytes, the most abundant cells of the CNS. Astrocytes mediate neuronal differentiation and homeostasis, and evidence is increasing that astrocytes interact with the immune system. The concept of immune privilege of the CNS may be weakening, but it is clear that astrocytes dampen inflammation and have beneficial, neuroprotective effects on the healthy brain. Astrocytes need activation by IFN-γ to unfold their anti-inflammatory potential, in forms such as IL-27 production [141]. Even when unable to prevent T-cell responses in the brain after prolonged provocation (e.g., by IFN-γ), their function does not become purely detrimental. When activated, astrocytes harbour mechanisms of damage limitation, such as production of neuroprotective NGF and preferential restimulation of Th2 over Th1 cells. When this is not sufficient to prevent autoimmune damage to the CNS, it may still control tissue damage to some extent. The overall picture of astrocytes is as CNS-intrinsic cells that combat local inflammation and maintain immune privilege, thus minimising damage.
Acknowledgments
This review was supported by SFB 854 TP14. The authors are grateful to Dr. J. Svoboda for helping with the immunohistochemical study of astrocytic activation in the rat model of Parkinson's disease. Scheme of brain (Figure 2, left) was kindly provided by Servier Medical Art.
References
- 1.Fields RD, Stevens-Graham B. Neuroscience: new insights into neuron-glia communication. Science. 2002;298(5593):556–562. doi: 10.1126/science.298.5593.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beattie EC, Stellwagen D, Morishita W, et al. Control of synaptic strength by glial TNFα . Science. 2002;295(5563):2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 3.Haydon PG. Glia: listening and talking to the synapse. Nature Reviews Neuroscience. 2001;2(3):185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- 4.Araque A, Sanzgiri RP, Parpura V, Haydon PG. Astrocyte-induced modulation of synaptic transmission. Canadian Journal of Physiology and Pharmacology. 1999;77(9):699–706. [PubMed] [Google Scholar]
- 5.Seifert G, Schilling K, Steinhäuser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nature Reviews Neuroscience. 2006;7(3):194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- 6.Strömberg I, Björklund H, Dahl D. Astrocytes responses to dopaminergic denervations by 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine as evidenced by glial fibrillary acidic protein immunohistochemistry. Brain Research Bulletin. 1986;17(2):225–236. doi: 10.1016/0361-9230(86)90119-x. [DOI] [PubMed] [Google Scholar]
- 7.Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Disease and Associated Disorders. 2000;14(supplement 1):S47–S53. doi: 10.1097/00002093-200000001-00008. [DOI] [PubMed] [Google Scholar]
- 8.Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer’s disease. Journal of Neuroimmunology. 2000;108(1-2):227–235. doi: 10.1016/s0165-5728(00)00285-x. [DOI] [PubMed] [Google Scholar]
- 9.Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP. The role of glial reaction and inflammation in Parkinson’s disease. Annals of the New York Academy of Sciences. 2003;991:214–228. doi: 10.1111/j.1749-6632.2003.tb07478.x. [DOI] [PubMed] [Google Scholar]
- 10.Henning J, Strauss U, Wree A, et al. Differential astroglial activation in 6-hydroxydopamine models of Parkinson’s disease. Neuroscience Research. 2008;62(4):246–253. doi: 10.1016/j.neures.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Hirsch EC, Hunot S, Hartmann A. Neuroinflammatory processes in Parkinson’s disease. Parkinsonism and Related Disorders. 2005;11(supplement 1):S9–S15. doi: 10.1016/j.parkreldis.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 12.Przedborski S, Goldman JE. Pathogenic role of glial cells in Parkinson's disease. In: Hertz L, editor. Non-Neuronal Cells of the Nervous System: Function and Dysfunction. New York, NY, USA: Elsevier; 2004. pp. 967–982. [Google Scholar]
- 13.Teismann P, Schulz JB. Cellular pathology of Parkinson’s disease: astrocytes, microglia and inflammation. Cell and Tissue Research. 2004;318(1):149–161. doi: 10.1007/s00441-004-0944-0. [DOI] [PubMed] [Google Scholar]
- 14.Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson’s disease. Journal of Neural Transmission, Supplement. 2000;(60):277–290. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
- 15.Chauhan NB, Siegel GJ, Lee JM. Depletion of glial cell line-derived neurotrophic factor in substantia nigra neurons of Parkinson’s disease brain. Journal of Chemical Neuroanatomy. 2001;21(4):277–288. doi: 10.1016/s0891-0618(01)00115-6. [DOI] [PubMed] [Google Scholar]
