

Abstract
Rationale
Mice lacking the EF-hand Ca2+ sensor S100A1 display endothelial dysfunction due to distorted Ca2+ activated NO generation.
Objective
To determine the pathophysiological role of S100A1 in endothelial cell (EC) function in experimental ischemic revascularization.
Methods and Results
Patients with chronic critical lower limb ischemia (CLI) showed almost complete loss of S100A1 expression in hypoxic tissue. Ensuing studies in S100A1 knockout (SKO) mice subjected to femoral artery resection (FAR) unveiled insufficient perfusion recovery and high rates of autoamputation. Defective in vivo angiogenesis prompted cellular studies in SKO ECs and human ECs with siRNA-mediated S100A1 knockdown demonstrating impaired in vitro and in vivo proangiogenic properties (proliferation, migration, tube formation), and attenuated vascular endothelial growth factor (VEGF)- and hypoxia-stimulated eNOS activity. Mechanistically, S100A1 deficiency compromised eNOS activity in ECs both by interrupted stimulatory S100A1/eNOS interaction and PKC hyperactivation that resulted in inhibitory eNOS phosphorylation and enhanced VEGF-receptor 2 (VEGFR2) degradation with attenuated VEGF signaling. Ischemic SKO tissue recapitulated the same molecular abnormalities with insufficient in vivo NO generation. Unresolved ischemia entailed excessive VEGF accumulation in SKO mice with aggravated VEGFR2 degradation and blunted in vivo signaling through the proangiogenic PI3K/Akt/eNOS cascade. NO supplementation strategies rescued defective angiogenesis and salvaged limbs in SKO mice post-FAR.
Conclusions
Our study shows for the first time downregulation of S100A1 expression in patients with CLI and identifies S100A1 as critical for EC function in postnatal ischemic angiogenesis. These findings link its pathological plasticity in CLI to impaired neovascularization prompting further studies to probe S100A1’s microvascular therapeutic potential.
Keywords: Angiogenesis, S100A1, edothelium, nitric oxide
INTRODUCTION
Chronic critical limb ischemia (CLI), the most advanced form of peripheral arterial occlusive disease (PAOD), is a common clinical problem with poor clinical prognosis.1 Without successful revascularization, up to 40% of patients will require major limb amputation within a year of diagnosis exceeding annual mortality rates of 20%.1 Though we have seen rapid evolution of endovascular revascularization and improvement in medical treatment over the past 15 years, therapeutic options to improve bridge-collateral (arteriogenesis) and/or capillary formation (angiogenesis) are limited.1 The prevailing lack of microvascular-targeted treatments fostered novel cell- and molecular-based strategies being tested in phase I and II clinical trials with promising preliminary results.2
In this regard, the most recent discovery of the EF-hand calcium (Ca2+) binding protein S100A1 as virtually indispensable for postnatal endothelial cell (EC) function3, 4 has sparked interest in its potential significance in vascular pathophysiology. Originally identified in the brain as a hydrophobic low molecular weight (22 kD) dimeric protein, S100A1 belongs to the largest subgroup of Ca2+ sensors within the EF-hand Ca2+ binding protein superfamily.5 The group, referred to as S100 proteins, comprises 21 isoforms each of which is characterized by a cell- and tissue-type specific expression pattern. S100A1, known for its high abundance in cardiomyocytes, controls a Ca2+-driven functional network orchestrating contractile performance, cell survival and metabolism.5
In ECs, S100A1 likewise seems to be an important part of the cells versatile molecular toolkit to relay intracellular Ca2+ oscillations that regulate vascular tone.3, 4 Lack of endothelial S100A1 attenuates in vitro and in vivo arterial vasorelaxation basally and in response to B2-kininergic and M2-muscarinic stimuli.3, 4 The Ca2+ sensor interacts with the inositol-triphosphate receptor (IP3R) and enhances intracellular Ca2+ release from the endoplasmic reticulum (ER) that indirectly accounts for enhanced endothelial nitric oxide synthase (eNOS) activation.3, 4 However, it is unclear whether the EF-hand Ca2+ sensor might also affect eNOS function directly. Hence, lack of S100A1 conveys endothelial dysfunction and suggests potential pathophysiological relevance in human vascular diseases.
In this study, we advance our understanding of S100A1’s role in vascular disease biology and identify the Ca2+ sensor as indispensable factor for postnatal ischemic neovascularization with potential clinical relevance. First time demonstration of a near complete loss of S100A1 expression in ischemic tissue from patients with chronic CLI spurred a comprehensive proof-of-concept study in S100A1 genetically-ablated (SKO) mice to determine its impact on postnatal ischemic neovascularization and underlying molecular mechanisms.
METHODS
A detailed description of the Materials and Methods used in this study is available in the expanded online Data Supplement. For the assessment of hindlimb ischemia and protein-protein interactions, we kindly refer to the online Data Supplement.
Patients
Briefly, in patients with chronic CLI and indication for surgical revascularization or major amputation, and in control patients, a surgical biopsy of the gastrocnemius muscle was taken. Patients gave their written informed consent to participate in the study. The study protocol has been approved by the ethics committee of the Medical Faculty at the Technische Universität Dresden.
Mice
SKO mice were derived on a C57Bl/6 background as described previously.6 All experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee and complied with the Guide for the Care and Use of Laboratory Animals. For femoral artery resection (FAR), mice were anesthetized and the right femoral artery was resected as described.7 In the course of the study, a modified minimal surgery was used. In some cases WT and SKO mice were treated with the NO donor diethylenetriamine/NO (DETA/NO, 2mg/kg) for three consecutive days immediately following FAR.8
In vivo matrigel plug assay
Mice were anesthetized, then two small subdermal pockets were created and each pocket received one matrigel plug. Plugs were harvested 16 days later. In some cases mice received injections of DETA/NO (2mg/kg) or vehicle (PBS).8 Recovered plugs were cryoemdedded, sliced, fixed, stained and subsequently imaged (Image J).
RNA isolation, reverse transcription and quantitative real-time PCR
The procedures for RNA isolation, reverse transcription and quantitative real-time PCR were essentially carried out as described previously.3
Endothelial cell preparation and siRNA-mediated endothelial S100A1 knockdown
Primary ECs from WT or SKO mice were prepared as described,9 with modifications mentioned in the Data Supplement. Human cardiac microvascular endothelial cells (HMVECs, CC-7030) (LONZA) were cultured according to the manufacturer’s instruction in EGM-2 MV BulletKit media (CC-3162). S100A1 knockdown was achieved transfecting cells with siRNA against human S100A1 (Sigma 231-791-2) using the Lipofectamine RNAiMAX reagent (Invitrogen).
Skeletal muscle cell isolation
Gastrocnemius muscle bundles of male WT mice (2-3 months old) were dissected.10 The isolated muscles bundles were treated with collagenase and muscle cells were then incubated under normoxic and hypoxic (2%O2) conditions.
[3H]Thymidine incorporation assay
Agonist-induced [3H]-thymidine incorporation into endothelial cells was assessed as described.11
Chemokinesis assay
EC migration was assessed according to a modified protocol from Liang et al.12 ECs were seeded and allowed to attach overnight. Following 24h of serum starvation (0.2%FBS), the cell monolayer was denuded with a pipette tip and further incubated with 1%FBS, ECs were then photographed to record a starting point. After 48h of incubation, cells were photographed again and “wound closure” was quantified.
