

Abstract
The goal of the present study was to explore, in our previously developed hybrid template, the effect of introduction of additional heterocyclic rings (mimicking catechol hydroxyl groups as bioisosteric replacement) on selectivity and affinity for the D3 versus D2 receptor. In addition, we wanted to explore the effect of derivatization of functional groups of the agonist binding moiety in compounds developed by us earlier from the hybrid template. Binding affinity (Ki) of the new compounds was measured with tritiated spiperone as the radioligand and HEK-293 cells expressing either D2 or D3 receptors. Functional activity of selected compounds was assessed in the GTPγS binding assay. In the imidazole series, compound 10a exhibited the highest D3 affinity whereas the indole derivative 13 exhibited similar high D3 affinity. Functionalization of the amino group in agonist (+)-9d with different sulfonamides derivatives improved the D3 affinity significantly with (+)-14f exhibiting the highest affinity. However, functionalization of the hydroxyl and amino groups of 15 and (+)-9d, known agonist and partial agonist, to sulfonate ester and amide in general modulated the affinity. In both cases loss of agonist potency resulted from such derivatization.
Keywords: Dopamine receptors, D2 receptor, D3 receptor, Agonist, Structure activity relationship study
1. Introduction
The dopamine (DA) receptor system has been targeted for drug development for a number of central nervous system (CNS) disorders, including drug abuse, schizophrenia, and Parkinson’s disease (PD).1,2 DA receptors are found throughout the CNS and periphery. So far five subtypes of DA receptors have been identified and are classified as being either D1-like or D2-like.3 These classifications are based on receptor pharmacology and function.4–7 The D1-like receptors, which include the D1 and D5 subtypes, activate adenylate cyclase activity upon receptor activation. The D2-like receptors, which include the D2, D3, and D4 subtypes, inhibit adenylate cyclase activity. The D3 receptor was found to have a distribution in the brain that is different from that of the D2 receptor.8,9 Recent study on the brain distribution of D3 receptors indicated highest density in the nucleus accumbens. In addition D3 receptors are also expressed at a higher level compared to D2 receptors in the extrastriatal regions and also in the thalamus.9 The D2 and D3 receptor subtypes posses 50% overall structural homology, and 75–80% in the agonist binding domains.10,11 It is important to mention that D3 receptor bound to an antagonist was recently crystallized to provide a detail molecular structure.12
Many compounds with various selectivities for the D3 versus D2 receptor have been developed.13,14 Due to high homology, development of selective agonists for D3 receptor is rather difficult as both receptors share nearly identical active binding sites for agonist interaction.15–17 Some of the well known D3 selective agonists include ropinirole and pramipexole, and these agonists were shown to exhibit a 4- to 10-fold higher affinity for the D3 than D2 receptor.18 In comparison, the field has faced fewer obstacles in developing highly selective D3 antagonists. In the majority of these compounds there is a piperazine ring connected to a suitable benzamide-type moiety via a variable-size linker, such as in BP 897 (Fig. 1).13,14,19
Figure 1.
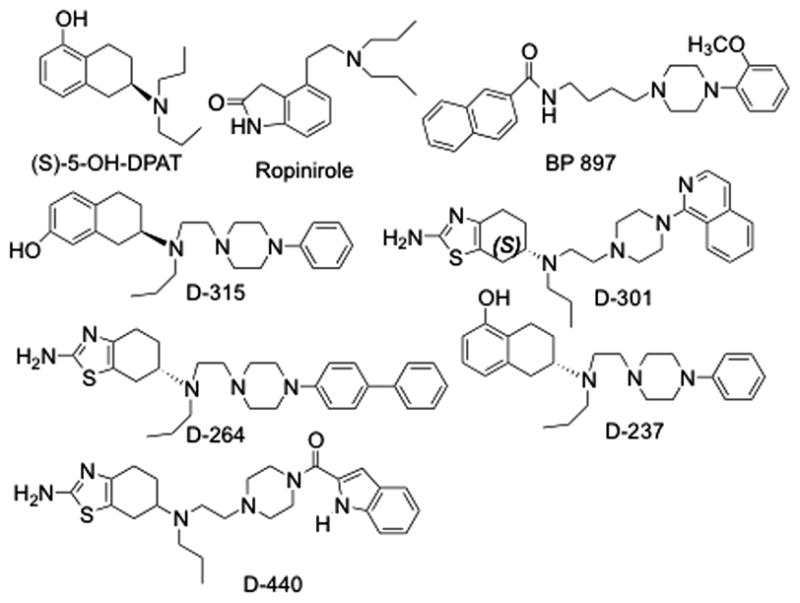
Based on our previously developed hybrid molecular template, we have recently generated highly selective D3 agonists such as D-264, D-301 and D-440 (see Fig. 1).20–24 Our subsequent structure-activity relationship (SAR) studies explored the effect of replacement of the catechol moiety by different heterocyclic rings and also the impact of various molecular modifications at the distal aromatic site connected to the piperazine ring.21–26 In our current SAR studies, we wanted to explore the effect of introduction of additional heterocyclic rings mimicking catechol hydroxyl groups as bioisosteric replacement, on selectivity and affinity for the D3 receptor. In addition, we wanted to explore the effect of substitutions in functional groups of the agonist binding moiety in our hybrid template.
2. Chemistry
Scheme 1 describes the synthesis of final targets 10a–f. Tetralone 1 was reacted with N-propylamine under reductive amination reaction conditions to give 2, which was resolved by chiral 4-(2-chlorophenyl)-2-hydroxy-5,5-dimethyl-1,3,2-dioxaphosphorinane 2-oxide, prepared according to the literature.22 Then, N-alkylation of (±)-2, (+)-2, and (−)-2 with ethylbromoacetate in acetonitrile in the presence of K2CO3 was achieved to produce compounds (±)-3, (+)-3, and (−)-3 in quantitative yield. Nitration of these compounds was achieved by using fuming HNO3 to yield 7-nitro derivative (±)-4, (+)-4, (−)-4 exclusively. Subsequent reduction of (±)-4 and acetylation of the resulting aniline yielded 5 quantitatively. Further nitration of 5 with fuming HNO3 resulted in isomeric mixture of 6- and 8-nitro derivatives 7 and 6, respectively and which were separated from one another by column chromatography. The acetate esters (±)-4, (+)-4, (−)-4, 6, and 7 were subjected to acid hydrolysis followed by coupling with (un)substituted piperazines, thus producing piperazine amides 8a–d, (+)-8d, and (−)-8d. Catalytic reduction of the nitro group followed by amide reduction using borane/THF complex gave intermediates 9a–d, (+)-9d, and (−)-9d which were used as the starting materials for preparation of the final compounds 10a–f. Phenylenediamine 9a was treated27 with formic acid to form imidazole 10a and under similar conditions compounds 9b and 9c gave isomeric imidazoles 10d and 10f, respectively.28 Compound 10e was obtained by reaction of compound 9b with CNBr.29 In another synthesis, compound 9a on reaction30 with CS2 and KOH gave imidazol-2-thione 10b, whereas reaction of 9a with 1,1′-carbonyldiimidazole resulted31 in imidazol-2-one 10c.
Scheme 1.

Reagents and conditions: (a) n-Propylamine, NaCNBH3, AcOH, 1,2-dichloroethane; (b) (+) or (−) chlocyphos, ethanol, recrystallized from isopropanol; (c) BrCH2 CO2 Et, K2 CO3, AcCN; (d) fuming HNO3; (e) (i) 10% Pd/C, H2, EtOH, (ii) AcCI, Et3N, CH2 CI2; (f) fuming HNO3; (g) (i) conc, HCI, (ii) EDCI, HtOH, 1-(2-substituted phenyl)piperazine, CH2CI2; (h) (i) 10% Pd/C, H2, CH3 OH, (ii) BH3, THF; (i) HCOOH; (j) CS2, KOH, EtOH; (k) 1,1′-carbonyldiimidazole, AcCN; (I)CNBr, H2O.
Scheme 2 outlines the synthesis of compounds 13 and 14a–f. In order to synthesize indole 13, compound 9d was reacted with bromoacetaldehyde diethyl acetal to give 11. N-Acetylation with subsequent hydrolysis and cyclization to indole intermediate of 11 was carried out using a mixture of trifluoroacetic acid and trifluoroacetic anhydride to yield 12 in moderate yield. Removal of N-trifluoroacetyl group was performed in refluxing MeOH leading to the final compound 13.32 In order to make sulfonamides and amides (14a–f), compound 9d and (+)-9d were reacted with corresponding sulfonyl chlorides and benzoyl/acetyl chlorides in the presence of pyridine to yield sulfonamides 14a–f and (+)-14f.33
Scheme 2.

Reagent and conditions: (a) Bromoacetaldehyde diethyl acetal, Na2CO3, EtOH, reflux; (b) (CF3CO)2/CF3 COOH, reflux; (c) MeOH, reflux; (d) YXCI, pyridine (for 14a, 4-CIC6H4SO2CI; for 14b, 4-CF3C6H4SO2Cl); for 14c, PhCOCI; for 14d, AcCI; for 14e, 4-CH3C6h4SO2Cl; for (±)14f and (+)14f, 4-CH3OC6h4SO2Cl).
Scheme 3 depicts the synthesis of compounds 16a, 16b, and 18. Compound 1522 was treated with 4-methyl benzenesulfonyl chloride and 4-methoxy benzenesulfonyl chloride in the presence of triethylamine in dichloromethane to afford compounds 16a and 16b, respectively. Finally, compound 18 was achieved by reaction of known compound 1720 with 4-methoxy benzenesulfonyl chloride.
