

Abstract
Fluorescence time-lapse microscopy has become a powerful tool in the study of many biological processes at the single-cell level. In particular, movies depicting the temporal dependence of gene expression provide insight into the dynamics of its regulation; however, there are many technical challenges to obtaining and analyzing fluorescence movies of single cells. We describe here a simple protocol using a commercially available microfluidic culture device to generate such data, and a MATLAB-based, graphical user interface (GUI) -based software package to quantify the fluorescence images. The software segments and tracks cells, enables the user to visually curate errors in the data, and automatically assigns lineage and division times. The GUI further analyzes the time series to produce whole cell traces as well as their first and second time derivatives. While the software was designed for S. cerevisiae, its modularity and versatility should allow it to serve as a platform for studying other cell types with few modifications.
Keywords: Microbiology, Issue 77, Cellular Biology, Molecular Biology, Genetics, Biophysics, Saccharomyces cerevisiae, Microscopy, Fluorescence, Cell Biology, microscopy/fluorescence and time-lapse, budding yeast, gene expression dynamics, segmentation, lineage tracking, image tracking, software, yeast, cells, imaging
Introduction
Single-cell analysis of gene expression has furthered our understanding of many aspects of gene regulation. Static snapshots of fluorescent reporter expression using flow cytometry or microscopy provide useful information on the distribution of single-cell expression, but lack the history and evolution of time series data required to directly inform gene expression dynamics. Fluorescence time-lapse microscopy presents a means to obtain both single-cell measurements and their history. Various experimental and analytical techniques have been developed to obtain and quantify movies of fluorescent reporter expression, thus imparting insights into gene regulation features (see 1 for a review) such as cell-to-cell variation 2,3, bacterial persister formation 4, transcription initiation and elongation 5, transcriptional bursting 6,7, cell-cycle dependence 8,9, and heritability 10. However, obtaining quality single-cell fluorescence time series involves significant technical challenges in culturing a monolayer of cells in a controllable environment and in high-throughput quantification of the acquired fluorescence movies. Here, we describe a procedure to obtain and analyze fluorescence movies of S. cerevisiae with no required experience in cell culture device manufacture or in software development (Figure 1).
First, we detail an example protocol to generate fluorescence time series movies for budding yeast expressing one or more fluorescent reporters. Though customized microfluidic culture chambers have been built and successfully employed previously 11-13, we use a commercially available microfluidic device from CellAsic (Hayward, CA). The system confines cells to monolayer growth and allows continual control of the perfusion environment. The microscopy protocol we present is a simple means to obtain fluorescence movies of budding yeast, but any modified experimental protocol (a customized culture device, alternative media conditions, etc.) yielding similar fluorescence movie data of single yeast cells may be substituted.
Next, we outline the analysis of the movies using a graphical user interface (GUI) -based software package in MATLAB (Mathworks, Natick, MA), dubbed the GUI for Rapid Analysis of Fluorescence Time Series (GRAFTS), to extract time series data for single cells. GRAFTS has similar features to the versatile, open-source software package Cell-ID 14 in segmenting and tracking cells and in extracting fluorescence intensity and geometric information. However, GRAFTS provides important additional features. First, it offers easy interactive editing of segmentation and tracking results to verify data accuracy, rather than just statistical gating of outlier region traces after analysis. Moreover, it extends the analysis to automatically designate lineage and cell-cycle points of interest of budding yeast. Determining when mother and daughter divide to form two independent cell regions is crucial to determining whole cell (mother including any connected bud) measurements throughout the cell cycle 8. The suite consists of three modules to accomplish these tasks. The first segments cell regions based on the contrast between focused and unfocused bright field images, and allows the user to define and visually test segmentation parameters. The second tracks (using Blair and Dufresne's MATLAB implementation of the Crocker et al. IDL routine, available at: http://physics.georgetown.edu/MATLAB/) and measures cell regions through time; automatically assigns lineages; and enables visual inspection and error correction. A simple plotting GUI is included to query single-cell properties. The third module ascribes bud emergence and division times, and outputs whole cell time series data as well as their first and second time derivatives (as discussed in 9). The analysis module outputs the data as a space-delimited text file for subsequent study in the statistical software of choice. Thus, the package enables the user to extract high quality time series data through a graphical interface. We have used this method to estimate real-time transcription rates in single budding yeast cells as a function of the cell cycle 9. While the modules have been optimized for budding yeast, the parameters or, if necessary, the freely available code may be adapted for other organisms and image types. Segmentation, tracking, and lineage assignment algorithms may be specific to types of imaging assigned and the organism in question. The existing algorithms could be replaced, but still retain the GUI interface that allows user-friendly visual inspection and correction of segmentation and tracking errors that invariably occur with any algorithm.
Protocol
1. Obtain Fluorescent Microscopy Movies of Single Yeast Cells Growing in a Microfluidic Chamber
Inoculate 1 ml SC media (synthetic defined media with 2% glucose and complete complement of amino acids) with cells from a freshly growing plate, and incubate the culture overnight ~16 hours on a roller drum at 30 °C. Prepare the culture such that the final OD600nm is ~0.1 for early log-phase growth.
Dilute the starter culture as needed and regrow 1 ml in a fresh test tube an additional 6-8 hours to OD600nm = 0.1. This provides cells growing at a nutrient-rich steady state at a density suitable for loading into the microfluidic device. (Alternatively, replace steps 1.1-1.2 with the procedure necessary to prepare cells as required for the experiment.)
Prepare a Y04C microfluidic culture plate from CellAsic: remove the shipping solution; rinse the wells with sterile water; and using the ONIX perfusion system flow water through wells 1-6 at 6 psi for 5 min to flush the channels. Then, flow from the loading well 8 at 6 psi for 10 sec (the loading channel has much higher flow rates and should be flushed separately).
Remove water from all wells and replace with 250 μl SC in the inlet wells 1-6 and 50 μl of the culture from step 1.2 in cell loading well 8. (Alternatively, place the desired media in the inlet wells 1-6 for experiments requiring switching between different conditions.)
Seal the ONIX manifold to the plate, and begin priming the channels and culture chamber by flowing from inlet wells 1-6 at 6 psi for 2 min followed by at least 5 min at 6 psi from only the inlet well holding the starting media for the experiment. Flow this continually until step 1.7.
