

Abstract
The discovery of endothelial derived relaxing factor (EDRF) and later nitric oxide (NO) as a biologically active substance led to intense focus on the vascular endothelium as a major site of physiological regulation and pathophysiological dysfunction. NO is clearly a potent vasodilator and plays a key role in establishing both whole body and regional “vascular tone.” In this context, skeletal muscle and human skin have the remarkable capacity to increase their blood flow 50–100 fold and this increase is due almost exclusively to local vasodilation. In general the mechanisms responsible for these vasodilator phenomena have been poorly understood. In the early 1990s, we and others started to ask if NO might explain the “unexplained” vasodilator responses seen in skeletal muscle and skin. We also asked how “NO tone” interacted with “sympathetic tone” and whether NO can override the vasoconstrictor responses normally generated when sympathetic nerves release norepinephrine. To our surprise we found that NO plays only a modest (non-obligatory) role in exercise hyperemia, reactive hyperemia and the neurally mediated rise in skin blood flow during whole body heat stress. By contrast, NO plays a major role in the skeletal muscle vasodilator responses to mental stress and the skin dilator responses to local heating. In animals, but not humans, NO can limit the ability of the sympathetic nerves to cause vasoconstriction in exercising muscles. Thus the role of NO in two of the most extreme dilator responses seen in nature is limited and in muscle the sympathetic nerves can restrain the dilation to defend arterial blood pressure.
Keywords: Endothelial function, vasodilation, muscle blood flow, skin blood flow, exercise
INTRODUCTION
The purpose of this talk is to summarize information presented at the 10th Mechanisms of Vasodilatation (MOVD) meeting held in Matsushima, Miyagi, Japan, in June 2009. The basis for this brief review is the MOVD Shepherd Lecture, which reviewed our work on the role of nitric oxide (NO) in physiological vasodilation in humans, and how NO might or might not limit sympathetic vasoconstriction in contracting skeletal muscles. In some forms of physiological vasodilation such as that seen in the forearm during mental stress or in human skin during local heating, endogenous NO plays a major role in the vasodilator responses (1,2). By contrast, it contributes but is not obligatory to the marked vasodilation seen during exercise, reactive hyperemia, and reflex cutaneous vasodilation associated with increases in core temperature (3,4,5). Additionally, in some models NO can limit the ability of the sympathetic nerves to cause vasoconstriction in active skeletal muscles, but this phenomenon, if it exists, is much less prominent in humans (6). However, before we get to the scientific aspects of the review, we would like to present some information about Dr. John T. Shepherd.
Who is John T. Shepherd
John T. Shepherd was born in Northern Ireland in 1919 (7). In high school he was a serious student but sometimes ran afoul of the school authorities and was subject to the harsh corporal punishment (caning) typical of elite boarding schools in the United Kingdom at that time. He also excelled in running and was highly competitive at the half-mile (800 meters). After high school, in the late 1930’s, we was apprenticed to the railroads but found that job unsatisfying and after a year or two joined his two older brothers in medical school at the Queen’s University in Belfast. Initially, his goal was to become a surgeon but he was convinced by the note physiologist Henry Barcroft that working in physiology research would improve his chances of obtaining surgical training. Subsequently, he joined the “Belfast Group” during World War II in which he remained affiliated with until moving to Mayo permanently in the late 1950’s. His first research focused on the collateral circulation around the hip and in the thigh in an effort to provide information relevant to surgical treatment of traumatic leg injuries suffered by soldiers during World War II.
During the early and middle 1950’s he and the group in Belfast conducted a remarkable series of studies on vasodilating and vasoconstricting factors in the human forearm using venous occlusion plethysmography and brachial artery infusions of drugs (8). They also studied exercise, reactive hyperemia, and mental stress. In the context of this talk, and the MOVD meeting, it is interesting to note that as early as 1953 they had clearly demonstrated that brachial artery infusions of acetylcholine caused marked vasodilation (Figure 1) (9). However, the mechanisms responsible for the dilation remained elusive until the discovery of an endothelial derived relaxing factor (EDRF)/NO in the 1980s.
Figure 1.

Forearm vasodilator responses to brachial artery infusions of acetylcholine. This was the first clear demonstration that the human forearm vasodilates and responds to acetylcholine. The mechanisms responsible for this vasodilation could not be explained until the emergence of EDRF/NO. (Figure from ref. 9)
Dr. Shepherd first came to Mayo in September of 1953 for one year to work with Earl H. Wood, a pioneer in cardiac catheterization. He then returned to Belfast and was invited back to Mayo to join the permanent staff as a consultant in physiology in 1957. At Mayo he developed a robust research program on local and reflex control of the circulation and branched out into studies in isolated blood vessels and animal models. He also held a variety of important institutional, national, and international leadership positions including serving as Dean of the Mayo Medical School, President of the American Heart Association, Director of Research and long term membership on the Board of Governors of Mayo Foundation. During the “cold war” he also facilitated research collaboration between the U.S. and Soviet space programs. Dr. Shepherd is also known for his many publications, masterful review articles, and several textbooks including The Human Cardiovascular System: Facts and Concepts (1979) that he coauthored with Paul Vanhoutte (10). At this writing, Dr. Shepherd is now 90 years old and unfortunately suffering from Alzheimer’s disease and is no longer able to participate in the international scientific exchanges he enjoyed immensely. With these vignettes as a background, we will now proceed to the main body of the review article.