- 16.Mena MA, García de Yébenes J. Glial cells as players in parkinsonism: the “good,” the “bad,” and the “mysterious” glia. Neuroscientist. 2008;14(6):544–560. doi: 10.1177/1073858408322839. [DOI] [PubMed] [Google Scholar]
- 17.Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends in Immunology. 2007;28(1):12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Prat A, Biernacki K, Wosik K, Antel JP. Glial cell influence on the human blood-brain barrier. Glia. 2001;36(2):145–155. doi: 10.1002/glia.1104. [DOI] [PubMed] [Google Scholar]
- 19.Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends in Immunology. 2007;28(1):5–11. doi: 10.1016/j.it.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 20.Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. Journal of Anatomy. 2002;200(6):629–638. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alvarez JI, Dodelet-Devillers A, Kebir H, et al. The hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334(6063):1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- 22.Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer's disease, role of cytokines. Scientific World Journal. 2012;2012 doi: 10.1100/2012/756357.756357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forloni G, Mangiarotti F, Angeretti N, Lucca E, de Simoni MG. β-amyloid fragment potentiates IL-6 and TNF-α secretion by LPS in astrocytes but not in microglia. Cytokine. 1997;9(10):759–762. doi: 10.1006/cyto.1997.0232. [DOI] [PubMed] [Google Scholar]
- 24.Lee SC, Liu W, Dickson DW, Brosnan CF, Berman JW. Cytokine production by human fetal microglia and astrocytes: differential induction by lipopolysaccharide and IL-1β . Journal of Immunology. 1993;150(7):2659–2667. [PubMed] [Google Scholar]
- 25.Meda L, Bonaiuto C, Baron P, Otvos L, Jr., Rossi F, Cassatella MA. Priming of monocyte respiratory burst by β-amyloid fragment (25-35) Neuroscience Letters. 1996;219(2):91–94. doi: 10.1016/s0304-3940(96)13177-3. [DOI] [PubMed] [Google Scholar]
- 26.Klegeris A, McGeer PL. beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. Journal of Neuroscience Research. 1997;49:229–235. doi: 10.1002/(sici)1097-4547(19970715)49:2<229::aid-jnr11>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 27.Smits HA, van Beelen AJ, de Vos NM, et al. Activation of human macrophages by amyloid-β is attenuated by astrocytes. Journal of Immunology. 2001;166(11):6869–6876. doi: 10.4049/jimmunol.166.11.6869. [DOI] [PubMed] [Google Scholar]
- 28.Font-Nieves M, Sans-Fons MG, Gorina R, et al. Induction of COX-2 enzyme and down-regulation of COX-1 expression by lipopolysaccharide (LPS) control prostaglandin E2 production in astrocytes. Journal of Biological Chemistry. 2012;287(9):6454–6468. doi: 10.1074/jbc.M111.327874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vincent VA, Tilders FJ, van Dam AM. Inhibition of endotoxin-induced nitric oxide synthase production in microglial cells by the presence of astroglial cells: a role for transforming growth factor beta. Glia. 1997;19:190–198. doi: 10.1002/(sici)1098-1136(199703)19:3<190::aid-glia2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 30.Frautschy SA, Yang F, Irrizarry M, et al. Microglial response to amyloid plaques in APPsw transgenic mice. American Journal of Pathology. 1998;152(1):307–317. [PMC free article] [PubMed] [Google Scholar]
- 31.Zaheer A, Zaheer S, Sahu SK, et al. A novel role of glia maturation factor: induction of granulocyte-macrophage colony-stimulating factor and pro-inflammatory cytokines. Journal of Neurochemistry. 2007;101(2):364–376. doi: 10.1111/j.1471-4159.2006.04385.x. [DOI] [PubMed] [Google Scholar]
- 32.Zaheer A, Sahu SK, Wu YH, et al. Diminished cytokine and chemokine expression in the central nervous system of GMF-deficient mice with experimental autoimmune encephalomyelitis. Brain Research. 2007;1144(1):239–247. doi: 10.1016/j.brainres.2007.01.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaheer A, Zaheer S, Sahu SK, Yang B, Lim R. Reduced severity of experimental autoimmune encephalomyelitis in GMF-deficient mice. Neurochemical Research. 2007;32(1):39–47. doi: 10.1007/s11064-006-9220-x. [DOI] [PubMed] [Google Scholar]
- 34.Zhu TZ, Xu YH, Dong B, et al. β-elemene inhibits proliferation of human glioblastoma cells through the activation of glia maturation factor β and induces sensitization to cisplatin. Oncology Reports. 2011;26(2):405–413. doi: 10.3892/or.2011.1276. [DOI] [PubMed] [Google Scholar]