Prepare the microscope for the experiment. We use a Zeiss Axio Observer.Z1 widefield microscope with a Cascade II back-illuminated EMCCD camera (Photometrics, Tuscon, AZ) and a Lumen 200 metal-halide arc lamp (PRIOR Scientific, Rockland, MA) for fluorescence excitation attenuated to 10% to prevent photobleaching. To minimize the switching time required to acquire in multiple fluorescent channels, we couple a single, triple-bandpass dichroic filter cube to appropriate excitation/emission filters for CFP, YFP, and RFP (Chroma Technology Corp, Bellows Falls, VT; set 89006) set in external, fast-switching filter wheels (Ludl Electronic Products, Novato, CA).
Place a few drops of immersion fluid on the objective (if needed, we use a Zeiss Plan-Apochromat 63X/1.40 Oil DIC), and mount the microfluidic device securely in the stage of the inverted microscope. Ensuring the plate does not shift at all in the stage throughout the experiment improves cell tracking during data analysis. We simply tape the plate to the stage mount to prevent its shifting.
Focus on the leftmost third of the first culture chamber (where the chamber height is smallest) using one of the embedded position markers. Turn off flow from the media inlet well, and flow from cell loading well 8 at 6 psi in 5 sec bursts. Move the stage to look around the culture chamber for cells. Increase loading flow time and pressure until desired cell density is achieved, but avoid overloading and clogging the leftmost barrier where fresh media enters the chamber.
Begin flowing from the media inlet well containing the starting media at 6 psi.
In MetaMorph (Molecular Devices, Sunnyvale, CA) or other microscopy automation and control software, setup a multi-dimensional acquisition to take multiple images at multiple stage positions over time. Set the number of and interval between time points. Select several stage positions with ~10 cells each (more will overcrowd quickly). We typically use 5 min intervals and image up to 16 positions, which each require ~11 seconds for acquisition at each time point.
Setup the acquisition so that at each stage position at each time point the program will: focus on the transmitted light bright field (BF) image using MetaMorph's built-in autofocus method (implementing the autofocus as a custom-written journal at the start of each stage position provides greater control over focus accuracy); acquire the BF image at +1 μm to the focal plane (f.p.); acquire a BF out of focus (BFOOF) image at -4 μm to the f.p. (use custom-written journals to change the focus as required between images); and acquire one grayscale image for each fluorescent reporter at the f.p. using settings optimized for rapid acquisition with minimal photobleaching.
If necessary, obtain background and shading correction images for each fluorescent reporter in an area of the culture chamber with no cells.
Prepare the flow program for the experiment in the ONIX control software. Simultaneously begin the acquisition program in MetaMorph and the flow program in the ONIX control software.
At the end of the experiment, correct uneven background and shading in the fluorescent images (if needed), and optionally combine the .tif files for each image channel at each stage position into a .stk movie file (e.g. create BF, BFOOF, CFP, YFP and RFP movies for position 1).
2. Format and Segment Data for Tracking Using the FormatData GUI in MATLAB (Figure 2)
Run the FormatData.m file in MATLAB to open the data input GUI. At the top, specify or browse to the folder containing the image files to set the data directory, and then use the GUI to specify the data types and image files recorded in the experiment.
To the right, select the radio button next to "Time-lapse" to indicate which analysis type to perform. "Time-lapse" will treat the image series as a time series (e.g. a movie of cells growing in a microfluidic chamber). "Static" will treat each image series as a set of snapshots taken from a population (e.g. multiple images taken at different positions on a single slide sample). In this GRAFTS version, time-lapse data for multiple positions can be processed but as a separate file for each position (see step 2.14).
Check the box for image registration to correct for imprecise stage movements by shifting each image in the movies to minimize apparent cell movement within the frame. We strongly recommend this as it greatly improves tracking (reducing manual track curation later), but will add random, "white noise" pixel values to fill in where images have been shifted at the border. It could also be selected after viewing segmented BF images in step 2.7 or at any point until step 2.13.
Check the "Bright Field" box in the "Data Channels" panel. Click "Select" to the right to load the BF images. For *.TIF files, select all BF images corresponding to a single movie by holding the Shift key while selecting, then click "Open". For *.STK files, just select the BF stack of interest. If the images are successfully recognized, the file names will be listed in the popup menu to the right in the "Data Recognized" panel. For *.TIF files, be sure the file names appear in the correct order (save files with sequential names during acquisition - BF_t001, etc.).
Check the "Bright Field (out of focus)" box and use the "Select" button as in step 2.4 to load the BFOOF files, which will be listed in the popup to the right. (BFOOF obtained as BF -4 μm relative to the f.p. at 63X segments well.)
Check the "Cell Mask" box. In the "Mask Source" panel, select the radio button next to "Segment BF". Push the "Parameters" button to open the segmentation GUI. (Alternatively, select a pre-segmented mask movie by selecting the radio button next to the "Select Mask File" button, pressing the latter, and selecting the movie as in step 2.4. In this case, a BFOOF movie is not needed, and can skip to step 2.8.)
Set the different segmentation parameters (descriptions of each can be viewed by pressing "Parameter descriptions"), and test the segmentation quality by clicking "Test" (Figure 3). Successfully segmented regions will be green with magenta borders. Be sure to use the slider to check multiple time points in the movie. When the segmentation is satisfactory, select "Done" to return the segmentation parameters to the FormatData GUI.
Check the "Color Images" box. Enter the color name for the first fluorescent image channel in the text edit box, and then press "Add and Select" to load the color files as in step 2.4. The color name will be added to the list box on the left, and the file names will be added to the table on the right. If a mistake is made in loading the color files, select the color name in the list box on the left and click "Remove Color" to remove the files. Repeat for each color channel.
If additional sub-region masks based on one of the fluorescent colors (e.g. fluorescently labeled nucleus → nuclear region mask) are desired, check the "Color Mask" box. If not, skip to step 2.12. In the "Color Mask Source" panel, enter the sub-region mask name in the text edit box. Choose a color from the popup menu in the "Color Mask Source" panel (requires the color to have been added in step 6) and push "Parameters" to open a threshold testing GUI.
Push the "Auto-threshold" button to automatically determine the threshold using Otsu's method 15, and the image will highlight the areas preserved above the threshold (Figure 4). The threshold can be manually adjusted using the edit box. Use the scroll bar to confirm the threshold is appropriate for all time points. When satisfied, push "Done" to return the threshold to the FormatData GUI.
Push "Add and Segment" to generate the sub-region mask using the threshold module applied to the selected color images. The color mask name and source will be displayed in the table to the left, with the relevant recognized file names appearing in the table on the right. (Alternatively, push "Add and Select" to load pre-made sub-region mask files.)
At the bottom, specify or browse to the desired folder to be used as the save directory.