Matrigel tube formation assay
ECs were seeded on matrigel-covered wells and stimulated with 30μg/ml ECGS in DMEM containing 0.2%FBS. After 24h of incubation, cells were photographed and tube formation was quantified by counting the branching points.
Immunoblot analysis
Assessment of protein expression and phosphorylation from tissue and cellular extracts was carried out as described in details elsewhere,3, 4 details can be found in the Data Supplement.
Measurement of NO and nitrite in cell culture supernatants and tissue homogenates
NO in medium from EC cultures subjected to VEGF stimulation or hypoxia was measured using a NO/Nitrite/Nitrate assay (R&D systems) according to manufacturer’s instructions. The PKC inhibitors chelerythrin (Tocris, Cat.No 1330) and calphostin-C (Tocris, Cat.No. 1626) were used at 1μM and 100nM, respectively. Nitrate levels in skeletal muscle tissue homogenates were measured with the Nitrate/Nitrite Colorimetric Assay Kit (Caymen Chemicals 780001).
Measurement of NOS/eNOS activity and PKC activity
Nitric oxide synthase activity from skeletal muscle tissue and ECs was measured with the Nitric Oxide Synthase Assay Kit (EMD) following manufacturer instructions. Protein Kinase C activity from WT and SKO skeletal muscle tissue was measured with the PepTag Protein Kinase Assay (Promega) following manufacturer’s instructions. PKC inhibitors were used as indicated.
Statistical analysis
Student’s t-test and one-way ANOVA with Tukey’s post-test for multiple comparisons were used to analyze the appropriate data using GraphPad PRISM software. Fisher’s exact test was used to compare clinical score data by comparing mice with no evidence of hindlimb necrosis to mice with any evidence of necrosis. Data are presented ±SEM in the Figures. A p-value <0.05 was considered statistically significant.
RESULTS
Loss of S100A1 expression in patients and mice with critical lower limb ischemia
Ischemic gastrocnemius muscle (GM) tissue biopsies from from five patients with advanced chronic CLI (Rutherford stages 4-6) exhibited decreased S100A1 mRNA amount to approximately 5% of S100A1 levels in control biopsies (Figure 1A). Control non-ischemic GM samples were derived from three patients with indication for elective knee arthroplasty and exclusion of peripheral arterial occlusive disease (PAOD; for detailed patient characteristics see Supplement methods Table I.). Surgically-induced hindlimb ischemia in C57BL/6 wild type (WT) mice due to right-sided femoral artery resection (FAR) mirrored the loss of S100A1 expression in ischemic GM tissue (Figure 1B). S100A1 expression levels in hypoxic ECs being relevant to ischemic neovascularization was determined next. Murine arterial ECs exposed to 2%O2 over 48 hours displayed rapid S100A1 mRNA downregulation (Figure 1C) similar to human iliac artery ECs (HIAECs, data not shown). S100A1 levels in isolated adult murine skeletal muscle (SM) fibers (Supplement Figure I.) involved in contractility regulation, however, exhibited lesser attenuation when treated with the same protocol (Figure 1C). Indicative of a greater susceptibility of ECs to hypoxia-induced loss of S100A1, our clinical observation prompted further investigation of the pathophysiological role of S100A1 in ECs in regenerative ischemic neovascularization taking advantage of S100A1 gene-ablated mice (SKO).
Figure 1. S100A1 is downregulated in critical limb ischemia.

(A) Digital subtraction angiography of a representative patient with peripheral arterial occlusive disease (PAOD) due to femoro-popliteal arterial obstruction (arrow points to site of proximal arterial obstruction). Gastrocnemius muscle biopsies (arrowhead) were analyzed for S100A1 mRNA expression using qRT-PCR (n=3 control, 5 CLI, *p<0.05) (Scale bar=5cm). (B) WT mice were subjected to FAR and S100A1 protein levels in GM muscle samples were assessed by immunoblot analysis before (NI) or 3 days post FAR (I) (n=3 each *p<0.05 vs NI). (C) Isolated primary murine WT EC or Skeletal muscle fibers were subjected to hypoxia for up to two days. mRNA was extracted and subjected to qRT-PCR for S100A1 (n=3, *p<0.05 vs EC).
High rate of autoamputation in SKO mice with experimental hindlimb ischemia
To determine the in vivo significance of our findings, we chose the model of experimentally-induced CLI due to FAR in 8-12 week-old WT and SKO mice. Recovery from hindlimb ischemia was classified employing a clinical score as previously described (Supplement methods).13 In WT mice, active use of the right foot declined after surgery but improved by day seven, and activity of the preserved limb ultimately returned to near-normal levels with an average score below one by day 15 (Figure 2A). A similar abrupt but persistent reduction in hindlimb use occurred in SKO mice that developed into partial or total autoamputation of the affected limb (average score greater than three) in 80% of SKO mice between seven and 14 days post-FAR (Figure 2A). Suggestive of a lack of regenerative reperfusion and/or revascularization, we next measured arterial blood flow recovery in ischemic SKO hindlimbs post-FAR.
Figure 2. S100A1 deficient mice undergo auto-amputation following femoral artery resection (FAR).

(A) WT or SKO mice underwent FAR of the right hind limb (red circles). Representative examples of animals 15 days post-op are shown. Clinical score was assessed 15 days post-op (n=19 for each genotype, see Methods section for description of clinical scores). * p< 0.05, Fisher's exact test. (B) Hind limb perfusion was assessed by determining the velocity time integral (VTI) ratio of the blood flow in the tibial posterior artery of the ischemic and the contra-lateral non-ischemic limb using a pulsed wave doppler (n=10 for each genotype, p<0.05 WT vs. SKO). (C) Perfusion ratio of the ischemic to non-ischemic hind limb was assessed in mice 15 days post FAR using colored microspheres (n=5 for each genotype, * p<0.05 ischemic SKO vs. ischemic WT)
Impaired reperfusion and neovascularization in ischemic hindlimbs of SKO mice
Arterial limb perfusion recovery was examined by pulsed-wave (PW) Doppler ultrasonography (Supplement Figure II.).14 SKO mice at risk for autoamputation never achieved more than 25-35% perfusion in the tibialis posterior artery of ischemic hindlimbs one and two weeks post-FAR (Figure 2B). Accordingly, direct measure of ischemic tissue perfusion using colored microspheres at day 15 post-FAR unveiled an 80% blood flow reduction in SKO ischemic limbs compared with controls (Figure 2C). Structural analysis of postischemic bridge-collateral formation by immunofluorescent smooth muscle actin (SMA) staining and infra-abdominal aortic corrosion cast, respectively, revealed no evidence for decreased arteriogenesis in SKO animals when compared with WT-FAR (Figure 3A, Supplement Figure III.). However, histological and immunofluorescent analyses of the EC marker von Willebrand factor (vWF) and CD31, respectively, yielded a significant reduction of the capillary/fiber ratio in ischemic SKO muscle tissue (Figure 3B). Indicative of a predominant defect in postischemic angiogenesis, we raised the question whether loss of S100A1 per se might compromise proangiogenic properties of ECs.
Figure 3. Impaired reperfusion and neovascularization in ischemic hindlimbs of SKO mice.