- 35.Lim R, Hicklin DJ, Ryken TC. Suppression of glioma growth in vitro and in vivo by glia maturation factor. Cancer Research. 1986;46(10):5241–5247. [PubMed] [Google Scholar]
- 36.Zaheer S, Thangavel R, Sahu SK, Zaheer A. Augmented expression of glia maturation factor in Alzheimer’s disease. Neuroscience. 2011;194:227–233. doi: 10.1016/j.neuroscience.2011.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thangavel R, Stolmeier D, Yang X, Anantharam P, Zaheer A. Expression of glia maturation factor in neuropathological lesions of Alzheimer's disease. Neuropathology and Applied Neurobiology. 2012;38:572–581. doi: 10.1111/j.1365-2990.2011.01232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerschensteiner M, Gallmeier E, Behrens L, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? Journal of Experimental Medicine. 1999;189(5):865–870. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lessmann V, Gottmann K, Malcangio M. Neurotrophin secretion: current facts and future prospects. Progress in Neurobiology. 2003;69(5):341–374. doi: 10.1016/s0301-0082(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 40.Flügel A, Matsumuro K, Neumann H, et al. Anti-inflammatory activity of nerve growth factor in experimental autoimmune encephalomyelitis: inhibition of monocyte transendothelial migration. European Journal of Immunology. 2001;31:11–22. doi: 10.1002/1521-4141(200101)31:1<11::AID-IMMU11>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 41.Villoslada P, Hauser SL, Bartke I, et al. Human nerve growth factor protects common marmosets against autoimmune encephalomyelitis by switching the balance of T helper cell type 1 and 2 cytokines within the central nervous system. Journal of Experimental Medicine. 2000;191(10):1799–1806. doi: 10.1084/jem.191.10.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tuszynski MH. Intraparenchymal NGF infusions rescue degenerating cholinergic neurons. Cell Transplantation. 2000;9(5):629–636. doi: 10.1177/096368970000900508. [DOI] [PubMed] [Google Scholar]
- 43.Hyman C, Hofer M, Barde Y-A, et al. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature. 1991;350(6315):230–232. doi: 10.1038/350230a0. [DOI] [PubMed] [Google Scholar]
- 44.Lingor P, Unsicker K, Krieglstein K. GDNF and NT-4 protect midbrain dopaminergic neurons from toxic damage by iron and nitric oxide. Experimental Neurology. 2000;163(1):55–62. doi: 10.1006/exnr.2000.7339. [DOI] [PubMed] [Google Scholar]
- 45.Eddelston M, Mucke L. Molecular profile of reactive astrocytes—implications for their role in neurologic disease. Neuroscience. 1993;54(1):15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Appel E, Kolman O, Kazimirsky G, Blumberg PM, Brodie C. Regulation of GDNF expression in cultured astrocytes by inflammatory stimuli. NeuroReport. 1997;8(15):3309–3312. doi: 10.1097/00001756-199710200-00023. [DOI] [PubMed] [Google Scholar]
- 47.Moretto G, Walker DG, Lanteri P, et al. Expression and regulation of glial-cell-line-derived neurotrophic factor (GDNF) mRNA in human astrocytes in vitro. Cell and Tissue Research. 1996;286(2):257–262. doi: 10.1007/s004410050695. [DOI] [PubMed] [Google Scholar]
- 48.Gill SS, Patel NK, Hotton GR, et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nature Medicine. 2003;9:589–595. doi: 10.1038/nm850. [DOI] [PubMed] [Google Scholar]
- 49.Patel NK, Bunnage M, Plaha P, Svendsen CN, Heywood P, Gill SS. Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: a two-year outcome study. Annals of Neurology. 2005;57(2):298–302. doi: 10.1002/ana.20374. [DOI] [PubMed] [Google Scholar]
- 50.Lang AE, Gill S, Patel NK, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Annals of Neurology. 2006;59(3):459–466. doi: 10.1002/ana.20737. [DOI] [PubMed] [Google Scholar]
- 51.Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. Journal of Molecular Medicine. 1997;75(3):165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- 52.Aloisi F, Care A, Borsellino G, et al. Production of hemolymphopoietic cytokines (IL-6, IL-8, colony-stimulating factors) by normal human astrocytes in response to IL-1β and tumor necrosis factor-α . Journal of Immunology. 1992;149(7):2358–2366. [PubMed] [Google Scholar]
- 53.Samoilova EB, Horton JL, Hilliard B, Liu T-ST, Chen Y. IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. Journal of Immunology. 1998;161(12):6480–6486. [PubMed] [Google Scholar]