Press "Format Data" to read the image files (using the tiffread2.m code developed by Francois Nedelec, available at: http://www.mathworks.com/MATLABcentral/fileexchange/10298), process the data, and create a *.mat file in the specified folder. This file will serve as the input to the ProcessTimeSeries GUI.
Repeat step 2.4-2.13 for the set of movies for each stage position.
3. Track Cells and Lineages through Time with ProcessTimeSeries GUI, and Curate ID and Lineage Assignments (Figure 5)
- Run the ProcessTimeSeries.m file in MATLAB to open the tracking GUI. Set the following parameters after opening the GUI:
- "Chamber height" ("File I/O" panel, upper left) - this indicates the height of the chamber in which the cells are trapped. It is used as a constraint in approximating cell volume as a 3D ellipsoid based on the major and minor axis of the cell region as in 8.
- "μm/pixel" ("File I/O" panel) - the conversion factor to calibrate pixel to μm distance in the images so cross-sectional area and volume can be calculated in μm2 and μm3, respectively.
- "Filters panel" (below "File I/O" panel) - set minimum and maximum parameters to filter out junk regions in the mask: "Area" - region area in pixels; "Ecc" - eccentricity factor, more circular as → 0; "SF" - shape factor, more circular as → 1.
Click "Load Images" in the "File I/O" panel, and select the *.mat data file of interest output by the FormatData GUI. The region mask (and any sub-region masks) is applied to each color channel, and a number of different measurements are made (Table 1). The data file is then saved. (If loading a file into ProcessTimeSeries from a previous session, it will ask if the analysis should be restarted from the beginning. CAUTION: This will erase any track curation already completed and treat the data as if it were fresh from the FormatData GUI. If accidentally restarted, quickly hit Ctrl+C in the MATLAB command window to prevent the previously curated *.mat file from being overwritten.)
- Next, track the cells. In the "Track Cells" panel (next to the "Filters" panel), set the following parameters:
- "Max Displacement" - the maximum distance in pixels a cell can travel between adjacent images before being labeled as a different cell. Higher displacement means the algorithm can account for cells moving more, but it also increases the complexity of matching cell regions in crowds. We start at 12 and decrease if an error occurs in tracking (see step 3.4).
- "Minimum Length" - minimum number of occurrences for a cell trace to be considered a valid data series. We use 1 to prevent removal of any real traces, but this can be increased if segmentation results in short-lived, spurious region traces.
- "Frame Memory" - maximum number of consecutive frames a cell region can disappear and be given the same ID when it returns. If it disappears for longer than "Frame Memory", it will be given a new ID upon returning. We use 2 to allow regions to be missed by segmentation for 2 frames before returning.
Click "Track Cells" to track regions from one frame to the next using the region centroids (Blair and Dufresne MATLAB implementation of Crocker, Grier, and Weeks IDL routine, available at: http://physics.georgetown.edu/MATLAB/), to assign lineages for newly appearing buds, and to save the data. Lineages are automatically assigned by minimizing distances between putative buds and potential mothers in each frame. (If an error appears in the MATLAB command window due to "difficult combinatorics", decrease "Max Displacement" and repeat tracking.) Change parameters in the popup windows as necessary for your data.
- Curate poorly segmented regions and mistakes in ID or lineage assignments using the various GUI tools or shortcut keys (shown in a popup window by pressing the button above "Selected Cell Information" panel).
- Slider, "Prev Image", "Next Image" (Ctrl+left/right) - use to display and move between frames in the image series.
- "Image #" - displays the frame number of the current image in the left and right axes. Move to a specified frame by typing its number in the right "Image #" edit box.
- "Delta frame" - shows the difference in frame number between the right and left axes (Δ = right - left).
- "Hide Text" (Ctrl+T) - toggles between displaying the cell IDs in the right axes or not.
- "Hide Labels" (Ctrl+L) - toggles between displaying the cell IDs in the left axes or not.
- "Mask" popup menu - choose between displaying no mask, the cellular mask, or any user-defined sub-masks for display in the left panel.
- "Overlay" popup menu (Ctrl+1-9) - choose to display BF and color layers in the left frame. Show BF is the only one that toggles the BF layer on or off in both axes. Toggle between display or not by selecting the overlay name in the menu again ("*" denotes overlay is displayed). Ctrl+1 toggles BF overlay, with 2-9 toggling the color overlays in order.
- "Set Colors" - use to determine color for overlay display. A popup window will query which fluorescence channel to set, and then the color palette will appear for user definition.
- "Modify Regions and Tracks" panel - these controls effect changes on the cell region mask and trace information:
- "Update tracks" (Ctrl+U) - use to reassign cell IDs. If no cells are selected in right axes, the GUI will prompt for a cell ID to change. When ID X is changed to Y, all future instances of X will be changed to Y, and any future instances of the current Y, will be switched to X. (Occasionally one cell will switch back and forth between 2 IDs, repeatedly. This is due to the tracking trying to accommodate multiple IDs in an oversegmented region rather than an error in the update ID function.) Multiple cells can be selected and their IDs changed simultaneously, but be sure to give each a unique ID. To assign a region to an ID that does not exist in the current file, enter "0" and one will be generated. Use Ctrl+W to swap the IDs of two highlighted cells.
- "Delete Selected Tracks" (Ctrl+D) - use to delete the selected cell or multiple cell regions in the current right axes frame. (If no cells are selected, will prompt for ID.) To permanently delete all instances of an ID trace, check the box next to the Delete Selected Tracks button (text will change to "Delete ALL"). This is useful for getting rid of persistent junk regions, but first check future frames to make sure the ID doesn't switch to a valid cell or bud region.
- "Merge Regions" (Ctrl+A or M) - smooths and combines cell regions. Click a cell or cells in the right axes to select (region will turn red if selected). Merging on one region will morphologically close to fill in cracks or small missing chunks using a disk-shaped element. Merging two or more nearby regions will combine them into one region and give the lowest ID to the new region. This is useful for over-segmented regions (often when cells have large vacuoles).
- "Draw region" - use the mouse to draw a region in the right axes where desired and double click to finish. Provide a unique ID not present in the data set (between 1 and 65535). Cancel by pressing delete on the keyboard.
- "Lineage Tracking" panel - after tracking, lineage assignments are automatically generated. When new ID regions appear, the new cell ID appears in pink and a red line is drawn to the mother region. New regions too large to be buds or too far away from potential mothers appear in green and are assigned a mother ID of "NaN" ("not a number", see Table 2). Regions existing at the first time point have a mother ID of "0". To fix individual assignments, enter the mother and bud IDs in the text edit fields and click "Fix". To erase a cell's mother, enter "0" as its mother. A faster method is to select two regions in the right image and press Ctrl+F. This will automatically assign the older region as the mother and the newer region as the daughter, while erasing any previously existing mother assignments for the daughter cell. CAUTION: Clicking "Calculate Lineages" at any time will recalculate all lineage assignments, including those the user has previously curated.