Scheme 3.

Reagents and conditions: (a) YXCI, Et3N, DCM, over night (For 16a, 4-CH3C6H4SO2Cl; for 16b and 18, 4-CH3OC6H4SO2Cl).
3. Results and discussion
In our goal to explore the effect of addition of an heterocyclic moiety mimicking catechol hydroxyl groups bioisosterically on a phenyl ring, compounds 10a–f and 13 were designed and synthesized. In this set, compound 10a exhibited relatively higher affinity for the D3 receptor with good selectivity (Ki, D3 = 19.7 nM, D2/D3 = 22.5). Compounds 10f and 10d were moderately potent at the D3 receptor. These three compounds are imidazole derivatives, and in the past such derivatives have shown high affinity for the D2 receptor.34 Similar to imidazole containing compounds, indole derivative 13 displayed similar modest affinity at D3 and weaker affinity at D2 receptors (Ki; D2 = 127 nM and D3 = 10.7 nM, Table 1). Benz[e]indole derivatives have been shown in the past to exhibit potent agonist activity for the dopamine D2 receptor.35 The remaining compounds in the series were weak to inactive. Collectively, the current results show that introduction of a heterocyclic moiety in our hybrid molecules are well to modestly tolerated.
Table 1.
Affinity for cloned rat D2L and D3 receptors expressed in HEK293 cells measured by inhibition of [3H]spiperone binding
| Compound | Ki (nM), rD2L [3H]spiperone | Ki (nM), rD3 [3H]spiperone | D2L/D3 |
|---|---|---|---|
| (−)-5-OH-DPAT | 58.8 ± 11.0 | 1.36 ± 0.28 | 43.2 |
| Ropinirole | 2,674 ± 305 | 29.3 ± 4.2 | 91.3 |
| (+)-17 D-315c | 40.6 ± 3.6 | 1.77 ± 0.42 | 22.9 |
| (−)-15 D-237a | 26.0 ± 7.5 | 0.825 ± 0.136 | 31.5 |
| D-264b | 264 ± 40 | 0.92 ± 0.23 | 253 |
| D-301 | 269 ± 16 | 2.23 ± 0.60 | 121 |
| D-440 | 1,073 ± 92 | 1.84 ± 0.51 | 583 |
| 9d | 638 ± 40 | 22.2 ± 3.8 | 28.7 |
| (+)-9d (D-515) | 601 ± 98 | 20.2 ± 2.1 | 29.8 |
| (−)-9d (D-516) | 2,707 ± 178 | 45.5 ± 8.8 | 59.6 |
| 10a (D-261) | 443 ± 57 | 19.7 ± 1.3 | 22.5 |
| 10b (D-262) | 230 ± 25 | 112 ± 41 | 2.05 |
| 10c (D-263) | 417 ± 93 | 195 ± 62 | 2.14 |
| 10f (D-311) | 520 ± 22 | 54.1 ± 12.8 | 9.61 |
| 10d (D-312) | 1,280 ± 218 | 60.0 ± 12.9 | 21.3 |
| 10e (D-314) | 15,922 ± 3,820 | 1,790 ± 215 | 8.89 |
| 13 (D-355) | 127 ± 2 | 10.7 ± 2.32 | 11.9 |
| 14a (D-357) | 72.4 ± 7.3 | 4.13 ± 0.90 | 17.5 |
| 14b (D-358) | 91.2 ± 6.1 | 8.64 ± 2.10 | 10.6 |
| 14c (D-359) | 624 ± 54 | 109 ± 37 | 5.72 |
| 14d (D-360) | 1,296 ± 322 | 174 ± 28 | 7.44 |
| 14e (D-397) | 39.2 ± 6.1 | 3.69 ± 0.61 | 10.6 |
| 14f (D-401) | 64.9 ± 12.0 | 2.22 ± 0.22 | 29.2 |
| (+)-14f (D-532) | 101 ± 10 | 2.32 ± 0.35 | 43.5 |
| (−)-16b (D-399) | 497 ± 214 | 17.6 ± 3.1 | 28.2 |
| 16a (D-400) | 632 ± 264 | 32.0 ± 12.6 | 19.8 |
| 18 (D-398) | 1,076 ± 375 | 26.9 ± 4.77 | 40.0 |
We next wanted to evaluate the effect of various substitutions on the amino and hydroxyl functional groups of 9d, 15 and 17. We previously characterized these molecules as potent and selective agonists for D3 receptors.22,25 Compounds 14a–f were designed to explore the effect of different electron withdrawing and donating groups on affinity and selectivity for the D3 receptor. In this effort, compound 9d was converted into 14a–f by derivatization of the amino group to various sulfonamides and amides. The sulfonamides 14e and 14f containing electron donating groups exhibited the highest affinity for D3 receptor (Ki, D3 = 3.69 and 2.22 nM for 14e and 14f, respectively, Table 1) in this series. In this regard, 14f was relatively more selective for D3 receptors compared to p-toluene sulfonamide derivative 14e (D2/D3, 29.2 vs 10.6 for 14f and 14e, respectively). Enantiomerically pure (+)-14f exhibited higher selectivity (D2/D3 = 43.5). Similarly, sulfonamides 14a–b showed an interesting trend of activity. Thus, 14a and 14b containing electronegative 4-Cl and 4-CF3 substitutions exhibited high affinity (Ki; D3 = 4.13 and 8.64 nM for 14a and 14b, respectively) although they were less potent compared to 14e and 14f. Interestingly, benzamide and acetamide compounds 14c–d were much weaker at D3 (Ki; D3 = 109 and 174 nM for 14c and 14d, respectively, Table 1).
Next in our effort to evaluate the effect on modification of functional OH group in D3 preferring potent agonists 1522 and D3 preferring compound 17, compounds 16a–b and 18 were designed. Previously, compound 15 was shown to exhibit potent agonist activity in vitro and in vivo experiments.22 Conversion of the hydroxyl group in this compound to sulfonate ester provided 16a, 16b and 18. Compound 16b, containing the 4-methoxy substituent, exhibited comparable activity to 4-methyl substituted 16a (Ki, D3 = 17.6 vs 32.0 nM for 16b and 16a, respectively). Overall, sulfonamide derivatives produced from 9d showed higher affinity for D2/D3 compared to sulfone ester derived from (−)-15 and (+)-17 (see Table 1).
Our recent work points to hydroxyl group functionality playing a critical role in agonist activity of hybrid compounds.36 Thus, compound 15 (D-237), a potent agonist for D2/D3 receptors, was shown to exhibit much lower affinity for the D3 receptor mutant S192A, indicating a critical role of the hydroxyl group in interacting with serine-192 in the agonist binding cavity of the receptor.36 In this regard, it has been demonstrated that serine-192 plays a critical role via H-bond formation in agonist activity at the D3 receptor. Thus, we wanted to evaluate whether functionalization of this hydroxyl group in 15 will lead to any changes in agonist activity. Indeed, compound (−)-16b which is derived from agonist (−)-15 (D-237), failed to show any agonist activity in the GTPγS binding assay (Table 2). Similarly, (+)-14f, derived from partial agonist (+)-9d, displays high affinity and selectivity for D3 selective molecule but was inactive in the functional assay (Table 2). These results reinforce the critical requirement of the hydroxyl and amino groups for interaction with the agonist binding domains e.g. S192A, in the D3 binding pocket, in order for the receptor to become activated. Finally, recently a crystal structure of human D3 receptor complexed with D2/D3 antagonist eticlopride was identified.12 Further docking of another D3 antagonist in D3 molecular structure identified a second binding pocket unique to D3 receptor, indicating the reason for selectivity for D3.12 This information should be useful for development of selective antagonist. In the current context of our work, which focuses on development of agonist, it would be interesting to see whether the D3 molecular structure could potentially be used to develop selective D3 agonist even though the agonist binding domains for both D2 and D3 receptors are very similar. Although, identification of involvement of any accessory binding sites for agonists unique to D3 receptor should provide selectivity for D3 preferring agonists.
Table 2.
Stimulation of [35S]GTPγS binding to cloned human D2 and D3 receptors expressed in CHO cells
| D-compound | hCHO-D2 | hCHO-D3 | |||
|---|---|---|---|---|---|
|
|
|
||||
| EC50 (nM) | Emax (%) | EC50 (nM) | Emax (%) | D2/D3 | |
| Dopamine | 227 ± 11a | 100 | 7.64 ± 0.56 | 100 | 26.6 |
| D-301a | 116 ± 16 | 88.4 ± 3.9 | 0.82 ± 0.20 | 102 ± 2 | 141 |
| D-440c | 114 ± 12 | 101 ± 5 | 0.26 ± 0.07 | 103 ± 10 | 438 |
| (−)-15 D-237b | 2.22 ± 0.27 | 63.4 ± 3.5 | |||
| (−)-16b D-399 | Not active | Not active | |||
| (+)-9d D-515 | 179 ± 47 | 36.8 ± 8.7 | 30.3 ± 6.4 | 58.6 ± 4.8 | |
| (+)-14f D-532 | Not active | Not active | |||
4. Conclusion
The results from our current SAR studies have provided additional insight on molecular structural requirements for introduction of an heterocyclic ring onto the phenyl ring of the hybrid template. Appropriate location of N-atoms in these heterocyclic ring can potentially provide H-bonding thereby acting as a bioisosteric replacement of hydroxyl group. Imidazole derivative 10a and the indole derivative 13 produced comparable affinity for D3 receptor, however, 13 exhibited higher affinity for D2 compared to 10a. Interestingly, the effect of substitutions on amino functionality in compound 9d was different compared to the effect of substitutions on the hydroxyl group of 15 and 17. Sulfonamide derivative (+)-14f derived from (+)-9d produced significant gain in affinity for both D2 and D3 receptors compared to the parent molecule but lost functional agonist activity.
