

Abstract
Tail-anchored (TA) membrane proteins destined for the endoplasmic reticulum are chaperoned by cytosolic targeting factors that deliver them to a membrane receptor for insertion. While a basic framework for TA protein recognition is now emerging, the decisive targeting and membrane insertion steps are not understood. Here we reconstitute the TA protein insertion cycle with purified components, present crystal structures of key complexes between these components, and perform mutational analyses based on the structures. We show that a committed targeting complex, formed by a TA protein bound to the chaperone ATPase Get3, is initially recruited to the membrane via an interaction with Get2. Once recruited, Get1 interacts with Get3 to drive TA protein release in an ATPase-dependent reaction. After releasing its TA protein cargo, the now vacant Get3 recycles back to the cytosol concomitant with ATP binding. This work provides a detailed structural and mechanistic framework for the minimal TA protein insertion cycle.
Approximately 5% of eukaryotic membrane proteins are anchored to the lipid bilayer by a single C-terminal transmembrane domain (TMD)1-4. These ‘tail-anchored’ (TA) proteins are found in virtually all cellular membranes and perform essential functions in processes including protein trafficking, degradation, cell death, and membrane biogenesis. TA proteins in compartments of the secretory and endocytic pathways are first targeted to and inserted into the endoplasmic reticulum (ER) membrane by a post-translational targeting pathway conserved across eukaryotes5-9 and archaea10,11.
This pathway begins with a ‘pre-targeting’ factor that captures newly synthesized TA proteins via their TMDs near the ribosome12,13. In yeast, the pre-targeting factor is Sgt2, which assembles with Get3, Get4, and Get5 to form a TMD recognition complex (TRC)12,14,15. Assembly of TRC permits substrates to be transferred from Sgt2 to Get3 in an ATP-dependent manner12. Get3 (termed TRC40 in mammals) is a homodimeric ATPase whose conformation is regulated by its nucleotide state16-20. Both crystallographic and functional analyses support a model in which an ATP-bound, ‘closed’ dimer of Get3 binds substrates in a large hydrophobic groove that spans both subunits16-18,20. This substrate-Get3-nucleotide complex is therefore the committed targeting complex (see Supplementary Discussion).
In yeast, genetic and physical interaction studies have identified the ER-localized membrane proteins Get1 and Get2 as potential receptors for Get37,21. Whether Get1/2/3 constitute the minimal targeting and insertion machinery, how they function, or what their essential roles are during TA protein insertion are not known. We now combine functional reconstitution of TA protein insertion with structural analysis of key intermediate complexes to provide a mechanistic framework for the TA protein insertion cycle.
The minimal insertion machinery
We first reconstituted the TA protein insertion cycle with purified recombinant factors. A functional TA protein targeting complex was assembled and purified from in vitro translation reactions (Sup. Fig. S1). The complex contained radiolabeled and epitope-tagged Sec61β (an ER-localized TA protein) bound to recombinant yeast Get3 in roughly the 2:1 ratio expected from structural studies. This recombinant targeting complex was functional as judged by membrane insertion of Sec61β into ER-derived yeast rough microsomes (yRM), but not protein-free liposomes (Fig. 1a). Microsomes from ΔGet1 and ΔGet2 yeast strains displayed little insertion activity, while ΔGet3 microsomes were similar to wild-type yRMs. Sec61β insertion efficiency with the purified targeting complex was ~2 fold higher than for Sec61β in crude translation reactions (data not shown), consistent with the observation that the latter contains a heterogeneous mixture of Sec61β complexes with other factors8,13,22. Thus, purified Get3-Sec61β is a committed targeting complex for Get1- and Get2-dependent membrane insertion.
Figure 1. Reconstitution of TA protein insertion with purified components.

(a) Yeast microsomes (yRM) from the indicated strains were tested for insertion of purified Get3-Sec61β targeting complex (top) or by immunoblotting (bottom). The protease-protected fragment (PF) is diagnostic of successful insertion. Liposomes are a negative control. (b) Quantification of Get1 and Get2 levels in yRM by immunoblotting. (c) Protein composition of yRM and proteoliposomes containing recombinant proteins. Twenty-fold relative excess of proteoliposomes were analyzed. (d) Insertion of purified targeting complexes into liposomes, yRM, or rGet1/2 proteoliposomes. VAMP2 and Sed5 indicate TMDs from rat VAMP2 or yeast Sed5. Get1/2 concentration is indicated. (e) Insertion of purified Get3-Sec61β targeting complex into different rGet1/2 proteoliposomes. Autoradiographs and quantified data are shown.
The TA insertion defect of ΔGet1 and ΔGet2 microsomes is due solely to loss of Get1 and/or Get2. To show this, E.coli-produced and purified recombinant Get1 and Get2 (rGet1 and rGet2; Sup. Fig. S2) were each added to detergent extracts prepared from ΔGet1 or ΔGet2 yRMs, reconstituted into proteoliposomes, and tested for function (Sup. Fig. S3). Proteoliposomes from ΔGet1 yRMs were inactive for TA protein insertion, but restored by replenishment with physiologic levels of rGet1, but not rGet2. ΔGet2 proteoliposomes required both rGet1 and rGet2 to restore insertion to near wild-type levels (Sup. Fig. S3), as expected since Get1 is absent from ΔGet2 yRMs (Fig. 1a). We also biochemically depleted Get1 and Get2 from wild-type yRM and showed that the resulting insertion defect could be replenished with rGet1 and rGet2, but neither individually (Sup. Fig. S4). Thus, rGet1 and rGet2 are fully functional in replacing their native counterparts during Get3 dependent TA protein insertion.
The lack of co-purifying membrane proteins with Get1 and Get2 (Sup. Fig. S5), and the absence of other membrane proteins uncovered in genetic studies15,23,24, suggested that Get1 and Get2 are sufficient for Get3-mediated TA protein insertion. Indeed, proteoliposomes containing physiologic concentrations of only rGet1 and/or rGet2 (Fig. 1b, c) were indistinguishable from yRM in mediating insertion of three different purified TA protein targeting complexes (Fig. 1d). Incorporating super-physiologic levels of rGet1/2 did not further improve insertion, while lower levels reduced overall insertion efficiency (Fig. 1e).
The recombinant system required both rGet1 and rGet2 (Fig. 1e), precisely mirroring the results in vivo7 and in crude proteoliposomes (Sup. Fig. S3 and S4). Interaction analysis confirmed that rGet1 and rGet2 form a complex via their membrane domains in detergent solution (Sup. Fig. S6), suggesting that during reconstitution, they are incorporated as a complex. Taken together, the dependence on rGet1 and rGet2, their interaction with each other, their functionality in replacing the endogenous proteins, and high efficiency insertion at native concentrations all argue strongly that we have reconstituted physiologically relevant TA protein insertion with a defined targeting complex and only two membrane proteins.