(A) WT and SKO mice were subjected to FAR and procured at times indicated. The arterial hindlimb circulation was corrosion casted to visualize developing collaterals. (n=4 each genotype, scale bar=10mm). (B) SKO and WT mice were subjected to FAR and gastrocnemius muscles from ischemic and contra-lateral non-ischemic limbs were procured 15 days later. Paraffin embedded cross-sections were immunohistochemically stained for vWF (arrows, counterstained with hematoxylin and eosin to visualize nuclei and fibers, respectively) or immunofluorescently stained for CD31 (CD31, green; nuclear DNA was visualized with Hoechst 33342, blue). Note decrease in CD31 staining in ischemic SKO muscle tissue. Representative cross sections are shown (Scale bar=200μm). Fibers and capillaries positive for vWF were counted in the paraffin embedded cross sections and the resulting ratio was graphed (n=5 for each genotype) * p< 0.05 vs. ischemic SKO.
Blunted proangiogenic properties of SKO ECs and rescue of defective capillary formation by NO
Isolated SKO ECs displayed significantly lower proliferation (Figure 4A) and migration rates (Figure 4B) than WT ECs under normoxic conditions. Likewise, formation of capillary-like tube networks on growth factor supplemented matrigel was compromised in SKO ECs (Figure 4C). We next determined whether these defects might translate into distorted angiogenesis in vivo. To this end, we employed subcutaneously implanted matrigel plugs in 12-week-old WT and SKO mice that were harvested 16 days later. Bases of matrigel plugs explanted from WT mice were densely populated by CD31-positive ECs and SMA-stabilized capillaries were traceable up to the plug’s center sections (Figure 4D). In contrast, matrigel plugs recovered from SKO mice unveiled a significantly lower number of organized capillaries at the bottom and fewer penetrations towards the center (Figure 4D). Importantly, intraperitoneal NO supplementation with diethylenetriamine-NO (DETA/NO) was sufficient to rescue this defect. A DETA/NO dosage was used that was previously described not to significantly change blood pressure or heart rate in mice (2mg/kg) but exert NO-dependent cardiac protection.8 Explanted matrigel plugs 16 days after DETA/NO treatment displayed marked improvement of capillary neoformation that was virtually indistinguishable from WT mice (Figure 4E). The NO-mediated rescue directed our attention to molecular abnormalities in S100A1-deficient ECs that contribute to distorted postischemic NO and capillary formation.
Figure 4. Blunted proangiogenic properties of SKO ECs in vitro and defective capillary formation in vivo.

Primary murine ECs were isolated from WT and SKO mice and were assayed for: (A) [3H] thymidine incorporation in response to different concentrations of FBS with or without endothelial cell growth supplement (ECGS). (B) The chemokinetic response to 1%FBS using an in vitro wound repair (Scratch-) assay (24h), (C) EC tube formation on Matrigel matrix. All experiments were repeated at least three times with different primary isolates and performed in duplicate. Representative images are shown in (B) and (C). The stippled area in (B) denotes the extent of the originally denuded area. *p<0.05 vs WT. (D) Matrigel plugs were implanted into WT and SKO mice (n=3) and harvested after 16 days. Recovered plugs (two for each animal) were cryoembedded and stained for the EC marker CD31 and smooth muscle actin (SMA). Nuclear DNA was visualized with Hoechst 33342 (10x magnification, scale bar=200μm, *p<0.05 WT vs SKO, n=6 plugs each genotype). Representative images (60x magnification, scale bar=20μm) are shown, CD31 (green), SMA (red), DNA (blue). (E) Matrigel plugs were implanted into WT or SKO mice. Mice received DETA/NO (2 mg/kg, i.p.) or saline three times weekly for two weeks. Plugs were harvested 16 days post implantation, cryoembedded and stained as in (D) at a depth of 1 mm. Scale bar=200μm. *p<0.05 vs SKO +DETA/NO.
Abrogated NO generation in ischemic hindlimbs of SKOs and abnormal eNOS regulation in S100A1-depleted ECs
SKO ischemic GM tissue samples showed blunted postischemic NO production compared to WT FAR mice (Figure 5A). Abnormalities in VEGF- and hypoxia-mediated eNOS function in SKO ECs are unknown though. Here, we demonstrate for the first time an attenuated NO generation in response to VEGF and hypoxia in cultured SKO ECs when compared with WT ECs (Figure 5B and Supplement Figure IV-B., resp.). To elaborate on potential mechanisms, human microvascular ECs (HMVECs) were subjected to siRNA-mediated knockdown of S100A1 (HMVEC-SKD) that resulted in an approximately 80% reduction of the protein after 48 hours (Supplement Figure IV-A.). Alike SKO-derived ECs, HMVEC-SKDs responded with abrogated NO generation to stimulation by VEGF or hypoxia (2%O2) (Figure 5C and Supplement Figure IV-C., resp.). Subsequent phosphorylation analysis of the inhibitory eNOS threonine 495 (p-T495) site revealed significantly augmented levels in HMVECs-SKDs, whereas control HMVECs demonstrated enhanced phosphorylation of the stimulatory serine 1177 (p-S1177) site (Figure 5D). The previously demonstrated ability of S100 proteins to attenuate PKC activity, which plays a critical role in p-T495 phosphorylation,15 prompted us to study PKC activity.16, 17 In line with abnormal eNOS regulation, HMVEC-SKDs exhibited significantly greater PKC activity under VEGF stimulation than control cells (Figure 5E). Inhibition of PKC with the chemical compound calphostin-C prevented the increase of eNOS pT-495 (Supplement Figure V-A.). Consequently, HMVEC-SKDs experienced a significantly greater relative increase in NO generation upon PKC inhibition than control cells (Supplement Figure V-B.). The partial restoration of NO generation by PKC inhibition suggested an additional mechanism and spurred the question whether S100A1 might directly interact and modulate eNOS activity in ECs and ischemic tissue.
Figure 5. Reduced VEGF-stimulated NO production in SKO ECs and hindlimbs.
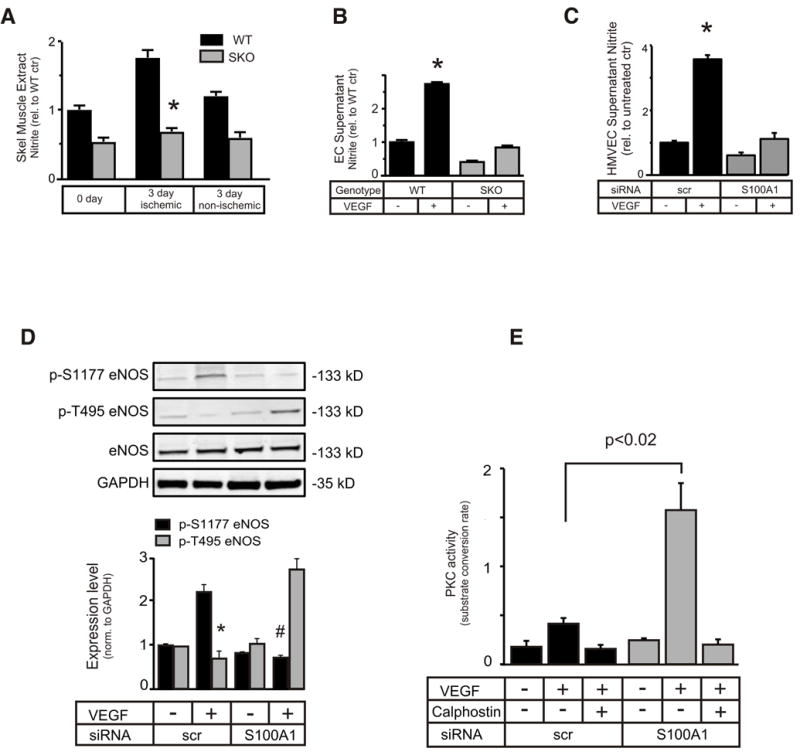
(A) NOS activity of skeletal muscle extracts procured at different times post FAR was assessed by measuring nitrite production (n=6, * p<0.05 SKO vs. WT). (B) Primary ECs derived from WT and SKO mice or (C) HMVECs subjected to siRNA-mediated knockdown of S100A1, were treated with 50 ng/ml VEGF for 16h. eNOS-derived nitrite was determined in the cell supernatant as described in the Methods section (*p<0.05 vs untreated WT or VEGF treated SKO (B) or S100A1 siRNA(C), resp., experiments were done in duplicate and repeated 3 times). (D) VEGF-dependent phosphorylation status of p-S1177 or p-T495 eNOS was examined in HMVECs subjected to S100A1 knockdown (*p<0.05 vs S100A1 kd pT495 eNOS, #p<0.05 vs scr pS1177 eNOS). (E) PKC activity was examined in HMVECs subjected to S100A1 knockdown and treated with 50 ng/ml VEGF for 15 min. (n=3 independent assays, performed in triplicate.) Calphostin-C (100ng/ml) was included to inhibit PKC, where indicated.