- 54.Mendel I, Katz A, Kozak N, Ben Nun A, Revel M. Interleukin-6 functions in autoimmune encephalomyelitis: a study in gene-targeted mice. European Journal of Immunology. 1998;28:1727–1737. doi: 10.1002/(SICI)1521-4141(199805)28:05<1727::AID-IMMU1727>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 55.Leech MD, Barr TA, Turner DG, et al. Cutting edge: IL-6-dependent autoimmune disease: dendritic cells as a sufficient, but transient, source. The Journal of Immunology. 2013;190:881–885. doi: 10.4049/jimmunol.1202925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giralt M, Ramos R, Quintana A, et al. Induction of atypical EAE mediated by transgenic production of IL-6 in astrocytes in the absence of systemic IL-6. Glia. 2013;61:587–600. doi: 10.1002/glia.22457. [DOI] [PubMed] [Google Scholar]
- 57.de Jong BA, Huizinga TWJ, Bollen ELEM, et al. Production of IL-1β and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. Journal of Neuroimmunology. 2002;126(1-2):172–179. doi: 10.1016/s0165-5728(02)00056-5. [DOI] [PubMed] [Google Scholar]
- 58.Schiffenbauer J, Streit WJ, Butfiloski E, Labow M, Edwards C, III, Moldawer LL. The induction of EAE is only partially dependent on TNF receptor signaling but requires the IL-1 type I receptor. Clinical Immunology. 2000;95(2):117–123. doi: 10.1006/clim.2000.4851. [DOI] [PubMed] [Google Scholar]
- 59.Sutton C, Brereton C, Keogh B, Mills KHG, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. Journal of Experimental Medicine. 2006;203(7):1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hofstetter HH, Ibrahim SM, Koczan D, et al. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cellular Immunology. 2005;237(2):123–130. doi: 10.1016/j.cellimm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 61.Komiyama Y, Nakae S, Matsuki T, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. Journal of Immunology. 2006;177(1):566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 62.Hebel K, Rudolph M, Kosak B, Chang H-D, Butzmann J, Brunner-Weinzierl MC. IL-1β and TGF-β act antagonistically in induction and differentially in propagation of human proinflammatory precursor CD4+ T cells. Journal of Immunology. 2011;187(11):5627–5635. doi: 10.4049/jimmunol.1003998. [DOI] [PubMed] [Google Scholar]
- 63.Choi C, Park JY, Lee J, et al. Fas ligand and Fas are expressed constitutively in human astrocytes and the expression increases with IL-1, IL-6, TNF-α, or IFN-γ . Journal of Immunology. 1999;162(4):1889–1895. [PubMed] [Google Scholar]
- 64.del Rey A, Klusman I, Besedovsky HO. Cytokines mediate protective stimulation of glucocorticoid output during autoimmunity: involvement of IL-1. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 1998;275(4):R1146–R1151. doi: 10.1152/ajpregu.1998.275.4.R1146. [DOI] [PubMed] [Google Scholar]
- 65.Wekerle H, Linington C, Lassmann H, Meyermann R. Cellular immune reactivity within the CNS. Trends in Neurosciences. 1986;9(6):271–277. [Google Scholar]
- 66.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. Journal of Neuroscience Research. 1991;28(2):254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 67.Tran EH, Hoekstra K, van Rooijen N, Dijkstra CD, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. Journal of Immunology. 1998;161(7):3767–3775. [PubMed] [Google Scholar]
- 68.Agrawal S, Anderson P, Durbeej M, et al. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. Journal of Cell Biology. 2006;173(2):1007–1019. doi: 10.1084/jem.20051342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gold R, Schmied M, Tontsch U, et al. Antigen presentation by astrocytes primes rat T lymphocytes for apoptotic cell death A model for T-cell apoptosis in vivo. Brain. 1996;119(2):651–659. doi: 10.1093/brain/119.2.651. [DOI] [PubMed] [Google Scholar]
- 70.Gold R, Hartung H-P, Lassmann H. T-cell apoptosis in autoimmune diseases: termination of inflammation in the nervous system and other sites with specialized immune-defense mechanisms. Trends in Neurosciences. 1997;20(9):399–404. doi: 10.1016/S0166-2236(97)01079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bechmann I, Steiner B, Gimsa U, et al. Astrocyte-induced T cell elimination is CD95 ligand dependent. Journal of Neuroimmunology. 2002;132(1-2):60–65. doi: 10.1016/s0165-5728(02)00311-9. [DOI] [PubMed] [Google Scholar]
- 72.Bechmann I, Mor G, Nilsen J, et al. FasL (CD95L, Apo1L) is expressed in the normal rat and human brain: evidence for the existence of an immunological brain barrier. Glia. 1999;27:62–74. doi: 10.1002/(sici)1098-1136(199907)27:1<62::aid-glia7>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 73.Bechmann I, Lossau S, Steiner B, et al. Reactive astrocytes upregulate Fas (CD95) and Fas ligand (CD95L) expression but do not undergo programmed cell death during the course of anterograde degeneration. Glia. 2000;32:25–41. doi: 10.1002/1098-1136(200010)32:1<25::aid-glia30>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 74.Kohji T, Tanuma N, Aikawa Y, et al. Interaction between apoptotic cells and reactive brain cells in the central nervous system of rats with autoimmune encephalomyelitis. Journal of Neuroimmunology. 1998;82(2):168–174. doi: 10.1016/s0165-5728(97)00198-7. [DOI] [PubMed] [Google Scholar]
