

Abstract
Miltefosine is an alkylphosphocholine that shows broad-spectrum in vitro antifungal activities and limited in vivo efficacy in mouse models of cryptococcosis. To further explore the potential of this class of compounds for the treatment of systemic mycoses, nine analogs (3a–3i) were synthesized by modifying the choline structural moiety and the alkyl chain length of miltefosine. In vitro testing of these compounds against the opportunistic fungal pathogens Candida albicans, Candida glabrata, Candida krusei, Aspergillus fumigatus, and Cryptococcus neoformans revealed that N-benzyl-N,N-dimethyl-2-{[(hexadecyloxy)hydroxyphosphinyl]oxy}ethanaminium inner salt (3a), N,N-dimethyl-N-(4-nitrobenzyl)-2-{[(hexadecyloxy)hydroxyphosphinyl]oxy}ethanaminium inner salt (3d), and N-(4-methoxybenzyl)-N,N-dimethyl-2-{[(hexadecyloxy)hydroxyphosphinyl]oxy}ethanaminium inner salt (3e) exhibited minimum inhibitory concentrations (MIC) of 2.5–5.0 μg/mL against all tested pathogens, when compared to miltefosine with MICs of 2.5–3.3 μg/mL. Compound 3a showed low in vitro cytotoxicity against three mammalian cell lines similar to miltefosine. In vivo testing of 3a and miltefosine against C. albicans in a mouse model of systemic infection did not demonstrate efficacy. The results of this study indicate that further investigation will be required to determine the potential usefulness of the alkylphosphocholines in the treatment of invasive fungal infections.
Keywords: alkylphosphocholine, miltefosine, antifungal, cryptococcosis, candidiasis
Miltefosine (hexadecylphosphocholine) is a synthetic alkylphosphocholine that belongs to the class of phospholipids. It was initially developed as an antineoplastic agent,1,2 and was later discovered to possess antileishmanial properties and registered as the first oral drug for the treatment of visceral leishmaniasis in India and Germany, and for the treatment of cutaneous leishmaniasis in Colombia.3 Pharmacokinetic studies indicate that miltefosine has good bioavailability and a long half-life in patients with leishmania (7 days for the first elimination and 31 days for the terminal elimination).4 This may be attributable to its improved in vivo antileishmanial activity relative to analogs with even more potent in vitro activities.3 Miltefosine also possesses antibacterial,5 antiprotozoal,6 and antiviral activities.7
Miltefosine was demonstrated to be active against Candida albicans and Cryptococcus neoformans in 1999.8 In recent years, miltefosine was found to exhibit broad-spectrum antifungal activities against clinically important fungal pathogens9 and dermytophytes10 in addition to inhibiting Candida albicans biofilm formation and maturation.11 Mechanistic studies indicated that miltefosine inhibits cytochrome c oxidase in the model organism Saccharomyces cerevisiae and phospholipase B in the fungal pathogen C. neoformans,9,12 while in human cells it inhibits activation of the protein kinase B pathway as well as phosphatidylcholine synthesis.13 However, none of these targets is essential for the survival of fungal cells according to what is known in S. cerevisiae. Therefore, miltefosine and analogs remain to be an intriguing class of compounds in terms of their precise antifungal target.
Miltefosine gained particular interest in antifungal therapy due to the reported in vivo efficacy in a mouse model of cryptococcosis.9 In a more recent study aimed at further evaluating its in vivo efficacy in mouse models of cryptococcal meningoencephalitis and disseminated cryptococcosis, miltefosine demonstrated limited effects in mice that were challenged with a low infecting inoculum.14 Meningoencephalitis requires the drug to cross the blood brain barrier to exert its action. Given that miltefosine has a higher distribution in the lung and kidney of mice than in brain,15 we hypothesized that it might be more active in vivo against systemic mycoses rather than infections in the brain. With chemical synthesis, new analogs could be prepared and included for testing this hypothesis. Therefore, we designed and synthesized several new miltefosine analogs and evaluated their antifungal activities in vitro and in vivo in a candidiasis mouse model.