History of the Vascular Endothelium as a Site of Vascular Regulation
Until the seminal observation of Furchgott and Zawadzki, the vascular endothelium was largely seen as having a protective barrier function separating the circulating factors in the blood from the tissues (11). This all changed with the demonstration that the vascular endothelium released a so-called “endothelial-derived relaxing factor” that mediated the vasodilator responses to acetylcholine. Later the main EDRF was identified as NO during the MOVD meeting held in Rochester, MN, in 1986. These observations also formed the basis of the Nobel Prize in Medicine and Physiology which was awarded to Furchgott, Ignarro, and Murad in 1998.
Does NO Explain Unexplained Vasodilation
With the emergence of NO as the “main” EDRF, a major question that arose and remains is “what role does NO play in physiological vasodilation and especially unexplained vasodilation?” We use the term “unexplained vasodilation” to highlight a number of forms of vasodilation that can occur in response to physiological stimuli but were unexplained in the middle- to late-1980’s and into the present. When EDRF/NO emerged there were five major forms of physiological vasodilation in humans that had been observed for many years but had defied clear-cut explanations. Therefore, we wondered if NO was the missing vasodilator substance. These areas included the rise in forearm blood flow seen during mental stress in humans, the rise in skin blood flow seen during local and reflex heating in humans, exercise hyperemia, and reactive hyperemia.
To study these phenomena we revivified many of the approaches that the Belfast group had mastered in the 1950’s and added a few new wrinkles of our own including laser Doppler studies of the skin, and the use of Doppler ultrasound to measure beat-to-beat changes in brachial artery and hence forearm blood flow (12). Figure 2 shows our general experimental set-up. Our studies were also facilitated by the development of arginine analogs that could be administered to humans to block nitric oxide synthase (NOS).
Figure 2.
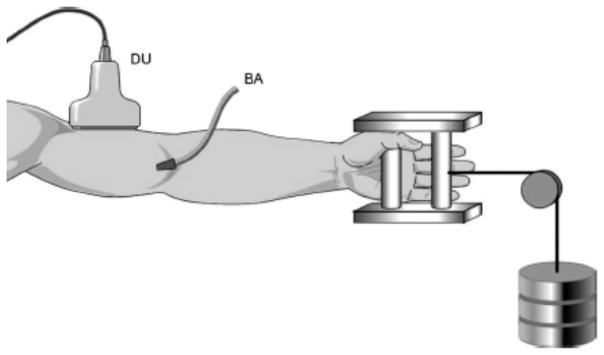
Schematic diagram of our general experimental set-up. We have used brachial artery drug infusions and measured forearm blood flow with plethysmography or Doppler ultrasound to explore the contributions of NO and other vasodilating factors to the regulation of forearm muscle and skin blood flow during mental stress, local and reflex heating, exercise, and reactive hyperemia. DU = Doppler ultrasound; BA = Brachial artery catheter
Mental Stress
Our first study on the role of NO as a mediator of physiological vasodilation tested the hypothesis that NO contributes to the forearm vasodilator responses to mental stress in humans (1). Prior to our study, it had been known for many years that mental stress can evoke marked forearm vasodilation. In animals the so-called “defense reaction” can also evoke a skeletal muscle vasodilation and is mediated by sympathetic cholinergic nerves (13). In humans the forearm blood flow response to mental stress can be blunted but not eliminated by atropine (14). Additionally, in humans it has been difficult to clearly establish whether or not there are sympathetic vasodilator nerves to skeletal muscle, but the preponderance of evidence suggests that in contrast to animals such as cats, dogs, and rodents, there are no sympathetic cholinergic nerves innervating skeletal muscle (15).
There are many interesting anecdotes concerning mental stress and the strategies used to evoke stress in the subjects. Prior to the advent of modern human subject protection committees, a variety of deceptive techniques were used in unsuspecting subjects and truly impressive levels of vasodilation were observed (13). In more recent times, mental arithmetic, the color word test, or other forms of stress have been used. Along these lines, it should also be noted that individual subjects’ responses to mental stress is highly variable. In some subjects there is a marked rise in blood pressure and heart rate, while other subjects demonstrate almost no response. Some subjects also respond more profoundly to one form of stress and are essentially non-responsive to other forms.
In our studies we have adopted the color word test to evoke mental stress (1). In this test, slides with the names of colors are shown to the subject. However, the font color of the letters used to spell the name of the color is different. For example, the word yellow is spelled with blue letters. The subject is then asked to say the color versus the word which evokes mental stress and the expected rise in heart rate and blood pressure. When this technique was used to cause mental stress in human volunteers who had had one forearm treated with the NOS inhibitor L-NMMA, the forearm vasodilator responses to mental stress were reduced by about 60–70% (Figure 3). We also demonstrated that at least some of the dilator response could also be blunted by atropine.
Figure 3.