5. Experimental section
Reagents and solvents were purchased from commercial suppliers and used as received unless otherwise noted. Dry solvents were obtained following the standard procedure. All reactions were performed under N2 atmosphere unless otherwise indicated. Analytical silica gel 60 F254-coated TLC plates were purchased from EMD Chemicals, Inc. and were visualized with UV light or by treatment with phosphomolybdic acid (PMA), Dragendorff’s reagent or ninhydrin. Whatman Purasil® 60A silica gel 230–400 mesh was used for flash column chromatographic purifications. The proton nuclear magnetic resonance (1H NMR) spectra were measured on Varian 400 MHz FT NMR spectrometer using tetramethylsilane (TMS) as an internal standard. The NMR solvent used was CDCl3 unless otherwise indicated. Mass spectra were recorded on Micromass QuattroLC triple quadrupole mass spectrometer. Melting points were recorded using MEL-TEMP II (Laboratory Devices Inc., USA) capillary melting point apparatus and were uncorrected. Elemental analyses were performed by Atlantic Microlab, Inc. and were within ±0.4% of the theoretical value.
5.1. N-Propyl-1,2,3,4-tetrahydronaphthalen-2-amine ((±)-2)
A mixture 2-tetralone 1 (5 g, 35.7 mmol), n-propylamine (4.22 g, 71.3 mmol) and glacial acetic acid (3 mL) in 1,2-dichloroethane (100 mL) was stirred at room temperature for 20 min. NaCNBH3 (6.72 g, 107 mmol) dissolved in methanol (15 mL) was added to the above reaction mixture. The reaction mixture was stirred at room temperature overnight. The solvent was evaporated and saturated NaHCO3 solution (30 mL) was added to the mixture, which was then extracted with ethyl acetate (3 × 100 mL). The combined organic layers were dried (Na2SO4) and the solvent evaporated in vacuo to afford the crude product, which was further purified by column chromatography (EtOAc/MeOH/Et3N 95:4:1) to give the product (±)-2 (6.14 g, 91%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.94 (t, J = 7.6 Hz, 3H), 1.28 (br s, 1H), 1.49–1.64 (m, 3H), 2.02–2.08 (m, 1H), 2.56–2.62 (m, 1H), 2.68 (t, J = 7.2 Hz, 2H), 2.81–2.87 (m, 2H), 2.89–2.92 (m, 1H), 3.02 (dd, J = 15.2, 4.8 Hz, 1H), 7.05–7.11 (m, 4H).
5.2. Resolution of N-propyl-1,2,3,4-tetrahydronaphthalen-2-amine ((±)-2)
Racemic (±)-2 was resolved into its (+)- and (−)-isomers by using both (−)- and (+)-isomers of the synthetic resolving agent 4-(2-chlorophenyl)-2-hydroxy-5,5-dimethyl-1,3,2-dioxaphosphorinane 2-oxide. This optically active resolving agent was prepared according to the published procedure. Compound (±)-2 (7.5 g, 39.62 mmol) and (+)-4-(2-chlorophenyl)-2-hydroxy-5,5-dimethyl-1,3,2-dioxaphosphorinane 2-oxide (10.85 g, 39.22 mmol) were dissolved by warming in 40 mL of ethanol. The solution was cooled to room temperature and then to 0 °C. The precipitated crystals were filtered off, washed with cold ether to yield 7.5 g of the salt ( (c 1, MeOH). Further recrystallization (two times) from hot isopropanol yielded the salt, 6.8 g ( (c 1, MeOH). Further crystallization of the salt from hot isopropanol did not change the optical rotation any further. The salt was then hydrolyzed in the presence of 20% NaOH solution in water under stirring conditions for 2 h at room temperature. The aqueous layer was extracted with dichloromethane (3 × 150 mL), dried over Na2SO4, and evaporated to dryness to yield (−)-2 (2.6 g, 35%). ( (c 1, MeOH).
(±)-2 (4.4 g, 23.24 mmol) was similarly treated by using (−)-4-(2-chlorophenyl)-2-hydroxy-5,5-dimethyl-1,3,2-dioxaphosphorinane 2-oxide (6.36 g, 23.01 mmol). Recrystallization from hot isopropanol yielded the salt, 3.8 g ( (c 1, MeOH). Further crystallization of the salt from hot isopropanol did not change the optical rotation to a significant extent. Hydrolysis of the salt following the above-mentioned procedure yielded (+)-2 (1.4 g, 32%). ( (c 1, MeOH).
5.3. Procedure A: ethyl 2-(propyl(1,2,3,4-tetrahydronaphthalen-2-yl)amino)acetate ((±)-3)
Into the stirred suspension of compound (±)-2 (9.0 g, 47.54 mmol), anhydrous K2CO3 (19.71 g, 143 mmol) in acetonitrile (200 mL) was added ethyl 2-bromoacetate (9.53 g, 57.05 mmol). The reaction mixture was refluxed overnight, cooled to room temperature and filtered. The residue was washed with ethyl acetate and the combined filtrate was evaporated to obtain the crude product, which was purified by column chromatography (hexanes/EtOAc 1:4) to afford (±)-3 (12.3 g, 94%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.86 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.47–1.65 (m, 3H), 2.04–2.11 (m, 1H), 2.64–2.69 (m, 2H), 2.69–2.97 (m, 4H), 3.06–3.13 (m, 1H), 3.40 (s, 2H), 4.17 (q, J = 7.2 Hz, 2H), 7.05–7.11 (m, 4H).
5.4. (R)-Ethyl 2-(propyl(1,2,3,4-tetrahydronaphthalen-2-yl)amino)acetate ((+)-3)
This compound was prepared following procedure A in which compound (+)-2 (1.2 g, 6.34 mmol), ethyl 2-bromoacetate (0.84 mL, 7.61 mmol), and anhydrous K2CO3 (2.63 g, 19.02 mmol) were used to afford compound (+)-3 (1.58 g, 90%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.86 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.47–1.65 (m, 3H), 2.04–2.11 (m, 1H), 2.64–2.69 (m, 2H), 2.69–2.97 (m, 4H), 3.06–3.13 (m, 1H), 3.40 (s, 2H), 4.17 (q, J = 7.2 Hz, 2H), 7.05–7.11 (m, 4H).
5.5. (S)-Ethyl 2-(propyl(1,2,3,4-tetrahydronaphthalen-2-yl)amino)acetate ((−)-3)
This compound was prepared following procedure A in which compound (−)-2 (0.6 g, 3.17 mmol), ethyl 2-bromoacetate (0.42 mL, 3.80 mmol), and anhydrous K2CO3 (1.31 g, 9.51 mmol) were used to afford compound (−)-3 (0.72 g, 88%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.86 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.47–1.65 (m, 3H), 2.04–2.11 (m, 1H), 2.64–2.69 (m, 2H), 2.69–2.97 (m, 4H), 3.06–3.13 (m, 1H), 3.40 (s, 2H), 4.17 (q, J = 7.2 Hz, 2H), 7.05–7.11 (m, 4H).
5.6. Procedure B: Ethyl 2-((7-nitro-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)acetate ((±)-4)
Fuming HNO3 (90%, 90 mL) was transferred through cannula into a 250-mL round bottom flask cooled to −50 °C containing compound (±)-3 (11.0 g, 39.97 mmol). The mixture was stirred at −20 °C for 30 min and poured onto ice. It was neutralized by 20% NaOH solution at 0 °C and extracted by ethyl acetate (3 × 150 mL). The combined organic layers were dried (Na2SO4) and the solvent evaporated to obtain the crude product, which was purified by column chromatography (hexanes/EtOAc 1:4) to afford (±)-4 (11.0 g, 86%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.45–1.54 (m, 2H), 1.58–1.72 (m, 1H), 2.11–2.15 (m, 1H), 2.66 (t, J = 7.6 Hz, 2H), 2.76–2.92 (m, 2H), 2.98–3.07 (m, 2H), 3.12–3.20 (m, 1H), 3.42 (s, 2H), 4.17 (q, J = 6.8 Hz, 2H), 7.19–7.23 (m, 1H), 7.92–7.96 (m, 2H).
5.7. (R)-Ethyl 2-((7-nitro-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)acetate ((+)-4)
This compound was prepared following procedure B in which compound (+)-3 (0.6 g, 2.18 mmol) and Fuming HNO3 (90%, 6 mL) were used to afford compound (+)-4 (0.61 g, 87%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.45–1.54 (m, 2H), 1.58–1.72 (m, 1H), 2.11–2.15 (m, 1H), 2.66 (t, J = 7.6 Hz, 2H), 2.76–2.92 (m, 2H), 2.98–3.07 (m, 2H), 3.12–3.20 (m, 1H), 3.42 (s, 2H), 4.17 (q, J = 6.8 Hz, 2H), 7.19–7.23 (m, 1H), 7.92–7.96 (m, 2H).
5.8. (S)-Ethyl 2-((7-nitro-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)acetate ((−)-4)
This compound was prepared following procedure B in which compound (−)-3 (0.2 g, 0.73 mmol) and Fuming HNO3 (90%, 2 mL) were used to afford compound (−)-4 (0.2 g, 87%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.45–1.54 (m, 2H), 1.58–1.72 (m, 1H), 2.11–2.15 (m, 1H), 2.66 (t, J = 7.6 Hz, 2H), 2.76–2.92 (m, 2H), 2.98–3.07 (m, 2H), 3.12–3.20 (m, 1H), 3.42 (s, 2H), 4.17 (q, J = 6.8 Hz, 2H), 7.19–7.23 (m, 1H), 7.92–7.96 (m, 2H).