The available structure–antifungal activity relationship (SAR) information on alkylphosphocholines was the basis for designing new compounds in this study. A hydrophobic chain in the miltefosine analogs with 16 to 18 carbon atoms is necessary for antifungal activity.16 Reduction of the alkyl chain length to 12 carbon atoms,16,17 increasing the chain length to 22 carbon atoms,8 or insertion of ester/amide functionalities in the middle of this chain16 significantly reduces the antifungal activity. Structurally more complex alkylglycerophosphocholines exhibit moderate activities against C. albicans and C. neoformans, when compared with alkylphosphocholine derivatives.16 Extensive modification of the N-substitution and the C2 unit of the choline moiety (head group) resulted in a large number of compounds,17-19 some of which showed activities more potent than erucylphosphocholine19 that is 8-fold less potent than miltefosine.16 Within this class, octadecylphosphocholine demonstrates as much as a 4-fold increase in in vitro potency against C. albicans when compared to miltefosine.16 It appears that the intact head group or the presence of at least two small N-methyl groups plays a key role for antifungal activity. Based on the above SAR information, we decided to synthesize compounds by slightly modifying the structure of miltefosine (Figure 1).
Figure 1.
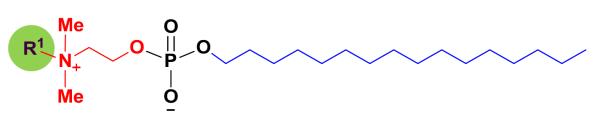
Miltefosine (R1 = Me) based synthetic template.
We first designed compound 3a with a benzyl group replacing one methyl group of the choline structural moiety in miltefosine, taking into consideration of the strong antimicrobial activity of benzalkonium chloride that possesses the benzyldimethylammonio structural moiety.20 However, the zwitterion nature of 3a makes it distinctly different from the cationic surface-acting benzalkonium chloride. While this design allows the compound to retain most of the structural features required for antifungal activity within the class, it also increases lipophilicity due to the introduction of an aromatic ring, as indicated by the calculated octane-water partition coefficient (cLogP) from 1.80 for miltefosine to 3.80 for 3a,21 which may improve antifungal properties. The synthetic method for the preparation of 3a is an adaption of the reported procedures18,22 and is depicted in Scheme 1. Quaternization of N,N-dimethylaminoethanol (1) with benzyl bromide afforded the quaternary ammonium salt 2, which was subject to phosphorylation reaction of n-hexadecanol with POCl3 followed by hydrolysis to afford the target compound.23
Scheme 1.

Reagents and conditions: (a) R1X, CH3CN, room temperature, 1-3 h; (b) (1) R2OH, POCl3, Et3N, CHCl3, 0°C, room temperature, 2 h; (2) pyridine, 2a-2i, 0°C, room temperature, 12 h; (3) H2O, room temperature, 1 h.
As shown in Table 1, in vitro antifungal testing by the method described previously24 indicated that compound 3a showed potent activities with minimum inhibitory concentrations (MICs) ranging from 2.5 to 5.0 μg/mL against the opportunistic fungal pathogens C. albicans, Candida glabrata, Candida krusei, Aspergillus fumigatus, and C. neoformans. The compound was also fungicidal against all tested fungal pathogens with minimum fungicidal concentrations (MFCs)25 from 2.5 to 15.0 μg/mL. Its antifungal potency is similar to that of miltefosine with MICs and MFCs of 2.1–3.3 and 2.1–9.2 g/mL, respectively, against the aforementioned pathogens.
Table 1.
In vitro antifungal activity and cytotoxicity of miltefosine and compounds 3a-3ia
| Antifungal activity (MIC/MFC, μg/mL)b |
Cytotoxicity (IC50, μg/mL)c |
|||||||
|---|---|---|---|---|---|---|---|---|
|
C. albicans ATCC 90028 |
C. glabrata ATCC 90030 |
C. krusei ATCC 6258 |
A. fumigatus ATCC 90906 |
C. neoformans ATCC 90113 |
Vero | HepG2 | LLC-PK11 | |
| Miltefosine | 2.5/2.5 | 3.3/4.2 | 2.5/3.3 | 2.9/9.2 | 2.1/2.1 | >25 | >25 | 2.7 |
| 3a | 2.5/2.5 | 3.3/3.3 | 5.0/15.0 | 2.5/2.5 | 3.3/3.3 | >25 | >25 | 4.8 |
| 3b | 6.7/8.3 | 8.3/8.3 | 10.0/10.0 | 10.0/– | 3.3/3.3 | >25 | >25 | 10.8 |
| 3c | – /– d | 4.2/4.2 | – /– | – /– | – /– | >25 | 13 | 4.7 |
| 3d | 2.5/2.5 | 2.5/2.5 | 2.5/3.3 | 2.5/7.5 | 2.5/2.5 | >25 | 11.5 | 1.6 |
| 3e | 4.2/4.2 | 2.5/3.3 | 2.0/3.3 | 2.5/7.5 | 4.2/4.2 | >25 | 12.9 | 1.9 |
| 3f | – /– | 11.2/11.2 | – /– | 12.5/12.5 | – /– | >25 | 21 | 3.2 |
| 3g | – /– | 10.8/10.8 | – /– | 20.0/20.0 | – /– | >25 | 21.3 | 5.5 |
| 3h | – /– | 2.5/3.3 | 3.3/3.3 | 2.5/5.0 | – /– | >25 | 19.6 | 2.1 |
| 3i | – /– | 16.6/16.6 | – /– | – /– | – /– | >25 | 13.2 | 4.1 |
| Amphotericin B | 0.9/1.0 | 1.3/1.5 | 1.7/1.7 | 2.5/5.0 | 0.5/0.5 | |||
| Doxorubicin | >5 | 0.9 | 0.7 | |||||
Both antifungal and cytotoxicity data are mean values based on three independent experiments except for the antifungal data of compound 3f with mean values from two independent experiments.
