

Abstract
Background
Long QT syndrome (LQTS) is the most common cardiac channelopathy with 15 elucidated LQTS-susceptibility genes. Approximately 20% of LQTS cases remain genetically elusive.
Methods and Results
We combined whole exome sequencing (WES) and bioinformatic/systems biology to identify the pathogenic substrate responsible for non-syndromic, genotype-negative, autosomal dominant LQTS in a multigenerational pedigree and established the spectrum and prevalence of variants in the elucidated gene among a cohort of 102 unrelated patients with “genotype-negative/phenotype-positive” LQTS. WES was utilized on three members within a genotype-negative/phenotype-positive family. Genomic triangulation combined with bioinformatic tools and ranking algorithms led to the identification of a CACNA1C mutation. This mutation, Pro857Arg-CACNA1C, co-segregated with the disease within the pedigree, was ranked by three disease-network algorithms as the most probable LQTS-susceptibility gene, and involves a conserved residue localizing to the PEST domain in the II–III linker. Functional studies reveal that Pro857Arg-CACNA1C leads to a gain-of-function with increased ICa,L and increased surface membrane expression of the channel compared to wildtype. Subsequent mutational analysis identified 3 additional variants within CACNA1C in our cohort of 102 unrelated cases of genotype-negative/phenotype-positive LQTS. Two of these variants also involve conserved residues within Cav1.2’s PEST domain.
Conclusions
This study provides evidence that coupling WES and bioinformatic/systems biology is an effective strategy for the identification of potential disease causing genes/mutations. The identification of a functional CACNA1C mutation co-segregating with disease in a single pedigree suggests that CACNA1C perturbations may underlie autosomal dominant LQTS in the absence of Timothy syndrome.
Keywords: arrhythmia, calcium, genetics, ion channel, long QT syndrome
Introduction
In the United States, sudden cardiac arrest (SCA) secondary to ventricular fibrillation (VF) accounts for over 300,000 deaths annually, including thousands of individuals younger than 35 years of age who die suddenly and unexpectedly.1, 2 VF stemming from congenital long QT syndrome (LQTS) is a leading cause for autopsy-negative sudden unexplained death.3
LQTS has a prevalence as high as 1 in 2000 persons4 and is most often characterized as an autosomal dominant inherited disorder of myocardial repolarization, defined by heart rate-corrected QT interval (QTc) prolongation on resting electrocardiogram and an increased predilection for syncope, seizures, and SCA in the absence of organic heart disease or other non-cardiac pathology (i.e. skeletal myopathy, dysmorphic features, or mental retardation).5 However, extremely rare, multi-system “syndromic” disorders associated with prolonged QT intervals, like Anderson-Tawil syndrome (ATS)6, 7 and Timothy syndrome (TS),8–11 have also been described.
To date, 15 LQTS-susceptibility genes have been discovered, with 75% of clinically robust LQTS attributed to mutations in three genes that encode for critical potassium (KCNQ1, 30% to 35%, LQT1; KCNH2, 25% to 30%, LQT2) or sodium (SCN5A, LQT3, ~ 5% to 10%) ion channel α-subunits largely responsible for the cardiac action potential12–14. The remaining LQTS-susceptibility genes (LQT4-15) encode for either cardiac channels, channel interacting proteins, or structural membrane scaffolding proteins that modulate channel function, and collectively contribute to < 5% of LQTS15, 16. Consequently, 20% of patients with a clinically strong diagnosis of LQTS remain genetically elusive and are labeled as “genotype-negative” LQTS.
With exception of the three major LQTS genes originally discovered in the mid 1990’s following multi-generational whole genome familial linkage studies and positional cloning, the majority of the minor LQTS-susceptibility genes have been discovered using a biological plausible, candidate gene approach. Major technological advances in DNA sequencing have emerged recently allowing for rapid whole genome or whole exome interrogation of patient samples for the identification of novel pathogenic mutations. In fact, several recent reports have utilized whole exome sequencing (WES) approaches targeting a “trio” of affected and/or unaffected members within a pedigree, to discover novel genetic substrates for a variety of non-cardiac, heritable diseases.17–20 In this study, we performed a WES “trio” analysis approach on a large multi-generational, “genotype-negative” LQTS pedigree to identify a novel cause for classical, “non-syndromic”, autosomal dominant LQTS followed by mutational analysis of the newly discovered genetic substrate in a large cohort of unrelated patients with robust clinical evidence for LQTS but a heretofore negative genetic test (i.e. genotype-negative/phenotype-positive-LQTS).
Methods
Study Subjects
A 15–member (8 affected, 5 unaffected, 2 unknown) multigenerational family, presenting with autosomal dominant inherited LQTS without syndactyly, cognitive impairments, facial dysmorphisms, or any other non-cardiac clinical characteristics suggestive of Timothy syndrome, (Figure 1A and Table 1) that remained genotype-negative following commercially available LQTS genetic testing, was referred to the Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory for further research-based genetic testing. Following written consent for this IRB-approved study, medical records, including 12-lead surface electrocardiograms, and peripheral blood lymphocytes were obtained for 12 family members. Genomic DNA was obtained using the Puregene DNA Isolation Kit (Qiagen, Inc, Valencia, CA). The symptomatic index case (QTc = 498 ms), unaffected father (QTc = 383 ms), and an affected maternal aunt (QTc = 479 ms) were selected for WES.
Figure 1.

Whole Exome Sequencing and Familial Genomic Triangulation for the Elucidation of a Novel Genetic Substrate for LQTS. (A) Black circles/squares are affected, grey are borderline, and white are unaffected with LQTS. Arrow identifies the proband. Asterisks represent those who were whole exome sequenced. Numbers in each circle/square represent QTc values (milliseconds) when available. Plus signs represent Pro857Arg-CACNA1C mutation positive family members. Minus signs represent mutation negative family members. NA denotes where DNA samples were not available. (B) Lead II of the electrocardiogram for the index case (III.2). The full electrocardiogram is available in Supplemental Figure 1A. (C) Schematic representation of our sequencing strategy. (D) DNA sequencing chromatogram showing the Pro857Arg mutation.
Table 1.