5.9. Ethyl 2-((7-acetamido-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)acetate (5)
To the stirred solution of (±)-4 (11.0 g, 34.36 mmol) in dry EtOH (100 mL) was added 10% Pd/C (1.10 g). The reaction was continued under 50 psi H2 for 3 h and the reaction mixture was filtered through celite. Solvent was evaporated to afford crude compound (9.8 g, 98%). Acetyl chloride (2.94 mL, 41.32 mmol) was added into a solution of the crude compound (9.8 g, 33.75 mmol) and Et3N (14.4 mL) in anhydrousCH2Cl2 at 0 °C and then stirred at room temperature for 4 h. The reaction mixture was diluted with CH2Cl2, washed with brine and the organic layer was dried (Na2SO4), evaporated in vacuo and the residue was purified by column chromatography (EtOAc/hexanes 1:1) to afford compound 5 (11.0 g, 98%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.89 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.44–1.52 (m, 2H), 1.54–1.60 (m, 1H), 2.04–2.10 (m, 1H), 2.15 (s, 3H), 2.64 (t, J = 7.2 Hz, 2H), 2.72–2.92 (m, 4H), 3.05–3.09 (m, 1H), 3.39 (s, 2H), 4.17 (q, J = 7.2 Hz, 2H), 6.99–7.01 (m, 1H), 7.16–7.08 (m, 1H), 7.26–7.27 (m, 1H).
5.10. Ethyl 2-((7-acetamido-8-nitro-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)acetate (6) and ethyl 2-((7-acetamido-6-nitro-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)acetate (7)
Compound 5 (6.5 g, 19.55 mmol) was placed in 250-mL flask and cooled down to −50 °C, 70 mL of 90% HNO3 was transferred dropwise through cannula into the flask. The mixture was stirred at −20 °C for 30 min and poured onto ice. The mixture was neutralized by 20% NaOH solution at 0 °C and extracted by EtOAc (3 × 100 mL). The combined organic layers were dried (Na2SO4) and the solvent evaporated to obtain the crude product which was purified by column chromatography (hexanes/EtOAc 1:1). The first eluent afforded 7 (4.0 g, 54%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.28 (t, J = 7.2 Hz, 3H), 1.45–1.57 (m, 2H), 1.59–1.69 (m, 1H), 2.10–2.12 (m, 1H), 2.27 (s, 3H), 2.68 (t, J = 7.2 Hz, 2H), 2.72–2.87 (m, 2H), 2.89–3.16 (m, 2H), 3.10–3.06 (m, 1H), 3.41 (s, 2H), 4.18 (q, J = 7.2 Hz, 2H), 7.90 (d, J = 6.0 Hz,1H), 8.45 (d, J = 5.2 Hz,1H), 10.21 (s, 1H). MS (ESI): 378.40 (C19H28N3O5, [M+H]+). The second eluent provided 6 (2.4 g, 33%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.91 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.42–1.55 (m, 2H), 1.57–1.67 (m, 1H), 2.06–2.10 (m, 1H), 2.16 (s, 3H), 2.62 (t, J = 7.2 Hz, 2H), 2.72–2.86 (m, 3H), 2.86–3.00 (m, 1H), 3.02–3.12 (m, 1H), 3.41 (s, 2H), 4.16 (q, J = 7.2 Hz, 2H), 7.19–7.22 (m, 1H), 7.79–7.83 (m, 1H), 8.23–8.25 (m, 1H). MS (ESI): 378.30 (C19H28N3O5, [M+H]+).
5.11. Procedure C: 2-((7-amino-6-nitro-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)-1-(4-(2-methoxyphenyl)-piperazin-1-yl)ethanone (8a)
Compound 7 (280 mg, 0.742 mmol) was refluxed in 12 N HCl overnight. The mixture was evaporated to dryness followed by overnight drying under vacuum at 70 °C. The yellow solid was dissolved in CH2Cl2 (40 mL). EDCI (242 mg, 1.26 mmol), HOBT (150 mg, 1.11 mmol) and Et3N (1.48 mL, 14.84 mmol) were added and stirred at room temperature for 1 h. Then, 1-(2-methoxylphenyl)-piperazine (339 mg, 1.48 mmol) was added into the reaction mixture and stirred for 24 h. The reaction mixture was diluted with CH2Cl2, washed with water, brine, the organic layer was dried (Na2SO4), and evaporated in vacuo. The residue was purified by column chromatography (hexanes/EtOAc/MeOH/Et3N 40:52:4:4) to afford 8a (300 mg, 84%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.46–1.55 (m, 2H), 1.57–1.68 (m, 1H), 2.01–2.04 (m, 1H), 2.52–2.57(m, 2H), 2.65–2.76 (m, 2H), 2.83–2.87 (m, 2H), 2.95–3.05 (m, 5H), 3.47 (s, 2H), 3.70–3.84 (m, 4H), 3.88 (s, 3H), 6.09 (br s, 2H), 6.52–6.54 (m, 1H), 6.88–6.95 (m, 3H), 7.01–7.05 (m, 1H), 7.77–8.00 (m,1H).
5.12. 2-((7-Amino-6-nitro-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)-1-(4-phenylpiperazin-1-yl)ethanone (8b)
This compound was prepared following procedure C in which compound 7 (1.5 g, 3.97 mmol) and 1-phenylpiperazine (1.32 g, 7.95 mmol) were used to afford compound 8b (0.92 g, 55%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.45–1.55 (m, 2H), 1.59–1.66 (m, 1H), 2.01 (br s, 1H), 2.51–2.55 (m, 2H), 2.69–2.75 (m, 2H), 2.78–2.87 (m, 2H), 3.00–3.04 (m, 1H), 3.13–3.19 (m, 4H), 3.45–3.46 (m, 2H), 3.69–3.83 (m, 4H), 5.83 (br s, 2H), 6.51–6.52 (m,1H), 6.90–6.95 (m, 3H), 7.29–7.31 (m, 2H), 7.84–7.86 (m,1H).
5.13. 2-((7-Amino-8-nitro-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)-1-(4-phenylpiperazin-1-yl)ethanone (8c)
This compound was prepared following procedure C in which compound 6 (2.35 g, 6.23 mmol) and 1-phenylpiperazine (2.07 g, 12.45 mmol) were used to afford compound 8c (1.50 g, 53%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.89 (t, J = 7.2 Hz, 3H), 1.46–1.56 (m, 2H), 1.59–1.64 (m, 1H), 2.01–2.05 (m, 1H), 2.53–2.58 (m, 2H), 2.69–3.04 (m, 4H), 3.13–3.18 (m, 4H), 3.39–3.52 (m, 2H), 3.70–3.83 (m, 4H), 4.84–4.87 (m, 2H), 6.59–6.61 (m,1H), 6.88–7.00 (m, 4H), 7.27–7.31 (m, 2H).
5.14. 2-((7-Nitro-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)-1-(4-phenylpiperazin-1-yl)ethanone ((±)8d)
This compound was prepared following procedure C in which compound (±)-4 (8.56 g, 26.73 mmol) and 1-phenylpiperazine (8.89 g, 53.47 mmol) were used to afford compound (±)-8d (8.17 g, 70%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.91 (t, J = 7.2 Hz, 3H), 1.47–1.56 (m, 2H), 1.65–1.75 (m, 1H), 2.09–2.13 (m, 1H), 2.55–2.59 (m, 2H), 2.83–2.90 (m, 2H,), 2.97–3.02 (m, 2H), 3.10–3.18 (m, 5H), 3.49 (s, 2H), 3.70–3.82 (m, 4H), 6.88–6.94 (m, 3H), 7.17–7.33 (m, 3H), 7.90–7.95 (m, 2H).
5.15. (R)-2-((7-Nitro-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)-1-(4-phenylpiperazin-1-yl)ethanone ((+)-8d)
This compound was prepared following procedure C in which compound (+)-4 (0.61 g, 1.90 mmol) and 1-phenylpiperazine (0.58 mL, 3.80 mmol) were used to afford compound (+)-8d (0.6 g, 72%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.91 (t, J = 7.2 Hz, 3H), 1.47–1.56 (m, 2H), 1.65–1.75 (m, 1H), 2.09–2.13 (m, 1H), 2.55–2.59 (m, 2H), 2.83–2.90 (m, 2H,), 2.97–3.02 (m, 2H), 3.10–3.18 (m, 5H), 3.49 (s, 2H), 3.70–3.82 (m, 4H), 6.88–6.94 (m, 3H), 7.17–7.33 (m, 3H), 7.90–7.95 (m, 2H).
5.16. (S)-2-((7-Nitro-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)-1-(4-phenylpiperazin-1-yl)ethanone ((−)-8d)
This compound was prepared following procedure C in which compound (−)-4 (0.35 g, 1.09 mmol) and 1-phenylpiperazine (0.33 mL, 2.18 mmol) were used to afford compound (−)-8d (0.31 g, 65%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.91 (t, J = 7.2 Hz, 3H), 1.47–1.56 (m, 2H), 1.65–1.75 (m, 1H), 2.09–2.13 (m, 1H), 2.55–2.59 (m, 2H), 2.83–2.90 (m, 2H,), 2.97–3.02 (m, 2H), 3.10–3.18 (m, 5H), 3.49 (s, 2H), 3.70–3.82 (m, 4H), 6.88–6.94 (m, 3H), 7.17–7.33 (m, 3H), 7.90–7.95 (m, 2H).