- "Selected Cell Information" panel - "Get Info" will display some measurements of the regions selected in the right axes. "Measurements…" allows the user to determine which measurements are displayed (as well as define the default list for other operations such as "Export Data"). Can be used to determine correct IDs and lineages (e.g. if cell X has a bud at time t, it most likely does not have another bud at time t + 5 min).
- "Save Changes" (Ctrl+S) - updates the *.mat file to reflect user changes. USE FREQUENTLY (there is no undo function)!
- "Generate Plots" - opens the GeneratePlots GUI. Used to quickly plot selected variable information and simple calculations for single-cell regions highlighted in the right axes of the ProcessTimeSeries GUI, their mean, or the mean of all regions in each frame. This GUI can calculate rough first derivatives, but be sure to specify the correct time interval at the top left. Plots may be output to a figure for saving by pressing "Generate Figure".
To properly curate data: merge any oversegmented regions; delete any regions not capturing the whole cell; be sure mother-bud relationships are properly assigned (a red line appears between them at the time of bud emergence, when the bud ID is pink, see Table 2); and eliminate any green IDs by fixing the ID, assigning the region a mother, assigning no mother ("0"), or deleting the region. Bud data will be added to that of the mother during the final analysis so do not merge a bud region to its mother (it will lead to inaccurate volume estimation).
Visually inspect data for each frame up to the last time of interest, but it is not required of the entire time series if not using the later frames.
Be sure to save changes.
Repeat steps 3.1-3.7 for the *.mat file for each stage position.
4. Exporting Data for Analysis
To simply export raw region measurements for each cell through time, push "Export Data". The measurements of interest can be selected in the GUI that opens, and they will be output in a space-delimited *.txt file with variable names as column headers.
To analyze the time series for whole cells over time, calculate cell-cycle information, and take derivatives, push "Time Series Analysis". A window will open allowing selection of the measurements of interest and input of analysis parameters (Figure 6).
Enter the last time point curated in the GUI (all subsequent frames will be ignored). The default smoothing and cell-cycle assignment parameters work well for our haploid and diploid strains imaged in 5 min intervals, but these may need to be varied for best results. (Check that the parameter values are appropriate by manually determining budding and division time for a test data set and comparing to the performance of the automated algorithm.)
- When all parameters are set, push "Analyze" to:
- Construct arrays for each raw measurement and subtract the background (measured as the average intensity of the non-cell area of the first frame in each fluorescent movie, or can be specified to account for average autofluorescence per cell pixel).
- Fit a smoothing spline to the selected measurements to eliminate high-frequency measurement noise (as discussed in 9).
- Automatically determine bud emergence and cell division times for each cell based on changes in the slope of the volume time series as discussed in 9. The bud measurements will then be added to the mother's measurements between emergence and division each cycle for a contiguous whole cell time series. A normalized cell-cycle progression will also be calculated ranging from 0 to 2 from division to division.
- Fit a smoothing spline to take stable 1st and 2nd derivatives of the selected measurements. (For the purpose of well-defined derivatives, time series are made continuous across division by shifting data for the following cell cycle to meet the endpoint for the previous cycle.)
- Output raw and smoothed time series for all regions, and whole cell smoothed, continuous, and differentiated time series as an array for each measurement. The first element is a color index for background subtraction, the rest of the first row holds cell IDs, the rest of the first column is the frame number, and the remaining elements hold the time series for each single cell in a column with each row corresponding to the frame in the first column. A similar array of cell-cycle progression time series and lineage information will also be output. The array "LineageArray" contains a table with each row representing a mother-bud relationship and the first through fifth columns are mother ID, bud ID, frame of first bud region, estimated budding frame, and estimated division frame. Data is exported to a *.mat file (MATLAB data file).
Representative Results
A successfully performed and analyzed experiment will yield mostly continuous time series for single whole cells with realistically assigned bud emergence and division times. As an example, we performed the above protocol with a haploid yeast strain expressing an integrated copy of Cerulean fluorescent protein (CFP) driven by the constitutive ADH1 promoter to observe how growth and global expression may vary over time in single cells (Table 4, Y47). We ran the time series analysis to obtain whole cell (Figure 7A) measurements over time, and a dependence on the cell cycle emerges for both volume and expression as found in 9. After bud formation, the total combined volume (mother + bud) increases more quickly than before bud emergence (consistent with 8,16) while protein concentration, or average intensity, decreases slightly (Figures 7B and 7C). The rise in combined integrated protein (volume x concentration in this case) also accelerates after budding (Figure 7D). Comparing to the mother region alone (Figure 7B&D), these results demonstrate the importance of properly incorporating bud contributions to the whole cell measurement and highlight the need for proper bud formation and division time assignments. As we have reported 9, we can use the differentiated time series to calculate an instantaneous relative mRNA level M(t) and transcription rate A(t), which both also increase after budding (Figure 7E and 7F):
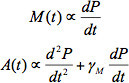 where P(t) is the total protein at time t and γM is the mRNA degradation rate 0.04 min-1
17. These are not absolute quantities as our protein time series are measured in an arbitrary fluorescent unit.
where P(t) is the total protein at time t and γM is the mRNA degradation rate 0.04 min-1
17. These are not absolute quantities as our protein time series are measured in an arbitrary fluorescent unit.
The ability to generate stable time derivatives of measurement time series also benefits kinetic studies. We use a yeast strain expressing an observable, chimerical transcription factor with switchable transcription activity (Table 4, Y962). The master transcription factor of the phosphate starvation pathway, Pho4p, is controlled through nucleo-cytoplasmic localization in response to extracellular phosphate concentratio n18. We replaced the DNA binding domain of Pho4p with tetR, which binds the tet operon (tetO), and C-terminally fused the new transcription factor to a yellow fluorescent protein to create Pho4-tetR-YFP 9. By switching the phosphate level in the perfusion media, we could then toggle expression of an integrated CFP driven by a synthetic promoter composed of 7 tetO binding sites and the CYC1 minimal promoter (7xtetOpr-CFP). A time series analysis shows when the transcription factor is localized to the nucleus (using a fusion of RFP and the nuclear protein Nhp2p to create an RFP-based nuclear submask as in step 2.9-10), and when the target gene starts and stops transcription (Figure 8). By analyzing the derivative series over time, activation and deactivation times of the target promoter in each single cell can be inferred even when the concentration of the stable fluorescent reporter is high.