Summary of Pedigree as Shown in Figure 1
| PATIENT | SYMPTOMS | QTc (ms) | THERAPY | Pro857Arg STATUS |
|---|---|---|---|---|
| I.1 | Died in late 60s during heart surgery | NA | NA | NA |
| I.2 | Status Unknown | NA | NA | NA |
| II.1† | Asymptomatic | 383 | None | − |
| II.2 | Asymptomatic | 486 | None | + |
| II.3 | Out of hospital cardiac arrest | NA | ICD Implantation | + |
| II.4 | Died during infancy | NA | NA | NA |
| II.5† | Syncope during pregnancy | 479 | None | + |
| II.6 | Asymptomatic | 454 | None | + |
| II.7 | Asymptomatic | 421 | None | − |
| III.1 | Asymptomatic | 406 | None | − |
| III.2*,† | Postpartum agonal breathing, syncope | 498 | ICD Implantation, sympathetic denervation | + |
| III.3 | Asymptomatic | 415 | None | − |
| IV.1 | Asymptomatic | 450 | Prophylactic beta blocker therapy | + |
| IV.2 | Asymptomatic | 455 | None | + |
| IV.3 | Asymptomatic | NA | None | − |
Index Case
Whole Exome Sequenced
NA Not available
+ Pro857Arg present
− Pro857Arg absent
QTc is the heart rate corrected QT interval and is shown in milliseconds
In addition, 102 unrelated patients (71 females, 98 % Caucasian, average age at diagnosis = 23 ± 16 years, and an average QTc of 516ms ± 6.6 S.E.M.) with robust clinical evidence for LQTS (QTc ≥ 480 ms and/or a Schwartz-Moss21 score ≥ 3.5) that were referred to our laboratory previously for genetic testing were included (see Table 2 for cohort demographics). All patients signed written consent for this IRB-approved study. All 102 patients were mutation negative following LQTS mutational analysis (by DHPLC and DNA sequencing) of the three major LQTS genes: KCNQ1, KCNH2, and SCN5A and eight minor LQTS genes: AKAP9, ANKB, CAV3, KCNE1, KCNE2, KCNJ2, SCN4B, and SNTA1. In addition, all patients were negative for large KCNQ1 and KCNH2 gene rearrangements (whole single or multiple exon deletions/duplications) following gene-specific copy number variation analysis using multiplex ligation-dependent probe amplification technique.
Table 2.
Demographics of the LQTS Genotype-negative/Phenotype Positive Cohort
| LQTS Genotype-negative/Phenotype-positive Cohort (N=102) | |
|---|---|
| Age at Diagnosis (yrs) ± standard deviation | 23 ± 16 |
| Sex (male/female) | 31/71 |
| Caucasian | 98% |
| QTc (ms) ± S.E.M. | 516 ± 6.6 |
| Syncope | 47% |
| Cardiac Arrest | 22% |
| Family History of Sudden Cardiac Death | 51% |
Whole Exome Sequencing (WES)
WES and subsequent variant annotation was performed on genomic DNA derived from the symptomatic index case (III.2, Figure 1A and Table 1, ECG Supplemental Figure 1A), unaffected father (II.1, Figure 1A and Table 1, ECG Supplemental Figure 1B), and affected maternal aunt (II.5, Figure 1A and Table 1, ECG Supplemental Figure 1C) by the Mayo Clinic Advanced Genomics Technology Center and Bioinformatics Core facilities. Paired-end indexed libraries were prepared using the manufacturer’s protocol (Agilent, Santa Clara, CA). The whole exome capture was carried out using SureSelect Human All Exon 50MB kit (Agilent; Santa Clara, CA). The flow cells were sequenced as 101x2 paired end reads on an Illumina HiSeq 2000 using TruSeq SBS sequencing kit version 3 and HiSeq data collection version 1.4.8 software (Illumina, Inc., San Diego, CA). Base-calling was performed using Illumina’s RTA version 1.12.4.2 (Illumina, Inc., San Diego, CA).
The sequence data was processed for alignment, variant calling and annotation was completed using Targeted RE-sequencing Annotation Tool (TREAT).22 The TREAT annotation included alignment to the human genome 37 (hg19) using BWA,23 duplicate read removal using PICARD (http://picard.sourceforge.net/) and GATK24 for local INDEL realignment and base quality score recalibration of aligned reads. Single nucleotide variants (SNVs) and insertion/deletions (INDELs) were then called using SNVMix25 and GATK24 respectively. The SNVs and INDEL annotations provided by TREAT analysis were then filtered and summarized using custom Perl and shell scripts. The custom scripts intersected the SNVs of the 2 samples in the affected group (affected index case [III.2] and affected maternal aunt [II.5]) and intersected the SNVs of the sample in the unaffected case (unaffected father [II.1]). The SNVs in the unaffected group were then subtracted from the SNVs in the affected group to identify the novel SNVs shared by the symptomatic index case and affected maternal aunt. SNVs in the affected samples at sites with less than 5x coverage and variants present in either dbSNP,26 the 1000 Genomes Project,27 (http://browser.1000genomes.org/index.html) or the 200 BGI28 (Beijing Genomics Institute) Danish exomes were excluded (See Supplemental Table 1).
Systems Biology-Aided Exome Filtering
The LQTS interactome,29 which features 1629 genes of the human genome that are considered to be the most likely genes that encode proteins which influence or regulate cardiac repolarization, thus representing possible candidate genes for LQTS, was utilized to determine which genes (harboring variants), shared between the index case and affected maternal aunt, could be potential LQTS candidates. Next, three systems biology ranking algorithms: Endeavor30 (http://homes.esat.kuleuven.be/~bioiuser/endeavour/tool/endeavourweb/php), SUSPECTS31 (http://www.genetics.med.ed.ac.uk.suspects/), and ToppGene32 (http://toppgene.cchmc.org/prioritization.jsp), were used to rank the putative disease causative genes and were accessed publicly through the internet. Default prioritization parameters were used for each algorithm. The three canonical LQTS-disease causing genes: KCNQ1, KCNH2, and SCN5A were used as the three algorithms’ training genes to rank identified mutation carrying genes. Subsequently, the rare, shared variants that localized within the published LQTS interactome: CACNA1C, DMRTB1, DSCAML1, GLTSCR, SLC26A1, MAP1A, MARCKS and PPP1R12C (listed alphabetically here) were prioritized in each of these algorithms.
DNA Sanger Sequencing for Variant Confirmation and Co-Segregation Analysis
Following systems biology-aided exome filtering, all 8 candidate variants (CACNA1C (NM_000719) c.2570C>G, p.Pro857Arg; DMRTB1 (NM_033067) c.248_252dupCCGCCC, p.Ala83_Ala84dup; DSCAML1 (NM_020692) c.6307G>A, p.Ala2103Thr; GLTSCR1 (NM_015711) c.91A>G, p.Ser31Gly; SLC26A1 (NM_00022042) c.2007C>G, p.Asp669Glu; MAP1A (NM_002373) c.2842G>A, p.Val948Ile; MARCKS (NM_002356) c.552_557dupTGAGGC, p.Glu185_Ala186dup) and PPP1R12C (NM_017607) c.188A>G, p.Asp83Gly) identified by WES were confirmed and pedigree analysis for LQTS phenotype/variant co-segregation in the multigenerational pedigree was completed independently for all 8 variants using standard DNA dye terminator cycle sequencing protocols and an ABI Prism 377 automated sequencer (Applied Biosystems Inc., Foster City, CA). DNA sequence chromatograms were analyzed using Chromas version 1.45 (Queensland, Australia).