The Get2c-Get3-ADP•AlF4- complex
Membrane targeting presumably involves an interaction between Get3 and the conserved cytosolic domain(s) of Get1 and/or Get2 (Fig. 2a, 3a and Sup. Fig. S10). These fragments (called ‘Get1c’ and ‘Get2c’) do not interact with each other (Sup. Fig. S6 and S7), but both bind tightly to Get3 (Sup. Fig. S7 and S8), and inhibit the ER-insertion of Sec61β (Sup. Fig. S8). The ability of Get3 to interact in two different ways with the Get1/2 receptor complex suggested that each interaction might serve a different purpose in the insertion cycle.
Figure 2. The Get2 fragment complex with ADP•AlF4--bound Get3.

(a) Predicted topology of S. cerevisiae Get2 with large cytosolic-facing region (yellow). (b) Structure of two Get2 fragments (yellow) bound to the closed Get3 dimer (green, blue). Two Mg2+ADP•AlF4- complexes and a zinc atom are indicated (spheres). An orthogonal view into the substrate-binding composite hydrophobic groove is shown on the right. (c) Get3 residues in the Get2 interface are indicated; most contacts are to one Get3 monomer (green). (d) Closeup of interactions along helix α1 of Get2, including R17, K20 and F21. (e) Closeup of interactions along helix α2 of Get2, including the conserved salt bridge between R29 and E253.
Figure 3. The Get1 fragment complex with Get3.

(a) Predicted topology of S. cerevisiae Get1 with large cytosolic-facing region (magenta). (b) Structure of two Get1 fragments (magenta) bound to the open dimer state of Get3 (green, blue). The composite hydrophobic groove is completely disrupted. (c) Get3 residues in the Get1 interface are indicated; significant contacts are made to both monomers (green, blue). (d) Closeup of interactions between Get1 helix α2 (magenta) and one Get3 monomer (green), including the conserved salt bridge between R73 and E253. This interface overlaps extensively with the Get2c binding surface (see Fig. 2e and S11). (e) Closeup of interactions between the Get1 hairpin loop and the active site of the adjacent Get3 monomer (blue).
The closed-dimer form of ADP•AlF4--bound Get3 probably mimics the TA substrate-bound conformation that targets to the membrane16-18,20. This Get3-ADP•AlF4- complex crystalized with Get2c, and the structure was determined to 2.1 Å resolution (Table S1; Sup. Fig. S9). The structure reveals Get3 in a ‘closed’ dimer conformation with ADP•AlF4- bound in each active site (Fig. 2b). Two Get2 fragments, each comprising two helices connected by a short linker, bind to equivalent sites on opposite faces of the symmetric Get3 homodimer. Each interface buries ~960 Å2 of surface area, largely restricted to a single Get3 monomer (Fig. 2c, green; Sup. Fig. S10). Get3 residues within the interface undergo little conformational change upon binding to Get2c (Sup. Fig. S11). The N-terminal helix of Get2 lies in a cleft defined on one end by short loops following helix α10 and α11 of Get3, and on the other by the loop following helix α9 and the extreme N-terminus of Get3 (Fig. 2d). Three conserved, negatively charged residues in Get3, D265, E307 and D308, make direct contact with Get2c. The second helix of Get2 lies in a cleft defined by Get3 helices α10 and α11 (Fig. 2e). This surface is largely hydrophobic except for a conserved salt-bridge between E253 (Get3) and R29 (Get2c). The C-terminal end of the Get2 fragment, which is not conserved, makes poorly ordered contacts with the adjacent Get3 monomer (Fig. 2c, blue).
The TA substrate binding site in Get3 comprises a large hydrophobic groove spanning the α-helical subdomains of both monomers16. In the Get2c-Get3 complex, this groove is intact (Fig. 2b and S20), suggesting that Get2 captures the closed Get3 targeting complex without disrupting the TA binding site. The long, flexible linker that tethers the helical N-terminus of Get2 to its first TMD would facilitate this process. Thus, we propose that the Get2c-Get3-ADP•AlF4- structure represents a snapshot of the initial encounter between the closed dimer targeting complex and the receptor.
The Get1c-Get3 complex
Get3 was also crystallized in the presence of Get1c. Whether or not ADP•AlF4- was present during crystallization, the Get3-Get1c crystals lacked nucleotide. The structure of this nucleotide-free complex was determined to 3.0 Å resolution (Table S1; Sup. Fig. S9) and revealed Get3 in an ‘open’ conformation, with two Get1 fragments bound to equivalent sites on opposite faces of the symmetric Get3 homodimer (Fig. 3b). Each Get1 fragment adopts an antiparallel coiled-coil structure and buries ~1,030 Å2 of surface area in a bipartite interface split evenly between the two Get3 subunits (Fig. 3c and S10). As observed in the Get2c complex, Get3 residues in the interface undergo little conformational change upon binding to Get1c (Sup. Fig. S11). Binding to one Get3 monomer is primarily mediated by hydrophobic contacts between helix α2 of Get1c and the cleft defined by helices α10 and α11 of Get3 (Fig. 3c,d, green). Binding to the other monomer is mediated by the first helix of Get1c, which interacts with Get3 helices α4 and α5, and by a six-residue loop in Get1c that directly contacts the ATP binding site (Fig. 3c,e, blue; described below).
Importantly, many of the Get3 residues that contact Get1c also mediate interactions with Get2c (Sup. Fig. S10 and S11). For example, the conserved R73(Get1c)–E253(Get3) salt-bridge almost perfectly mimics the R29(Get2c)–E253(Get3) interaction (Fig. 2e and 3d). The presence of overlapping binding sites suggests that Get1 and Get2 cannot simultaneously occupy the same site on Get3, as illustrated by dissociation of the Get3-Get2c complex by Get1c (Sup. Fig. S11). Previous work underscores the functional significance of this region of Get3: alanine substitutions within the shared interface, including F246A, Y250A, E253A and Y298A, exhibit a strong loss-of-function phenotype in yeast18. Moreover, two of these positions, Y250 and E253, have been implicated in the ATP-dependent binding of Get425. Thus, the α10/α11 region of Get3 is a binding hotspot that likely plays an important regulatory role at different stages of the targeting cycle.