- 75.Kohji T, Matsumoto Y. Coexpression of Fas/FasL and Bax on brain and infiltrating T cells in the central nervous system is closely associated with apoptotic cell death during autoimmune encephalomyelitis. Journal of Neuroimmunology. 2000;106(1-2):165–171. doi: 10.1016/s0165-5728(00)00238-1. [DOI] [PubMed] [Google Scholar]
- 76.Mor G, Gutierrez LS, Eliza M, Kahyaoglu F, Arici A. Fas-Fas ligand system-induced apoptosis in human placenta and gestational trophoblastic disease. American Journal of Reproductive Immunology. 1998;40(2):89–94. doi: 10.1111/j.1600-0897.1998.tb00396.x. [DOI] [PubMed] [Google Scholar]
- 77.Kauma SW, Huff TF, Hayes N, Nilkaeo A. Placental Fas ligand expression is a mechanism for maternal immune tolerance to the fetus. Journal of Clinical Endocrinology and Metabolism. 1999;84(6):2188–2194. doi: 10.1210/jcem.84.6.5730. [DOI] [PubMed] [Google Scholar]
- 78.Guller S, LaChapelle L. The role of placental fas ligand in maintaining immune privilege at maternal-fetal interfaces. Seminars in Reproductive Medicine. 1999;17(1):39–44. doi: 10.1055/s-2007-1016210. [DOI] [PubMed] [Google Scholar]
- 79.Aschkenazi S, Straszewski S, Verwer KMA, Foellmer H, Rutherford T, Mor G. Differential regulation and function of the Fas/Fas ligand system in human trophoblast cells. Biology of Reproduction. 2002;66(6):1853–1861. doi: 10.1095/biolreprod66.6.1853. [DOI] [PubMed] [Google Scholar]
- 80.Ferguson TA, Griffith TS. A vision of cell death: insights into immune privilege. Immunological Reviews. 1997;156:167–184. doi: 10.1111/j.1600-065x.1997.tb00967.x. [DOI] [PubMed] [Google Scholar]
- 81.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270(5239):1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 82.Ichinose M, Masuoka J, Shiraishi T, Mineta T, Tabuchi K. Fas ligand expression and depletion of T-cell infiltration in astrocytic tumors. Brain Tumor Pathology. 2001;18(1):37–42. doi: 10.1007/BF02478923. [DOI] [PubMed] [Google Scholar]
- 83.Saas P, Walker PR, Hahne M, et al. Fas ligand expression by astrocytoma in vivo: maintaining immune privilege in the brain? Journal of Clinical Investigation. 1997;99(6):1173–1178. doi: 10.1172/JCI119273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aloisi F, Ria F, Adorini L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunology Today. 2000;21(3):141–148. doi: 10.1016/s0167-5699(99)01512-1. [DOI] [PubMed] [Google Scholar]
- 85.Xiao B-G, Link H. Is there a balance between microglia and astrocytes in regulating Th1/Th2-cell responses and neuropathologies? Immunology Today. 1999;20(11):477–479. doi: 10.1016/s0167-5699(99)01501-7. [DOI] [PubMed] [Google Scholar]
- 86.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends in Neurosciences. 1996;19(8):312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 87.Aloisi F, Ria F, Columba-Cabezas S, Hess H, Penna G, Adorini L. Relative efficiency of microglia, astrocytes, dendritic cells and B cells in naive CD4+ T cell priming and Th1/Th2 cell restimulation. European Journal of Immunology. 1999;29(9):2705–2714. doi: 10.1002/(SICI)1521-4141(199909)29:09<2705::AID-IMMU2705>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 88.Tontsch U, Rott O. Cortical neurons selectively inhibit MHC class II induction in astrocytes but not in microglial cells. International Immunology. 1993;5(3):249–254. doi: 10.1093/intimm/5.3.249. [DOI] [PubMed] [Google Scholar]
- 89.Lee SC, Collins M, Vanguri P, Shin ML. Glutamate differentially inhibits the expression of class II MHC antigens on astrocytes and microglia. Journal of Immunology. 1992;148(11):3391–3397. [PubMed] [Google Scholar]
- 90.Neumann H, Boucraut J, Hahnel C, Misgeld T, Wekerle H. Neuronal control of MHC class II inducibility in rat astrocytes and microglia. European Journal of Neuroscience. 1996;8(12):2582–2590. doi: 10.1111/j.1460-9568.1996.tb01552.x. [DOI] [PubMed] [Google Scholar]
- 91.Zeinstra E, Wilczak N, de Keyser J. Reactive astrocytes in chronic active lesions of multiple sclerosis express co-stimulatory molecules B7-1 and B7-2. Journal of Neuroimmunology. 2003;135(1-2):166–171. doi: 10.1016/s0165-5728(02)00462-9. [DOI] [PubMed] [Google Scholar]