5.17. Procedure D: N6-(2-(4-(2-methoxyphenyl)piperazin-1-yl)ethyl)-N6-propyl-5,6,7,8-tetrahydronaphthalene-2,3,6-triamine (9a)
To a solution of 8a (290 mg, 0.603 mmol) in dry MeOH (20 mL) was added 10% Pd/C (58 mg). The reaction was continued under 70 psi H2 for 36 h and the reaction mixture filtered through celite. Solvent was evaporated to afford crude compound 250 mg (92%). Into the solution of this crude product (220 mg, 0.487 mmol) in dry THF was added 2.4 mL solution of BH3–THF complex (1 M). The reaction mixture was refluxed overnight, cooled to room temperature and then quenched with MeOH at 0 °C. The solvent was evaporated and the solid complex was suspended in 6 N HCl in methanol. After refluxing for 5 h, MeOH was evaporated in vacuo. Reaction mixture was made alkaline using saturated K2CO3 solution. The aqueous layer was extracted with EtOAc (3 × 20 mL), the combined organic layers were dried (Na2SO4) and concentrated in vacuo. The crude product was purified by column chromatography (Hex/EtOAc/MeOH/Et3N 40:52:4:4) to yield pure compound 9a (138 mg, 65%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.91 (t, J = 7.2 Hz, 3H), 1.49–1.57 (m, 2H), 1.64–1.74 (m, 1H), 2.07–2.11 (m, 1H), 2.56–2.64 (m, 6H), 2.73–2.83 (m, 6H), 2.87–3.0 (m, 7H), 3.83 (s, 3H), 6.84–6.86 (m, 1H), 6.89–6.96 (m, 2H), 6.97–7.02 (m, 1H), 7.21 (br s, 2H).
5.18. N6-(2-(4-Phenylpiperazin-1-yl)ethyl)-N6-propyl-5,6,7,8-tetrahydronaphthalene-2,3,6-triamine (9b)
This compound was prepared from compound 8b (0.9 g, 1.99 mmol) following procedure D to give compound 9b (0.73 g, 84%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.92 (t, J = 7.2 Hz, 3H), 1.46–1.56 (m, 2H), 1.57–1.65 (m, 1H), 1.99–2.01 (m, 1H), 2.52–2.56 (m, 2H), 2.59–2.77 (m, 8H), 2.91–2.99 (m, 1H), 3.21–3.24 (m, 4H), 3.32 (br s, 1H), 6.43 (s, 1H), 6.44 (s, 1H), 6.87 (t, J = 7.2 Hz, 1H), 6.94–6.96 (m, 2H), 7.26–7.30 (m, 2H).
5.19. N7-[2-(4-Phenylpiperazin-1-yl)-ethyl]-N7-propyl-5,6,7,8-tetrahydronaphthalene-1,2,7-triamine (9c)
This compound was prepared from compound 8c (0.9 g, 1.99 mmol) following procedure D to give compound 9c (0.73 g, 84%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.47–1.59 (m, 2H), 1.65–1.68 (m, 1H), 1.99 (br s, 1H), 2.52–2.59 (m, 5H), 2.66 (br s, 4H), 2.71–2.80 (m, 5H), 3.20–3.22 (m, 4H), 3.65–3.68 (m, 1H), 6.47–6.49 (m, 1H), 6.56–6.60 (m, 1H), 6.86 (t, J = 7.2 Hz, 1H), 6.2–6.94 (m, 2H), 7.25–7.29 (m, 2H).
5.20. N2-[2-(4-Phenylpiperazin-1-yl)-ethyl]-N2-propyl-1,2,3,4-tetrahydronaphthalene-2,7-diamine ((±)-9d)
This compound was prepared from compound (±)-8d (7.2 g, 17.04 mmol) following procedure D to give compound (±)-9d (6.27 g, 97%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.46–1.54 (m, 2H), 1.55–1.64 (m, 1H), 1.99 (br s, 1H), 2.50–2.59 (m, 4H), 2.64–2.67 (m, 4H), 2.70–2.81 (m, 4H), 3.19–3.22 (m, 4H), 3.51–3.60 (m, 3H), 6.44–6.57 (m, 2H), 6.83–6.98 (m, 4H), 7.21–7.28 (m, 2H).
5.21. (R)-N2-(2-(4-Phenylpiperazin-1-yl)ethyl)-N2-propyl-1,2,3,4-tetrahydronaphthalene-2,7-diamine ((+)-9d)
This compound was prepared from compound (+)-8d (0.55 g, 1.26 mmol) following procedure D to give compound (+)-9d (0.32 g, 65%) as a brown viscous liquid. (c 0.5, MeOH). 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.46–1.54 (m, 2H), 1.55–1.64 (m, 1H), 1.99 (br s, 1H), 2.50–2.59 (m, 4H), 2.64–2.67 (m, 4H), 2.70–2.81 (m, 4H), 3.19–3.22 (m, 4H), 3.51–3.60 (m, 3H), 6.44–6.57 (m, 2H), 6.83–6.98 (m, 4H), 7.21–7.28 (m, 2H). The free base was converted into HCl salt, mp: 202–204 °C Anal. [C25H36N4·4HCl·H2O] C, H, N.
5.22. (S)-N2-(2-(4-Phenylpiperazin-1-yl)ethyl)-N2-propyl-1,2,3,4-tetrahydronaphthalene-2,7-diamine ((−)-9d)
This compound was prepared from compound (−)-8d (0.3 g, 0.69 mmol) following procedure D to give compound (−)-9d (0.17 g, 63%) as a brown viscous liquid. (c 0.5, MeOH). 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.46–1.54 (m, 2H), 1.55–1.64 (m, 1H), 1.99 (br s, 1H), 2.50–2.59 (m, 4H), 2.64–2.67 (m, 4H), 2.70–2.81 (m, 4H), 3.19–3.22 (m, 4H), 3.51–3.60 (m, 3H), 6.44–6.57 (m, 2H), 6.83–6.98 (m, 4H), 7.21–7.28 (m, 2H). The free base was converted into HCl salt, mp: 200–202 °C Anal. [C25H36N4·4HCl] C, H, N.
5.23. Procedure E: N-(2-(4-(2-methoxyphenyl)piperazin-1-yl)ethyl)-N-propyl-5,6,7,8-tetrahydro-1H-naphtho[2,3-d]imidazol-6-amine (10a)
Diamine 9a (65 mg, 0.149 mmol) and 98% HCOOH (22 μL, 0.357 mmol) were heated under N2 at 140 °C for 5 h. After cooling to room temperature, 5 mL of saturated NaHCO3 were added and extracted with EtOAc (3 × 10 mL). The combined organic layers were dried (Na2SO4), concentrated in vacuo and purified by column chromatography (Hex/EtOAc/MeOH/Et3N 40:52:4:4) to yield 10a (45 mg, 67%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.92 (t, J = 7.2 Hz, 3H), 1.53–1.62 (m, 2H), 1.66–1.76 (m, 1H), 2.09–2.12 (m, 1H), 2.63–2.69 (m, 4H), 2.6 (br s, 4H), 2.85–3.17 (m, 11H), 3.84 (s, 3H), 6.84–6.86 (m, 1H), 6.88–6.92 (m, 2H), 6.97–7.02 (m, 1H), 7.34 (s, 2H), 7.99 (s, 2H). MS (ESI): 448.50 (C27H38N5O, [M+H]+). The free base was converted into HCl salt, mp: 90–93 °C. Anal. [C27H37N5O·4HCl ·1.5H2O·0.4CH3CH2OH] C, H, N.
5.24. 6-((2-(4-(2-Methoxyphenyl)piperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydro-1H-naphtho-[2,3-d]imidazole-2(3H)-thione (10b)
A mixture of diamine 9a (100 mg, 0.228 mmol), CS2 (51 mg, 0.663 mmol), KOH (32 mg, 0.570 mmol), H2O (0.1 mL) and EtOH (2 mL) was refluxed for 3 h. The reaction mixture was evaporated to dryness and purified by column chromatography (Hex/EtOAc/MeOH/Et3N 40:52:4:4) offered 10b (92 mg, 84%) as a brown viscous liquid. 1H NMR (400 MHz, DMSO) δ ppm 0.86 (t, J = 7.2 Hz, 3H), 1.37–1.46 (m, 2H), 1.54–1.58 (m, 1H), 1.91–1.97 (m, 1H), 2.41–2.64 (m, 10H), 2.74–2.93 (m, 9H), 3.76 (s, 3H), 6.82–6.85 (m, 6H), 12.30 (s, 2H). MS (ESI): 480.50 (C27H38N5OS, [M+H]+). The free base was converted into HCl salt, mp: 162–166 °C. Anal. [C27H37N5OS·3HCl·0.7H2O·0.3CH3CH2OH] C, H, N.
5.25. 6-((2-(4-(2-Methoxyphenyl)piperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydro-1H-naphtho-[2,3-d] imidazol-2(3H)-one (10c)
Diamine 9a (160 mg, 0.366 mmol) was added to 1,1′-carbonyldiimidazole (118 mg, 1.574 mmol) in AcCN (5 mL), the mixture was stirred at room temperature for 2 h and then refluxed overnight. The solvent was removed and the residue was purified by column chromatography (Hex/EtOAc/MeOH/Et3N 40:52:4:4) offered 10c (140 mg, 83%) as a brown viscous liquid. 1H NMR (400 MHz, DMSO) δ ppm 0.89 (t, J = 7.2 Hz, 3H), 1.43–1.51 (m, 2H), 1.52–1.61 (m, 1H), 1.97–1.99 (m, 1H), 2.48–2.66 (m, 4H), 2.69–2.76 (m, 10H), 2.89–3.12 (m, 5H), 3.83 (s, 3H), 6.69–6.71 (m, 2H), 6.84–6.89 (m, 1H), 6.91–6.94 (m, 2H), 6.95–7.01 (m, 1H), 7.34 (s, 2H), 7.68 (s, 2H), 9.20 (s, 1H), 9.27 (s, 1H). MS (ESI): 464.50 (C27H38N5O2, [M+H]+). The free base was converted into HCl salt, mp: 112–116 °C. Anal. [C27H37N5O2·3HCl·1.4H2O·0.3CH3CH2OH] C, H, N.