The most striking aspect of the Get3-Get1c structure is how the Get1 coiled-coil wedges between the Get3 subunits, completely disrupting the hydrophobic TA substrate binding site (Fig. 3b). Such an interaction could effect substrate release from the Get3 targeting complex. However, parts of the bipartite Get1 binding site on Get3 – including the ATPase motifs and portions of helices α4 and α5 (Fig. 3c, blue) – are buried in the ATP-bound, fully closed dimer conformation. By contrast, the bipartite Get1 binding site is largely exposed to solvent in the Mg2+ADP-bound state (Sup. Fig. S12)17,20. This implies that ATP hydrolysis by the targeting complex is needed to expose the Get1 binding site on Get3 (Fig. 3c and S12, green and blue). Once exposed, Get1 would complete the Get3 transition from closed-to-open, disrupting the hydrophobic groove to promote release of the TA substrate and ADP (which binds weakly to substrate-free Get3; Sup. Fig. S18). Importantly, the rigid Get1 coiled-coil is perpendicular to the plane of the membrane, thereby positioning the hydrophobic groove of Get3 parallel to the membrane. This implies that the TMD of a TA protein is precisely released along the membrane surface, presumably facilitating its subsequent insertion.
Targeting and substrate release
Conserved contacts between Get3-Get2 and Get3-Get1 were disrupted with point mutations (R17E and R73E, respectively), verified to prevent binding (Sup. Fig. S13), and shown to sharply reduce insertion in the reconstituted system (Fig. 4a). When the substrate-Get3 interaction was monitored by crosslinking (Sup. Fig. S14), Get1c, but not Get2c, was found to release TA substrate from Get3 (>50% at 500 nM) (Fig. 4b). This activity was abolished by the R73E mutation that disrupts Get3-Get1c interactions (Fig. 4c). Thus, Get1c and Get2c both inhibit insertion (Sup. Fig. S8), but for different reasons: Get1c causes premature substrate release, while Get2c competitively precludes targeting.
Figure 4. Mutational analysis of Get1/2/3 function.

(a) Insertion assay with purified Get3-Sec61β targeting complex and proteoliposomes containing the indicated purified proteins. Liposomes and yRM are controls. Get1* and Get2* indicate mutants inactive in Get3 interaction (R73E and R17E, respectively). (b) Substrate-release from targeting complexes incubated with Get1c or Get2c; release was monitored by loss of the crosslink between radiolabeled substrate and Get3. (c) As in (b), with wild-type and mutant fragments at 0.5 μM. (d) Substrate interaction with Get3 or the ATPase-deficient Get3(D57N) was assessed by crosslinking after incubation with liposomes or proteoliposomes containing the indicated recombinant proteins. (e) As in (d), but comparing wild-type and mutant Get1/2 complexes. (f) Relative insertion efficiency into rGet1/2 proteoliposomes of targeting complexes prepared with wild-type Get3 or Get3(D57N).
When reconstituted into proteoliposomes at more physiologic concentrations, neither rGet1 nor rGet2 was able to effect substrate release, while the complete rGet1/2 complex was active (Fig. 4d). Importantly, disrupting the Get3-Get1 interaction (with R73E) or the Get3-Get2 interaction (with R17E) abolished the ability of Get1/2 to stimulate substrate release (Fig. 4e). Thus, while Get1c at super-physiologic concentrations can drive substrate release on its own, full-length Get1 in the membrane is unable to do so at physiologic levels. In this context, Get1 requires Get2 (and specifically, its ability to bind Get3) to release substrate from Get3.
Based on the Get3-Get1c structure, ATP hydrolysis by the Get3 targeting complex is likely to be necessary for its interaction with Get1. Indeed, targeting complexes containing an ATPase-deficient Get3 mutant (D57N) were poorly inserted into rGet1/2 proteoliposomes (Fig. 4f) despite no impairment of Get3(D57N) interaction with substrate or rGet1/2 (Sup. Fig. S15 and data not shown). Analysis of TA substrate interaction with Get3(D57N) revealed that rGet1/2 was unable to induce release (Fig. 4d, 4e). Taken together, the functional analysis indicates that the Get3-Get2 interaction is important for targeting, and that this step is a pre-requisite for substrate release. Substrate release, in turn, depends on both ATP hydrolysis by Get3 and its ability to interact with Get1.
ATP-dependent recycling
The ATP that Get3 hydrolyzes prior to substrate release is apparently acquired from the in vitro translation reaction (and maintained during purification) since insertion proceeds efficiently without additional ATP in the purified system (Fig. 5a). This is consistent with structural analysis suggesting that nucleotide is shielded from bulk solvent in the fully closed Get3-ATP-substrate ternary complex (see Supplementary Discussion). Yet, we (Sup. Fig. S16) and others6,17 have puzzlingly found that insertion reactions into crude yRMs, but not rGet1/2 proteoliposomes, are stimulated by ATP, non-hydrolyzable ATP analogues or ADP. The explanation for this discrepancy proved to be the near-stoichiometric presence of Get3 on Get1/2 in yRMs (Sup. Fig. S5), but not rGet1/2 proteoliposomes. Accordingly, binding Get3 to rGet1/2 proteoliposomes restored ATP-dependence (Fig. 5b), while removing Get3 from yRM (by using ΔGet3 yeast) eliminated the ATP requirement for maximal insertion (Fig. 5c).
Figure 5. ATP-dependent recycling of empty Get3 from Get1.

(a) Insertion of purified Get3-Sec61β targeting complex into the indicated vesicles with or without an ATP regenerating system. (b) Proteoliposomes containing rGet1/2, or rGet1/2 bound with Get3 (left panel), were tested for insertion activity of purified targeting complex in the presence or absence of ATP (right panel). (c) Purified targeting complex was tested for insertion into wild-type yRMs or those from a ΔGet3 strain, with or without ATP. (d) Closeup of the Get1c-Get3 complex (magenta and blue) modeled onto the active site of the closed, ADP•AlF4--bound Get3 dimer (grey). Steric (dashed lines) and electrostatic clashes between conserved residues in Get1 and the nucleotide $-phosphate are apparent. (e) Dissociation of Get3-Get1c, monitored by FRET, upon titration with the indicated nucleotides. Curve fits of triplicate measurements (mean +/- s.e.m.) are shown. The reaction contained 10 nM Get3(D57N) and 100 nM Get1c. (f) As in (e), but with 10 nM Get3(D57N) and 200 nM Get2c.
These results indicate that after TA substrate release, Get3 remains bound to microsomal membranes. In the nucleotide-free Get3-Get1c structure, which mimics this ‘post-insertion’ complex, residues within the conserved loop of Get1 (59ISAQDN64) insert into the Get3 active site (Fig. 3e) and deform it relative to the ADP•AlF4--bound conformation (Fig. 5d). Modeling ATP into the active site reveals steric and electrostatic clashes between Get1 and ATP, suggesting that free ATP should displace Get3 from Get1. Indeed, the Get3-Get1c interaction was quantitatively disrupted by micromolar concentrations of ATP (Fig. 5e). ADP was far less effective, while AMP failed to disrupt the Get3-Get1c complex. This ATP-dependent Get3 dissociation was also verified with full-length Get1 using pull-down assays (Sup. Fig. S19). By contrast, none of the tested nucleotides disrupted Get2c binding to Get3 (Fig. 5f). Thus, free ATP binding dissociates the Get1-Get3 complex to recycle Get3 from the membrane following TA substrate release.