Cell Culture and Transfection
HEK293 cells were transfected with plasmids encoding either WT- or Pro857Arg-Cav1.2 with YFP fused at the N-terminus (Antzelevitch et al. 200733), β2cN4, and α2δ1 in a 1:1:1 molar ration using Lipofectamine 2000 and cultured in DMEM + 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C + 5% CO2 as previously described (Best et al. 201134). HEK293 cell experiments were performed 40–48 hours post-transfection.
Electrophysiology
Whole cell ruptured patch clamp experiments were performed in HEK293 cells at 21–24°C. The pipette solution consisted of (in mM) 114 CsCl, 10 EGTA, 10 HEPES, 5 Mg-ATP, and 0.1 Li-GTP (to pH 7.2 with CsOH). The recording solution consisted of (in mM) 147 CsCl, 5 CaCl2, and 10 HEPES (to pH 7.4 with CsOH). Upon electrical access, capacitive transients were obtained by applying a 5-mV hyperpolarizing pulse. Whole cell Ca2+ currents (ICa,L) were elicited at 10 mV increments from a holding potential of −80 mV and recorded using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). Data were sampled at 25 kHz and filtered at 5 kHz, and leak and capacitive currents were subtracted using a P/4 protocol. Data are presented as current density by normalizing ICa,L to cell capacitance. Data are plotted in Figure 2 as mean ± SEM and significance at each voltage (defined as P <0.05) was determined by Student’s t-test using Origin 7.5 software (OriginLab, Northhampton, MA).
Figure 2.

Functional Analysis of Pro857Arg-Cav1.2 Mutation Using Whole Cell Patch Clamp Technique. (A) Representative ICa,L traces recorded from HEK293 cells expressing WT- or Pro857Arg-Cav1.2 along with auxiliary β2cN4 and α2δ1 subunits. (B) Current-voltage (I–V) relations constructed from WT-Cav1.2 (■, n = 10) or Pro857Arg-Cav1.2 expressing cells (●, n = 10). Data were compared using Student’s unpaired t-test and * denotes p < 0.05 when WT-Cav1.2 was compared with Pro857Arg-Cav1.2.
Cell Surface Biotinylation
HEK293 cells grown in 10 cm culture dishes were cooled to 4°C, washed three times in ice-cold PBS, and biotinylated for 30 minutes with 0.5–1.0 mg/ml EZ Link Sulfo-NHS-LC-Biotin (Thermo Scientific Pierce Protein Research Products, Rockford, IL) at 4°C. Cells were then washed twice in ice-cold Tris-buffered saline (TBS) followed by an additional wash in PBS to remove biotinylation reagent. Cells were harvested directly by scraping and lysed in buffer containing 150 mM NaCl, 20 mM Tris-HCl (pH 7.4), 1% Triton X-100, 0.5 deoxycholic acid, and protease inhibitor cocktail containing 2 mM phenylmethylsulfonyl fluoride, 50 μg/ml aprotinin, 50 μg/ml benzamidin, 50 μg/ml leupeptin, and 5 μM pepstatin A. For capture of cell surface biotinylated proteins, 0.5–1.0 mg soluble protein lysate was incubated with 75 μl High Capacity NeutrAvidin agarose beads (Thermo Scientific Pierce Protein Research Products) for 1–2 hours at 4°C. NeutrAvidin beads were washed five times and proteins were eluted by heating for 5 minutes at 95°C with SDS-PAGE sample buffer.
SDS-PAGE and Western blotting
Protein samples were separated using SDS-PAGE and transferred to polyvinylidene difluoride membranes. Nonspecific binding sites were blocked using 5% (wt/vol) dried skim milk in TBS with 0.05% Tween-20 (TBST). Membranes were probed with the following antibodies: rabbit anti-Cav1.2 (#AAC-003, Alomone Labs, Jerusalem, Israel), mouse anti-transferrin receptor (clone H68.4, Invitrogen), and rabbit anti-GAPDH (#sc-25778, Santa Cruz Biotechnology, Santa Cruz, CA) overnight at room temperature. Membranes were washed subsequently four times for 10 minutes before incubation in appropriate horseradish peroxidase-conjugated secondary antibody for one hour. After additional washes in TBST, immunoreactivity was visualized using ECL Plus (GE Healthcare Life Sciences, Pittsburgh, PA). Membrane stripping was performed at 55°C for 30 minutes in buffer containing 62.5 mM Tris-HCl, pH 6.8, 2% SDS, 100 mM β–mercaptoethanol, and 100 mM DTT. Densitometric analysis was achieved using ImageJ (NIH, Bethesda, MD).
CACNA1C Mutational Analysis
Comprehensive open reading frame and splice junction mutational analysis of the entire coding region (57 amplicons that encode 2139 amino acids) of CACNA1C (GenBank accession number NM_000719) was performed on genomic DNA derived from 102 unrelated LQTS diagnosed patients (described above) using PCR, denaturing high performance liquid chromatography (DHPLC; WAVE DNA Fragment Analysis System, Transgenomic Inc., Omaha, NE), and DNA sequencing (ABI Prism 377; Applied Biosystems Inc., Foster City, CA). Primer Sequences, PCR conditions, and DHPLC conditions are available upon request.
To be considered as a potential LQTS-causing variant, the CACNA1C variant had to i) be a non-synonymous (substitution of a nucleotide which leads to a residue change) variant (i.e. synonymous single-nucleotide polymorphisms were excluded), ii) involve a highly conserved residue, iii) be absent in at least 680 ethnically-matched “in house” controls (1360 reference alleles) and all publicly available databases including the 1000 Genomes Project27 (n=1094 subjects; 381 Caucasian, 246 African-American, 286 Asians, and 181 Hispanics) the NHLBI GO Exome Sequencing Project35 (n=5379 subjects; 3510 Caucasians and 1869 African-Americans) (http://evs.gs.washington.edu/EVS/), and the 12000 Exome Chip36 (n=12000 subjects) (http://genome.sph.umich.edu/wiki/Exome_Chip_Design). Control genomic DNA was obtained from the European Collection of Cell Cultures (HPA Culture Collections, UK), the Human Genetic Cell Repository sponsored by the National Institute of General Medical Sciences and the Coriell Institute for Medical Research (Camden, New Jersey).