S100A1 directly interacts with eNOS in ECs in a Ca2+-dependent manner
In support of this argument. S100A1 co-precipitated with eNOS in a Ca2+-dependent manner using murine EC lysates (Figure 6A). Abrogated eNOS/S100A1 interaction by EGTA corroborated Ca2+- dependency of their binding. Increasing cytosolic Ca2+ concentrations in these cells either using bradykinin or the Ca2+ ionophore A23187 prior to extract preparation and immunoprecipitation further enhanced S100A1-eNOS interaction (Figure 6A). Addition of increasing concentrations of recombinant human S100A1 protein (rhS100A1) to homogenates of human umbilical vein ECs (HUVEC) showed dose-dependency of the Ca2+-dependent binding between both human proteins, and similar results were obtained with adenovirally-overexpressed S100A1 in HUVECs (data not shown). Brief hypoxia (2%O2, for 2 hours), which does not appreciably result in loss of S100A1 protein, similarly augmented S100A1/eNOS binding in control HMVECs (Supplement Figure VI.). When examined by confocal microscopy both in murine WT ECs as well as HIAECs, S100A1 displayed a granular cytosolic pattern and co-localized with eNOS throughout the cell (Figure 6B). Employing the in situ duolink assay, the distinct S100A1/eNOS interaction pattern could be confirmed in intact HUVECs that corroborated the immunoprecipitation studies (Figure 6B).
Figure 6. S100A1 interacts with eNOS and regulates its activity.
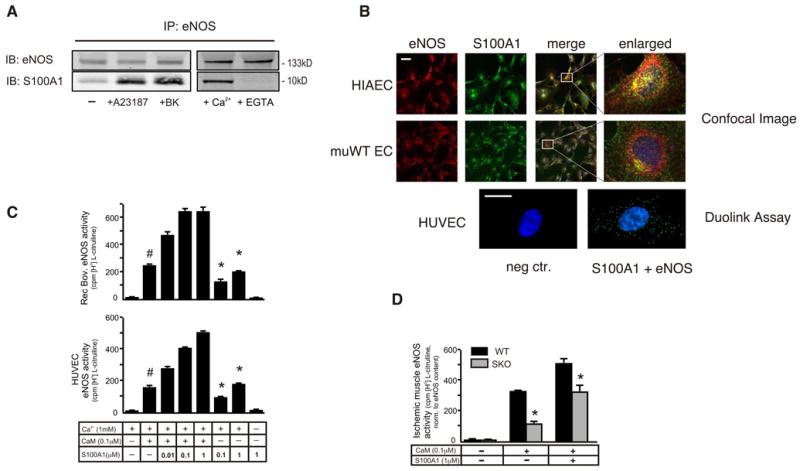
(A) Lysates were prepared from ECs pretreated, or not, with A23187 or bradykinin. In a separate experiment immunoprecipitations were performed in the presence of Ca2+ or EGTA. Immunoblots were probed for the presence of eNOS or S100A1. Representative blots are shown, the experiment was repeated at least three times with similar results. (B) ECs were immunofluorescently labeled with an antibody to eNOS (red) or S100A1 (green) and assayed by confocal microscopy (40x magnification, scale bar=40μm); enlarged merged images show partial co-localization (yellow). Close protein contact between S100A1 and eNOS was also shown using the duolink assay (60x magnification, scale bar=10μm). (C) Recombinant bovine eNOS (upper panel) or eNOS immunoprecipitated from HUVEC cell extract (lower panel) was incubated with 1 mM Ca2+ in the presence or absence of CaM and increasing amounts of rhS100A1, as indicated. eNOS activity was determined by measuring the conversion of [3H] L-arginine to L-citrulline. # p<0.05 vs reaction in the absence of added S100A1. *p<0.05 vs reaction in the absence of added CaM and rhS100A1. The experiment was performed in duplicate and repeated 3 times. (D) WT and SKO mice were subjected to FAR and extracts from ischemic muscles were prepared 3 days later and used to immunoprecipitate eNOS. eNOS activity in the immunoprecipitates was assayed with or without added CaM and/or S100A1 and adjusted to eNOS protein content (n=6, * p<0.05 SKO vs WT).
S100A1 protein positively modulates activity of eNOS
To determine whether this interaction actually modulates function of the enzyme, we carried out reconstituted eNOS activity assays both using recombinant bovine eNOS and immunoprecipitated eNOS from EC lysates. As shown in Figure 6C, addition of increasing concentrations of rhS100A1 dose-dependently augmented the Ca2+/CaM-dependent arginine-citrulline conversion rate both of bovine recombinant and EC–derived eNOS. Interestingly, quantitative assessment of the CaM-eNOS interaction in S100A1-overexpressing and control HUVECs applying the duolink assay provided no evidence for enhanced CaM-eNOS binding through increased cellular S100A1 protein levels (Supplement Figure VII.). Vice versa, increasing concentrations of recombinant CaM did not attenuate S100A1 co-precipitating with eNOS (data not shown). These results indicate an additive activation of eNOS through both Ca2+-sensors rather than mutually competitive or facilitated binding. Incubation of immunoprecipitated eNOS from both groups with recombinant CaM resulted in a significantly greater increase in eNOS activity from S100A1-competent WT tissue than SKO. Ensuing administration of rhS100A1 protein further increased eNOS activity from SKO, corroborating the additive effect of both Ca2+-sensors (Figure 6D). Indicating that S100A1 deficiency both directly and indirectly impairs eNOS function, we continued to investigate how these molecular abnormalities might affect in vivo responsiveness and efficiency of the VEGF-PI3K-Akt-eNOS signaling axis in ischemic tissue.