A model for the insertion cycle
Figure 6 illustrates our working framework for the insertion cycle. Substrate-bound Get3 in the closed conformation and loaded with nucleotide (either ATP or ADP; see Supplementary Discussion) is captured at the membrane by the cytosolic domain of Get2. The apparently long and flexible Get2 tether may facilitate this initial encounter and bring the intact targeting complex near to the site of insertion. After this targeting step, Get1 mediates the post-targeting reactions of substrate release and insertion. Get1 binding to the targeting complex would be facilitated by partial destabilization of the closed dimer following ATP hydrolysis, and by the high local concentration of Get3 achieved by its recruitment via Get2. Binding to the rigid Get1 coiled-coil would orient Get3 so that the substrate is now in close proximity to the membrane. Moreover, by stabilizing the open conformation, Get1 binding would disrupt the Get3 hydrophobic groove and promote release of substrate and ADP. At present, we do not know whether Get1/2 functions as a heterodimer or heterotetramer, although we favor the latter given the symmetric structure of the Get3 dimer. The released substrate would insert unassisted into the lipid bilayer directly26,27, or be chaperoned by the TM domains of the Get1/2 complex. Finally, the empty Get3 would release from Get1 concomitant with ATP binding. This Get3 would now be primed to accept the next substrate from the cytosolic pre-targeting complex for another round of targeting.
Figure 6. Model for TA protein insertion.
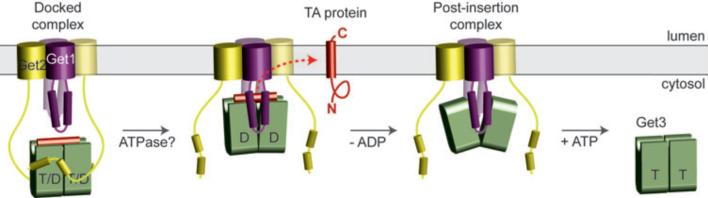
Nucleotide- and TA substrate-bound Get3 in a closed-dimer conformation forms the ‘docked complex’ by association with Get2. Following ATP hydrolysis, Get1 interacts with and orients Get3 along the membrane surface. This stabilizes the open-dimer conformation of Get3, disrupts the composite hydrophobic groove, and promotes TA substrate release for membrane insertion. The Get3-Get1 ‘post-insertion complex’ is dissociated by ATP binding, recycling Get3 back to the cytosol. See Supplementary Discussion for more details.
METHODS
Reagents and basic procedures
Antibodies against Get1 (residues 61-74) and Get2 (residues 2-12) were generated against synthetic peptides conjugated to KLH via terminal cysteines. Antibody against yeast Get3 was against the whole recombinant protein. Antibody production was by Lampire Biological Laboratories. The antibodies against the 3F4 tag and Sec61β have been described8. The Sec61α antibody was a generous gift of Tom Rapoport (Harvard University). DeoxyBigCHAP (DBC) was obtained from Calbiochem. Yeast strains were from Open Biosystems collections and generously provided by Tom Dever. The following lipids were obtained from Avanti Polar Lipids: Egg Phosphatidylcholine (PC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (PE), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-lissamine rhodamine B (Rhodamine-PE). Each lipid was dissolved and stored in Chloroform at -20 °C or -80 °C. Protease inhibitor cocktail was from Roche (EDTA-free Complete tablets) and dissolved as a 25X stock in aqueous buffer just prior to use. In vitro translation, chemical crosslinking and immunoprecipitations were as before8,13,28.
Preparation of proteins for functional analysis
The genes encoding full-length or cytosolic fragments of S. cerevisiae Get1, Get2 and Get3 were amplified by PCR from genomic DNA. Site-directed mutants were obtained by QuickChange mutagenesis (Stratagene). Unless otherwise noted, all constructs were subcloned into a pET28 derivative (Novagen) modified to incorporate a tobacco etch virus (TEV) protease cleavage site between an N-terminal 6×His tag and the polylinker. All constructs were verified by DNA sequencing.
Expression and purification of full-length Get3 (wild-type and D57N) was carried out as described previously16. Full-length Get1 and Get2 (wild-type and mutants) were expressed in E. coli Rosetta2/pLysS (Novagen) using the Overnight Express Autoinduction System 1 (Novagen). Cells were disrupted in buffer A (50 mM HEPES, pH 8.0, 500 mM NaCl, 10 mM imidazole, 5% glycerol) with 1 mM PMSF using a high-pressure microfluidizer (Avestin), and the insoluble pellet was isolated by centrifugation. This pellet was washed in buffer A, re-centrifuged, and solubilized for 1 h at 4 °C in buffer A containing 0.5% n-dodecyl-N,N-dimethylamine-N-oxide (LDAO). The detergent-soluble fraction was then subjected to nickel-affinity chromatography (Ni-NTA agarose, Qiagen) in buffer A containing 30 mM imidazole and 0.1% LDAO. Protein was eluted at ~1 mg/ml in buffer A containing 200 mM imidazole and 0.1% LDAO, and stored in aliquots at -80 °C. Protein concentrations were determined using calculated A280 extinction coefficients.
The cytosolic Get1 fragment (residues 21-104) was expressed for 3 h at 37 °C (wild-type) or overnight at 25 °C (R73E mutant) in E. coli BL21(DE3)/pRIL (Novagen), following induction with 0.1 mM IPTG. Cells were disrupted in buffer B (50 mM Tris, pH 7.5, 500 mM NaCl, 10 mM imidazole, 5% glycerol, 5 mM !-mercaptoethanol) with 1 mM PMSF using a microfluidizer. After clearing by centrifugation, the supernatant was batch-purified by nickel-affinity chromatography. Protein was eluted in buffer B containing 200 mM imidazole, dialyzed into 10 mM Tris pH 7.5, 250 mM NaCl and 40% glycerol, and then stored at -80 °C. This was typically followed by gel filtration (Superdex 200 10/300 GL, GE Healthcare) in 10 mM Tris pH 7.5, 200 mM NaCl. Fractions were pooled and stored in aliquots at -80 °C. Protein concentrations were determined using calculated A280 extinction coefficients.
The cytosolic Get2 fragment (residues 1-38 or 1-106; wild-type and R17E) was expressed with an N- or C-terminal 6×His tag overnight at 25 °C and purified by nickel-affinity chromatography as described above for the Get1 fragment. After dialysis against 10 mM Tris, pH 7.5 and 200 mM NaCl, proteins were further purified by gel filtration in 10 mM Tris, pH 7.5 and 150 mM NaCl. Fractions were pooled, concentrated, and stored in aliquots at -80 °C. Protein concentration was determined by BCA (Pierce).