- 92.Soos JM, Ashley TA, Morrow J, Patarroyo JC, Szente BE, Zamvil SS. Differential expression of B7 co-stimulatory molecules by astrocytes correlates with T cell activation and cytokine production. International Immunology. 1999;11(7):1169–1179. doi: 10.1093/intimm/11.7.1169. [DOI] [PubMed] [Google Scholar]
- 93.Nikcevich KM, Gordon KB, Tan L, et al. IFN-γ-activated primary murine astrocytes express B7 costimulatory molecules and prime naive antigen-specific T cells. Journal of Immunology. 1997;158(2):614–621. [PubMed] [Google Scholar]
- 94.Cornet A, Bettelli E, Oukka M, et al. Role of astrocytes in antigen presentation and naive T-cell activation. Journal of Neuroimmunology. 2000;106(1-2):69–77. doi: 10.1016/s0165-5728(99)00215-5. [DOI] [PubMed] [Google Scholar]
- 95.Cross AH, Ku G. Astrocytes and central nervous system endothelial cells do not express B7-1 (CD80) or B7-2 (CD86) immunoreactivity during experimental autoimmune encephalomyelitis. Journal of Neuroimmunology. 2000;110(1-2):76–82. doi: 10.1016/s0165-5728(00)00327-1. [DOI] [PubMed] [Google Scholar]
- 96.Satoh J, Lee YB, Kim SU. T-cell costimulatory molecules B7-1 (CD80) and B7-2 (CD86) are expressed in human microglia but not in astrocytes in culture. Brain Research. 1995;704(1):92–96. doi: 10.1016/0006-8993(95)01177-3. [DOI] [PubMed] [Google Scholar]
- 97.Aloisi F, Ria F, Penna G, Adorini L. Microglia are more efficient than astrocytes in antigen processing and in Th1 but not Th2 cell activation. Journal of Immunology. 1998;160(10):4671–4680. [PubMed] [Google Scholar]
- 98.Kort JJ, Kawamura K, Fugger L, Weissert R, Forsthuber TG. Efficient presentation of myelin oligodendrocyte glycoprotein peptides but not protein by astrocytes from HLA-DR2 and HLA-DR4 transgenic mice. Journal of Neuroimmunology. 2006;173(1-2):23–34. doi: 10.1016/j.jneuroim.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 99.Owens T, Renno T, Taupin V, Krakowski M. Inflammatory cytokines in the brain: does the CNS shape immune responses? Immunology Today. 1994;15(12):566–571. doi: 10.1016/0167-5699(94)90218-6. [DOI] [PubMed] [Google Scholar]
- 100.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annual Review of Immunology. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 101.Constant SL, Bottomly K. Induction of TH1 and TH2 CD4+ T cell responses: the alternative approaches. Annual Review of Immunology. 1997;15:297–322. doi: 10.1146/annurev.immunol.15.1.297. [DOI] [PubMed] [Google Scholar]
- 102.Baumgart M, Moos V, Schuhbauer D, Müller B. Differential expression of major histocompatibility complex class II genes on murine macrophages associated with T cell cytokine profile and protective/suppressive effects. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(12):6936–6940. doi: 10.1073/pnas.95.12.6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ohmori K, Hong Y, Fujiwara M, Matsumoto Y. In situ demonstration of proliferating cells in the rat central nervous system during experimental autoimmune encephalomyelitis: evidence suggesting that most infiltrating T cells do not proliferate in the target organ. Laboratory Investigation. 1992;66(1):54–62. [PubMed] [Google Scholar]
- 104.Matsumoto Y, Hanawa H, Tsuchida M, Abo T. In situ inactivation of infiltrating T cells in the central nervous system with autoimmune encephalomyelitis. The role of astrocytes. Immunology. 1993;79(3):381–390. [PMC free article] [PubMed] [Google Scholar]
- 105.Sun D, Coleclough C, Whitaker JN. Nonactivated astrocytes downregulate T cell receptor expression and reduce antigen-specific proliferation and cytokine production of myelin basic protein (MBP)-reactive T cells. Journal of Neuroimmunology. 1997;78(1-2):69–78. doi: 10.1016/s0165-5728(97)00083-0. [DOI] [PubMed] [Google Scholar]
- 106.Xiao B-G, Diab A, Zhu J, van der Meide P, Link H. Astrocytes induce hyporesponses of myelin basic protein-reactive T and B cell function. Journal of Neuroimmunology. 1998;89(1-2):113–121. doi: 10.1016/s0165-5728(98)00123-4. [DOI] [PubMed] [Google Scholar]
- 107.Gimsa U, Øren A, Pandiyan P, et al. Astrocytes protect the CNS: antigen-specific T helper cell responses are inhibited by astrocyte-induced upregulation of CTLA-4 (CD152) Journal of Molecular Medicine. 2004;82(6):364–372. doi: 10.1007/s00109-004-0531-6. [DOI] [PubMed] [Google Scholar]
- 108.Nair A, Frederick TJ, Miller SD. Astrocytes in multiple sclerosis: a product of their environment. Cellular and Molecular Life Sciences. 2008;65(17):2702–2720. doi: 10.1007/s00018-008-8059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Körmendy D, Hoff H, Hoff P, et al. Impact of the CTLA-4/CD28 axis on the processes of joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2013;65:81–87. doi: 10.1002/art.37714. [DOI] [PubMed] [Google Scholar]