Statistical Analysis
Data in Figures 2 and 3 are plotted as mean ± SEM and significance (defined as p ≤ 0.05) was determined by Student’s t-test using Origin 7.5 software (OriginLab, Northhampton, MA). The normality was confirmed for each t-test using the Sharpiro-Wilk test.
Figure 3.
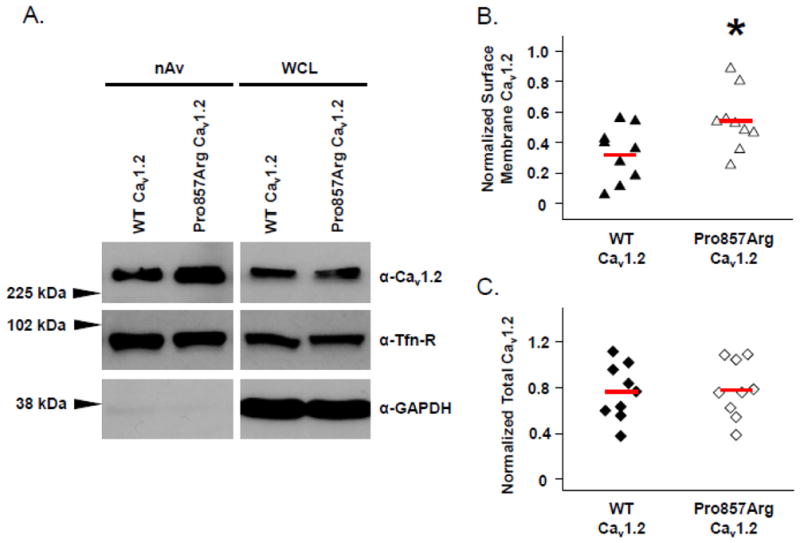
Increased Surface Membrane Density of Cav1.2 Pro857Arg Channels. HEK293 cells expressing either YFP-tagged Cav1.2 WT or Cav1.2-Pro857Arg along with auxiliary β2cN4 and α2δ1 subunits were cell surface biotinylated at 4°C. (A) NeutrAvidin (nAv)-captured surface membrane proteins (left panels) and whole cell lysates (WCL, right panels) were analyzed by SDS-PAGE and western blotting. Densitometric analysis of surface membrane Cav1.2 signal (B) or total cellular Cav1.2 (C) was performed to summarize the results of nine independent experiments. In both cases, normalization of Cav1.2 signal was achieved using the corresponding signal of the endogenous surface membrane protein transferrin receptor (Tfn-R). Solid red lines through distributions indicate population means. Data were compared using Student’s unpaired t-test and * denotes p ≤ 0.05 when WT-Cav1.2 was compared with Pro857Arg-Cav1.2.
Results
Genotype-negative/Phenotype-positive Autosomal Dominant LQTS Pedigree
The index case is a 33-year-old Caucasian female (III.2 Figure 1A and Table 1) who presented at the age of 27 years with a postpartum agonal breathing event. Subsequent medical history revealed multiple startle-triggered and exercise-induced syncopal events starting at the age of 13 years. Her electrocardiogram revealed a prolonged QTc of 498 ms (Figure 1B and Supplemental Figure 1A). Her medical therapy included ICD placement and a left cardiac sympathetic denervation. Due to complications, she had a brief period in which her ICD was removed. Subsequent to ICD removal, she had one more event while sleeping that included shaking and agonal breathing, from which she spontaneously recovered. Following this event, an ICD was re-implanted. Her family history is positive for LQTS and a sudden unexplained infant death.
Screening electrocardiograms were performed on 11 of the 12 available family members, and genomic samples were gathered from 12 family members. Based on the ECG findings, 7 family members have borderline/prolonged QTc and/or a significant history of cardiac events. The 5 family members with normal ECGs were asymptomatic (Figure 1A and Table 1).
Overall, the family history and inheritance pattern indicated autosomal dominant LQTS. One of the index case’s maternal aunts, II.3, suffered out of hospital cardiac arrest at the age of 44 which resulted in significant neurological damage. A second maternal aunt, II.4, died during infancy of unknown cause. A third maternal aunt, II.5, suffered one syncopal event during pregnancy (QTc = 479 ms, ECG available Supplemental Figure 1C). The index case’s daughter, IV.1, has been treated with prophylactic beta block therapy since the age of 8 years (QTc = 450 ms). The index case’s asymptomatic mother, II.2, has a prolonged QTc of 486 ms. The electrocardiogram of the index case’s asymptomatic maternal uncle, II.6, shows sinus bradycardia, early repolarization, and a QTc of 454 ms (Table 1).
Whole Exome Sequencing (WES) for Novel Pathogenic Substrate Identification in a Large Multigenerational LQTS Pedigree
Genomic DNA samples of the “trio” of participants in the pedigree, index case (III.2), her affected maternal aunt (II.5), and unaffected father (II.1), were analyzed by WES. Between the three samples, approximately 98% of the reads were mapped, and approximately 44% of the reads were within the target region (Supplemental Table 1).
The detected single nucleotide variations (SNVs) and insertion/deletions (INDELs) were configured in a spreadsheet so subsequent filtering strategies could be utilized to identify potential pathogenic variants. After alignment, and SNV calling, 46,307 genetic variants were present in the exome of the index case (Supplemental Table 1). Following exclusion of “common” variants present in 1000 Genomes Project,27 dbSNP,26 and 200 Beijing Genomics Institute (BGI) exomes28 as well as all synonymous SNVs, 263 variants remained (Supplemental Table 1). Variants shared between the index case (III.2) and the unaffected father (II.1) and unshared variants between the index case (III.2) and the affected maternal aunt (II.5) were subtracted leaving 110 rare variants present in both the index case and the maternal aunt (data not shown).
Of these 110 remaining variants, only 8 variants, 6 SNVs and 2 InDels, (CACNA1C c.2570C>G, p.Pro857Arg; DMRTB1 c.248_252dupCCGCCC, p.Ala83_Ala84dup; DSCAML1 c.6307G>A, p.Ala2103Thr; GLTSCR1 c.91A>G, p.Ser31Gly; SLC26A1 c.2007C>G, p.Asp669Glu; MAP1A c.2842G>A, p.Val948Ile; MARCKS c.552_557dupTGAGGC, p.Glu185_Ala186dup) and PPP1R12C c.188A>G, p.Asp83Gly) resided in genes that were present in the previously published 1629 node LQTS interactome29, suggesting these 8 variants as the best candidates for pathogenicity in this multi-generational LQTS pedigree.