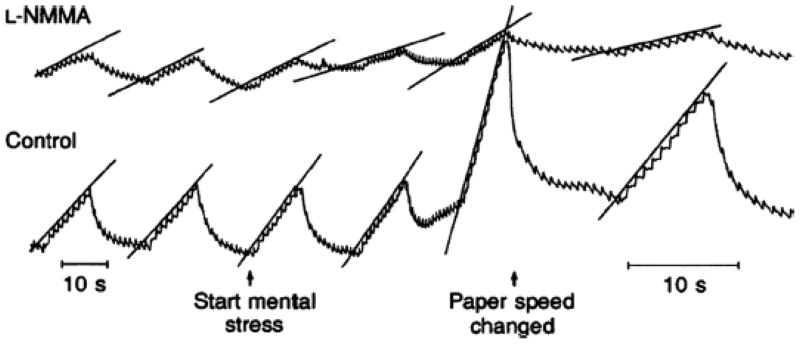
Effects of NOS inhibition via brachial artery infusions of L-NMMA on the forearm vasodilator responses to mental stress. This was one of the first demonstrations that NO might explain “unexplained” vasodilation in humans. (Figure from ref. 1)
Since there is little evidence for sympathetic cholinergic nerves in the human forearm, we hypothesized that local release of acetylcholine from endothelial cells might have contributed to the dilation. Additionally, the increase in blood pressure and heart rate associated with the mental stress could have caused shear or mechanically-mediated release of NO from the vascular endothelium. Mental stress can also cause a rise in circulating epinephrine which can evoke beta-2 mediated vasodilation (in part due to NO release) in skeletal muscle of humans (15). In summary, in our first study on NO and unexplained vasodilation in humans, we found strong evidence that NO is a major contributor to the vasodilator responses to mental stress.
Skin Blood Flow: Whole Body Heating
At rest in a thermoneutral or cool environment skin blood flow is quite low. However, in response to either local heating or increases in core temperature, there can be marked vasodilation in the cutaneous circulation. During whole body heating, when core temperature rises 1–2°C, skin blood flow can rise from a total of 200–300 ml/min all the way to values of 6–8 l/min (16). Additionally, when cool, skin blood flow can approach zero. Thus, the ability of the skin to vasodilate in response to either local or whole body heating is dramatic.
In our studies on the role of NO in skin blood flow, we evaluated the effects of NOS inhibition on the reflex increases in skin blood flow that occur during whole body heating. It has been known for many years that when core temperature rises more than ~.5°C, sweating begins and there is a marked rise in skin blood flow. This increase in flow is neurally mediated. Since the rise in flow occurs at about the same time that sweating commences, one idea is that the acetylcholine released from the sympathetic cholinergic nerves that innervate sweat glands also cause an increase in skin blood flow. However, studies as early as the 1950’s demonstrated that while atropine can delay the onset of cutaneous vasodilation during whole body heating it has little overall impact on the magnitude of the rise in flow (17). More recently it has been shown that while sympathetic cholinergic nerves play an obligatory role in the cutaneous dilator responses seen during whole body heating, the substance that causes the dilation is not acetylcholine, but instead some unknown vasodilating co-transmitter (18).
When one forearm is treated with NOS inhibitors via brachial artery infusions of arginine analogs, the rise in skin blood flow during whole body heating is only marginally affected (19). When the NOS inhibitors are given via microdialysis catheters inserted just below the skin and laser Doppler devices are used to measure the rise in flow it appears as if NO contributes about 30% of the total vasodilation seen in the skin during whole body heating (Figure 4) (4). In this context, NOS inhibition can frequently cause a 20–30% reduction in flow during many forms of vasodilation because many vasodilating substances act on both vascular smooth muscle and endothelial cells. This general observation has been made by numerous laboratories and suggests that NO contributes to but is not the major factor responsible for the cutaneous vasodilation seen in whole body heating in humans.
Figure 4.

Effects of NOS inhibition delivered via microdialysis to the skin on the cutaneous vasodilator responses to whole body heating in humans. Microdialysis of atropine had little effect on this response even though local sweating was abolished. By contrast, NOS inhibition caused ~30% reduction in skin blood flow on the affected site. When whole body cooling began blood flow on both the treated and untreated sites fell dramatically. (Figure from ref. 4)
A variety of additional candidate dilator substances have been suggested as contributing to the neurally-mediated rise in skin blood flow seen during body heating in humans (20,21). However, the vasodilating factor or factors that mediate the skin blood flow responses to whole body heating remains one of the great mysteries of integrative physiology in humans.
Skin Blood Flow: Local Heating
Local heating of a small area of skin can also evoke vasodilation. Depending on the absolute heat applied and the rate of heating, the pattern of cutaneous vasodilation can vary. However, with a step increase in local heating there is typically a bimodal skin vasodilator response (2). There is an initial peak response that is largely dependent on an axon reflex. This peak is followed by a “nadir” a few minutes later and then a prolonged rise in skin blood flow as heating continues. To address the mechanisms responsible for both the pattern and magnitude of the vasodilation we used a combination of nerve blocks, topically applied local anesthesia, and NOS inhibition with microdialysis to “pharmacodissect” this response (Figure 5). The initial response was only minimally affected by cutaneous nerve block but dramatically blunted by topical anesthesia, suggesting that an axon reflex was critical in its generation. This initial response was also only minimally influenced by NOS inhibition. By contrast, the more prolonged “plateau” response was highly NO-dependent. Therefore, NO appears to be the main dilating substance associated with the sustained rise in cutaneous blood flow seen during local heating. It is also interesting to note that NO plays a different role in these two forms of cutaneous vasodilation.
Figure 5.

Effects of NOS inhibition delivered via microdialysis to the skin on the cutaneous vasodilator responses to local heating. NOS inhibition has little effect on the initial peak response to local heating, but markedly blunts the sustained response. (Figure from ref. 2)
Exercise
As is the case with the marked rise in skin blood flow seen during body heating, the increase in blood flow to contracting skeletal muscles can be marked. In fact, blood flow to a maximally perfused small mass of active muscle can increase 50- to 100-fold. In other words, blood flow at rest can be on the order of 2–3 ml/100g/tissue/min and increase to 200–300 ml/100g/min (22,23). In some skeletal muscle vascular beds, values close to 400–500 ml/100g/min have been reported.