- 110.Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nature Immunology. 2002;3(7):611–618. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- 111.Allison J. Immunotherapy. Current Opinion in Immunology. 2002;14:631–632. [Google Scholar]
- 112.Chambers CA, Krummel MF, Boitel B, et al. The role of CTLA-4 in the regulation and initiation of T-Cell responses. Immunological Reviews. 1996;(153):27–46. doi: 10.1111/j.1600-065x.1996.tb00919.x. [DOI] [PubMed] [Google Scholar]
- 113.Brunner MC, Chambers CA, Chan FK-M, Hanke J, Winoto A, Allison JP. CTLA-4-mediated inhibition of early events of T cell proliferation. Journal of Immunology. 1999;162(10):5813–5820. [PubMed] [Google Scholar]
- 114.Maszyna F, Hoff H, Kunkel D, Radbruch A, Brunner-Weinzierl MC. Diversity of clonal T cell proliferation is mediated by differential expression of CD152 (CTLA-4) on the cell surface of activated individual T lymphocytes. Journal of Immunology. 2003;171(7):3459–3466. doi: 10.4049/jimmunol.171.7.3459. [DOI] [PubMed] [Google Scholar]
- 115.Pandiyan P, Gärtner D, Soezeri O, Radbruch A, Schulze-Osthoff K, Brunner-Weinzierl MC. CD152 (CTLA-4) determines the unequal resistance of Th1 and Th2 cells against activation-induced cell death by a mechanism requiring PI3 kinase function. Journal of Experimental Medicine. 2004;199(6):831–842. doi: 10.1084/jem.20031058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hurwitz AA, Sullivan TJ, Sobel RA, Allison JP. Cytotoxic T lymphocyte antigen-4 (CTLA-4) limits the expansion of encephalitogenic T cells in experimental autoimmune encephalomyelitis (EAE)-resistant BALB/c mice. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(5):3013–3017. doi: 10.1073/pnas.042684699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rudolph M, Hebel K, Miyamura Y, Maverakis E, Brunner-Weinzierl MC. Blockade of CTLA-4 decreases the generation of multifunctional memory CD4+ T cells in vivo. Journal of Immunology. 2011;186(10):5580–5589. doi: 10.4049/jimmunol.1003381. [DOI] [PubMed] [Google Scholar]
- 118.Gärtner D, Hoff H, Gimsa U, Burmester G-R, Brunner-Weinzierl MC. CD25 regulatory T cells determine secondary but not primary remission in EAE: impact on long-term disease progression. Journal of Neuroimmunology. 2006;172(1-2):73–84. doi: 10.1016/j.jneuroim.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 119.Yang JF, Tao HQ, Liu YM, et al. Characterization of the interaction between astrocytes and encephalitogenic lymphocytes during the development of experimental autoimmune encephalitomyelitis (EAE) in mice. Clinical & Experimental Immunology. 2012;170:254–265. doi: 10.1111/j.1365-2249.2012.04661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fitzgerald DC, Ciric B, Touil T, et al. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. Journal of Immunology. 2007;179(5):3268–3275. doi: 10.4049/jimmunol.179.5.3268. [DOI] [PubMed] [Google Scholar]
- 121.Batten M, Li J, Yi S, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nature Immunology. 2006;7(9):929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 122.Stumhofer JS, Laurence A, Wilson EH, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nature Immunology. 2006;7(9):937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 123.Jehs T, Faber C, Juel HB, Nissen MH. Astrocytoma cells upregulate expression of pro-inflammatory cytokines after co-culture with activated peripheral blood mononuclear cells. APMIS. 2011;119(8):551–561. doi: 10.1111/j.1600-0463.2011.02773.x. [DOI] [PubMed] [Google Scholar]
- 124.Meinl E, Aloisi F, Ertl B, et al. Multiple sclerosis. Immunomodulatory effects of human astrocytes on T cells. Brain. 1994;117(6):1323–1332. doi: 10.1093/brain/117.6.1323. [DOI] [PubMed] [Google Scholar]
- 125.Gimsa U, Iglič A, Fiedler S, et al. Actin is not required for nanotubular protrusions of primary astrocytes grown on metal nano-lawn. Molecular Membrane Biology. 2007;24(3):243–255. doi: 10.1080/09687860601141730. [DOI] [PubMed] [Google Scholar]
- 126.Munn DH, Zhou M, Attwood JT, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 127.Kwidzinski E, Bunse J, Kovac AD, et al. IDO (indolamine 2,3-dioxygenase) expression and function in the CNS. Advances in Experimental Medicine and Biology. 2003;527:113–118. doi: 10.1007/978-1-4615-0135-0_13. [DOI] [PubMed] [Google Scholar]
- 128.Kwidzinski E, Bunse J, Aktas O, et al. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. The FASEB Journal. 2005;19(10):1347–1349. doi: 10.1096/fj.04-3228fje. [DOI] [PubMed] [Google Scholar]