Preparation of liposomes
The standard liposome mixture typically contained PC:PE:Rhodamine PE at a mass ratio of 8:1.9:0.1. Rhodamine-PE serves as a tracer to follow the lipid recovery. Lipid solutions were mixed in the above ratios as chloroform stocks, adjusted to 10 mM DTT, and dried in a glass tube by centrifugation under vacuum (SpeedVac, Eppendorf) for 12 h. Lipid films were hydrated to a final concentration of 20 mg/ml in lipid buffer (50 mM HEPES-KOH, pH 7.4, 15% glycerol) and mixed end-toend for 6 h at 25 °C with intermittent vortexing. The milky and uniform suspension was subjected to three freeze-thaw cycles (freeze in liquid nitrogen; thaw at 37°C) and extruded at 65 °C 11 times through 100-nm polycarbonate membranes using an Avanti mini-extruder27,29. 100 μl single-use aliquots of the final clear liposome solution were flash frozen in liquid nitrogen and stored at -80 °C.
Purification of recombinant targeting complex
The DNA template for the double-Strep-tagged human Sec61β was generated by PCR using a 5’ oligo that encodes the T7 promoter, start codon, and tag. This template was transcribed and translated in RRL as described28, but with 0.15 mg/ml His-Get3 (added from a 20 mg/ml stock in 10 mM Tris-HCl pH 7.5, 100 mM NaCl and 40% glycerol). A 2 ml translation reaction was diluted two-fold with ice-cold column buffer (20 mM HEPES-KOH, pH 7.4, 100 mM potassium acetate, 2 mM magnesium acetate, 1 mM DTT) and centrifuged for 30 min at 100,000 rpm in a TLA100.3 rotor at 4 °C. The post-ribosomal supernatant was bound to a 400 μl DEAE-sepharose fast flow column at 4 °C, washed with column buffer, and eluted with a buffer containing 50 mM HEPES-KOH, pH7.4, 320 mM potassium acetate, 7 mM magnesium acetate, and 1 mM DTT. The elution was passed over 200 μl Strep-Tactin agarose (IBA, Germany) one to three times. After washing with four column volumes of Strep-Tactin buffer (SB: 50 mM HEPES-KOH, pH 7.4, 10% glycerol, 150 mM potassium acetate, 7 mM magnesium acetate, 1 mM DTT) at 4 °C and eluted five times with 150 μl of SB containing 10 mM Desthiobiotin (Novagen). The peak fractions, measured by counting radioactivity, were pooled. The final sample contained ~10,000 cpm/μl. The concentration of Get3 in the final sample was estimated to be ~ 80 nM. Thus, the targeting complex in our typical preparation is ~40 nM, assuming a 2:1 ratio of Get3 to Sec61β. This was either used immediately, or frozen in aliquots in liquid nitrogen and stored at -80 °C. Targeting complexes containing the TMDs of rat VAMP2 and S. cerevisiae Sed5 in place of the Sec61β TMD were made similarly.
Insertion assay
Posttranslational insertion assay was performed as described before8, with the following minor modifications. For a standard reaction, 8 μl of purified targeting complex was mixed with 1 μl of ATP regenerating system (ARS: 2 mM ATP, 10 mM creatine phosphate, and 40 μg/ml creatine kinase) and 1 μl of either yeast rough microsomes (yRM), liposomes, reconstituted proteoliposomes, or a matched buffer. ARS was omitted in some reactions as indicated in the figure legends. After incubation at 32 °C for 30 min, the samples were treated with proteinase K (0.5 mg/ml) for 60 min on ice, and the protease digestion terminated with 5 mM PMSF and transfered to 10-volumes of boiling 1% SDS as described8. The protease-protected fragment (PF) was then immunoprecipitated using the 3F4 antibody directed against the C-terminus of the Sec61β construct. Immunoprecipitated products were analyzed by SDS-PAGE and quantified by phosphorimaging.
Preparation of rough microsomes from yeast
Yeast microsomes were prepared by modifications of the methods previously described30-32,38. TAP-Get1 or Get deletion strains (obtained from the respective yeast collections available from Open Biosystems) were grown at 30 °C to a density of 2 A600 U in 1 liter of YPD medium containing 2% glucose. Cells were collected by centrifugation at 3000 X g for 5 min, and washed twice with ice-cold distilled water. All subsequent steps were on ice or at 4 °C. The cell pellet was resuspended in 50 ml of homogenization buffer (20 mM HEPES-KOH, pH 7.4, 100 mM potassium acetate, 2 mM magnesium acetate) and centrifuged for 5 min at 3000 X g. The resulting cell pellet was resuspended in homogenization buffer containing 2 mM DTT and protease inhibitor cocktail (Roche) at a concentration of 1 ml per gram of cell pellet. Pre-chilled glass beads were added (3 g/ml of suspension), and cell lysis was induced as follows: the tube was vigorously shaken up and down through 50 cm path length at ~1-2 cycles per second for three 1-min periods separated by 1 min chilling on ice. Approximately 50% of the cells were broken by this method as visualized by microscopy. The fluid phase was drained off through a fine nylon mesh into a JA17 tubes and spun at 10,000 X g for 10 min. The post-mitochondrial supernatant was briefly centrifuged in a MLA80 rotor at 70,000 rpm for 8 min. Each 2 ml of the clear supernatant was layered on 1 ml of 0.67M sucrose cushion in homogenization buffer and centrifuged for 30 min in a TLA100.3 rotor at 70,000 rpm. The resulting membrane pellet was resuspended in homogenization buffer containing 250 mM sucrose and 2 mM DTT to a final standard concentration of 100 A280 (measured after solubilization in 1% SDS). 1 μl of yRM at this concentration is defined as two equivalents (eq). One liter of culture yielded about 2400 eq. Aliquots were frozen in liquid nitrogen and stored at -80 °C.