MIC: minimum inhibitory concentration (lowest concentration that allows no detectable growth). MFC: minimum fungicidal concentration (the lowest concentration that kills the fungus), which was determined by removing 5 μL from each assay well with no visible growth, transferring to fresh media and incubating at the appropriate temperature for 2-3 days. The highest test concentration for compounds 3a-3i and miltefosine was 20 μg/mL; the highest test concentration for amphotericin B was 5 μg/mL.
IC50: 50% growth inhibition. The highest test concentration for compounds 3a-3i and miltefosine was 25 μg/mL; the highest test concentration for doxorubicin was 5 μg/mL.
Not active at 20 μg/mL.
To investigate the influence of the chain length to the antifungal activity within this series, analogs 3b and 3c with the same head group but an alkyl chain length of C14 and C18, respectively, were prepared by a synthetic method similar for 3a. However, compound 3b showed decreased activity against C. albicans, C. glabrata, C. krusei, and A. fumigatus in terms of MICs and MFCs when compared with 3a (Table 1), and compound 3c was only active against C. glabrata with an MIC/MFC of 4.2/4.2 μg/mL, indicating that C16 is an optimal alkyl chain length.
Keeping a constant C16 alkyl chain, we next synthesized six analogs (3d–3i) with different head groups by replacing one methyl group of the choline moiety in miltefosine with p-nitrobenzyl, p-chlorobenzyl, p-bromobenzyl, p-methoxybenzyl, cinnamyl, and allyl groups. Among these, compound 3d with an N-4-nitrobenzyl substitution produced the best in vitro activity profiles, exhibiting slightly improved potency against C. glabrata and A. fumigatus when compared to miltefosine (Table 1). Compound 3e with an N-4-methoxybenzyl substitution also showed good activities similar to 3a and 3d (Table 1). Compounds 3f and 3g with a halogen-substituted aromatic ring were only active against C. glabrata and A. fumigatus, and 3i with an N-allylic substitution, the only compound without an aromatic ring in this series, was only active against C. glabrata with an MIC/MFC of 16.6/16.6 μg/mL. It appears that among the five tested fungal species, C. glabrata is most susceptible to this series of compounds. Evidently, the minor structural differences of these compounds, especially for compounds 3a and 3d–3g, have a significant effect on their activity profiles. In addition, the permeability of the compounds towards different fungal cells, which may be associated with their lipophilicities, may play a role in the observed activities. Coincidentally, the three compounds 3a, 3d, and 3e with close chemical structures that showed excellent activity profiles have similar calculated LogP values ranging from 3.69 to 3.80 (Scheme 1).
The in vitro antifungal activity data of miltefosine obtained in this study (Table 1) are similar to those reported in the literature.9 The potent activities of the three synthetic analogs (3a, 3d, and 3e) are further evident by comparison with the ‘gold standard’ clinical drug amphotericin B. Compounds 3a and 3d that showed strong activity against C. albicans with the same MIC/MFC of 2.5/2.5 μg/mL are equally potent as miltefosine, and are about 2- to 3-fold less potent than amphotericin B which has an MIC/MFC of 0.9/1.0 μg/mL (Table 1).