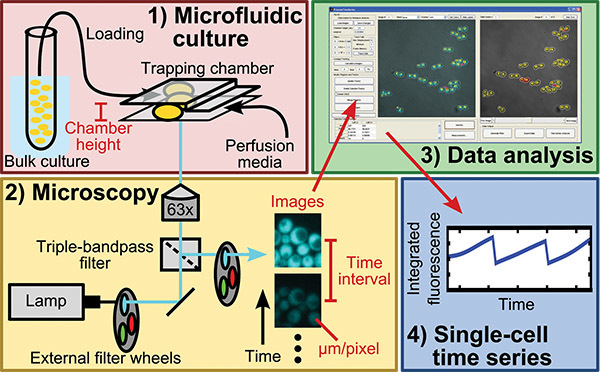 Figure 1. Schematic of protocol. The protocol describes steps for 1) microfluidic culture, 2) fluorescence, time-lapse microscopy, 3) data analysis, and 4) output of single-cell time series of cell measurements. Text in red indicates information required for data analysis.
Figure 1. Schematic of protocol. The protocol describes steps for 1) microfluidic culture, 2) fluorescence, time-lapse microscopy, 3) data analysis, and 4) output of single-cell time series of cell measurements. Text in red indicates information required for data analysis.
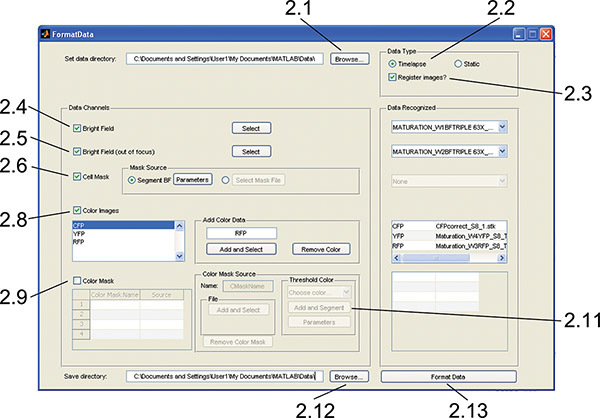 Figure 2. FormatData GUI. The interface is used to import, format, and segment image data for later calculations and visual inspection. Numbers indicate the protocol step corresponding to each component. Click here to view larger figure.
Figure 2. FormatData GUI. The interface is used to import, format, and segment image data for later calculations and visual inspection. Numbers indicate the protocol step corresponding to each component. Click here to view larger figure.
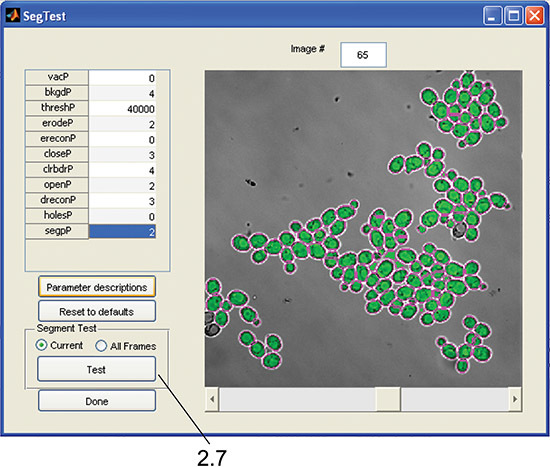 Figure 3. SegTest GUI. The interface is used to test cell segmentation parameters and visually confirm segmentation accuracy as in step 2.7.
Figure 3. SegTest GUI. The interface is used to test cell segmentation parameters and visually confirm segmentation accuracy as in step 2.7.
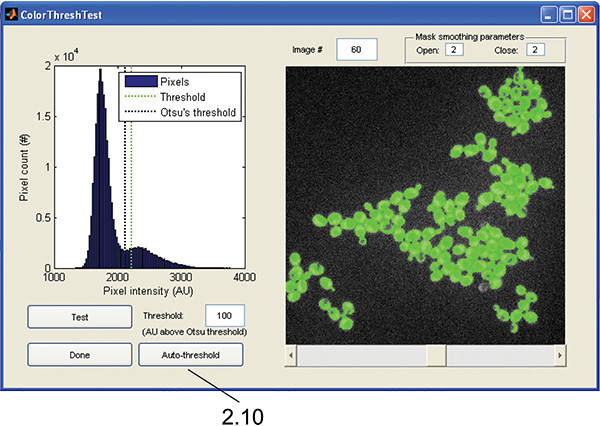 Figure 4. ColorThreshTest GUI. The interface is used to test the intensity threshold needed to create a subcellular mask from the chosen fluorescence channel as in step 2.10.
Figure 4. ColorThreshTest GUI. The interface is used to test the intensity threshold needed to create a subcellular mask from the chosen fluorescence channel as in step 2.10.
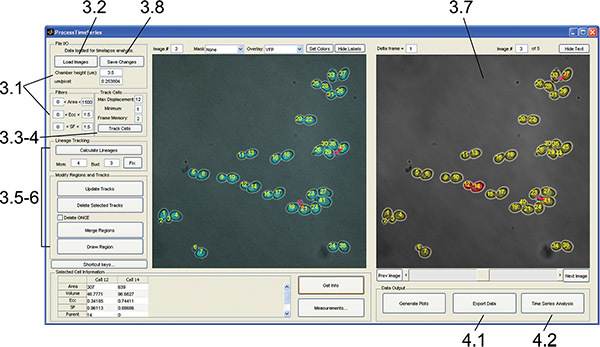 Figure 5. ProcessTimeSeries GUI. The interface is used to measure, track, visually inspect, and edit cell regions and lineage assignments. Numbers indicate the protocol step corresponding to each component. Click here to view larger figure.
Figure 5. ProcessTimeSeries GUI. The interface is used to measure, track, visually inspect, and edit cell regions and lineage assignments. Numbers indicate the protocol step corresponding to each component. Click here to view larger figure.