Excessive postischemic VEGF levels and attenuated VEGFR2 signal relay in SKO ischemic hindlimbs
Analysis of the oxygen sensor hypoxia-inducible factor 1α (HIF-1α), which is known to drive vascular endothelial growth factor (VEGF) expression,18 revealed significantly greater protein levels in ischemic SKO than ischemic WT GM (Figure 7A). Concordantly, postischemic abundance of VEGF protein in SKO hindlimbs exceeded the VEGF rise in WT tissue by approximately 10-fold (Figure 7B). Similar abnormalities were found for other hypoxia-regulated growth factors including hepatocyte growth factor (HGF), placental growth factor (PlGF) and insulin-like growth factor (IGF) (Supplement Figure VIII.) indicating unresolved ischemia in SKO hindlimbs. Previous studies that reported rapid degradation of VEGF receptor-2 (VEGFR2) in response to sustained VEGF stimulation via a PKC-dependent mechanism19, 20 prompted investigation of abundance and phosphorylation status of key effectors along the VEGFR2 signaling axis. Both total and phosphorylated VEGFR2 protein levels were hardly detectable in ischemic SKO tissue extracts compared with only a slight decrease in ischemic WT (Figure 7C). When HMVEC-SKDs were exposed to VEGF, faster in vitro VEGF2 degradation was observed compared with controls and this effect was blocked by the PKC inhibitor calphostin-C (Supplement Figure IX.). Interestingly, ischemic SKO tissue unveiled significantly higher postischemic PKC activity (Supplement Figure X.), akin to enhanced PKC activity in hypoxic HMVEC-SKDs. Accordingly, relative active phosphorylation status of VEGFR2 downstream effectors such as PI3Kγ, PDK and Akt was significantly lower in ischemic SKO–derived GM compared with ischemic WT, whereas total levels were significantly higher (Figure 7D, Supplement Figure XI-A.). VEGFR1 in vivo levels were indeed unchanged in both groups (Figure 7C) while downstream activity of the PLCγ-ERK1/2 axis was significantly higher in ischemic SKO than WT tissue (Supplement Figure XI-B.). In line with published reports,19 we confirmed that VEGR1 remains unaltered in ECs in response to prolonged VEGF stimulation (Supplement Figure XII.). Besides evidence for blunted postischemic VEGFR2 signaling in SKO, the postischemic VEGF overshoot might also entail dysbalanced VEGFR1 activity.
Figure 7. Augmented activation of HIF-1α and VEGF in the ischemic hind limb of SKO mice disturbs VEGFR2 signaling, leading to eNOS inhibition.

SKO and WT mice were subjected to FAR and gastrocnemius muscles of the ischemic and contra-lateral non-ischemic hind limb were procured and homogenized after three days (except for VEGF). Representative samples are shown for all immunoblots. Muscle extracts were immunoblotted for (A) HIF-1α, (B) VEGF (at times indicated), (C) total and p-Y1175 VEGFR2 and VEGFR1, (D) total and p-S473-Akt. All expression levels were normalized to GAPDH (n=4 for each genotype and condition, * p< 0.05 vs ischemic WT).
Inhibitory eNOS phosphorylation pattern in SKO ischemic hindlimbs
Driven by abnormal in vitro eNOS regulation and attenuated in vivo VEGFR2-PI3K-Akt signaling,21 we next raised the question whether eNOS phosphorylation pattern and expression was similarly modified in ischemic tissue from SKO mice. Interestingly, ischemic SKO GM tissue unveiled an approximately two-fold increase in total eNOS protein expression over WT (Figure 8A). In line with our in vitro results, the enzyme exhibited a more than 7-fold increase in phosphorylation of the inhibitory phospho-threonine 495 (p-T495) over the stimulatory phospho-serine 1177 (p-S1177) site (Figure 8A) when compared with WT. Among PKC isoforms that are known to attenuate Ca2+/CaM-dependent eNOS activity through p-T495 phosphorylation,15 expression analysis showed a marked increase in PKCε protein and phosphorylation levels (Figure 8B) in accordance with elevated PKC activity in ischemic SKO tissue homogenates (Supplement Figure X.). Expression analysis of PP1, which dephosphorylates the p-T495 site,15 showed no difference between groups (data not shown).
Figure 8. NO donor administration prevents auto-amputation of the ischemic SKO hind limb and restores angiogenesis.

(A) Muscle extracts from WT and SKO mice were prepared 3 days post FAR and immunoblotted for eNOS (total, p-T495 and p-S1177) and (B) total and p-PKC-ε.(C) SKO mice underwent FAR and received the NO donor DETA/NO (2 mg/kg, i.p.) or saline daily for the following three days. Clinical score was assessed at day 15 post-op (n=6, * p< 0.05 vs. non-ischemic saline). *p<0.05 vs SKO +DETA/NO.
Direct NO substitution salvages ischemic limb loss in SKO mice
Given the fact that NO administration is sufficient to rescue defective in vivo angiogenesis in SKOs, we finally sought to answer the question whether the observed lack of NO bioavailability is actually causative for tissue necrosis in SKO FAR mice. To this end, WT and SKO animals were treated with DETA/NO for three consecutive days immediately post-FAR. Using the same dosage that rescued in vivo angiogenesis in SKO mice, NO treatment completely prevented autoamputation in all SKO mice (clinical score below one), while five out of six saline injected SKO mice still displayed evidence of limb necrosis 15 days post-FAR (Figure 8C). Our in vivo rescue corroborates defective NO bioavailability as key for the postischemic SKO phenotype and NO supplementation as sufficient to bypass abnormal eNOS regulation and restore defective angiogenic activity.
DISCUSSION
This is the first report providing direct genetic evidence for a critical role of S100A1 in postnatal ischemic angiogenesis. Most salient findings of this study, that originates from the clinical finding of diminished S100A1 expression in patients with CLI, are the identification of endothelial S100A1 as i) an indispensable factor for mounting an adaptive angiogenic response to ischemia, and ii) a stimulatory eNOS-interacting protein critical for postischemic NO generation. To determine S100A1’s role in postischemic revascularization and decipher underlying cellular and molecular mechanisms, we took advantage of the hindlimb ischemia model in SKO mice. Among the relevant differentiated cell types expressing S100A1 in muscle tisse, ECs emerged as particular susceptibility to a hypoxia-induced loss of S100A1 expression. Our focus was thus drawn on S100A1’s impact on in vitro and in vivo endothelial proangiogenic properties being essential to reparative neovascularization.
After surgical blood flow interruption,7, 13 SKO mice exhibited a high rate of tissue necrosis and subsequent auto-amputation of ischemic limbs due to insufficient NO generation. Similarly severe phenotypes have been observed in mice with homozygous ablation of Akt1 and eNOS,7, 13 which groups S100A1 alongside genes that are crucial for postnatal neovascularization through the modulation of NO homeostasis. In support of this notion, assessment of changes in postischemic blood flow revealed failure of arterial perfusion recovery in SKO mice akin to Akt1- and eNOS-knockout mice undergoing FAR.7, 13 However, ensuing investigation of bridge-collateral formation that fuels distal blood flow and occurs under normoxic conditions22 even showed a trend towards augmented collateral wall formation in SKO. This implicates at least normal smooth muscle cell (SMC) expansion compared with WT, and is consistent with reports on unchanged function of S100A1-deficient SMCs.3, 4 But, as shown previously, S100A1-deficient endothelium blunted normoxic arterial NO generation and dilatation in response to endogenous vasorelaxing factors.3, 4 It is thus conceivable that normal amounts of collateral arteries grew in SKOs post FAR but were unable to dilate adequately due to dysfunctional S100A1-deficient endothelium.22 The endothelial defect could explain in part diminished arterial blood flow recovery in SKO mice. Notwithstanding, evaluation of exercise- and shear-stress-based vasodilation of SKO arteries is needed to fully understand the role of S100A1 in EC-dependent arterial reserve mechanisms.22
Turning our attention to angiogenesis then unveiled a striking difference in postischemic microvasculature formation that clearly distinguished post-FAR SKO from WT mice. Ischemic limbs of WT mice exhibited the expected hypoxia-driven enhancement of capillary formation.13 In contrast, SKO mice failed to mount an adaptive angiogenic response. Hence, SKO that suffered autoamputation may not have even reached the critical threshold of newly formed microvasculature necessary to preserve their limbs. In line with this argument, essential steps of the fine-tuned sequence of postischemic capillary formation including EC proliferation, migration and capillary-like tube network formation23, 24 were all severely compromised in SKO ECs. Use of an integrative in vivo angiogenesis assay24 substantiated the postnatal inability of SKO mice to create three-dimensional capillary networks. Although these assays are not representative of the site and environment where postischemic angiogenesis occurs,24 they nevertheless unearth a basic defect in SKO ECs that might explain in part defective postischemic angiogenesis seen in vivo. These results further imply a pathophysiological role of the hypoxia-driven loss of endothelial S100A1 expression for impaired revascularization seen in patients with chronic CLI. Molecular mechanisms attenuating S100A1 expression in hypoxic ECs remain to be determined.