Along these lines, the factors responsible for this marked vasodilation have been of interest to physiologists since at least the 1870’s. At that time Gaskell identified a number of potential mechanisms that could cause this marked hyperemia including the mechanical effects of contraction on the vessels, substances transported in the blood, substances released by nerves, or substances (metabolites) released by the contracting skeletal muscles (24).
In spite of >130 years of almost continuous investigation on the mechanisms responsible for the marked vasodilation in skeletal muscle during exercise, no single factor or substance has emerged. To address the role of NO in exercise hyperemia, we used our standard techniques along with rhythmic forearm handgripping. Brachial artery infusions of NOS inhibitors have been given before and during rhythmic handgripping. Blood flow has been measured using Doppler ultrasound and depending on the circumstances NOS inhibition causes up to a 10–20% reduction in forearm blood flow during exercise (5,25). However, analysis of the data is confounded by the fact that NOS inhibition also reduces resting blood flow so that when the rise in flow above baseline is considered, the decrement in flow is on the order of perhaps 10%.
In general, investigators using other models including the leg during one-leg exercise or during whole body exercise have demonstrated a modest role for NO in exercise hyperemia. NOS inhibition during whole body exercise can also cause interpretative challenges as a result of the changes in systemic arterial pressure. Since whole body NOS inhibition causes a 10–20 mm increase in arterial pressure it can activate baroreflexes and cause a concurrent reduction in sympathetic outflow to the skeletal muscles. This means that while the NOS inhibition is clearly blocking a vasodilator signal (NO), it is also indirectly removing a tonic vasoconstrictor signal (norepinephrine released by sympathetic traffic). This combination of events makes interpretation of whole body NOS inhibition data more challenging especially during conditions like exercise.
With the limited effects of NOS inhibition on exercise hyperemia (whatever model is used) there is also a powerful indirect argument against a major role for adenosine and adenosine-related metabolites as major factors contributing to exercise hyperemia under normal free-flow conditions. This assertion is made because when exogenous adenosine and ATP are infused into the brachial (adenosine) or femoral (ATP) arteries the vasodilator responses to these drugs can be markedly attenuated in the presence of NOS inhibitors (26–28). If these compounds were obligatory for exercise hyperemia under normal conditions, one would anticipate a greater blunting of the blood flow responses to exercise by NOS inhibitors.
In contrast to the responses seen during exercise where NOS inhibition typically has only a modest effect, the recovery of blood flow following exercise is also faster after NOS inhibition in almost every model that has ever been studied in humans. Finally, one idea is that it is difficult to evaluate the role of NO in exercise hyperemia because redundant dilator systems govern this response so that if one pathway is blocked other dilating mechanisms become more prominent leading to a normal dilator response. In this context, when NOS inhibition is given along with inhibitors of prostaglandins and other vasodilating factors that might contribute to exercise hyperemia, it has been difficult to show that NO plays a major role in exercise hyperemia (Figure 6). What is remarkable about exercise hyperemia is that it continues to exist even after multiple drugs affecting multiple vasodilator pathways are administered (26). This suggests that either some unknown factor or factors are responsible for the dilation or it is an example of a super redundant biological system.
Figure 6.
Effects of NOS inhibition followed by cyclooxygenase inhibition on blood flow to rhythmically contracting forearm muscles. NOS inhibition has a modest but sustained effect on this response, cyclooxygenase inhibition a less sustained response. (Figure from ref. 5)
Reactive Hyperemia
Following periods of ischemia to almost any organ there is an immediate and marked hyperemia that occurs after the relief of the ischemia and the restoration of perfusion pressure. This is followed by an exponential decay of the hyperemia as the flow returns to baseline. In the human forearm, as the period of ischemia is increased from 1–5 min there is an increase in the peak blood flow, and the time required for the flow to return to baseline is also longer. When the interruption of blood flow is extended to 10–20 min the peak blood flow response does not increase much, but rather than a prompt exponential recovery of the flow, the peak response is maintained for a period of minutes prior to recovery (29).
Like exercise, reactive hyperemia has been an area of intense research focus for more than a century. The main ideas are that either unloading of the blood vessels during the period of ischemia when perfusion pressure is essentially zero causes a mechanically-initiated vasodilation, or that vasodilating substances released from the ischemic tissue (metabolites) cause the hyperemia. However, as is the case with exercise, it has been difficult to show that any specific substance is absolutely obligatory in this response. At the time of the discovery of EDRF/NO the only substance that had been clearly established to participate in reactive hyperemia in the human forearm were vasodilating prostanoids. Cyclooxygenase inhibitors could reduce the peak hyperemic response, and depending on the experiment and drug used, also alter the recovery of flow (30).
Using our standard techniques we evaluated the contribution of NO to forearm reactive hyperemia after 5 min of ischemia (3). When the data was analyzed in terms of absolute flow, NOS inhibition caused a modest reduction in the peak blood flow response and the recovery of flow was faster. However, NOS inhibition also reduced baseline flow and when the data was analyzed as change in flow above baseline, the peak flow responses were no longer different, but the recovery was still faster during NOS inhibition. This faster recovery may be due to less flow mediated NO release during the recovery period. In general, studies reporting that NOS inhibition reduced peak blood flow during reactive hyperemia have not considered the impact of altered baseline flow on the hyperemic response.