- 129.Johnson TS, Munn DH, Maria BL. Modulation of tumor tolerance in primary central nervous system malignancies. Clinical and Developmental Immunology. 2012;2012:14 pages. doi: 10.1155/2012/937253.937253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.von Bergwelt-Baildon MS, Popov A, Saric T, et al. CD25 and indoleamine 2,3-dioxygenase are up-regulated by prostaglandin E2 and expressed by tumor-associated dendritic cells in vivo: additional mechanisms of T-cell inhibition. Blood. 2006;108(1):228–237. doi: 10.1182/blood-2005-08-3507. [DOI] [PubMed] [Google Scholar]
- 131.Braun D, Longman RS, Albert ML. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood. 2005;106(7):2375–2381. doi: 10.1182/blood-2005-03-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Baban B, Chandler PR, Sharma MD, et al. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. Journal of Immunology. 2009;183(4):2475–2483. doi: 10.4049/jimmunol.0900986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yan Y, Zhang G-X, Gran B, et al. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in Experimental autoimmune encephalomyelitis. Journal of Immunology. 2010;185(10):5953–5961. doi: 10.4049/jimmunol.1001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Krause D, Suh H-S, Tarassishin L, et al. The tryptophan metabolite 3-hydroxyanthranilic acid plays anti-inflammatory and neuroprotective roles during inflammation: role of hemeoxygenase-1. American Journal of Pathology. 2011;179(3):1360–1372. doi: 10.1016/j.ajpath.2011.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ruddick JP, Evans AK, Nutt DJ, Lightman SL, Rook GAW, Lowry CA. Tryptophan metabolism in the central nervous system: medical implications. Expert Reviews in Molecular Medicine. 2006;8(20):1–27. doi: 10.1017/S1462399406000068. [DOI] [PubMed] [Google Scholar]
- 136.Nemeth H, Toldi J, Vecsei L. Role of kynurenines in the central and peripheral nervous systems. Current Neurovascular Research. 2005;2:249–260. doi: 10.2174/1567202054368326. [DOI] [PubMed] [Google Scholar]
- 137.Guillemin GJ, Smith DG, Kerr SJ, et al. Characterisation of kynurenine pathway metabolism in human astrocytes and implications in neuropathogenesis. Redox Report. 2000;5(2-3):108–111. doi: 10.1179/135100000101535375. [DOI] [PubMed] [Google Scholar]
- 138.Trajkovic V, Vuckovic O, Stosic-Grujicic S, et al. Astrocyte-induced regulatory T cells mitigate CNS autoimmunity. Glia. 2004;47(2):168–179. doi: 10.1002/glia.20046. [DOI] [PubMed] [Google Scholar]
- 139.Magnus T, Schreiner B, Korn T, et al. Microglial expression of the B7 family member B7 homolog 1 confers strong immune inhibition: implications for immune responses and autoimmunity in the CNS. Journal of Neuroscience. 2005;25(10):2537–2546. doi: 10.1523/JNEUROSCI.4794-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lipp M, Brandt C, Dehghani F, Kwidzinski E, Bechmann I. PD-L1 (B7-H1) regulation in zones of axonal degeneration. Neuroscience Letters. 2007;425(3):156–161. doi: 10.1016/j.neulet.2007.07.053. [DOI] [PubMed] [Google Scholar]
- 141.Hindinger C, Bergmann CC, Hinton DR, et al. IFN-gamma signaling to astrocytes protects from autoimmune mediated neurological disability. PLoS One. 2012;7 doi: 10.1371/journal.pone.0042088.e42088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Øren A, Falk K, Rötzschke O, Bechmann I, Nitsch R, Gimsa U. Production of neuroprotective NGF in astrocyte-T helper cell cocultures is upregulated following antigen recognition. Journal of Neuroimmunology. 2004;149(1-2):59–65. doi: 10.1016/j.jneuroim.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 143.Levi-Montalcini R, Skaper SD, dal Toso R, Petrelli L, Leon A. Nerve growth factor: from neurotrophin to neurokine. Trends in Neurosciences. 1996;19(11):514–520. doi: 10.1016/S0166-2236(96)10058-8. [DOI] [PubMed] [Google Scholar]
- 144.Arredondo LR, Deng C, Ratts RB, et al. Role of nerve growth factor in experimental autoimmune encephalomyelitis. European Journal of Immunology. 2001;31:625–633. doi: 10.1002/1521-4141(200102)31:2<625::aid-immu625>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 145.Villoslada P, Genain CP. Role of nerve growth factor and other trophic factors in brain inflammation. Progress in Brain Research. 2004;146:403–414. doi: 10.1016/S0079-6123(03)46025-1. [DOI] [PubMed] [Google Scholar]
- 146.Neumann H, Misgeld T, Matsumuro K, Wekerle H. Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(10):5779–5784. doi: 10.1073/pnas.95.10.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]