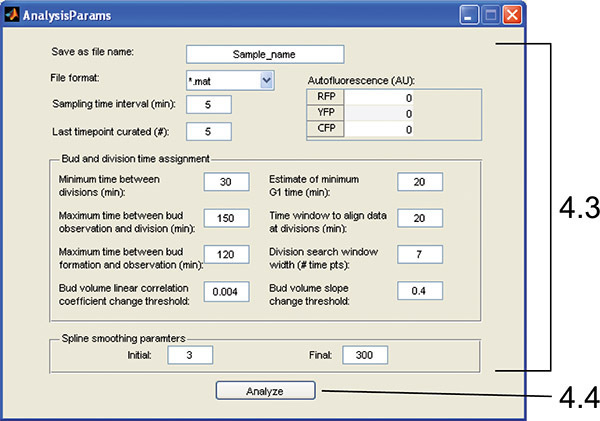 Figure 6. AnalysisParams GUI. The interface is used to enter parameters required for the time series analysis as in steps 4.3 and 4.4.
Figure 6. AnalysisParams GUI. The interface is used to enter parameters required for the time series analysis as in steps 4.3 and 4.4.
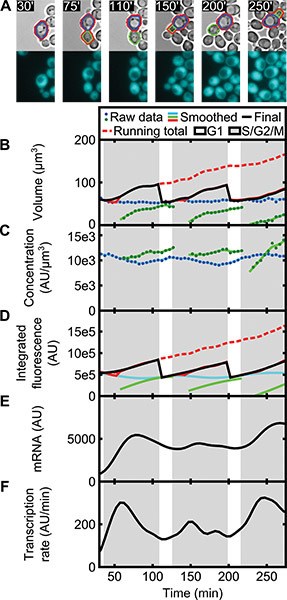 Figure 7. Single-cell time series of a haploid yeast expressing ADH1pr-CFP in microfluidic culture over several generations.(A) Bright field (top row) and CFP (bottom row) micrographs are shown at the indicated time points for a cell throughout the experiment. For the segmented mother cell (blue outline) and its buds (green outline), the contiguous whole cell is outlined in red. Raw mother and bud (B) volume and (C) protein concentration time series were smoothed to remove measurement noise, and (D) integrated CFP fluorescence was calculated as the product of volume and concentration. The whole cell (red) trace is extended past division to keep a running total that is easy to fit to a differentiable smoothing spline across divisions (B and D). The (E) relative mRNA level and (F) instantaneous transcription rate are calculated using equations (1-2) and the spline fit in (D).
Figure 7. Single-cell time series of a haploid yeast expressing ADH1pr-CFP in microfluidic culture over several generations.(A) Bright field (top row) and CFP (bottom row) micrographs are shown at the indicated time points for a cell throughout the experiment. For the segmented mother cell (blue outline) and its buds (green outline), the contiguous whole cell is outlined in red. Raw mother and bud (B) volume and (C) protein concentration time series were smoothed to remove measurement noise, and (D) integrated CFP fluorescence was calculated as the product of volume and concentration. The whole cell (red) trace is extended past division to keep a running total that is easy to fit to a differentiable smoothing spline across divisions (B and D). The (E) relative mRNA level and (F) instantaneous transcription rate are calculated using equations (1-2) and the spline fit in (D).
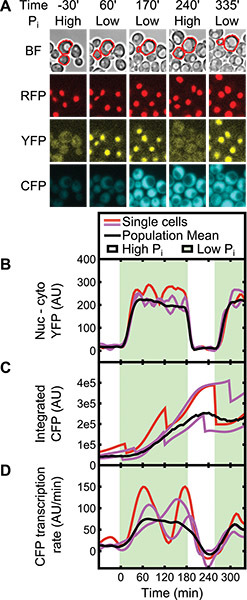 Figure 8. Promoter transcription rate response to nucleo-cytoplasmic shuttling of a Pho4-YFP fusion in single yeast cells. (A) Micrographs from the four indicated acquisition channels are shown for a cell with a switchable YFP-fused Pho4-tetR transactivator that (B) localizes to the RFP-marked nucleus in response to low phosphate and (C) drives expression of a 7xtetOpr-CFP reporter. (D) The inferred transcription rate changes with localization on average (black line) and at the single-cell level (red and magenta lines, where red represents the contiguous cell region outlined in red in A). Sharp drops in CFP expression in B coincide with cell division when some protein is lost to the daughter cell.
Figure 8. Promoter transcription rate response to nucleo-cytoplasmic shuttling of a Pho4-YFP fusion in single yeast cells. (A) Micrographs from the four indicated acquisition channels are shown for a cell with a switchable YFP-fused Pho4-tetR transactivator that (B) localizes to the RFP-marked nucleus in response to low phosphate and (C) drives expression of a 7xtetOpr-CFP reporter. (D) The inferred transcription rate changes with localization on average (black line) and at the single-cell level (red and magenta lines, where red represents the contiguous cell region outlined in red in A). Sharp drops in CFP expression in B coincide with cell division when some protein is lost to the daughter cell.
| Measurement Name | Description |
| Time | Image frame number |
| Area | Total number of pixels comprising the region |
| Volume | (4/3)πabc, where a = ½ major axis length of region, b = ½ minor axis length, and c = b or ½ "Chamber height" (whichever is smaller) |
| (X)_(Y)mean | Mean pixel intensity for the region mask (Y) in color channel (X) |
| (X)_(Y)median | Median pixel intensity for the region mask (Y) in color channel (X) |
| (X)_(Y)std | Standard deviation of pixel intensities for the region mask (Y) in color channel (X) |
| (X)_(Y)iqr | Interquartile range of pixel intensities for the region mask (Y) in color channel (X), 75th percentile - 25th percentile value |
| (X)_(Y)T20pct | Mean intensity for the top 20% of pixel intensities for region mask (Y) in color channel (X). Useful in comparison with the next measurement to determine subcellular localization without a submask. |
| (X)_(Y)B80pct | Mean intensity for the bottom 80% of pixel intensities for region mask (Y) in color channel (X). Useful in comparison with the previous measurement to determine subcellular localization without a submask. |
| (X)_(Y)M50pct | Mean intensity for the middle 50% of pixel intensities for region mask (Y) in color channel (X) |
| (X)_cellint | Integrated fluorescence of color channel (X) (mean pixel intensity x volume) for the whole cell (mother and any connected bud) |
| (Z)Raw | Raw time series data output by "Time Series Analysis" for variable (Z) |
| (Z)Smooth | Spline-smoothed raw time series data output by "Time Series Analysis" for variable (Z) |
| (Z)Whole | Whole cell (mother + attached bud (Z)Rawsmooth) time series data output by "Time Series Analysis" for variable (Z) |
| (Z)Cont | Whole cell time series data made continuous at divisions ("running total") output by "Time Series Analysis" for variable (Z) |
| (Z)WhSmooth | Spline-smoothed (Z)Whole cell time series data output by "Time Series Analysis" for variable (Z) |
| (Z)ddt | First time-derivative of spline-smoothed (Z)Whole cell time series data output by "Time Series Analysis" for variable (Z) |
| (Z)d2dt2 | Second time-derivative of spline-smoothed (Z)Whole cell time series data output by "Time Series Analysis" for variable (Z) |
Table 1. Measurement variable nomenclature in ProcessTimeSeries GUI.