When we performed NOS inhibition after subjects had taken 1200 mg of ibuprofen to study potential NO/prostaglandin interactions during reactive hyperemia we found that the peak blood flow response following ischemia was clearly blunted when both NOS inhibitors and cyclooxygenase inhibitors were given (Figure 7). By contrast, the recovery of blood flow seemed to be prolonged during dual inhibition.
Figure 7.

Effects of NOS inhibition alone and in combination with cyclooxygenase inhibition reactive hyperemia after 5 min of brachial artery occlusion. The upper panel shows the absolute blood flow values and the lower panel shows the change from baseline, an important consideration because NOS inhibition reduces baseline FBF. NOS inhibition had little impact on the rise in forearm blood flow above baseline immediately following the restoration of flow to the forearm. However, it did speed the recovery of flow to baseline. (Figure from ref. 3)
Thus, it appears that NO is not an obligatory component of the reactive hyperemia response in the human forearm. Since most of the blood flow responses in the forearm during reactive hyperemia occur in the skin and skeletal muscle, this suggests that NO is not essential to see a nearly normal reactive hyperemia response in those tissues.
Summary of Unexplained Vasodilation and NO
In the above section we have shown that NO is a key dilating substance in two forms of vasodilation that were “unexplained” prior to the discovery of EDRF/NO. NO clearly has a major role in the forearm vasodilator responses to mental stress, but its release is probably evoked by a number of convergent mechanisms including the mechanical stimuli associated with the increases in heart rate and blood pressure seen during mental stress, epinephrine acting via beta-2 receptors, and perhaps local release of acetylcholine from selected endothelial cells (4,15). At this time, there is no convincing evidence that human muscle is innervated by sympathetic cholinergic nerves as is the case in a number of animal species. NO also appears to play a major role in the sustained vasodilation seen during local heating (2). Again, the mechanism responsible for the NO release is unknown. By contrast, NO has only a modest role in the hyperemic responses seen during reflex increases in skin blood flow during whole body heating, exercise hyperemia, and reactive hyperemia. Whether these vasodilator phenomena are caused primarily by some currently “unknown” vasodilating substance or mechanism, or whether a host of “super” redundant mechanisms govern them is currently not known.
Nitric Oxide and Blood Pressure in Humans
In the earlier sections of this review we have focused on NO and specific vasodilator responses in humans. An equally important question is what role does NO play in whole body blood pressure regulation in humans? In this context, a number of investigators have given systemic doses of NOS inhibitors (usually L-NMMA or L-NAME) to humans. In some studies doses of NOS inhibitors that reduce whole body NO synthesis by ~70% have been given. With these doses mean arterial pressure typically rises 15–25 mmHg (31,32).
In the context of the above observations, we became interested in developing a better understanding of how NO contributes to systemic hemodynamics in humans. One problem with systemic doses of NOS inhibition is that the rise in blood pressure they evoke can stimulate the baroreflexes and cause a reflex reduction in sympathetic outflow. This means what while a powerful vasodilating substance (NO) is inhibited, an equally powerful constricting mechanism is withdrawn. Along these lines, when cardiac output is measured during NOS inhibition, the combined rise in mean arterial pressure and fall in cardiac output (from ~6–7 L/min to ~4 L/min) means that the vascular resistance almost doubles. Additionally, when NOS inhibition occurs after systemic α-adrenergic blockade the rise in vascular resistance is greater than seen without systemic α-adrenergic blockade confirming the competing roles of NO and the sympathetic nervous system in blood pressure regulation.
In summary, measurements of cardiac output during whole body NOS inhibition suggests that the contribution of NO to baseline vascular tone in young healthy subjects is even greater than that suggested by the increases in blood pressure seen during NOS inhibition (31).
To better understand the interactions between sympathetic tone, blood pressure, and NOS inhibition, we performed systemic NOS inhibition in normotensive subjects with varying levels of baseline sympathetic outflow as assessed by direct measures of muscle sympathetic nerve activity (MSNA) (32,33). The idea is that since baseline MSNA can vary markedly in young normotensive subjects, the vasoconstricting actions of high MSNA are offset by NO in a way that “buffers” the effects of high levels of MSNA on blood pressure. The main finding of this study was that during low and moderate doses of systemic NOS inhibition, individuals with high levels of resting MSNA had larger increases in blood pressure than subjects with low levels of resting MSNA (Figure 8). While the relationships are complex, they suggest that in subjects with high levels of MSNA NO plays a critical role in limiting the blood pressure raising effects of the high levels of MSNA (33,34). This finding has implications for hypertension, and raises the hypothesis that individuals with high levels of resting sympathetic nerve activity may be more susceptible to the development of hypertension if their “endothelial function” becomes impaired. This may account for the relationships between MSNA and blood pressure in younger vs. older adults (35,36).
Figure 8.
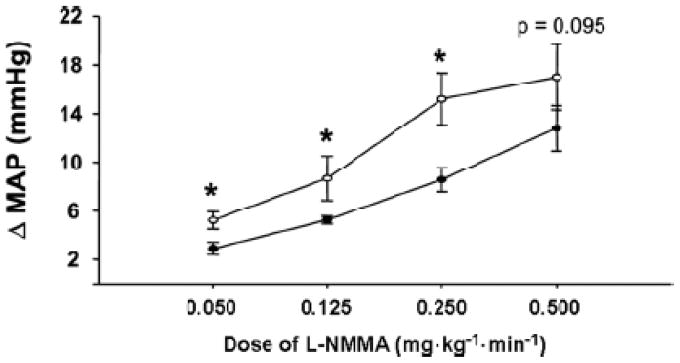
Interactions between differing levels of baseline MSNA in young healthy subjects and the blood pressure responses to whole body NOS inhibition. Individuals with high levels of sympathetic activity have a greater increase in blood pressure during low and moderate dose NOS inhibition.