Depletion of Get1 and Get2 from microsomal extract
1.5 ml (1500 eq) of TAP-Get1 yRMs were adjusted to 1% DeoxyBigCHAP (DBC) in solubilization buffer (50 mM HEPES-KOH, pH 7.4, 500 mM potassium acetate, 5 mM magnesium acetate, 250 mM sucrose, 1 mM DTT and protease inhibitor cocktail). After 10 min incubation on ice, the detergent extract was centrifuged at 100,000 rpm for 30 min in a TLA100.3 rotor at 4 °C. The supernatant (yRM extract) was incubated with 0.1 ml of IgG-Sepharose (GE Healthcare) for 1 h at 25 °C. The unbound fraction was incubated with 0.1 ml of anti-Get2 antibodies coupled to Protein-A agarose for 1 h at 25 °C. The flow-through was finally incubated with a mixture of 0.1 ml of each anti-Get1 and anti-Get2-antibodies coupled to Protein A agarose for 1 hr at 25 °C. The flow-through from this column was used for reconstitution studies. It should be noted that a residual amount of Get1/2 is sufficient to achieve the maximal insertion under in vitro conditions. Therefore, multiple rounds of depletion of Get1/2 (with at least ~95% depletion) were necessary to fully deplete insertion activity. For purification of the TAP-tagged Get1 (and associated proteins), the IgG-Sepharose resin from above was washed with low salt buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 10% glycerol, 0.25% DBC and 1 mM DTT) and eluted with 70 U TEV-protease (Invitrogen) overnight at 4 °C. The TEV elution was adjusted to 2.5 mM CaCl2, and incubated with calmodulin sepharose (GE Helathcare) for 90 min at 4 °C. The beads were washed with low salt buffer containing CaCl2 and eluted with low salt buffer containing 5 mM EGTA. The eluted proteins were precipitated with TCA and analyzed by SDS-PAGE.
Reconstitution of proteoliposomes from microsome extracts
Following earlier methods30,33,34, yRM were adjusted to a concentration of 1 eq/μl to the following conditions: 50 mM HEPES-KOH, pH 7.4, 500 mM potassium acetate, 5 mM magnesium actetate, 250 mM sucrose, 1mM DTT, 1% DBC and protease inhibitor cocktail. After 10 min on ice, the ribosomes were removed by centrifugation at 100,000 rpm for 30 min in a TLA100.3 rotor at 4°C. Typically, 100 μl of this clarified yRM extract was mixed with 10 μl of liposomes (200 μg) and 50 mg of Biobeads SM2 (Bio-Rad). The Biobeads were prewashed extensively ahead of time with methanol and water. The mixture was incubated 12-16 hr with gentle overhead mixing at 4°C. The fluid phase was separated from the beads, diluted with five volumes of ice-cold distilled water, and sedimented in a TLA100.3 rotor in micro-test tubes at 75,000 rpm for 30 min at 4°C. The proteoliposomes were resuspended in 25 μl of membrane buffer (MB: 50 mM HEPES-KOH, pH 7.4, 100 mM potassium acetate, 5 mM magnesium acetate, 250 mM sucrose, and 1 mM DTT).
Reconstitution of proteoliposomes with purified proteins
The optimum method for reconstitution of purified Get1/2 was empirically determined after testing various detergents and reconstitution methods (Sup. Fig. S17) The precise method of reconstitution proved to be important for obtaining maximally functional proteoliposomes. The incorporation and activity of Get1/2 varied with different detergents. Of those tested, DBC worked the best to achieve the maximal activity of Get1/2. Every batch of DBC requires some degree of optimization with respect to the amount of Biobeads used for detergent removal. For a standard reconstitution reaction, 100 μl of reconstitution buffer (RB: 50 mM HEPES-KOH, pH 7.4, 500 mM potassium acetate, 5 mM magnesium actetate, 250 mM sucrose, 1 mM DTT, 0.25% DBC) was mixed with 10 μl of liposome (200 μg) and purified Get1/2 at the desired concentration. For preparation of liposomes or Ni2+-NTA liposomes used as controls in the assays, purified proteins were omitted. This mixture was added to between 25 mg and 30 mg of Biobeads (optimized for each batch of DBC), and incubated with overhead mixing for 12 hr at 4 °C. The fluid phase was separated and diluted with five volume of ice-cold water. In some instances, the proteoliposomes were mixed with Get3, incubated for 15 min at 25 °C, followed by 30 min at 4 °C with shaking, to allow binding. After dilution, the liposomes were sedimented in a TLA100.3 rotor in micro-test tubes at 75,000 rpm for 30 min at 4 °C. The proteoliposomes were resuspended in 25 μl of membrane buffer (MB: 50 mM HEPES-KOH, pH 7.4, 100 mM potassium acetate, 5 mM magnesium acetate, 250 mM sucrose, and 1 mM DTT). SDS-PAGE Coomassie staining and immunoblots were performed to assess the efficiency of protein incorporation; the Rhodamine-PE served as a marker for lipid recovery. Typical recovery for Get1 and Get2 reconstitution was ~50%.
Multi-angle light scattering
The absolute molecular masses of individual proteins and complexes were measured by static multi-angle light scattering (MALS). Purified samples were injected onto a Superdex 200 HR 10/30 gel-filtration column (GE Healthcare) equilibrated with 10 mM Tris pH 7.5, 150 mM NaCl and 2 mM DTT. The purification system was coupled to an online static light-scattering detector (Dawn HELEOS II, Wyatt Technology), a refractive-index detector (Optilab rEX; Wyatt Technology) and a UV detector (UPC-900, GE Healthcare). Absolute weight-average molar masses were calculated using the ASTRA software (Wyatt Technology).
Receptor fragment binding assays
Gel-filtration purified, 6×His-tagged Get1(21-104), Get2(1-106) and Get3 (wild-type and D57N) proteins were labeled with amine-reactive succinimidyl esters of Alexa488 or Alexa594 (Invitrogen). Labeling reactions were carried out by incubating ~150 μM protein and ~600 μM dye for 1 hr at room temperature in 100 mM NaBicarbonate, pH 8.3, 200 mM NaCl. After labeling, proteins were desalted and concentrated in Amicon Ultra filtration units (Millipore) to ~100 μM in 20 mM HEPES, pH 7.5, 200 mM NaCl (receptor fragments) or 20 mM HEPES, pH 7.5, 200 mM NaCl, 2 mM DTT (Get3), and stored in aliquots at -80 °C. Protein concentration was determined using calculated A280 extinction coefficients after correcting for dye absorbance. Under these labeling conditions we typically observed ~0.5-1.5 mols of dye per mol of protein.
Dissociation constants (Kd) were determined by titrating a fixed amount of labeled, nucleotide-free Get3 with labeled Get1c or Get2c. Fluorescence measurements were made in 96-well format using a Safire2 (Tecan) plate reader. Alexa594-labeled fragment was excited by FRET from Alexa488-labeled Get3 (wild-type or D57N), using excitation and emission wavelengths of 495 and 615 nM. All experiments were carried out in 150 μl of 50 mM HEPES, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 5% glycerol, 0.02% Tween20 and 2 mM DTT. Blank titrations were carried out in the absence of labeled Get3 and were subtracted from the respective titration curves obtained in the presence of labeled Get3. The difference curves were evaluated by nonlinear regression using the following quadratic binding equation: ΔY=0.5* Bmax/P*(Kd+P+X-sqrt((Kd+P+X)2-4*P*X)), where Bmax is the amplitude, P is the total concentration of labeled Get3, X is total concentration of labeled Get1c or Get2c, and Kd is the dissociation constant.