Candidate gene priority ranking using three in silico bioinformatic/systems biology algorithms (ToppGene32, Endeavour30, and Suspects31) with KCNQ1, KCNH2, and SCN5A serving as the network training genes, unanimously ranked the L-type calcium channel encoded by CACNA1C as the number one LQTS-causing candidate gene, followed by SLC26A1, MAP1A, DMRTB1, DSCAML1, GLTSCR1, PPP1R12C, and then MARCKS (Table 3). In addition, none of the 102 shared variants that resided outside the LQTS interactome scored higher than CACNA1C with any of the in silico tools (data not shown).
Table 3.
Gene Ranking Using Bioinformatic/Systems Biology Algorithms
In addition to the in silico analysis which ranked CACNA1C unanimously as the most likely LQTS-associated candidate gene within the exomes of these patients, we analyzed the genomic DNA from all 12 family members (7 affected and 5 unaffected) for the presence/absence of all 8 variants. Consistent with the in silico prediction, Pro857Arg-CACNA1C was the only variant with complete co-segregation with the LQTS phenotype (i.e. present in all affected and absent in all unaffected family members) in this 12 member multi-generational pedigree (Figure 1A, and Table 1).
The Pro857Arg-CACNA1C missense mutation is a single nucleotide genetic variant (c. 2570 C>G) resulting in the substitution of a neutral, non-polar amino acid (proline, pro) for a positively charged amino acid (arginine, arg) at position 857 of the L-type calcium channel (LTCC, also annotated as Cav1.2). This putative pathogenic mutation was absent among 1094 (1000 Genomes Project27), 5379 (NHLBI GO Exome Sequencing Project35), 12000 (12000 exome chip36), 200 individuals (200 Danish BGI exomes),28 and an additional 680 ethnically matched controls analyzed in our laboratory. Pro857Arg localizes to Cav1.2’s II–III linker that contains the critical PEST domain, a sequence motif rich in the amino acids proline [P], glutamic acid [E], serine [S], and threonine [T]) that provides as an important signal for rapid protein degradation and LTCC protein stability37, 38.
In Vitro Functional Analysis of the Pro857Arg-CACNA1C
To further understand the effect that Pro857Arg has on the CACNA1C channel, we used the ruptured whole cell patch clamp technique to determine if there were electrophysiological differences between Pro857Arg- and WT-CACNA1C currents. Functional characterization of the mutation utilizing whole cell patch clamp technique in a heterologous HEK293 expression system revealed a gain-of-function phenotype with a 113% increase in peak ICa,L at +10 mV compared to wildtype (Figure 2), which may lengthen the cardiac action potential and prolong the QT interval.
Because the Pro857Arg-CACNA1C mutation lies within a PEST domain, we hypothesized that the Pro857Arg mutation may impair the normal degradation of Cav1.2, thus leading to an increase in surface membrane channels and functional ICa,L. To test this, we performed cell surface biotinylation experiments using HEK293 cells transiently expressing WT-CACNA1C or Pro857Arg-CACNA1C along with auxiliary β2cN4 and α2δ1 subunits. Figure 3 shows a representative western blot demonstrating that the Pro857Arg mutation leads to an increase in NeutrAvidin-isolated (and thus plasma membrane localized) Cav1.2 signal compared to WT (Figure 3A, top left panel). Detection of endogenous transferrin receptor was included to show that expressing the Pro857Arg-CACNA1C mutant did not lead to a nonspecific increase in surface membrane proteins (Figure 3A, middle left panel). Densitometric analysis of nine independent experiments showed a mean 64% increase in surface membrane expression of Pro857Arg-CACNA1C relative to WT-CACNA1C (Figure 3B, P = 0.05). No significant difference was detected in the total cellular Cav1.2 signal between Pro857Arg and WT (Figure 3A, top right panel and 3C). Probing for the cytosolic protein GAPDH ensured that the biotinylation reagent did not significantly cross the cell membrane (Figure 3A, bottom panels).
Spectrum and Prevalence of CACNA1C Variants in a Large Cohort of Unrelated Patients with “Genotype-Negative/Phenotype-Positive” LQTS
Following comprehensive mutational analysis of CACNA1C, we identified 3 novel, putative pathogenic missense variants in 3/102 (2.9%) unrelated “genotype-negative/phenotype-positive” LQTS patients (Table 4 and Figure 4). Two of these three CACNA1C variant-positive patients were females. All 3 missense variants (c.2500A>G, p.Lys834Glu; c.2570C>T, p.Pro857Leu; and c.5717G>A, p.Arg1906Gln) were absent in 680 ethnically matched (Caucasian) controls, 1094 subjects from the 1000 Genomes Project27, 5379 subjects from the NHLBI GO Exome Sequencing Project35 and the 12000 Exome Chip.39 Interestingly, 2 of the missense variants (Lys834Glu and Pro857Leu) localize to the same critical PEST domain where the original mutation (Pro857Arg) discovered in the multigenerational LQTS pedigree resides (Figure 4). In fact, one of the variants (Pro857Leu) affects the same amino acid residue (Proline 857) as the LQTS-causing mutation identified in the pedigree. A third variant (Arg1906Gln) localizes very closely to the recently described STIM1 (stromal interaction molecule 1) binding domain (amino acid residues 1806 – 1905) in Cav1.2’s C-terminus, where STIM1 is a Ca2+ sensor of the endoplasmic reticulum, and interacts with the CACNA1C resulting in a decrease of LTCC mediated current as well as chronically triggers LTCC internalization, suppressing channel function.40–42 Importantly, like the affected subjects within the multigenerational pedigree, none of these 3 CACNA1C variant-positive subjects exhibit any clinical characteristics suggestive of Timothy syndrome.
Table 4.
Summary of CACNA1C Variants
| Case # | Nucleotide Change | Protein Change | Location in CACNA1C | Age (years) | Sex | QTc (ms) | Symptoms | Family History LQTS | Family History SCD |
|---|---|---|---|---|---|---|---|---|---|
| 1 | c.2500 A>G | p.Lys834Glu | II–III | 15 | F | 475 | Syncope | No | No |
| 2* | c.2570 C>G | p.Pro857Arg | II–III | 27 | F | 498 | Agonal breathing | Yes | No |
| 3 | c.2570 C>T | p.Pro857Leu | II–III | 15 | M | 514 | None | Yes | Yes |
| 4 | c.5717 G>A | p.Arg1906Gln | C-terminal | 39 | F | 513 | Syncope, palpitations | No | No |
Figure 4.