| Cell ID color | Lineage status |
| yellow | mother assigned |
| pink | new bud (red line drawn to assigned mother) |
| green | no mother assigned |
Table 2. Cell ID color code for lineage assignment status.
| File Name | Description |
| FormatData.m | Data input GUI that formats movies and segments BF images for processing by ProcessTimeSeries.m |
| FormatData.fig | FormatData GUI window layout |
| BFsegment.m | BF segmentation module, with extractregions2.m and segmentregions.m |
| SegTest.m | Segmentation quality testing GUI |
| SegTest.fig | SegTest GUI window layout |
| extractregions2.m | First component of BF segmentation module to identify regions |
| segmentregions.m | Second component of BF segmentation module to separate and clean cell regions |
| color2submask.m | Fluorescence-based submask segmentation module |
| ColorThreshTest.m | Fluorescence-based threshold mask testing GUI |
| ColorThreshTest.fig | ColorThreshTest GUI window layout |
| tiffread2.m | Extracts data *.TIF or *.STK files for use in MATLAB |
| imalign.m | Image registration using 2D cross correlation |
| ProcessTimeSeries.m | Data processing GUI that tracks regions, assigns lineages, and enables visual correction of errors. Also provides GUI-based plotting capability and outputs whole-cell time series data |
| ProcessTimeSeries.fig | ProcessTimeSeries GUI window layout |
| cleanMask.m | Eliminates mask regions based on parameters in ProcessTimeSeries GUI |
| track.m | Tracks cell regions based on region centroids |
| OptimizeLineages.m | Lineage assignment module determines optimal mother-bud relationships based on physical distance and past and future budding events |
| getClosestObjects.m | Finds neighbor cells for each new cell ID (bud) |
| SelectVariables.m | Measurement selection GUI for choosing variables of interest |
| SelectVariables.fig | SelectVariables GUI window layout |
| GeneratePlots.m | Plotting GUI for data in ProcessTimeSeries window |
| GeneratePlots.fig | GeneratePlots GUI window layout |
| AnalyzeCellSeries.m | Time series analysis module determines cell division time and outputs whole cell measurements as described in the text |
| assignDivisionsInf2.m | Assigns bud sprout and cell division times |
| AnalysisParams.m | Cell time series analysis parameter input |
| AnalysisParams.fig | AnalysisParams GUI window layout |
Table 3. GRAFTS file descriptions.
| Strain ID | Genotype (W303-based) | Reference |
| Y47 | MATa leu2::ADH1pr-CFP-hisG::URA3::kanR::hisG | 19 |
| Y962 | MATa ADE+ NHP2::RFP-NAT spl2Δ::LEU2 pho4::TRP1 pho84Δ::klura3 phm4::HIS3MX6 leu2Δ::TEF1m7pr-PHO4ΔP2-tetR-cYFP URA3::7xtetOpr-CFP | 9 |
Table 4. Yeast strains used in this study.
Discussion
The above protocol describes a simple method to obtain and analyze fluorescence time series data with limited experience in microfluidics or in software development. It allows one to obtain time-lapse fluorescence movies of single yeast cells; extract relevant cell size and expression measurements; curate tracking and lineage assignments; and analyze the behavior of whole cells over time using a commercially available microfluidic culture device and a versatile graphical user interface (GUI). While the experimental, segmentation, and tracking steps have been approached in various ways previously 11,13,14, the above procedure is designed to make these techniques more accessible to a wider subset of the biology community. While the above procedure is optimized for a particular microfluidic culture device, the overall analysis can be adapted to similar devices as off-the-shelf microfluidic culture technologies become cheaper and more customizable. Fundamentally, the segmentation and region measurement algorithms we employ are similar to existing methods 13,14, but the GRAFTS software adds the ability to visually curate region tracking and lineage assignments and to assign cell-cycle phases accurately. Both of these features are crucial to accurately calculating the time-derivatives of data series.
There are several steps in the protocol which significantly impact the quality of the resulting time series data. Initially overloading the culture chamber with cells will decrease perfusion in the chamber (especially noticeable in kinetic experiments where chamber refresh time is important), and overgrowth will occur earlier in the experiment time course. This can best be avoided by starting with lower cell loading pressure for shorter times and gradually increasing one or both until an appropriate cell density is achieved. Stage position selection for imaging is a related and equally important consideration. Knowing the doubling time of the strain, plan how many cells will be in the image frame over time and anticipate how crowding will affect media diffusion. Ten dispersed cells will grow into ten microcolonies, but ten cells initially grouped together will grow into a single, large colony (we have noticed nutrient diffusion limitations at 6 psi to the center of a colony when it is greater than ~14 cells in diameter). Extra effort in the upstream protocol steps also facilitates manual data curation later, and improves the output time series. Ensure the plate is firmly mounted in the stage and use the image registration option in the FormatData GUI to vastly improve the accuracy in automatically tracking single cells through time. Spend time in choosing the best possible segmentation parameters also to reduce the number of errors which require attention. The tracking software works best with slight oversegmentation of cell regions rather than undersegmentation because remerging a split cell is a simpler task than drawing lines to divide regions; however, increased oversegmentation increases errors in lineage assignments due to the presence of false, new regions. Furthermore, be sure that all mother-bud relationships are properly assigned so that measurements for the whole cell will be accurate. If a bud is missed by the segmentation or grows outside of the image boundary, consider ending the mother's data series to prevent spurious results. This is easily done by changing the mother region to a unique ID (enter ID of "0") at the erroneous frame, and using the "Delete ALL" feature to remove all future instances of the new ID. Close attention to these steps will greatly increase the accuracy of the raw time series data.