Chase titrations were carried out by measuring FRET between Alexa488-labeled Get3 (wild-type or D57N) and Alexa594-labeled fragment in the presence of increasing concentrations of unlabeled fragment or nucleotide. Blank titrations were performed in the absence of labeled Get3, and were subtracted from the respective titration curves obtained in the presence of labeled Get3. The difference curves were evaluated by nonlinear regression using the following equation: ΔY=Fend+Bmax*P/(P+Kd_labeled*(1+X/Kd)), where Fend is the signal at saturation, Bmax is the amplitude of the signal change, P is the total concentration of labeled fragment, Kd_labeled is the dissociation constant of the Get3-fragment complex, X is the total concentration of the unlabeled component, and Kd is the dissociation constant of the unlabeled component.
Nucleotide binding assays
Fluorescence measurements were made in 96-well format using a Safire2 plate reader with excitation and emission wavelengths of 285 and 446 nm (Sup. Fig. S18). All experiments were carried out with gel-filtration purified, 6×His-tagged Get3(D57N) in 150 μl of 50 mM HEPES, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 5% glycerol, 0.02% Tween20 and 2 mM DTT. The dissociation constant for mant-ATP was measured by incubating 1 μM of Get3(D57N) with increasing concentrations of mant-ATP (Molecular Probes). Dissociation constants for unlabeled nucleotides were determined by incubating 1 μM Get3(D57N) with 1 μM mant-ATP and chasing with increasing concentrations of the corresponding unlabeled nucleotide. In each case, blank titrations were performed in the absence of Get3, and were subtracted from titration curves obtained in the presence of labeled Get3. ATP and ADP concentrations were determined by absorbance (ε259 = 15,400 M-1 cm-1). Dissociation constants were determined by curve fitting as described above.
TA substrate release assay
Get3-substrate complexes were assembled by in vitro translation as follows. First, the translation extract was passed through Phenyl Sepharose to remove endogenous TA binding proteins (particularly TRC40 and Bag6), then complemented with His6-Get3 at ~2 ug/ml. Translation of the TA substrate in this system was verified to result in Get3-substrate complexes by crosslinking, and was functional as judged by Get1/2-dependent insertion (data not shown). Complexes generated by this method were mixed with the fragments or proteoliposomes as indicated in the figure legends, incubated for 30 min at 32°C, and subjected to crosslinking with disuccinimidyl suberate (DSS) as before8. The samples were denatured in 1% SDS, diluted 10-fold in 0.5% Triton X-100 buffer, and subjected to pulldowns of His6-Get3 with immobilized Co2+ bound to chelating sepharose (GE). The Get3-substrate crosslink was visualized by autoradiography.
Preparation of Get3-receptor fragment complexes for crystallization
The gene encoding native, full-length S. cerevisiae Get3 was subcloned into pET19b (Novagen). For co-expression with N-terminal 6×His-tagged Get1(21-104), plasmids were co-transformed into E. coli BL21(DE3)/pRIL (Novagen). Proteins were expressed at 37 °C for 3 h by induction with 0.1 mM IPTG after the cells reached an A600 of ~0.6. Cells were disrupted and purified by nickel-affinity chromatography as described above for the Get1 and Get2 fragments. Protein was eluted in buffer B containing 200 mM imidazole, and then dialyzed into 10 mM Tris, pH 7.5, 100 mM NaCl, 2 mM DTT and 40% glycerol. This was followed by cleavage with 6×His-tagged TEV protease and removal of residual uncleaved Get1 fragment and the 6×His-tagged TEV protease by subtractive Ni-NTA purification. Finally, the complex was separated from excess Get1 fragment by gel filtration. Fractions were pooled, concentrated to ~10 mg ml-1 in 10 mM Tris, pH 7.5, 100 mM NaCl and 2 mM DTT, and stored in aliquots at -80 °C.
Co-expression of native Get3 and N-terminal 6×His-tagged Get2(1-106) or Get2(1-38) was performed as above, except that proteins were expressed at 25 °C for 6-8 h after induction. Following cell lysis and purification by nickel-affinity chromatography, the protein was dialyzed into 10 mM Tris, pH 7.5, 200 mM NaCl, 2 mM DTT. This was followed by cleavage with 6)His-tagged TEV protease and subtractive Ni-NTA purification. Finally, the complex was separated from excess Get2 fragment by gel filtration. Fractions were pooled, concentrated to ~15-20 mg ml-1 in 10 mM Tris, pH 7.5, 150 mM NaCl and 2 mM DTT, and stored in aliquots at -80%°C.
Crystallization
Crystals of S. cerevisiae Get1(21-104) in complex with S. cerevisiae Get3 were grown at room temperature using hanging drop vapour diffusion by mixing equal volumes of a protein solution with a reservoir solution containing 0.2 M K/Na tartrate, 16% PEG 3350, 0.1 M HEPES, pH 7.2 and 6% Polypropylene glycol P400. Crystals were cryoprotected in mother liquor supplemented with 20% ethylene glycol, and flash frozen in liquid nitrogen.
Crystals of S. cerevisiae Get2(1-38) in complex with S. cerevisiae Get3 and ADP•AlF4- were grown at room temperature using hanging drop vapour diffusion by mixing equal volumes of a protein solution containing 2 mM ADP, 2 mM MgCl2, 2 mM AlCl3 and 8 mM NaF with a reservoir solution containing 30% PEG 3350, 0.3 M ammonium acetate and 0.1 M Bis-Tris pH 6.0. Crystals were briefly soaked in mother liquor supplemented with 20% ethylene glycol and flash frozen in liquid nitrogen.
Structure determination and refinement
All data were collected at 100 K at APS beamline 21-IDG (λ=0.97856) and processed with HKL2000 (HKL Research). Data collection and refinement statistics are listed in Supplementary Table 1.
The structure of the Get1(21-104) complex with Get3 was determined to 3.0 Å by molecular replacement with PHASER35 using the open dimer (nucleotide-free) form of S. cerevisiae Get3 (PDB accession 3H8420, with the α-helical subdomain removed) as the search model. No solution could be obtained using the closed-dimer form of S. cerevisiae Get3 as the search model. Clear density was observed for the helical Get1 fragment and portions of the Get3 α-helical subdomain in the initial electron density maps. Model building and refinement were carried out in PHENIX36 and COOT37. The final model contains one Get3 homodimer (chains A and B), two Get1 fragments (chains C and D) and one zinc atom, and was refined to an R-factor (Rfree) of 22.4% (28.2%). 94.3% of the residues are in favored regions of the Ramachandran plot, and 0.9% are outliers. Side-chain density is generally weakest in the α-helical subdomains, and no interpretable density was observed for residues 1-4, 97-134, 155-157, 198-219, 280-284 and 352-354 in chain A; 1-4, 99-125, 191-210, 280-284 and 352-354 in chain B; 21-35 and 103-104 in chain C; and 21-36 and 99-104 in chain D.