Topology Diagram of the CACNA1C Channel Alpha-Subunit. Each variant is represented as a black circle followed by the amino acid position and change. Asterisk represents the mutation identified by whole exome sequencing in the pedigree (Figure 1). Patient history is displayed for each variant. The PEST domain sequence is highlighted showing the position of the three variants residing within this domain as well as the species conservation. STIM1 binding region, 1806–1905 is shown in the C-terminal tail of CACNA1C. STIM1 interacts with the LTCC to normally suppress channel function 40, 41.
Instead, the Lys834Glu variant was identified in a 15-year-old Caucasian female with a personal history of syncope, QTc of 475 ms, and a negative family history. The Arg1906Gln variant was identified in a 39-year-old Caucasian female who presented with palpitations, syncope, a QTc of 513 ms, and had a negative family history.
Similar to the multigenerational pedigree that yielded our CACNA1C discovery with its Pro857Arg mutation, the Pro857Leu missense variant was identified in an asymptomatic 15-year-old Caucasian male who was diagnosed with LQTS (QTc of 514 ms) following the sudden unexplained death of his 12 year-old sister during sleep. His mother, maternal grandmother, maternal great uncle, and maternal great aunt all have a previous history of syncopal events during childhood. Genomic DNA was available for the 12-year-old sister’s unaffected father, affected mother, and affected maternal grandmother. Both the affected mother and affected maternal grandmother were positive for Pro857Leu while the unaffected father was negative (data not shown). DNA was not available for either the affected maternal great uncle or affected maternal great aunt.
Discussion
Whole exome sequencing (WES) is a highly efficient means of interrogating the DNA sequence of the entire protein-encoding region of virtually every gene in the human genome for a given patient sample in order to potentially elucidate the underlying genetic mechanism of disease. However, in doing so, an investigator is often faced with tens of thousands of genetic variants, per patient DNA sample, that must be sifted through in order to identify the possible disease-causative variant. In order to identify potential candidate genes, we utilized publicly available internet-based gene prioritization tools like ToppGene,32 Endeavour,30 and SUSPECTS31 to bioinformatically rank candidate genes20 by training network algorithms to search a user defined gene list and prioritize genes for disease candidacy based on similarities to a set of user defined “training” genes (i.e. known disease causing genes like KCNQ1, KCNH2, or SCN5A for LQTS). Specifically for LQTS analysis, the previously published 1629 LQTS interactome29 (encompassing a set of genes with the greatest potential to regulate cardiac repolarization) proved to be a very useful tool in further filtering variants down to a very manageable number of less than 10 candidates.
Here, our study is the first to utilize a next generation WES “trio” approach17 followed by “LQTS interactome”29 variant filtration and disease-network analysis based gene ranking prioritization to discover a novel putative LQTS-disease causative missense mutation, Pro857Arg-CACNA1C, that properly co-segregated with the LQTS phenotype in a large, multigenerational pedigree with autosomal dominant “genotype-negative/phenotype-positive” LQTS. In addition to co-segregation, this mutation produced a gain-of-function effect on heterologously expressed Pro857Arg Cav1.2 channels with a uniform increase in ICa,L density. The electrophysiological findings are consistent with the increase in surface membrane channels identified by biotinylation experiments. Because ICa,L contributes to phase 2 and 3 of the action potential, a scaled increase in this depolarizing current might increase action potential duration and the associated QT interval. Transgenic overexpression of Cav1.2 channels in the mouse heart has demonstrated an increase in action potential duration, but this is accompanied by complex electrical remodeling of multiple ionic currents as well as hypertrophy.43 Thus, the extensive feedback regulation of Cav1.2 channels and possible electrical remodeling of multiple currents in the heart limit the ability to precisely extrapolate the increase in ICa,L observed in the heterologous expression studies to native human heart.
CACNA1C encodes for the α-subunit of the L-type calcium channel (LTCC, Cav1.2) which is critical for cellular excitability and the cardiac action potential, excitation-contraction coupling, and regulation of gene expression.44, 45 Interestingly, three (Lys834Glu, Pro857Leu, and Pro587Arg) of the four variants discovered in our study localize to a key PEST domain of the II–III linker of the LTCC α-subunit. PEST sequences, rich in proline (P), glutamic acid (E), serine (S), and threonine (T), act to signal rapid protein degradation through the cell’s quality control system.37, 38 A recent study has demonstrated that Akt (also known as protein kinase B, PKB) regulates LTCC channel protein stability by preventing Cav1.2 PEST sequence recognition and hence promoting stability of the channel complex resulting in increased macroscopic currents.46
However, mechanisms regulating the membrane stability of the LTCC are understood poorly, and how the unmasked PEST sequence leads to channel proteolysis is unknown. Our functional studies revealed that the mutation, Pro857Arg-CACNA1C precipitates a 113% increase in peak ICa,L at +10 mV compared to wildtype. We have also shown that Pro857Arg leads to a 64% increase of surface membrane expression compared to wildtype. The combination of these findings lead to our hypothesis that Lys834Glu-, Pro857Leu- and Pro857Arg-Cav1.2 may disrupt the functional PEST sequence and lead to increased channel protein stability, increased number of surface membrane L-type calcium channels, as seen through biotinylation experiments, and increased ICa,L, as seen through our electrophysiological studies of Pro857Arg-CACNA1C. The increase in ICa,L might increase the action potential duration and contribute to the genesis of early after depolarizations and potentially Ca2+ overload related delayed after depolarizations, resulting in an LQTS phenotype with the propensity for potentially lethal arrhythmias.
The Arg1906Gln variant, resides one amino acid away from the Cav1.2 C-terminus STIM1 (stromal interaction molecule 1) binding domain. STIM1, the main activator of store-operated Ca2+ channels, directly suppresses depolarization-induced opening of the voltage-gated Ca2+ channel Cav1.2. STIM1 binds to the C-terminus of Cav1.2 through its Ca2+ release–activated Ca2+ activation domain, acutely inhibits gating, and causes long-term internalization of the channel from the membrane.40–42 Due to its proximity, Arg1906Gln could alter STIM1 binding to CACNA1C, preventing the internalization of the channel, leading to an increased number of channels on the membrane, and a gain-of-function in Cav1.2 currents. All of the identified variants (Pro857Arg, Pro857Leu, Lys834Glu and Arg1906Gln) appear to potentially involve channel internalization and protein degradation pathways. Further characterization of these variants may give a deeper understanding of CACNA1C protein regulation.