5.26. N-(2-(4-Phenylpiperazin-1-yl)ethyl)-N-propyl-5,6,7,8-tetrahydro-1H-naphtho[2,3-d]imidazol-6-amine (10d)
This compound was prepared from compound 9b (0.13 mg, 0.33 mmol) following procedure E to give compound 10d (110 mg, 81%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.92 (t, J = 7.2 Hz, 3H), 1.58–1.62 (m, 2H), 1.63–1.80 (m, 1H), 2.27 (br s, 1H), 2.66–2.69 (m, 7H), 2.85–3.13 (m, 8H), 3.18–3.20 (m, 4H), 6.83–6.92 (m, 3H), 7.23–7.27 (m, 4H), 7.35 (s, 2H), 7.97 (s, 1H). The free base was converted into oxalate salt, mp: 101–103 °C. Anal. [C26H35N5·3(COOH)2·H2O] C, H, N.
5.27. N6-(2-(4-Phenylpiperazin-1-yl)ethyl)-N6-propyl-5,6,7,8-tetrahydro-1H-naphtho[2,3-d]imidazole-2,6-diamine (10e)
CNBr (68 mg, 0.638 mmol) was added to a solution of 9b (260 mg, 0.638 mmol) in H2O/EtOH (30 mL, 2:1). The mixture was stirred overnight and neutralized by ammonia. After extraction by CH2Cl2 (3 × 30 mL), the organic layer was evaporated and the residue was purified by column chromatography (Hex/EtOAc/MeOH/Et3N 40:52:4:4) offered 10e (130 mg, 47%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.91 (t, J = 7.2 Hz, 3H), 1.54–1.60 (m, 2H), 1.60–1.69 (m, 1H), 1.96 (br s, 1H), 2.60–2.74 (m, 7H), 3.16–3.29 (m, 8H), 3.36–3.38 (m, 2H), 3.66–3.70 (m, 2H), 5.58 (br s, 2H), 5.80 (br s, 2H), 6.86–6.94 (m, 3H), 7.24–7.28 (m, 2H). The free base was converted into oxalate salt, mp: 105–108 °C. Anal. [C28H36N4O2·3(COOH)2·3H2O] C, H, N.
5.28. N-(2-(4-Phenylpiperazin-1-yl)ethyl)-N-propyl-6,7,8,9-tetrahydro-1H-naphtho[1,2-d]imidazol-8-amine (10f)
This compound was prepared from compound 9c (0.43 mg, 1.05 mmol) following procedure E to give compound 10f (300 mg, 68%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.91 (t, J = 7.2 Hz, 3H), 1.49–1.55 (m, 2H), 1.72–1.77 (m, 1H), 2.10–2.17 (m, 1H), 2.58–2.66 (m, 8H), 2.2.77–2.81 (m, 2H), 2.87–3.01 (m, 4H), 3.19–3.20 (m, 5H), 6.83–6.87 (m, 2H), 6.84–6.89 (m, 1H), 6.91–6.93 (m, 2H), 7.02–7.04 (m, 1H), 7.24–7.28 (m, 3H), 7.99–8.0 (m, 1H). The free base was converted into oxalate salt, mp: 98–100 °C. Anal. [C26H35N5·3(COOH)2·H2O] C, H, N.
5.29. N7-(2,2-Diethoxyethyl)-N2-(2-(4-phenylpiperazin-1-yl)ethyl)-N2-propyl-1,2,3,4-tetrahydronaphthalene-2,7-diamine (11)
A solution of 9d (0.3 g, 0.764 mmol), Na2CO3 (81 mg, 0.764 mmol), and bromoacetaldehyde diethyl acetal (0.166 g, 0.84 mmol) in EtOH (3 mL), was heated at reflux for 1.5 days. The solvent was removed in vacuo and the residue was purified by column chromatography (CH2Cl2/MeOH 5:0.25) to yield pure compound 11 (0.23 g, 60%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.92 (t, J = 7.2 Hz, 3H), 1.24 (t, J = 7.2 Hz, 6H), 1.52–1.64 (m, 3H), 2.03 (m, 1H), 2.58 (m, 5H), 2.65–2.68 (m, 5H), 2.77 (m, 5H), 3.04 (br s, 1H), 3.19–3.22 (m, 6H), 3.53–3.60 (m, 2H), 3.68–3.76 (m, 2H), 4.67 (t, J = 6.8 Hz, 1H), 6.38 (m, 1H), 6.43–6.47 (m, 1H), 6.83–6.94 (m, 4H), 7.25 (d, J = 8.0 Hz, 1H), 7.27 (d, J = 8.0 Hz, 1H).
5.30. 2,2,2-Trifluoro-1-(7-((2-(4-phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydro-1H-benzo[f]indol-1-yl) ethanone (12)
A cooled mixture of trifluoroacetic anhydride in trifluoroacetic acid (1:1) was added to compound 11 (0.135 g, 0.27 mmol) at 0 °C. After stirring for 30 min, the cold mixture was diluted with trifluoroacetic acid (3 mL) and heated at reflux for 2.5 days. After removal of the solvent under reduced pressure, the residue was taken into CH2Cl2 (15 mL) and washed with saturated NaHCO3, dried (Na2SO4) and the solvent evaporated to get crude 12 which was purified by column chromatography (CH2Cl2/MeOH 5:0.25) to afford pure 12 (50 mg, 37%) as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 1.04 (t, J = 6.4 Hz, 3H), 1.90 (m, 3H), 2.44 (m, 1H), 3.03–3.26 (m, 6H), 3.43–3.48 (m, 8H), 3.80 (m, 5H), 6.67–6.70 (m, 1H), 6.95–6.96 (d, J = 7.2 Hz, 2H), 7.02 (t, J = 7.2 Hz, 1H), 7.29–7.33 (m, 3H), 7.43 (br s, 1H), 8.21–8.22 (br s, 1H).
5.31. N-(2-(4-Phenylpiperazin-1-yl)ethyl)-N-propyl-5,6,7,8-tetrahydro-1H-benzo[f]indol-7-amine (13)
A solution of compound 12 (25 mg, 0.05 mmol) in dry MeOH (5 mL) was heated at reflux for 24 h. Solvent was evaporated in vacuo and the residue was purified by column chromatography (CH2Cl2/MeOH 9:1) to yield pure compound 13 (10 mg, 50%) as a light brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.98 (t, J = 7.2 Hz, 3H), 1.90 (m, 3H), 2.37 (m, 1H), 2.71–2.73 (m, 4H), 2.83–3.36 (m, 14H), 3.78 (br s, 1H), 6.43 (br s, 1H), 6.85–6.91 (m, 3H), 7.15–7.19 (m, 2H), 7.24–7.27 (m, 2H), 7.34 (s, 1H). MS (ESI): 417.1 (C27H37N4, [M+H]+). The free base was converted into HCl salt, mp: 158–160 °C.
Anal. [C27H36N4·3HCl·0.6H2O·0.1C2H5OC2H5)] C, H, N.
5.32. Procedure F for the syntheses of compounds 14a–f
4-Substituted benzenesulfonyl chloride/benzoyl chloride/acetyl chloride (1 equiv) was added to cooled solution (0 °C) of 9d (1 equiv) in pyridine (5 mL). The mixture was stirred at RT overnight. The solvent was removed in reduced pressure and the residue was purified by column chromatography (CH2Cl2/MeOH 9:1) to yield pure 14a–f.
5.33. 4-Chloro-N-(7-((2-(4-phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-2-yl)benzenesulfonamide (14a)
This compound was prepared following procedure F in which compound (±)-9d (37 mg, 0.09 mmol) and 4-chloro benzenesulfonyl chloride (20 mg, 0.09 mmol) were used to afford compound 14a (42 mg, 80%) as a light brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.96 (t, J = 7.2 Hz, 3H), 1.76 (m, 3H), 2.36 (m, 1H), 2.67–2.99 (13H, m), 3.24 (br s, 4H), 6.87–6.92 (m, 6H), 7.27 (t, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 8.8 Hz, 2H). The free base was converted into HCl salt, mp: 164–166 °C. Anal. [C31H39ClN4O2S·2HCl·H2O] C, H, N.
5.34. N-(7-((2-(4-Phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-2-yl)-4-(trifluoromethyl)benzenesulfonamide (14b)
This compound was prepared following procedure F in which compound (±)-9d (54 mg, 0.14 mmol) and 4-(trifluoromethyl) benzenesulfonyl chloride (34 mg, 0.14 mmol) were used to afford compound 14b (58 mg, 71%) as a light brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.95 (t, J = 7.2 Hz, 3H), 1.72 (m, 3H), 2.18 (m, 1H), 2.65–2.97 (13H, m), 3.23 (br s, 4H), 6.85–6.92 (m, 6H), 7.27 (t, J = 8.0 Hz, 2H), 7.63 (d, J = 7.6 Hz, 2H), 7.93 (d, J = 8.0 Hz, 2H). The free base was converted into HCl salt, mp: 172–174 °C. Anal. [C32H39F3N4O2S·3HCl] C, H, N.