NO versus Sympathetic Nervous System during Exercise
In an earlier section of this review we discussed the potential role of NO in exercise hyperemia and concluded that at best, NO plays a modest role in exercise hyperemia and is clearly not obligatory. However, there are important potential interactions between NO and sympathetic vasoconstriction that might occur during exercise. Along these lines, the marked increase in skeletal muscle blood flow seen during exercise is a potential threat to whole body blood pressure regulation in humans. If peak skeletal muscle blood flow can be 2–3 L/kg/min, and if 10 kg of skeletal muscle is active, it is possible that the demand for blood flow by the skeletal muscles might outstrip the supply available by cardiac output which is ~20 L/min in healthy young male subjects (24). The threat posed by exercising skeletal muscle to systemic blood pressure regulation is seen in patients with various forms of autonomic failure or after surgical sympathectomy for hypertension (37). When these subjects exercise, even while supine or in the head-down position, blood pressure falls during exercise because the sympathetic nervous system is apparently unavailable to restrain blood flow to active skeletal muscle.
At the same time, there is evidence that the vasodilating factors released by skeletal muscle limit the ability of the sympathetic nerves to cause vasoconstriction in active skeletal muscles (38). Thus, during exercise there is a balance among vasodilation in skeletal muscle, the effects of the vasodilating substances on sympathetic vasoconstriction, and whole body blood pressure regulation. In animal species, there is evidence that NO plays a critical role in limiting sympathetic vasoconstriction via a process known as functional sympatholysis (39). In this context, we tested the ability of NO to blunt sympathetic vasoconstriction using our brachial artery drug infusion, forearm exercise, and Doppler blood flow model. We demonstrated that while exercise blunts sympathetic vasoconstriction in active skeletal muscle, it does not (in contrast to animal studies) eliminate it (Figure 9) (6). More importantly, in a series of studies we demonstrated that NO (again, in contrast to animal studies) was not the substance responsible for the blunting of sympathetic vasoconstriction in active muscles (40,41). Since the vast majority of cardiac output is directed to active skeletal muscles during exercise, modest vasoconstriction in these beds can have a marked affect on arterial pressure. When these results are viewed in the context of competition between the active skeletal muscles demand for blood flow during exercise and systemic arterial pressure regulation, it appears as if just enough sympathetic vasoconstriction remains in active skeletal muscles to permit blood pressure to be regulated.
Figure 9.
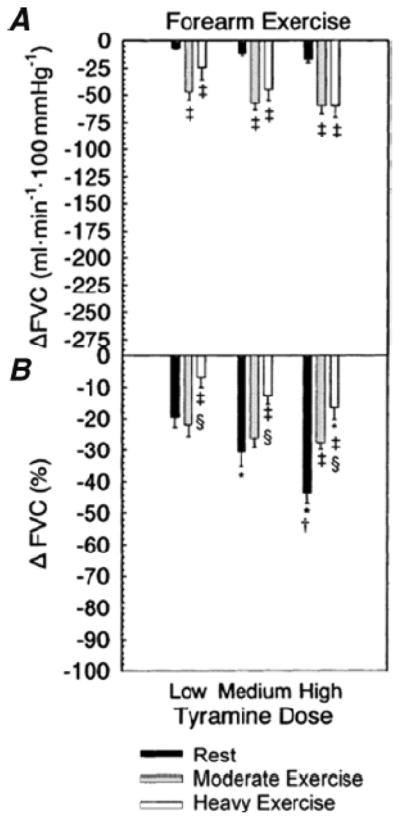
Effects of exercise on alpha-adrenergic vasoconstrictor responses in the human forearm. Vasoconstriction was evoked by stimulating endogenous norepinephrine released from the forearm sympathetic nerves by infusions of tyramine. Doses of tyramine were adjusted to account for the differences in blood flow at rest and during exercise. These data show that exercise blunts but does not eliminate the vasoconstrictor responses evoked by tyramine. Because the vast majority of cardiac output is directed to the contracting skeletal muscles during whole body exercise in humans, the remaining vasoconstrictor responses seen during exercise can have a profound effect on whole body blood pressure. (Figure from ref. 6)
Summary Closing Remarks
Over the last 15 or so years, we have attempted to understand the role that NO plays in explaining “unexplained” vasodilation, and we more generally have attempted to understand its role in integrative circulatory control of the human cardiovascular system. In this context, NO is a major physiological vasodilator in response to some hyperemic stimuli but not others. It also plays an important role in blood pressure regulation. After 1980, thinking about the contributions of vascular smooth muscle to blood flow or blood pressure regulation without thinking about the vascular endothelium was incomplete. In 2009 thinking about the role of NO without thinking about how it might interact with cardiovascular reflexes and the sympathetic nervous system is also incomplete. The studies of Dr. John T. Shepherd set the stage for many of the findings outlined in this review article and the conceptual framework he advocated continues to guide our thinking.