Despite the complexity of postischemic capillary neoformation, NO emerged as an ultimate signaling molecule governing this process.25 In line with this notion, we found impaired NO generation both in S100A1-deficient ischemic tissue, as well as in hypoxic and VEGF-stimulated S100A1-depleted ECs. Previous reports have already attributed abnormal Ca2+-mobilization from intracellular stores in SKO ECs to diminished Ca2+-dependent NO generation in response to B2-kininergic and M2-muscarinic stimuli.3, 4 Although VEGF-mediated eNOS activation relies in part on Ca2+-mobilization,25 the severity of the SKO phenotype suggested an even more substantial role of S100A1 in postischemic eNOS regulation. Ensuing molecular and imaging studies actually revealed first evidence for a direct Ca2+-dependent stimulatory interaction of S100A1 with eNOS in ECs. S100A1 protein dose-dependently augmented eNOS activity and potentiated the effect of Ca2+/CaM-dependent stimulation without evidence for facilitated CaM-eNOS interaction in ECs. These novel results support the notion that S100A1 might be a direct and positive modulator of eNOS function and advance our current understanding of S100A1’s molecular actions in ECs. These data provide the rationale for continued studies determining the relevant binding domains between eNOS and S100A1 and its impact on other accessory eNOS modulators such as Hsp90 or caveolin-1 in greater detail.15
In addition, our study unveiled another novel regulatory mechanism by which S100A1 can indirectly modulate eNOS function that involved PKC. S100 proteins have been reported to attenuate PKC activity by interfering directly with PKC, or indirectly with PKC substrates.16, 17 The kinase has previously been described to negatively interfere with eNOS activity by p-T495 phosphorylation.15 Here, we firstly demonstrate that S100A1 is necessary to attenuate PKC-dependent inhibitory phosphorylation of eNOS in VEGF-stimulated ECs. Partial restoration of NO generation in S100A1-depleted ECs by chemical PKC inhibition corroborates the significance of this finding. Since it is still not entirely clear how S100 proteins can mitigate PKC activity, more work is required to uncover the mechanism that confers S100A1-mediated inhibition of PKC in ECs. Nonetheless, from the deepended mechanistic insight, we conclude that compromised postischemic eNOS function in SKO and S100A1-depleted ECs is most likely orchestrated both by interrupted stimulatory S100A1/eNOS interaction and augmented p-T495 eNOS phosphorylation due to PKC hyperactivation.
Both molecular defects seem to have the potential to negatively amplify each other and might essentially contribute to blunted in vivo proangiogenic properties of S100A1-deficient ECs. We therefore focused next on in vivo consequences by examining the VEGF signaling-axis and eNOS posttranslational modifications. Reflecting unresolved tissue ischemia,18 protein levels of the main oxygen sensor HIF-1α were significantly higher in ischemic SKO tissue, and ensuing VEGF accumulation in operated SKO limbs exceeded the ischemic response in WT mice by more than 10-fold. But excessive VEGF production is not a genuine defect of SKO ECs since in vitro hypoxic VEGF production was indistinguishable from WT ECs (data not shown). Interestingly, eNOS-/- mice subjected to FAR responded with a postischemic decrease in VEGF mRNA expression due to a proposed concurrent genetic defect,26 indicating that the SKO phenotype might differ substantially with respect to adaptive proangiogenic pathways. The concomitant increase of the angiogenically active cytokines HGF, IGF and PlGF in SKO post-FAR further substantiates the view of a rather futile secondary VEGF overshoot that might in turn disturb the finely regulated proangiogenic cross-talk between growth factors.19
This is important to note since sustained VEGF stimulation can result in rapid downregulation and degradation of its receptor tyrosine kinase VEGFR2.19 In contrast to WT, assessment of VEGFR2 protein and phosphorylation uncovered virtually undetectable levels in ischemic SKO tissue. A potentially causal role for endothelial S100A1 herein emerged from our in vitro results showing accelerated VEGFR2 degradation in VEGF-stimulated S100A1-depleted ECs. Attenuation of this defect by a chemical PKC inhibitor once more implies a role for PKC, which is involved in VEGF-mediated VEGFR2 downregulation and degradation.19, 20 Enhanced PKC activity in S100A1-depleted ECs might therefore contribute to in vivo VEGFR2 degradation in SKO mice. In line with prevailing literature,19, 27 it is tempting to speculate that loss of S100A1 feeds into a vicious cycle of reduced in vivo VEGF-responsiveness nurtured in part by endothelial VEGFR2 degradation, which again fuels a secondary VEGF overshoot and compromises residual angiogenic capacity in SKOs. Accordingly, the downstream PI3K-Akt-eNOS signaling cascade, which accounts for many of the VEGF actions, including EC proliferation, migration, tube formation, and promotion of the release of NO23 yielded marked abnormalities in ischemic SKO tissue. Relative active site phosphorylation levels for PDK and Akt were reduced supporting the notion of abrogated activation. In this regard, permanent upregulation of PI3K, PDK, Akt and eNOS protein expression might be viewed as compensatory attempts to uphold efficacy. Interestingly, eNOS-/- mice subjected to FAR displayed similarly decreased postischemic Akt activation, although opposed to SKOs, total Akt levels were reported to be unchanged.28
Most importantly, however, the balance between activating and inhibitory phosphorylation of eNOS in ischemic SKO samples was significantly shifted in favor of the p-T495 site. This observation is consistent with diminished in vivo ischemic NO bioavailability and mirrors our in vitro results of an abnormal eNOS phosphorylation pattern in S100A1-depleted ECs upon VEGF stimulation.15, 21, 25 In view of our in vitro data, enhanced in vivo activity of PKC in ischemic SKO tissue might also be in part accountable for inhibitory eNOS phosphorylation. In support of this notion, indistinguishable expression and phosphorylation of PP1 between groups rather excludes a role for abnormal activity of the phosphatase assigned to the p-T495 site.15 The role of other phosphatases potentially regulated by S100A1 such as PP5,29 however, requires further evaluation. Increased PLCγ expression and ERK1/2 activity might finally result from a relative increase in VEGFR1 signaling19 due to imbalances between VEGFR2 and VEGFR1 levels in ischemic SKO tissue. This might add to signaling abnormalities in ischemic SKO tissue.