5.35. N-(7-((2-(4-Phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-2-yl)benzamide (14c)
This compound was prepared following procedure F in which compound (±)-9d (34 mg, 0.09 mmol) and benzoyl chloride (10 μL, 0.09 mmol) were used to afford compound 14c (33 mg, 78%) as a light brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.96 (t, J = 7.6 Hz, 3H), 1.82–1.84 (m, 3H), 2.36 (m, 1H), 2.73–3.19 (m, 18H), 3.51 (br s, 1H), 6.87–6.91 (m, 3H), 6.97–7.03 (m, 1H), 7.24–7.29 (m, 2H), 7.41–7.58 (m, 5H), 7.97 (d, J = 7.2 Hz, 2H), 8.50 (br s, 1H). The free base was converted into HCl salt, mp: 162–164 °C. Anal. [C32H40N4O·3HCl] C, H, N.
5.36. N-(7-((2-(4-Phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-2-yl)-acetamide (14d)
This compound was prepared following procedure F in which compound (±)-9d (50 mg, 0.13 mmol) and acetyl chloride (9 μL, 0.13 mmol) were used to afford compound 14d (30 mg, 55%) as a light brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.99 (t, J = 7.2 Hz, 3H), 1.86 (m, 3H), 2.18 (s, 3H), 2.34 (m, 1H), 2.77–3.22 (m, 18H), 3.59 (br s, 1H), 6.87–6.93 (m, 3H), 7.00 (d, J = 8.4 Hz, 1H), 7.21–7.29 (m, 3H), 7.39 (s, 1H), 7.67 (br s, 1H). MS (ESI): 435.1 (C27H39N4O, [M+H]+). The free base was converted into HCl salt, mp: 172–174 °C. Anal. [C27H38N4O·3HCl·0.3C2H5OC2H5] C, H, N.
5.37. 4-Methyl-N-(7-((2-(4-phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-2-yl)benzenesulfonamide (14e)
This compound was prepared following procedure F in which compound (±)-9d (50 mg, 0.13 mmol) and 4-methyl benzenesulfonyl chloride (25 mg, 0.13 mmol) were used to afford compound 14e (45 mg, 65%) as a light brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.93 (t, J = 6.4 Hz, 3H), 1.52–1.82 (m, 3H), 2.37 (s, 3H), 2.39 (m, 1H), 2.50–3.18 (m, 16H), 3.22 (br s, 4H), 6.80–6.96 (m, 6H), 7.22 (t, J = 8.0 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H). 13C NMR (400 MHz, CDCl3) δ ppm 11.88, 21.80, 25.31, 28.84, 29.94, 47.88, 49.04, 53.44, 53.68, 58.78, 116.38, 119.66, 119.95, 120.20, 121.66, 122.68, 127.50, 129.39, 129.83, 130.27, 134.96, 136.69, 143.74, 151.19. The free base was converted into HCl salt, mp: 225–227 °C. Anal. [C32H42N4O2S·2HCl·2H2O] C, H, N.
5.38. 4-Methoxy-N-(7-((2-(4-phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-2-yl)benzenesulfonamide ((±)-14f)
This compound was prepared following procedure F in which compound (±)-9d (50 mg, 0.13 mmol) and 4-methoxy benzenesulfonyl chloride (26 mg, 0.13 mmol) were used to afford compound (±)-14f (45 mg, 63%) as a light brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ ppm 0.91 (t, J = 7.2 Hz, 3H), 1.52–1.70 (m, 3H), 2.08 (m, 1H), 2.58–2.92 (m, 16H), 3.21 (br s, 4H), 3.81 (s, 3H), 6.80–6.96 (m, 8H), 7.26 (t, J = 7.6 Hz, 2H), 7.72 (d, J = 6.8 Hz, 2H). 13C NMR (400 MHz, CDCl3) δ ppm 11.98, 21.80, 25.41, 29.16, 29.93, 48.10, 49.14, 53.53, 53.84, 57.77, 58.08, 114.36, 116.31, 119.71, 119.86, 120.07, 121.95, 122.80, 129.37, 129.51, 129.65, 130.33, 131.18, 134.64, 151.33, 163.17. The free base was converted into HCl salt, mp: 230–232 °C. Anal. [C32H42N4O3S·2HCl·2H2O] C, H, N.
5.39. (R)-4-Methoxy-N-(7-((2-(4-phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-2-yl)benzenesulfonamide ((+)-14f)
This compound was prepared following procedure F in which compound (+)-9d (35 mg, 0.09 mmol) and 4-methoxy benzenesulfonyl chloride (19 mg, 0.09 mmol) were used to afford compound (+)-14f (35 mg, 70%) as a light brown viscous liquid. (c 0.5, MeOH). 1H NMR (400 MHz, CDCl3) δ ppm 0.91 (t, J = 7.2 Hz, 3H), 1.52–1.70 (m, 3H), 2.08 (m, 1H), 2.58–2.92 (m, 16H), 3.21 (br s, 4H), 3.81 (s, 3H), 6.80–6.96 (m, 8H), 7.26 (t, J = 7.6 Hz, 2H), 7.72 (d, J = 6.8 Hz, 2H). 13C NMR (400 MHz, CDCl3) δ ppm 11.98, 21.80, 25.41, 29.16, 29.93, 48.10, 49.14, 53.53, 53.84, 57.77, 58.08, 114.36, 116.31, 119.71, 119.86, 120.07, 121.95, 122.80, 129.37, 129.51, 129.65, 130.33, 131.18, 134.64, 151.33, 163.17. The free base was converted into HCl salt, mp: 232–234 °C. Anal. [C32H42N4O3S·2HCl·2H2O] C, H, N.
5.40. Procedure G for the syntheses of 16a, 16b, and 18
To cooled solution (0 °C) of 15 or 17 (1 equiv) in DCM (5 mL), were added Et3N (3 equiv) and 4-Substituted benzenesulfonyl chloride (1.5 equiv). The mixture was stirred at room temperature overnight. The reaction mixture was extracted with DCM and H2O. Solvent was removed in reduced pressure and the residue was purified by column chromatography (CH2Cl2/MeOH 9:1) to yield pure products.
5.41. (S)-6-((2-(4-Phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-1-yl 4-methylbenzenesulfonate (16a)
This compound was prepared following procedure G in which compound 15 (50 mg, 0.13 mmol), triethyl amine (53 μL, 0.38 mmol), and 4-methyl benzenesulfonyl chloride (36 mg, 0.19 mmol) were used to afford compound 16a (52 mg, 74%) as a light brown viscous liquid. (c 0.5, MeOH). 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.50 (m, 3H), 2.00 (m, 1H), 2.47 (s, 3H), 2.48–3.00 (m, 15H), 3.21 (m, 4H), 6.74 (d, J = 7.2 Hz, 1H), 6.86 (t, J = 7.2 Hz, 1H), 6.93 (d, J = 8.4 Hz, 2H), 6.98–7.40 (m, 2H), 7.27 (t, J = 7.2 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.77 (d, J = 7.6 Hz, 2H). 13C NMR (400 MHz, CDCl3) δ ppm 12.07, 21.99, 24.49, 25.15, 32.31, 48.27, 49.25, 53.52, 53.97, 57.20, 58.34, 116.28, 119.47, 119.97, 126.56, 128.42, 128.59, 129.35, 130.05, 130.57, 133.58, 139.33, 145.53, 148.16, 151.47. The free base was converted into HCl salt, mp: 220–222 °C. Anal. [C32H41N3O3S·2HCl·2H2O] C, H, N.
5.42. (S)-6-((2-(4-Phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-1-yl 4-methoxybenzenesulfonate (16b)
This compound was prepared following procedure G in which compound 15 (45 mg, 0.11 mmol), triethyl amine (48 μL, 0.34 mmol), and 4-methoxy benzenesulfonyl chloride (35 mg, 0.17 mmol) were used to afford compound 16b (47 mg, 73%) as a light brown viscous liquid. (c 0.5, MeOH). 1H NMR (400 MHz, CDCl3) δ ppm 0.91 (t, J = 7.2 Hz, 3H), 1.53 (m, 3H), 2.04 (m, 1H), 2.42–3.04 (m, 15H), 3.21 (m, 4H), 3.90 (s, 3H), 6.74 (d, J = 7.6 Hz, 1H), 6.86 (t, J = 7.6 Hz, 1H), 6.93 (d, J = 8.0 Hz, 2H), 6.96–7.04 (m, 4H), 7.26 (t, J = 7.6 Hz, 2H), 7.81 (d, J = 8.8 Hz, 2H). 13C NMR (400 MHz, CDCl3) δ ppm 12.02, 21.80, 24.42, 25.05, 32.18, 48.20, 49.22, 53.50, 53.92, 55.99, 57.33, 57.93, 114.60, 116.29, 119.58, 119.99, 126.58, 127.86, 128.36, 129.35, 130.51, 130.82, 139.08, 148.16, 151.43, 164.32. The free base was converted into HCl salt, mp: 225–227 °C. Anal. [C32H41N3O4S·2HCl·2H2O] C, H, N.