The in vitro cytotoxicity testing of compounds 3a–3i and miltefosine were performed by the method described previously26 against HepG2 (human hepatic carcinoma), Vero (African green monkey kidney fibroblast) and LLC-PK11 (pig kidney epithelial) cells in comparison with the cytotoxic drug doxorubicin. These compounds were not cytotoxic against Vero cells up to the highest tested concentration of 25 μg/mL. However, they showed cytotoxicity towards HepG2 cells with IC50 values in the range of 11.5 to >25 μg/mL and LLC-PK11 cells with IC50 values ranging from 1.6 to 10.8 μg/mL (Table 1). It is important to note that the in vitro antifungal activity of these compounds was not correlated with the in vitro cytotoxicity. For example, the antifungal activity of compounds 3c and 3h was less potent than compound 3a but they were more cytotoxic than 3a (Table 1). Among the three potent antifungal compounds (3a, 3d, and 3e), 3a was least cytotoxic, which was similar to miltefosine. Therefore, compound 3a was selected, along with miltefosine, for in vivo efficacy studies in a mouse model of candidiasis.
Taking advantage of the reported dosing values of miltefosine used in the mouse model of cryptococcosis (1.8 to 45 mg/kg orally)9,14 and the 50% effective dose of miltefosine produced in the mouse model of leishamniasis (7.72 mg/kg intraperitoneally),3 the mice were treated with 3a and miltefosine at 5 and 10 mg/kg via intraperitoneal administration at 4, 24, 48, 72, and 96 hours post inoculation with 1 × 106 cells of C. albicans SC5314 via tail-vein injection. The results showed that all compound-treated mice died on or before day 12, which is similar to the control mice treated with phosphate buffered saline (PBS) (Figure 2A). In a separate experiment with reduced dosing for 3a at 1 and 5 mg/kg and miltefosine at 5 mg/kg via the same treatment, the mice died on or before day 8, same as the control group treated with PBS. Oral administration of compound 3a at 5 and 25 mg/kg also did not enhance the survival rate of mice when compared with the control mice treated with PBS (Figure 2B). This indicated that miltefosine and compound 3a did not exhibit in vivo efficacy against C. albicans in a mouse model of systemic infection.
Figure 2.

In vivo antifungal efficacy of compound 3a and miltefosine (MTF) against Candida albicans infection. Five male CD1 mice (6-7 weeks old) per group were infected with 1 × 106 cells of C. albicans SC5314, followed by treatment with phosphate buffered saline (PBS) or compound by intraperitoneal injection (panel A) and by oral route (panel B) at 4, 24, 48, 72, and 96 hours post inoculation. Survival of the animals was monitored for 12 days. For the intraperitoneal administration experiment, P values evaluated by a Log-rank test are as follows: 0.8758 for PBS vs 3a 5 mg/kg, 0.0179 for PBS vs MTF 5 mg/kg, 0.1021 for PBS vs MTF 10 mg/kg, and 0.1822 for 3a 5 mg/kg vs MTF 5 mg/kg.
Since miltefosine as an antileishmanial drug has proven favorable pharmacokinetic profiles in mice15 and human,4 the failure of demonstrating in vivo antifungal efficacy in our candidiasis mouse model cannot be attributed to its chemical instability within bloodstream and infection sites. The previous study by Wiederhold et al14 suggested that protein binding could be associated with the ineffectiveness of miltefosine in the mouse model of disseminated cryptococcosis. While the reasons for the lack of the in vivo antifungal activity of miltefosine and analog 3a may be worth investigating, we should not exclude the possibility that chemical optimization may afford novel miltefosine-based derivatives that are highly selective and permeable to fungal cells and ultimately exert in vivo efficacy. Another potential therapeutic area for this class of compounds is the treatment of fungal biofilm-related infections on medical devices such as catheters, given that miltefosine can inhibit C. albicans biofilm formation and maturation.11 Thus, further work will be required to determine the potential usefulness of miltefosine and its analogs in the treatment of invasive or device-related fungal infections.
Supplementary Material
Acknowledgements
The authors thank Ms. Marsha Wright for in vitro antifungal testing, Mr. John Trott for in vitro cytotoxicity testing, and Dr. Bharthi Avula for high resolution mass spectrometry analyses. This work was supported by the NIH, NIAID, Division of AIDS, Grant No. AI 27094, the USDA Agricultural Research Service Specific Cooperative Agreement No. 58-6408-2-0009, the Duke University Center for AIDS Research (CFAR) with assistance of the NIH funded program (2P30 AI064518-06 to Y.-L.C.) and NIH/NIAID R01 grant AI50438 (J. H.), and NIH/NHGRI R01 grant HG004840 (X. P.).