Ultimate proof for the pathophysiological key role of compromised eNOS function in the SKO phenotype came from rescue experiments using pharmacological NO supplementation. NO was sufficient both to salvage ischemic SKO limbs and to restore in vivo microvasculature formation. Interestingly, a previous study showed that DETA/NO did not improve angiogenesis in ischemic hindlimbs of eNOS-/- mice.13 But this study used a 25-fold higher DETA-NO concentration, which suppressed in vivo angiogenesis in WT mice in our hands (data not shown). Hence, it seems reasonable to assume that endothelial S100A1-deficiency might blunt in vivo ischemic NO generation and angiogenesis both through interrupted S100A1-facilitated eNOS activation, unopposed PKC-mediated p-T495 phosphorylation and VEGFR2 degradation.
Despite the potential relevance of these novel mechanistic findings, important limitations of our study are noteworthy. First, FAR-induced hindlimb ischemia in otherwise healthy mice is not equivalent to CLI in humans which is frequently associated with comorbid illnesses30 and thereby warrants evaluation of S100A1’s role in the context of hyperglycemia and hypercholesterolemia. Second, in light of previously reported functions of S100A1 in endothelial Ca2+ homeostasis,3, 4 we expect dysfunctional eNOS not to be the sole mediator of the postischemic SKO phenotype.23 Besides NO, Akt suppresses various pro-apoptotic pathways and given attenuated Akt activity extended analysis of the potential susceptibility of SKO ECs to cell death is certainly needed. Third, oxidative stress resulting from inflammation and potential eNOS uncoupling may also play a role in defective angiogenesis in SKO mice.25 Fourth, NO generation in cellular assays was measured only indirectly by assessing NO2-/NO3- levels. More sensitive assays are certainly needed in subsequent studies deciphering the impact of the S100A1/eNOS interaction on NO homeostasis. Finally, a contribution of altered S100A1 skeletal muscle expression to ischemic limb loss is possible. The observed marginal attenuation of S100A1 expression in differentiated hypoxic skeletal muscle fibers and barely detectable S100A1 expression in WT skeletal myoblasts (Supplement Figure XIII.) nevertheless weakens the notion of a relevant role of skeletal muscle S100A1, at least in angiogenesis.
Overall, our clinically driven experimental study identifies S100A1 as critical for postnatal ischemic angiogenesis and eNOS function. Our molecular results, summarized in Supplement Figure XIV., provide advanced mechanistic insight demonstrating that the lack of endothelial S100A1 might attenuate in vivo eNOS activity by at least three distinct but additive/synergistic pathomechanisms – firstly, interrupted stimulatory S100A1/eNOS interaction; secondly, PKC hyperactivity that triggers inhibitory eNOS phosphorylation and abrogates VEGFR2 signal relay; and thirdly, as previously shown,3 insufficient IP3R-mediated Ca2+ release required for eNOS activation. These findings will prompt further studies to probe the microvascular therapeutic potential of restoring S100A1 levels in ischemic vascular disease.
Supplementary Material
Novelty and Significance.
What Is Known?
The EF-hand calcium (Ca2+) sensor protein S100A1, expressed in endothelial cells (ECs), facilitates endoplasmatic reticulum (ER) mediated Ca2+ release which promotes Ca2+-dependent eNOS activity.
Under normoxic conditions, the lack of endothelial S100A1 results in endothelial dysfunction by attenuating NO generation and agonist-induced vascular relaxation.
In ischemic tissue, the endothelial NO synthase (eNOS) is generated as a component of the vascular endothelial growth factor (VEGF) signaling pathway that is involved in postischemic revascularization.
What New Information Does This Article Contribute?
S100A1-deficient mice demonstrate postischemic hindlimb loss after arterial blood flow interuption due to defective postischemic angiogenesis. The latter related to defective VEGF-mediated eNOS activation and proangiogenic EC properties.
S100A1 promotes postischemic NO generation in ECs by directly activating eNOS via a Ca2+-dependent mechanism as well as by preventing the protein kinase C (PKC) mediated phosphorylation of the inhibitory threonine-495 eNOS site and VEGF receptor 2 degradation.
Molecular insights into defective vascular repair in S100A1-deficient mice links downregulated S100A1 expression in patients with advanced critical lower limb ischemia to impaired ischemic angiogenesis.
Here we describe a novel molecular pathomechanism with potential clinical significance for chronic critical limb ischemia (CLI), the most advanced form of peripheral arterial occlusive disease (PAOD). From our study, endothelial S100A1 emerged as an indispensable molecular factor for postischemic eNOS activation and angiogenesis, and which is therefore necessary for blood flow restoration and prevention of tissue necrosis, at least, in mice, and potentially in men. From a biological perspective, the central role of eNOS in angiogenic signaling in cardiovascular biology and disease highlights the importance of S100A1 as a direct regulator of eNOS activity. Its dual actions on Ca2+ and PKC activity, which can prevent the phosphorylation of eNOS on an inhibitors site and VEGF signaling, appear equally important. From a clinical standpoint, these observations link dysfunctional human ECs with hypoxia-induced loss of S100A1 and disturbed NO generation with insufficient reparative angiogenesis in patients with CLI and S100A1 expression loss in ischemic tissue. The advanced developmental stage of genetically-targeted therapies against heart failure that exploit S100A1’s pleiotropic actions in cardiomyocytes, will prompt translational research to probe the microvascular therapeutic potential of preserving or restoring endothelial S100A1 expression.
Acknowledgments
SOURCES OF FUNDING
This work was funded by NIH grants R01 HL 07842 (to KP), R01 HL 092130-01 and HL 092130-02S1 (to PM), and R01 HL56205 (to WJK), a postdoctoral fellowship from the American Heart Association (Great Rivers Affiliate) (to GR), the Rahel Goitein-Straus Scholarship from the Heidelberg University Medical Faculty (to CL), and grants from the Deutsche Forschungsgemeinschaft (#562 to PM and SP) and the German Cardiovascular Research Center (DZHK to PM and HAK).
Non-standard Abbreviations
- Akt
v-Akt murine thymoma viral oncogene homolog
- CaM
calmodulin
- CLI
critical limb ischemia
- DETA/NO
diethylenetriamine/NO
- EC
endothelial cell
- eNOS
endothelial nitric oxide synthase
- FAR
femoral artery resection
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GM
gastrocnemius muscle
- HGF
hepatocyte growth factor
- HIF1α
hypoxia inducible factor 1 alpha
- IGF
insulin-like growth factor
- IP
immunoprecipitation
- PAOD
peripheral arterial occlusive disease
- IP3(R)
inositol-1,4,5 trisphosphate (receptor)
- PDK
phosphoinositide dependent protein kinase
- PIP3
phosphatidylinositol 1,4,5 trisphosphate
- PI3K
phosphoinositide-3-kinase
- PKCɛ
protein kinase C epsilon
- PlGF
placental growth factor
- PP-1
Protein phosphatase 1
- SERCA
sarco/endoplasmic reticulum calcium-ATPase
- SKO
S100A1 knockout
- SKD
S100A1 knockdown
- SMA
smooth muscle actin
- TGFβ
transforming growth factor beta
- VEGF
vascular endothelial growth factor
- VEGFR2
vascular endothelial growth factor receptor 2
- vWF
von Willebrand Factor
- WB
western blot
- WT
wild-type
Footnotes
DISCLOSURES
None of the authors have any financial interests to disclose.