5.43. (R)-7-((2-(4-Phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-2-yl 4-methoxybenzenesulfonate (18)
This compound was prepared following procedure G in which compound 17 (35 mg, 0.09 mmol), triethyl amine (37 μL, 0.27 mmol), and 4-methoxy benzenesulfonyl chloride (28 mg, 0.13 mmol) were used to afford compound 18 (37 mg, 74%) as a light brown viscous liquid. (c 0.5, MeOH). 1H NMR (400 MHz, CDCl3) δ ppm 0.92 (t, J = 7.2 Hz, 3H), 1.50–1.72 (m, 3H), 2.15 (m, 1H), 2.60–2.88 (m, 14H), 3.20 (m, 5H), 3.88 (s, 3H), 6.64 (d, J = 8.0 Hz, 1H), 6.74–7.02 (m, 7H), 7.26 (t, J = 8.0 Hz, 2H), 7.75 (d, J = 8.4 Hz, 2H). 13C NMR (400 MHz, CDCl3) δ ppm 11.97, 21.35, 25.43, 29.20, 31.73, 48.06, 49.13, 53.45, 53.82, 55.98, 57.84, 114.55, 116.34, 120.05, 120.12, 123.16, 127.11, 129.37, 129.79, 130.92, 135.17, 147.79, 151.30, 164.28. The free base was converted into HCl salt, mp: 195–197 °C. Anal. [C32H41N3O4S·2HCl·2H2O] C, H, N.
5.44. Assessment of affinity for and activation of dopamine D2 and D3 receptors
Binding potency was monitored by inhibition of [3H]spiroperidol (16.2 Ci/mmole, Perkin-Elmer) binding to dopamine rD2 and rD3 receptors expressed in HEK293 cells, in a buffer containing 0.9% NaCl under conditions corresponding to our ‘high [radioligand] protocol’ as described by us previously.23,37 Observed IC50 values were converted to inhibition constants (Ki) by the Cheng–Prusoff equation.23 Functional activity of test compounds in activating dopamine hD2 and hD3 receptors expressed in CHO cells was measured by stimulation of [35S]GTPγS (1250 Ci/mmole, Perkin–Elmer) binding in comparison to stimulation by the full agonist dopamine as described by us previously.23
Supplementary Material
Acknowledgments
This work is supported by National Institute of Neurological Disorders and Stroke/National Institute of Health (NS047198, A.K.D.). We are grateful to Dr. K. Neve, Oregon Health and Science University, Portland, USA, for rD2L and D3 expressing HEK cells. We are also grateful to Dr. J. Shine, Garvan Institute for Medical Research, Sydney, Australia, for hD2L expressing CHO cells.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2013.03.059.
References and notes
- 1.Emilien G, Maloteaux JM, Geurts M, Hoogenberg K, Cragg S. Pharmacol Ther. 1999;84:133. doi: 10.1016/s0163-7258(99)00029-7. [DOI] [PubMed] [Google Scholar]
- 2.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Physiol Rev. 1998;78:189. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 3.Kebabian JW, Calne DB. Nature. 1979;277:93. doi: 10.1038/277093a0. [DOI] [PubMed] [Google Scholar]
- 4.Giros B, Martres MP, Sokoloff P, Schwartz JC. C R Biol. 1990;311:501. [PubMed] [Google Scholar]
- 5.Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. Nature. 1990;347:146. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- 6.Sunahara RK, Guan HC, O’Dowd BF, Seeman P, Laurier LG, Ng G, George SR, Torchia J, Van Tol HH, Niznik HB. Nature. 1991;350:614. doi: 10.1038/350614a0. [DOI] [PubMed] [Google Scholar]
- 7.Van Tol HH, Bunzow JR, Guan HC, Sunahara RK, Seeman P, Niznik HB, Civelli O. Nature. 1991;350:610. doi: 10.1038/350610a0. [DOI] [PubMed] [Google Scholar]
- 8.Gurevich EV, Joyce JN. Neuropsychopharmacology. 1999;20:60. doi: 10.1016/S0893-133X(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 9.Sun J, Xu J, Cairns NJ, Perlmutter JS, Mach RH. PLoS One. 2012;7:e49483. doi: 10.1371/journal.pone.0049483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Physiol Rev. 1998;78:189. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 11.Park BH, Fishburn CS, Carmon S, Accili D, Fuchs S. J Neurochem. 1995;64:482. doi: 10.1046/j.1471-4159.1995.64020482.x. [DOI] [PubMed] [Google Scholar]
- 12.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Science. 2010;330:1091. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boeckler F, Gmeiner P. Pharmacol Ther. 2006;112:281. doi: 10.1016/j.pharmthera.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Luedtkea RR, Mach RH. Curr Pharm Des. 2003;9:643. doi: 10.2174/1381612033391199. [DOI] [PubMed] [Google Scholar]
- 15.Woodward R, Coley C, Daniell S, Naylor LH, Strange PG. J Neurochem. 1996;66:394. doi: 10.1046/j.1471-4159.1996.66010394.x. [DOI] [PubMed] [Google Scholar]
- 16.Sartania N, Strange PG. J Neurochem. 1999;72:2621. doi: 10.1046/j.1471-4159.1999.0722621.x. [DOI] [PubMed] [Google Scholar]
- 17.Varady J, Wu X, Fang X, Min J, Hu Z, Levant B, Wang S. J Med Chem. 2003;46:4377. doi: 10.1021/jm030085p. [DOI] [PubMed] [Google Scholar]
- 18.Perachon S, Schwartz JC, Sokoloff P. Eur J Pharmacol. 1999;366:293. doi: 10.1016/s0014-2999(98)00896-6. [DOI] [PubMed] [Google Scholar]
- 19.Elsner J, Boeckler F, Heinemann FW, Hubner H, Gmeiner P. J Med Chem. 2005;48:5771. doi: 10.1021/jm0503805. [DOI] [PubMed] [Google Scholar]
- 20.Dutta AK, Venkataraman SK, Fei XS, Kolhatkar R, Zhang S, Reith ME. Bioorg Med Chem. 2004;12:4361. doi: 10.1016/j.bmc.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 21.Biswas S, Hazeldine S, Ghosh B, Parrington I, Kuzhikandathil E, Reith ME, Dutta AK. J Med Chem. 2008;51:3005. doi: 10.1021/jm701524h. [DOI] [PubMed] [Google Scholar]
- 22.Biswas S, Zhang S, Fernandez F, Ghosh B, Zhen J, Kuzhikandathil E, Reith ME, Dutta AK. J Med Chem. 2008;51:101. doi: 10.1021/jm070860r. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh B, Antonio T, Zhen J, Kharkar P, Reith ME, Dutta AK. J Med Chem. 2010;53:1023. doi: 10.1021/jm901184n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson M, Antonio T, Reith ME, Dutta AK. J Med Chem. 2012;55:5826. doi: 10.1021/jm300268s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown DA, Kharkar PS, Parrington I, Reith ME, Dutta AK. J Med Chem. 2008;51:7806. doi: 10.1021/jm8008629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh B, Antonio T, Gopishetty B, Reith M, Dutta A. Bioorg Med Chem. 2010;18:5661. doi: 10.1016/j.bmc.2010.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ansari KF, Lal C. Eur J Med Chem. 2009;44:4028. doi: 10.1016/j.ejmech.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 28.Dragovic DSV, Joksimovic J. Pharmazie. 1995;50:678. [PubMed] [Google Scholar]
- 29.Charifson PS, Grillot AL, Grossman TH, Parsons JD, Badia M, Bellon S, Deininger DD, Drumm JE, Gross CH, LeTiran A, Liao Y, Mani N, Nicolau DP, Perola E, Ronkin S, Shannon D, Swenson LL, Tang Q, Tessier PR, Tian SK, Trudeau M, Wang T, Wei Y, Zhang H, Stamos D. J Med Chem. 2008;51:5243. doi: 10.1021/jm800318d. [DOI] [PubMed] [Google Scholar]
- 30.Yang YH, Cheng MS, Wang QH, Nie H, Liao N, Wang J, Chen H. Eur J Med Chem. 2009;44:1808. doi: 10.1016/j.ejmech.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 31.Pelletier JC, Chengalvala M, Cottom J, Feingold I, Garrick L, Green D, Hauze D, Huselton C, Jetter J, Kao WL, Kopf GS, Lundquist JT, Mann C, Mehlmann J, Rogers J, Shanno L, Wrobel J. Bioorg Med Chem. 2008;16:6617. doi: 10.1016/j.bmc.2008.05.024. [DOI] [PubMed] [Google Scholar]
- 32.Wentland MP, Ye Y, Cioffi CL, Lou R, Zhou Q, Xu G, Duan W, Dehnhardt CM, Sun X, Cohen DJ, Bidlack JM. J Med Chem. 2003;46:838. doi: 10.1021/jm020429w. [DOI] [PubMed] [Google Scholar]
- 33.Zheng X, Oda H, Takamatsu K, Sugimoto Y, Tai A, Akaho E, Ali HI, Oshiki T, Kakuta H, Sasaki K. Bioorg Med Chem. 2007;15:1014. doi: 10.1016/j.bmc.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 34.Sukalovic V, Andric D, Roglic G, Kostic-Rajacic S, Schrattenholz A, Soskic V. Eur J Med Chem. 2005;40:481. doi: 10.1016/j.ejmech.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 35.Stjernlof P, Ennis MD, Hansson LO, Hoffman RL, Ghazal NB, Sundell S, Smith MW, Svensson K, Carlsson A, Wikstrom H. J Med Chem. 1995;38:2202. doi: 10.1021/jm00012a021. [DOI] [PubMed] [Google Scholar]
- 36.Kortagere S, Cheng SY, Antonio T, Zhen J, Reith ME, Dutta AK. Biochem Pharmacol. 2011;81:157. doi: 10.1016/j.bcp.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhen J, Antonio T, Dutta AK, Reith ME. J Neurosci Methods. 2010;188:32. doi: 10.1016/j.jneumeth.2010.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.