Interestingly, three unique CACNA1C gain-of-function missense mutations (Gly406Arg in CACNA1C’s alternative exon 8a, Gly406Arg in exon 8, and Gly402Ser in exon 8) have been described in Timothy syndrome (TS), an extremely rare (fewer than 20 people world-wide have been reported), multisystem disorder associated with extreme QT prolongation, syndactyly, autism spectrum disorder, and a myriad of other organ system abnormalities which often lead to early childhood death8–11. Importantly, none of the CACNA1C variant-positive individuals in the pedigree or the cohort study exhibit syndactyly, cognitive impairments, facial dysmorphisms, autism spectrum disorders or any other non-cardiac clinical characteristics suggestive of TS. Additionally a majority of the members within the pedigree harboring the Pro857Arg-CACNA1C mutation have survived into adulthood, which is extremely unlikely in patients with Timothy syndrome.
Although previously described in TS, no CACNA1C mutations have been reported for non-syndromic LQTS. With our functional studies confirming Cav1.2 gain-of-function as the cellular basis for the CACNA1C mutation-positive patient’s LQTS phenotype, a paradox has emerged. Namely, how can some gain-of-function mutations, like Gly406Arg in the alternatively spliced exon 8A, result in the multi-organ system phenotype of TS while other missense mutations, like Pro857Arg, yield a cardiac-only phenotype of autosomal dominant LQTS? With this being a new discovery, it is not surprising that the answer is elusive.
Several potential mechanisms have been proposed, but the most likely explanation is that CACNA1C is expressed as many different isoforms throughout the body. Therefore, variable splice isoforms, as well as differences in the corresponding accessory subunits (such as the β-subunit) vary between skeletal, cardiac, and neuronal tissues. In fact, a single nonsynonymous CACNA1C variant (Ala39Val), identified in patients with Brugada Syndrome, results in a trafficking defect of the channel that is apparent only in cells expressing the cardiac isoforms of CACAN1C, while being normal in the context of the neuronal rat isoform.47 Therefore, it is possible that the variants identified here will perturb only the cardiac isoforms of the channel or auxiliary subunits, and will have minimal or no effect on CACNA1C expressed in other tissues, thereby leading exclusively to cardiac disease.
Given that all unrelated TS patients described to date have either the same exact sporadic de novo Gly406Arg (16 cases) mutation in exon 8a, Gly406Arg in exon 8 (1 case) or a mutation residing only 4 amino acids away (Gly402Ser, 1 case), suggests that this specific amino acid residue or region of CACNA1C may have a unique functional effect on the LTCC in multiple organ systems, whereas mutations in other regions of CACNA1C may produce a cardiac-only phenotype. All three TS mutations lead to an impaired open-state voltage-dependent inactivation of the LTCC whereas Pro857Arg leads to increase in peak ICa,L current presumably through an increase in cell surface membrane expression of LTCCs without perturbing either channel kinetics or gating properties.
Limitations
Only one of the four identified CACNA1C variants have been characterized functionally thus far. Further functional studies will be necessary to definitively understand the pathogenic mechanisms underlying these CACNA1C variants and their implications in LQTS. HEK293 cells have specific current/kinetic properties and trafficking properties that may not be reflective of the properties of cardiomyoctes. Alternative approaches such as adenoviral transduction of primary cardiac myocytes or patient specific induced pluripotent stem cell (iPS)-derived cardiomyocytes may overcome some of the limitations of the heterologous expression system, but these approaches are beyond the scope of this study.
Conclusions
The combination of the 4 identified variants (absent in nearly 15,000 non-LQTS individuals) as well as familial co-segregation with disease for two variants (Pro857Arg and Pro857Leu) suggests that perturbations in the CACNA1C-encoded L-type calcium channel (LTCC) are bona fide pathogenic substrates for classical, non-syndromic, autosomal dominant LQTS with implications for LQTS genetic testing. If this frequency is validated in subsequent studies, CACNA1C may fall after KCNQ1, KCNH2, SCN5A, and KCNE1 and account for approximately 1% of LQTS.
Supplementary Material
Acknowledgments
We would like to acknowledge the services of the Medical Genome Facility, supported by the Mayo Center for Individualized Medicine.
Funding Sources: This work was supported by NIH PPG HL94291 (M.J.A and T.J.K) and by NIH R01 HL078878 (T.J.K). M.J.A is supported by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program. N.J.B is supported by an individual PhD predoctoral fellowship from the American Heart Association (12PRE11340005). J.R.G is supported by an individual MD/PhD predoctoral fellowship from the National Institutes of Health (F30-HL106993).
Footnotes
Conflict of Interest Disclosures: M.J.A is a consultant for Transgenomic. Intellectual Property derived from M.J.A’s research program resulted in license agreements in 2004 between Mayo Clinic Health Solutions (formerly Mayo Medical Ventures) and PGxHealth (formerly Genaissance Pharmaceuticals and now Transgenomic). However, Transgenomic did not contribute directly to this study in any manner. NJB, JMB, DJT, JRG, SM, JME, TJK and MJA have no conflicts of interest.