References
- 1.Lau JF, Weinberg MD, Olin JW. Peripheral artery disease. Part 1: Clinical evaluation and noninvasive diagnosis. Nat Rev Cardiol. 2011;8:405–418. doi: 10.1038/nrcardio.2011.66. [DOI] [PubMed] [Google Scholar]
- 2.Gupta R, Tongers J, Losordo DW. Human studies of angiogenic gene therapy. Circulation Research. 2009;105:724–736. doi: 10.1161/CIRCRESAHA.109.200386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pleger ST, Harris DM, Shan C, Vinge LE, Chuprun JK, Berzins B, Pleger W, Druckman C, Volkers M, Heierhorst J, Oie E, Remppis A, Katus HA, Scalia R, Eckhart AD, Koch WJ, Most P. Endothelial s100a1 modulates vascular function via nitric oxide. Circulation Research. 2008;102:786–794. doi: 10.1161/CIRCRESAHA.108.172031. [DOI] [PubMed] [Google Scholar]
- 4.Desjardins JF, Pourdjabbar A, Quan A, Leong-Poi H, Teichert-Kuliszewska K, Verma S, Parker TG. Lack of s100a1 in mice confers a gender-dependent hypertensive phenotype and increased mortality after myocardial infarction. American Journal of Physiology Heart and Circulatory Physiology. 2009;296:H1457–1465. doi: 10.1152/ajpheart.00088.2008. [DOI] [PubMed] [Google Scholar]
- 5.Kraus C, Rohde D, Weidenhammer C, Qiu G, Pleger ST, Voelkers M, Boerries M, Remppis A, Katus HA, Most P. S100a1 in cardiovascular health and disease: Closing the gap between basic science and clinical therapy. Journal of Molecular and Cellular Cardiology. 2009;47:445–455. doi: 10.1016/j.yjmcc.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Du XJ, Cole TJ, Tenis N, Gao XM, Kontgen F, Kemp BE, Heierhorst J. Impaired cardiac contractility response to hemodynamic stress in s100a1- deficient mice. Molecular and Cellular Biology. 2002;22:2821–2829. doi: 10.1128/MCB.22.8.2821-2829.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, Walsh K, Sessa WC. Akt1/protein kinase balpha is critical for ischemic and vegf-mediated angiogenesis. The Journal of Clinical Investigation. 2005;115:2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo Y, Stein AB, Wu WJ, Zhu X, Tan W, Li Q, Bolli R. Late preconditioning induced by no donors, adenosine a1 receptor agonists, and delta1-opioid receptor agonists is mediated by inos. American Journal of Physiology. Heart and Circulatory Physiology. 2005;289:H2251–2257. doi: 10.1152/ajpheart.00341.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi M, Inoue K, Warabi E, Minami T, Kodama T. A simple method of isolating mouse aortic endothelial cells. Journal of Atherosclerosis and Thrombosis. 2005;12:138–142. doi: 10.5551/jat.12.138. [DOI] [PubMed] [Google Scholar]
- 10.Friedrich O, Ehmer T, Fink RH. Calcium currents during contraction and shortening in enzymatically isolated murine skeletal muscle fibres. Journal of Physiology. 1999;517((Pt 3)):757–770. doi: 10.1111/j.1469-7793.1999.0757s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peppel K, Jacobson A, Huang X, Murray JP, Oppermann M, Freedman NJ. Overexpression of g protein-coupled receptor kinase-2 in smooth muscle cells attenuates mitogenic signaling via g protein-coupled and platelet-derived growth factor receptors. Circulation. 2000;102:793–799. doi: 10.1161/01.cir.102.7.793. [DOI] [PubMed] [Google Scholar]
- 12.Liang CC, Park AY, Guan JL. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nature Protocols. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 13.Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. The Journal of Clinical Investigation. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartley CJ, Reddy AK, Madala S, Entman ML, Michael LH, Taffet GE. Doppler velocity measurements from large and small arteries of mice. American Journal of Physiology. Heart and Circulatory Physiology. 2011;301:H269–278. doi: 10.1152/ajpheart.00320.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of thr(495) regulates ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circulation Research. 2001;88:E68–75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- 16.Sheu FS, Azmitia EC, Marshak DR, Parker PJ, Routtenberg A. Glial-derived s100b protein selectively inhibits recombinant beta protein kinase c (PKC) phosphorylation of neuron-specific protein f1/gap43. Brain Research Molecular Brain Research. 1994;21:62–66. doi: 10.1016/0169-328x(94)90378-6. [DOI] [PubMed] [Google Scholar]
- 17.Lin LH, Van Eldik LJ, Osheroff N, Norden JJ. Inhibition of protein kinase c- and casein kinase ii-mediated phosphorylation of gap-43 by s100 beta. Brain Research. Molecular Brain Research. 1994;25:297–304. doi: 10.1016/0169-328x(94)90165-1. [DOI] [PubMed] [Google Scholar]
- 18.Fraisl P, Mazzone M, Schmidt T, Carmeliet P. Regulation of angiogenesis by oxygen and metabolism. Developmental Cell. 2009;16:167–179. doi: 10.1016/j.devcel.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. Vegf receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 20.Singh AJ, Meyer RD, Band H, Rahimi N. The carboxyl terminus of vegfr-2 is required for pkc-mediated down-regulation. Molecular Biolology of the Cell. 2005;16:2106–2118. doi: 10.1091/mbc.E04-08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 22.Heil M, Schaper W. Influence of mechanical, cellular, and molecular factors on collateral artery growth (arteriogenesis) Circulation Research. 2004;95:449–458. doi: 10.1161/01.RES.0000141145.78900.44. [DOI] [PubMed] [Google Scholar]
- 23.Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circulation Research. 2007;100:782–794. doi: 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- 24.Staton CA, Reed MW, Brown NJ. A critical analysis of current in vitro and in vivo angiogenesis assays. International Journal of Experimental Pathology. 2009;90:195–221. doi: 10.1111/j.1365-2613.2008.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forstermann U, Sessa WC. Nitric oxide synthases: Regulation and function. European Heart Journal. 2011 doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu J, deMuinck ED, Zhuang Z, Drinane M, Kauser K, Rubanyi GM, Qian HS, Murata T, Escalante B, Sessa WC. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10999–11004. doi: 10.1073/pnas.0501444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsurumi Y, Murohara T, Krasinski K, Chen D, Witzenbichler B, Kearney M, Couffinhal T, Isner JM. Reciprocal relation between vegf and no in the regulation of endothelial integrity. Nature Medicine. 1997;3:879–886. doi: 10.1038/nm0897-879. [DOI] [PubMed] [Google Scholar]
- 28.Ouchi N, Oshima Y, Ohashi K, Higuchi A, Ikegami C, Izumiya Y, Walsh K. Follistatin-like 1, a secreted muscle protein, promotes endothelial cell function and revascularization in ischemic tissue through a nitric-oxide synthase-dependent mechanism. Journal of Biological Chemistry. 2008;283:32802–32811. doi: 10.1074/jbc.M803440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamaguchi F, Umeda Y, Shimamoto S, Tsuchiya M, Tokumitsu H, Tokuda M, Kobayashi R. S100 proteins modulate protein phosphatase 5 function: A link between ca2+ signal transduction and protein dephosphorylation. Journal of Biological Chemistry. 2012 doi: 10.1074/jbc.M111.329771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waters RE, Terjung RL, Peters KG, Annex BH. Preclinical models of human peripheral arterial occlusive disease: Implications for investigation of therapeutic agents. Journal of Applied Physiology. 2004;97:773–780. doi: 10.1152/japplphysiol.00107.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.