Acknowledgments
Supported by
National Institutes of Health research grants HL46493, HL83947, NS32352, and AR55819; and by the Frank R. and Shari Caywood Professorship, Mayo Foundation
References
- 1.Dietz NM, Rivera JM, Eggener SE, Fix RT, Warner DO, Joyner MJ. Nitric oxide contributes to the rise in forearm blood flow during mental stress in humans. J Physiol. 1994;480:361–368. doi: 10.1113/jphysiol.1994.sp020366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minson CT, Berry LT, Joyner MJ. Nitric oxide and neurally mediated regulation of skin blood flow during local heating. J Appl Physiol. 2001;91:1619–1626. doi: 10.1152/jappl.2001.91.4.1619. [DOI] [PubMed] [Google Scholar]
- 3.Engelke KA, Halliwill JR, Proctor DN, Dietz NM, Joyner MJ. Contribution of nitric oxide and prostaglandins to reactive hyperemia in human forearm. J Appl Physiol. 1996;81:1807–1814. doi: 10.1152/jappl.1996.81.4.1807. [DOI] [PubMed] [Google Scholar]
- 4.Shastry S, Minson CT, Wilson SA, Dietz NM, Joyner MJ. Effects of atropine and L-NAME on cutaneous blood flow during body heating in humans. J Appl Physiol. 2000;88:467–472. doi: 10.1152/jappl.2000.88.2.467. [DOI] [PubMed] [Google Scholar]
- 5.Schrage WG, Joyner MJ, Dinenno FA. Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol. 2004;557:599–611. doi: 10.1113/jphysiol.2004.061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tschakovsky ME, Sujirattanawimol K, Ruble SB, Valic Z, Joyner MJ. Is sympathetic neural vasoconstriction blunted in the vascular bed of exercising human muscle? J Physiol. 2003;541:623–635. doi: 10.1113/jphysiol.2001.014431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shepherd JT. Inside the Mayo Clinic: A Memoir. Afton Historical Society Press; 2004. [Google Scholar]
- 8.Shepherd JT. Physiology of the Circulation in Human Limbs in Health and Disease. WB Saunders Company; Philadelphia: 1963. [Google Scholar]
- 9.Duff F, Greenfield ADM, Shepherd JT, Thompson ID. A quantitative study of the response to acetylcholine and histamine of the blood vessels of the human hand and forearm. J Physiol. 1953;120:160–170. doi: 10.1113/jphysiol.1953.sp004883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shepherd ST, Vanhoutte PM. The Human Cardiovascular System. Facts and Concepts. Raven Press; New York: 1979. [Google Scholar]
- 11.Furchgott RF. The discovery of endothelium-derived relaxing factor and its importance in the identification of nitric oxide. JAMA. 1996;276:1186–1188. [PubMed] [Google Scholar]
- 12.Joyner MJ, Dietz NM, Shepherd JT. From Belfast to Mayo and beyond: the use and future of plethysmography to study blood flow in human limbs. J Appl Physiol. 2001;91:2431–2441. doi: 10.1152/jappl.2001.91.6.2431. [DOI] [PubMed] [Google Scholar]
- 13.Roddie IC. Human responses to emotional stress. Ir J Med Sci. 1977;146:395–417. doi: 10.1007/BF03030998. [DOI] [PubMed] [Google Scholar]
- 14.Blair DA, Glover WE, Greenfield ADM, Roddie IC. Excitation of cholinergic vasodilator nerves to human skeletal muscles during emotional stress. J Physiol. 1959;148:633–647. doi: 10.1113/jphysiol.1959.sp006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joyner MJ, Dietz NM. Sympathetic vasodilation in human muscle. Acta Physiol Scand. 2003;177:329–336. doi: 10.1046/j.1365-201X.2003.01090.x. [DOI] [PubMed] [Google Scholar]
- 16.Koroxenidis GT, Shepherd JT, Marshall RJ. Cardiovascular response to acute heat stress. J Appl Physiol. 1961;16:869–872. doi: 10.1152/jappl.1961.16.5.869. [DOI] [PubMed] [Google Scholar]
- 17.Roddie IC, Shepherd JT, Whelan RF. The contribution of constrictor and dilator nerves to the skin vasodilatation during body heating. J Physiol. 1957;136:489–497. doi: 10.1113/jphysiol.1957.sp005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kellogg DL, Jr, Pergola PE, Piest KL, Kosiba WA, Crandall CG, Grossmann M, Johnson JM. Cutaneous active vasodilation in humans is mediated by cholinergic nerve cotransmission. Circ Res. 1995;77:1222–1228. doi: 10.1161/01.res.77.6.1222. [DOI] [PubMed] [Google Scholar]
- 19.Dietz NM, Rivera JM, Warner DO, Joyner MJ. Is nitric oxide involved in cutaneous vasodilation during body heating in humans? J Appl Physiol. 1994;76:2047–2053. doi: 10.1152/jappl.1994.76.5.2047. [DOI] [PubMed] [Google Scholar]
- 20.Bennett LA, Johnson JM, Stephens DP, Saad AR, Kellogg DL., Jr Evidence for a role for vasoactive intestinal peptide in active vasodilatation in the cutaneous vasculature of humans. J Physiol. 2003;552:223–232. doi: 10.1113/jphysiol.2003.042135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong BJ, Minson CT. Neurokinin-1 receptor desensitization attenuates cutaneous active vasodilatation in humans. J Physiol. 2006;577:1043–1051. doi: 10.1113/jphysiol.2006.112508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersen P, Saltin B. Maximal perfusion of skeletal muscle in man. J Physiol. 1985;366:223–249. doi: 10.1113/jphysiol.1985.sp015794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Armstrong RB, Laughlin MH. Rat muscle blood flows during high-speed locomotion. J Appl Physiol. 1985;59:1322–1328. doi: 10.1152/jappl.1985.59.4.1322. [DOI] [PubMed] [Google Scholar]