To establish valid interpretation of the data output by pressing "Time Series Analysis", we recommend a few additional inquiries. Verify bud emergence and division times are accurately determined based on the user-input parameters. Manually record budding and division times for a test movie set and compare to the algorithm results. To visually estimate the time cytokinesis completes between a mother and daughter, look for their boundary to narrow and darken. (NB: A test strain with a fluorescently-marked nucleus aids in manual observation of division time. Another method would be to fluorescently-mark the plasma membrane and look for the gap between mother and daughter cells to close.). Also, check whether total fluorescence for a cell is better calculated as the average region intensity multiplied by the volume (when captured light originates from a thin cross-section of the cell) or as the integrated fluorescence over the region (when captured light originates from the entire cell). If the fluorescence profile across a cell matches the curve of a semi-ellipse described by the major and minor axes of the region 7, captured light originates from the entire cell, and total fluorescence should be calculated as (mean intensity) x (area). In our case at 63X with a depth of field much less than the cell height, the fluorescence profile is relatively flat and total fluorescence is calculated as (mean intensity) x (volume). Another consideration is the photobleaching of the fluorophore. The software does not consider photobleaching in the analysis, and thus the fluorescence time series output represents only the observable reporter. Photobleaching processes depend heavily on the acquisition settings and time interval used to obtain fluorescence images, the nature of the fluorophore, and various other experiment-specific parameters. Photobleaching also may not be well-described by a simple, first-order rate expression, which prevents a general algorithm for its treatment. We therefore do not account for photobleaching in the time series output, but the analytical method may be used to measure the kinetics of this process to aid the user's interpretation. Lastly, choose smoothing parameters suitable for the observed time series. Ideally, the smoothing splines will eliminate high-frequency measurement noise while preserving real features in the data. The parameters required to achieve this depend on the characteristics of the particular time series and may vary between experiments.
The software was intended to be flexible for analysis of various time-lapse fluorescence microscopy data. Any number of color channels can be included and any may be used to create a sub-region mask. While the algorithms work well for movies of budding yeast cells, many of the functionalities should extend to movies of other organisms as well. Region measurement (excepting volume), tracking, and curation depend only on the cell mask and are thus adaptable. The segmentation, lineage assignment, cell division determination, and time series analysis modules can be independently modified to suit specific needs. In addition, rather than using the included segmentation module, a pre-segmented mask movie can be input, and phase-contrast or differential interference contrast images can be substituted for bright field. The GUIs can then be used primarily to track and curate ID and lineage assignments.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Emily Jackson, Joshua Zeidman, and Nicholas Wren for comments on the software. This work was funded by GM95733 (to N.M.), BBBE 103316 and MIT startup funds (to N.M.).
References
- Locke JCW, Elowitz MB. Using movies to analyse gene circuit dynamics in single cells. Nature Reviews. Microbiology. 2009;7(5):383–392. doi: 10.1038/nrmicro2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld N, Young JW, Alon U, Swain PS, Elowitz MB. Gene Regulation at the Single-Cell Level. Science. 2005;307(5717):1962–1965. doi: 10.1126/science.1106914. [DOI] [PubMed] [Google Scholar]
- Colman-Lerner A, Gordon A, et al. Regulated cell-to-cell variation in a cell-fate decision system. Nature. 2005;437(7059):699–706. doi: 10.1038/nature03998. [DOI] [PubMed] [Google Scholar]
- Vega NM, Allison KR, Khalil AS, Collins JJ. Signaling-mediated bacterial persister formation. Nature Chemical Biology. 2012;8(5):431–433. doi: 10.1038/nchembio.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DR, Zenklusen D, Wu B, Chao JA, Singer RH. Real-Time Observation of Transcription Initiation and Elongation on an Endogenous Yeast Gene. Science. 2011;332(6028):475–478. doi: 10.1126/science.1202142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding I, Paulsson J, Zawilski S, Cox E. Real-time kinetics of gene activity in individual bacteria. Cell. 2005;123(6):1025–1061. doi: 10.1016/j.cell.2005.09.031. [DOI] [PubMed] [Google Scholar]
- Suter DM, Molina N, Gatfield D, Schneider K, Schibler U, Naef F. Mammalian Genes Are Transcribed with Widely Different Bursting Kinetics. Science. 2011;332(6028):472–474. doi: 10.1126/science.1198817. [DOI] [PubMed] [Google Scholar]
- Cookson NA, Cookson SW, Tsimring LS, Hasty J. Cell cycle-dependent variations in protein concentration. Nucleic Acids Research. 2010;38(8):2676–2681. doi: 10.1093/nar/gkp1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zopf CJ, Quinn K, Zeidman J, Maheshri N. Cell-cycle dependence of transcription dominates noise in gene expression. 2013. Submitted. [DOI] [PMC free article] [PubMed]
- Kaufmann BB, Yang Q, Mettetal JT, Van Oudenaarden A. Heritable stochastic switching revealed by single-cell genealogy. PLoS Biology. 2007;5(9):e239. doi: 10.1371/journal.pbio.0050239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson S, Ostroff N, Pang WL, Volfson D, Hasty J. Monitoring dynamics of single-cell gene expression over multiple cell cycles. Molecular Systems Biology. 2005;1(1):2005.0024. doi: 10.1038/msb4100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliwal S, Iglesias PA, Campbell K, Hilioti Z, Groisman A, Levchenko A. MAPK-mediated bimodal gene expression and adaptive gradient sensing in yeast. Nature. 2007;446(7131):46–51. doi: 10.1038/nature05561. [DOI] [PubMed] [Google Scholar]
- Charvin G, Cross FR, Siggia ED. A microfluidic device for temporally controlled gene expression and long-term fluorescent imaging in unperturbed dividing yeast cells. PloS One. 2008;3(1):e1468. doi: 10.1371/journal.pone.0001468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon A, Colman-Lerner A, Chin TE, Benjamin KR, Yu RC, Brent R. Single-cell quantification of molecules and rates using open-source microscope-based cytometry. Nat. Meth. 2007;4(2):175–181. doi: 10.1038/nmeth1008. [DOI] [PubMed] [Google Scholar]
- Otsu N. A Threshold Selection Method from Gray-Level Histograms. IEEE Trans. Sys. Man. Cyber. 1979;9(1):62–66. [Google Scholar]
- Goranov AI, Cook M, et al. The rate of cell growth is governed by cell cycle stage. Genes & Development. 2009;23(12):1408–1422. doi: 10.1101/gad.1777309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To T-L, Maheshri N. Noise Can Induce Bimodality in Positive Transcriptional Feedback Loops Without Bistability. Science. 2010;327(5969):1142–1145. doi: 10.1126/science.1178962. [DOI] [PubMed] [Google Scholar]
- O'Neill EM, Kaffman A, Jolly ER, O'Shea EK. Regulation of PHO4 nuclear localization by the PHO80-PHO85 cyclin-CDK complex. Science. 1996;271(5246):209–212. doi: 10.1126/science.271.5246.209. [DOI] [PubMed] [Google Scholar]
- Raser JM, O'Shea EK. Control of stochasticity in eukaryotic gene expression. Science. 2004;304(5678):1811–1814. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]