Footnotes
Supplementary data Experimental procedures and spectroscopic data for compounds 3a-3i. This material is available free of charge via the Internet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Vink SR, Van Blitterswijk WJ, Schellns JH, Verheij M. Cancer Treat. Rev. 2007;33:191. doi: 10.1016/j.ctrv.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 2(a).Eibl H, Unger C. Cancer Treat. Rev. 1990;17:233. doi: 10.1016/0305-7372(90)90053-i. [DOI] [PubMed] [Google Scholar]; (b) Houlihan WJ, Lohmeyer M, Workman P, Cheon SH. Med. Res. Rev. 1995;15:157. doi: 10.1002/med.2610150302. [DOI] [PubMed] [Google Scholar]
- 3.Seifert K, Lemke A, Croft SL, Kayser O. Antimicrob. Agents Chemother. 2007;51:4525. doi: 10.1128/AAC.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dorlo TP, van Thiel PP, Huitema AD, Keizer RJ, de Vries HJ, Beijnen JH, de Vries PJ. Antimicrob. Agents Chemother. 2008;52:2855. doi: 10.1128/AAC.00014-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Llull D, Rivas L, Garcia E. Antimicrob. Agents Chemother. 2007;51:1844. doi: 10.1128/AAC.01428-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Croft SL, Seifert K, Duchene M. Mol. Biochem. Parasit. 2003;126:165. doi: 10.1016/s0166-6851(02)00283-9. [DOI] [PubMed] [Google Scholar]
- 7.Chugh P, Bradel-Tretheway B, Monteiro-Filho CMR, Planelles V, Maggirwar SB, Dewhurst S, Kim B. Retrovirology. 2008;5:11. doi: 10.1186/1742-4690-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu Q, Ubillas RP, Zhou Y, Dubenko LG, Dener JM, Litvak J, Phuan PW, Flores M, Ye Z, Gerber RE, Truong T, Bierer DE. J. Nat. Prod. 1999;62:824. doi: 10.1021/np980425n. [DOI] [PubMed] [Google Scholar]
- 9.Widmer F, Wright LC, Obando D, Handke R, Ganendren R, Ellis DH, Sorrell TC. Antimicrob. Agents Chemother. 2006;50:414. doi: 10.1128/AAC.50.2.414-421.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong Z, Widmer F, Sorrell TC, Guse Z, Jolliffe KA, Halliday C, Lee OC, Kong F, Wright LC, Chen SCA. Antimicrob. Agents Chemother. 2007;51:2219. doi: 10.1128/AAC.01382-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vila TVM, Ishida K, de Souza W, Prousis K, Calogeropoulou T, Rozental S. J. Antimicrob. Chemother. 2013;68:113. doi: 10.1093/jac/dks353. [DOI] [PubMed] [Google Scholar]
- 12.Zuo X, Djordjevic JT, Bijosono Oei J, Desmarini D, Schibeci SD, Jolliffe KA, Sorrell TC. Mol. Pharmacol. 2011;80:476. doi: 10.1124/mol.111.072322. [DOI] [PubMed] [Google Scholar]
- 13(a).Berkovic D, Sievers S, Haase D, Fleer EA, Binder C. J. Exp. Ther. Oncol. 2002;2:85. doi: 10.1046/j.1359-4117.2002.01014.x. [DOI] [PubMed] [Google Scholar]; (b) Carrasco MP, Jimenez-Lopez JM, Segovia JL, Marco C. FEBS J. 2008;275:1675. doi: 10.1111/j.1742-4658.2008.06322.x. [DOI] [PubMed] [Google Scholar]; (c) Ruiter GA, Zerp SF, Bartelink H, van Blitterswijk WJ, Verheij M. Anticancer Drugs. 2003;14:167. doi: 10.1097/00001813-200302000-00011. [DOI] [PubMed] [Google Scholar]; (d) van der Luit AH, Budde M, Ruurs P, Verheij M, van Blitterswijk WJ. J. Biol. Chem. 2002;277:39541. doi: 10.1074/jbc.M203176200. [DOI] [PubMed] [Google Scholar]