- 24.Rowell LB. Ideas about control of skeletal and cardiac muscle blood flow (1876–2003): cycles of revision and new vision. J Appl Physiol. 2004;97:384–392. doi: 10.1152/japplphysiol.01220.2003. [DOI] [PubMed] [Google Scholar]
- 25.Schrage WG, Eisenach JH, Joyner MJ. Ageing reduces nitric-oxide- and prostaglandin-mediated vasodilatation in exercising humans. J Physiol. 2007;579:227–236. doi: 10.1113/jphysiol.2006.124313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joyner MJ, Wilkins BW. Exercise hyperaemia: is anything obligatory but the hyperaemia? J Physiol. 2007;583:855–860. doi: 10.1113/jphysiol.2007.135889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mortensen SP, Gonzales-Alonso J, Bune LT, Saltin B, Pilegaard H, Hellsten Y. ATP-induced vasodilation and purinergic receptors in the human leg: roles of nitric oxide, prostaglandins, and adenosine. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1140–R1148. doi: 10.1152/ajpregu.90822.2008. [DOI] [PubMed] [Google Scholar]
- 28.Mortensen SP, Nyberg M, Thaning P, Saltin B, Hellsten Y. Adenosine contributes to blood flow regulation in the exercising human leg by increasing prostaglandin and nitric oxide formation. Hypertension. 53:993–999. doi: 10.1161/HYPERTENSIONAHA.109.130880. [DOI] [PubMed] [Google Scholar]
- 29.Patterson GC, Whelan RF. The measurement of blood flow during reactive hyperaemia in man. J Physiol. 1955;127:13–14P. [PubMed] [Google Scholar]
- 30.Carlsson I, Wennmalm A. Effect of different prostaglandin synthesis inhibitors on post-occlusive blood flow in human forearm. Prostaglandins. 1983;26:241–252. doi: 10.1016/0090-6980(83)90092-8. [DOI] [PubMed] [Google Scholar]
- 31.Halliwill JR, Minson CT, Joyner MJ. Effect of systemic nitric oxide synthase inhibition on postexercise hypotension in humans. J Appl Physiol. 2000;89:1830–1836. doi: 10.1152/jappl.2000.89.5.1830. [DOI] [PubMed] [Google Scholar]
- 32.Charkoudian N, Joyner MJ, Barnes SA, Johnson CP, Eisenach JH, Dietz NM, Wallin BG. Relationship between muscle sympathetic nerve activity and systemic hemodynamics during nitric oxide synthase inhibition in humans. Am J Physiol Heart Circ Physiol. 2006;291:H1378–H1383. doi: 10.1152/ajpheart.00234.2006. [DOI] [PubMed] [Google Scholar]
- 33.Charkoudian N, Joyner MJ, Johnson CP, Eisenach JH, Dietz NM, Wallin BG. Balance between cardiac output and sympathetic nerve activity in resting humans: role in arterial pressure regulation. J Physiol. 2005;568:315–321. doi: 10.1113/jphysiol.2005.090076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hart EC, Charkoudian N, Wallin BG, Curry TB, Eisenach JH, Joyner MJ. Sex differences in sympathetic neural-hemodynamic balance: implications for human blood pressure regulation. Hypertension. 2009;53:571–576. doi: 10.1161/HYPERTENSIONAHA.108.126391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Narkiewicz K, Phillips BG, Kato M, Hering D, Bieniaszewski L, Somers VK. Gender-selective interaction between aging, blood pressure, and sympathetic nerve activity. Hypertension. 2005;45:522–525. doi: 10.1161/01.HYP.0000160318.46725.46. [DOI] [PubMed] [Google Scholar]
- 36.Joyner MJ, Charkoudian N, Wallin BG. A sympathetic view of the sympathetic nervous system and human blood pressure regulation. Exp Physiol. 2008;93:715–724. doi: 10.1113/expphysiol.2007.039545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marshall RJ, Schirger A, Shepherd JT. Blood pressure during supine exercise in idiopathic orthostatic hypotension. Circulation. 1961;24:76–81. doi: 10.1161/01.cir.24.1.76. [DOI] [PubMed] [Google Scholar]
- 38.Remensnyder JP, Mitchell JH, Sarnoff SJ. Functional sympatholysis during muscular activity. Circ Res. 1962;11:370–380. doi: 10.1161/01.res.11.3.370. [DOI] [PubMed] [Google Scholar]
- 39.Thomas GD, Victor RG. Nitric oxide mediates contraction-induced attenuation of sympathetic vasoconstriction in rat skeletal muscle. J Physiol. 1997;506:817–826. doi: 10.1111/j.1469-7793.1998.817bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dinenno FA, Joyner MJ. Blunted sympathetic vasoconstriction in contracting skeletal muscle of healthy humans: Is nitric oxide obligatory? J Physiol. 2003;553:281–292. doi: 10.1113/jphysiol.2003.049940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenmeier JB, Dinenno FA, Fritzlar SJ, Joyner MJ. Alpha1- and alpha2-adrenergic vasoconstriction is blunted in contracting human muscle. J Physiol. 2003;547:971–976. doi: 10.1113/jphysiol.2002.037937. [DOI] [PMC free article] [PubMed] [Google Scholar]