References
- 1.Liberthson RR. Sudden death from cardiac causes in children and young adults. N Engl J Med. 1996;334:1039–1044. doi: 10.1056/NEJM199604183341607. [DOI] [PubMed] [Google Scholar]
- 2.Chugh SS, Reinier K, Teodorescu C, Evanado A, Kehr E, Al Samara M, et al. Epidemiology of sudden cardiac death: Clinical and research implications. Prog Cardiovasc Dis. 2008;51:213–228. doi: 10.1016/j.pcad.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tester DJ, Ackerman MJ. Postmortem long qt syndrome genetic testing for sudden unexplained death in the young. J Am Coll Cardiol. 2007;49:240–246. doi: 10.1016/j.jacc.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-qt syndrome. Circulation. 2009;120:1761–1767. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moss AJ, Kass RS. Long qt syndrome: From channels to cardiac arrhythmias. J Clin Invest. 2005;115:2018–2024. doi: 10.1172/JCI25537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tristani-Firouzi M, Etheridge SP. Kir 2.1 channelopathies: The andersen-tawil syndrome. Pflugers Arch. 2010;460:289–294. doi: 10.1007/s00424-010-0820-6. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Benson DW, Tristani-Firouzi M, Ptacek LJ, Tawil R, Schwartz PJ, et al. Electrocardiographic features in andersen-tawil syndrome patients with kcnj2 mutations: Characteristic t-u-wave patterns predict the kcnj2 genotype. Circulation. 2005;111:2720–2726. doi: 10.1161/CIRCULATIONAHA.104.472498. [DOI] [PubMed] [Google Scholar]
- 8.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 9.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Severe arrhythmia disorder caused by cardiac l-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yarotskyy V, Gao G, Peterson BZ, Elmslie KS. The timothy syndrome mutation of cardiac cav1.2 (l-type) channels: Multiple altered gating mechanisms and pharmacological restoration of inactivation. J Physiol. 2009;587:551–565. doi: 10.1113/jphysiol.2008.161737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, et al. Long qt, syndactyly, joint contractures, stroke and novel cacna1c mutation: Expanding the spectrum of timothy syndrome. Am J Med Genet Part A. 2011;158A:182–187. doi: 10.1002/ajmg.a.34355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long qt syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 13.Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the familion® long qt syndrome genetic test. Heart Rhythm. 2011;6:1297–1303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Genetic testing in the long qt syndrome: Development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005;294:2975–2980. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 15.Gollob MH, Blier L, Brugada R, Champagne J, Chauhan V, Connors S, et al. Recommendations for the use of genetic testing in the clinical evaluation of inherited cardiac arrhythmias associated with sudden cardiac death: Canadian cardiovascular society/canadian heart rhythm society joint position paper. Canadian Journal of Cardiology. 2011;27:232–245. doi: 10.1016/j.cjca.2010.12.078. [DOI] [PubMed] [Google Scholar]
- 16.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. Hrs/ehra expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the heart rhythm society (hrs) and the european heart rhythm association (ehra) Heart Rhythm. 2011;8:1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 17.Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 18.Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in bag3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–282. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szperl AM, Ricano-Ponce I, Li JK, Deelen P, Kanterakis A, Plagnol V, et al. Exome sequencing in a family segregating for celiac disease. Clin Genet. 2011;80:138–147. doi: 10.1111/j.1399-0004.2011.01714.x. [DOI] [PubMed] [Google Scholar]
- 20.Erlich Y, Edvardson S, Hodges E, Zenvirt S, Thekkat P, Shaag A, et al. Exome sequencing and disease-network analysis of a single family implicate a mutation in kif1a in hereditary spastic paraparesis. Genome Res. 2011;21:658–664. doi: 10.1101/gr.117143.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crotti L, Celano G, Dagradi F, Schwartz PJ. Congenital long qt syndrome. Orphanet J Rare Dis. 2008;3:18. doi: 10.1186/1750-1172-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asmann YW, Middha S, Hossain A, Baheti S, Li Y, Chai HS, et al. Treat: A bioinformatics tool for variant annotations and visualizations in targeted and exome sequencing data. Bioinformatics. 2011;28:277–278. doi: 10.1093/bioinformatics/btr612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goya R, Sun MGF, Morin RD, Leung G, Ha G, Wiegand KC, et al. Snvmix: Predicting single nucleotide variants from next-generation sequencing of tumors. Bioinformatics. 2010;26:730–736. doi: 10.1093/bioinformatics/btq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smigielski EM, Sirotkin K, Ward M, Sherry ST. Dbsnp: A database of single nucleotide polymorphisms. Nucl Acids Res. 2000;28:352–355. doi: 10.1093/nar/28.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Via M, Gignoux C, Burchard E. The 1000 genomes project: New opportunities for research and social challenges. Genome Med. 2010;2:3. doi: 10.1186/gm124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 29.Berger SI, Ma’ayan A, Iyengar R. Systems pharmacology of arrhythmias. Sci Signal. 2010;3:ra30. doi: 10.1126/scisignal.2000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aerts A, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, et al. Gene prioritization through genomic data fusion. Nat Biotechnol. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- 31.Adie EJ, Adams RR, Evans KL, Porteous DJ, Pickard BS. Speeding disease gene discovery by sequence based candidate prioritization. BMC Bioinformatics. 2005;6:55. doi: 10.1186/1471-2105-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J, Xu H, Aronow BJ, Jegga AG. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics. 2007;8:392. doi: 10.1186/1471-2105-8-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by st-segment elevation, short qt intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Best JM, Foell JD, Buss CR, Delisle BP, Balijepalli RC, January CT, et al. Small gtpase rab11b regulates degradation of surface membrane l-type cav1.2 channels. Am J Physiol Cell Physiol. 2011;300:C1023–1033. doi: 10.1152/ajpcell.00288.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Exome Variant Server NESPE. Seattle, WA: [Date of Access [August 2012]]. URL: http://evs.gs.washington.edu/EVS/ [Google Scholar]
- 36.Abecasis G, Neale B. [Date of Access [August 2012]];Exome chip design. 2011 URL: Http://genome.Sph.Umich.Edu/wiki/exome_chip_design.
- 37.Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: The pest hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 38.Rechsteiner M, Rogers S. Pest sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:261–271. [PubMed] [Google Scholar]
- 39.Abecasis G, Neale B. Exome chip design. 2011. [Google Scholar]
- 40.Park CY, Shcheglovitov A, Dolmetsch R. The crac channel activator stim1 binds and inhibits l-type voltage-gated calcium channels. Science. 2010;330:101–105. doi: 10.1126/science.1191027. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, et al. The calcium store sensor, stim1, reciprocally controls orai and cav1.2 channels. Science. 2010;330:105–109. doi: 10.1126/science.1191086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuccotti A, Clementi S, Reinbothe T, Torrente A, Vandael DH, Pirone A. Structural and functional differences between l-type calcium channels: Crucial issues for future selective targeting. Trends Pharmacol Sci. 2011;32:366–375. doi: 10.1016/j.tips.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 43.Bodi I, Muth J, Hahn H, Petrashevskaya N, Rubio M, Koch S, et al. Electrical remodeling in hearts from a calcium-dependent mouse model of hypertrophy and failure: Complex nature of k+ current changes and action potential duration. J Am Coll Cardiol. 2003;41:1611–1622. doi: 10.1016/s0735-1097(03)00244-4. [DOI] [PubMed] [Google Scholar]
- 44.Benitah JP, Alvarez JL, Gomez AM. L-type ca(2+) current in ventricular cardiomyocytes. J Mol Cell Cardiol. 2010;48:26–36. doi: 10.1016/j.yjmcc.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 45.Dai S, Hall DD, Hell JW. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 2009;89:411–452. doi: 10.1152/physrev.00029.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Catalucci D, Zhang DH, DeSantiago J, Aimond F, Barbara G, Chemin J, et al. Akt regulates l-type ca2+ channel activity by modulating cavalpha1 protein stability. J Cell Biol. 2009;184:923–933. doi: 10.1083/jcb.200805063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sul JH, Han B, He D, Eskin E. An optimal weighted aggregated association test for identification of rare variants involved in common diseases. Genetics. 2011;188:181–188. doi: 10.1534/genetics.110.125070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.