- 14.Wiederhold NP, Najvar LK, Bocanegra R, Kirkpatrick WR, Sorrell TC, Patterson TF. Antimicrob. Agents Chemother. 2013;57:745. doi: 10.1128/AAC.01624-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15(a).Breiser A, Kim D-J, Fleer EA, Damenz W, Drube A, Berger M, Nagel GA, Eibl H, Unger C. Lipids. 1987;22:925. doi: 10.1007/BF02535556. [DOI] [PubMed] [Google Scholar]; (b) Arndt D, Zeisig R, Fichtner I, Teppke AD, Fahr A. Breast Cancer Res. Treat. 1999;58:71. doi: 10.1023/a:1006224611505. [DOI] [PubMed] [Google Scholar]
- 16.Obando D, Widmer F, Wright LC, Sorrell TC, Jolliffe KA. Bioorg. Med. Chem. 2007;15:5158. doi: 10.1016/j.bmc.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 17.Lukáč M, Mrva M, Fischer-Fodor E, Lacko I, Bukovský M, Miklášová N, Ondriska F, Devínsky F. Bioorg. Med. Chem. Lett. 2009;19:6346. doi: 10.1016/j.bmcl.2009.09.079. [DOI] [PubMed] [Google Scholar]
- 18.Lukáč M, Mojžiš J, Mojžišová G, Mrva M, Ondriska F, Valentová J, Lacko I, Bukovský M, Devínsky F, Karlovská J. Eur. J. Med. Chem. 2009;44:4970. doi: 10.1016/j.ejmech.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 19.Perrissoud D, Pietras M, Engel J. PCT Int. Appl. 2007 WO 2007071658 A2 20070628. [Google Scholar]
- 20(a).Karabit MS, Juneskans OT, Lundgren P. Int. J. Pharm. 1988;46:141. [Google Scholar]; (b) Mosca A, Russo F, Miragliotta GJ. Antimicrob. Chemother. 2006;57:566. doi: 10.1093/jac/dki474. [DOI] [PubMed] [Google Scholar]
- 21.Advanced Chemistry Development, Inc . ACD/Structure Designer Suite 12.0. Toronto, ON, Canada: http://www.acdlabs.com/products/pc_admet/physchem/sds/ [Google Scholar]
- 22.Salmon A, Jutzi P. J. Organomet. Chem. 2001;637–639:595. [Google Scholar]
- 23.Spectroscopic data for N-benzyl-N,N-dimethyl-2-{[(hexadecyloxy)hydroxyphosphinyl]oxy}-ethanaminium inner salt (3a): 1H NMR (400 MHz, CD3OD) δ 7.59 (d, 2H, J = 7.2 Hz, aromatic H), 7.51 (m, 3H, aromatic H), 4.62 (br s, 2H, N-CH2-Ph), 4.30 (br s, 2H, N-CH2CH2-O), 3.85 (q, 2H, J = 6.4 Hz, −OCH2-), 3.62 (m, 2H, N-CH2CH2-O), 3.11 (s, 6H, 2×N-CH3), 1.57 (m, 2H, −CH2), 1.39–1.20 (m, 26H, 13×CH2), 0.86 (t, 3H, J = 6.7 Hz, CH3) (the assignment of the 1H NMR resonances was facilitated by 2D NMR experiments of HMQC and HMBC); 13C NMR (100 MHz, CD3OD) δ 134.4 (2C), 131.8, 130.2 (2C), 128.8, 70.2, 66.9 (d), 65.3 (m), 60.1 (d), 51.2 (t, 2C), 33.1, 31.9 (d), 30.8 (6C), 30.8 (2C), 30.5 (2C), 26.9, 23.7, 14.6; HRESIMS m/z 482.3395 (calcd for [C27H50NO4P–H]−, 482.3405).
- 24.Li X-C, Jacob MR, Khan SI, Ashfaq MK, Babu KS, Agarwal AK, ElSohly HN, Manly SP, Clark AM. Antimicrob. Agents Chemother. 2008;52:2442. doi: 10.1128/AAC.01297-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minimum fungicidal concentration (MFC) was determined by removing 5 μL from each clear well, transferring to agar, and incubating at 35°C for 48 h for Candida spp. and Aspergillus fumigatus and 72 h for Cryptococcus neopformans. The MFC was defined as the lowest test concentration that allows no growth of the organism on agar.
- 26.Yang C-R, Zhang Y, Jacob MR, Khan SI, Zhang Y-J, Li X-C. Antimicrob. Agents Chemother. 2006;50:1710. doi: 10.1128/AAC.50.5.1710-1714.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.