The structure of the Get2(1-38) complex with Get3 was determined to 2.1 Å by molecular replacement with PHASER using a monomer of S. cerevisiae Get3 (PDB accession 2WOJ16, with the α-helical subdomain and ligands removed) as the search model. Density for the two helices of Get2(1-38) and portions of the Get3 α-helical subdomain was clearly visible in the initial electron density maps. Model building and refinement were carried out in PHENIX and COOT. The final model contains one Get3 homodimer (chains A and B), two Get2 fragments (chains C and D), two Mg2+ADP•AlF4- complexes, one zinc atom and 231 water molecules and was refined to an R-factor (Rfree) of 18.8% (23.3%). 98.0% of the residues are in favored regions of the Ramachandran plot, and 0.8% are outliers. No interpretable electron density was observed for residues 1-4, 101-126, 188-211, 280-284 and 353-354 in chain A; 1-3, 102-125, 154-158, 199-211, 280-282 and 351-354 in chain B; 1-3 and 35-38 in chain C; and 1-3 in chain D.
Miscellaneous
SDS-PAGE was with 15% Tris-glycine or 12% Tris-tricine gels. Quantification was by phosphorimaging using a Typhoon system with accompanying software. Most images for the figures were generated by exposure to Kodak MR X-ray film. Films were digitized by scanning. Structure figures were generated with Pymol39 and all figures were assembled using Adobe Photoshop and Illustrator.
Supplementary Material
Acknowledgements
Data were collected at beamline 21-IDG at the Advanced Photon Source (APS), Argonne National Laboratory, and we thank the beamline staff for support. We thank T. Dever for yeast strains, T. Rapoport for the Sec61α antibody, M. Downing for technical assistance, members of the Hegde, Keenan and E. Perozo labs and D. Freymann for advice, and A. Shiau and S. Shao for useful discussions and comments on the manuscript. Use of the APS, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. This work was supported by the Intramural Research Program of the NIH (to R.S.H.), an Edward Mallinckrodt, Jr. Foundation Grant (to R.J.K.), and NIH Grant R01 GM086487 (to R.J.K.).
Footnotes
Author Contributions. A.M, M.D. and E.B. produced, purified and characterized recombinant Get1, Get2 (full-length and fragments) and Get3. M.M. and R.S.H. performed the reconstitution experiments, including the substrate release and membrane insertion assays. A.M., M.D. and R.J.K. carried out crystallization and structure determination as well as the interaction analyses. R.S.H. and R.J.K. conceived the project. M.M., R.S.H. and R.J.K. wrote the paper. All authors discussed the results and commented on the manuscript.
Author Information. Atomic coordinates and structure factors for S. cerevisiae Get3 in complex with Get1(21-104) and for Mg2+ADP•AlF4--bound S. cerevisiae Get3 in complex with Get2(1-38) are deposited in the Protein Data Bank under ID code 3ZS8 and 3ZS9, respectively.
REFERENCES
- 1.Kutay U, Hartmann E, Rapoport TA. A class of membrane proteins with a C-terminal anchor. Trends Cell Biol. 1993;3:72–75. doi: 10.1016/0962-8924(93)90066-a. [DOI] [PubMed] [Google Scholar]
- 2.Beilharz T, Egan B, Silver PA, Hofmann K, Lithgow T. Bipartite signals mediate subcellular targeting of tail-anchored membrane proteins in Saccharomyces cerevisiae. J Biol Chem. 2003;278:8219–8223. doi: 10.1074/jbc.M212725200. [DOI] [PubMed] [Google Scholar]
- 3.Kalbfleisch T, Cambon A, Wattenberg BW. A bioinformatics approach to identifying tail-anchored proteins in the human genome. Traffic. 2007;8:1687–1694. doi: 10.1111/j.1600-0854.2007.00661.x. [DOI] [PubMed] [Google Scholar]
- 4.Kriechbaumer V, et al. Subcellular distribution of tail-anchored proteins in Arabidopsis. Traffic. 2009;10:1753–1764. doi: 10.1111/j.1600-0854.2009.00991.x. [DOI] [PubMed] [Google Scholar]
- 5.Favaloro V, Spasic M, Schwappach B, Dobberstein B. Distinct targeting pathways for the membrane insertion of tail-anchored (TA) proteins. J Cell Sci. 2008;121:1832–1840. doi: 10.1242/jcs.020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Favaloro V, Vilardi F, Schlecht R, Mayer MP, Dobberstein B. Asna1/TRC40-mediated membrane insertion of tail-anchored proteins. Journal of Cell Science. 2010 doi: 10.1242/jcs.055970. [DOI] [PubMed] [Google Scholar]
- 7.Schuldiner M, et al. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell. 2008;134:634–645. doi: 10.1016/j.cell.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stefanovic S, Hegde RS. Identification of a targeting factor for posttranslational membrane protein insertion into the ER. Cell. 2007;128:1147–1159. doi: 10.1016/j.cell.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 9.Borgese N, Fasana E. Targeting pathways of C-tail-anchored proteins. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbamem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 10.Sherrill J, Mariappan M, Dominik P, Hegde RS, Keenan RJ. A conserved archaeal pathway for tail-anchored membrane protein insertion. Traffic. 2011 doi: 10.1111/j.1600-0854.2011.01229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borgese N, Righi M. Remote origins of tail-anchored proteins. Traffic. 2010;11:877–885. doi: 10.1111/j.1600-0854.2010.01068.x. [DOI] [PubMed] [Google Scholar]
- 12.Wang F, Brown EC, Mak G, Zhuang J, Denic V. A chaperone cascade sorts proteins for posttranslational membrane insertion into the endoplasmic reticulum. Mol Cell. 2010;40:159–171. doi: 10.1016/j.molcel.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mariappan M, et al. A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature. 2010;466:1120–1124. doi: 10.1038/nature09296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang Y-W, et al. Crystal structure of Get4-Get5 complex and its interactions with Sgt2, Get3, and Ydj1. J Biol Chem. 2010;285:9962–9970. doi: 10.1074/jbc.M109.087098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jonikas MC, et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009;323:1693–1697. doi: 10.1126/science.1167983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mateja A, et al. The structural basis of tail-anchored membrane protein recognition by Get3. Nature. 2009;461:361–366. doi: 10.1038/nature08319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bozkurt G, et al. Structural insights into tail-anchored protein binding and membrane insertion by Get3. Proc Natl Acad Sci USA. 2009;106:21131–21136. doi: 10.1073/pnas.0910223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suloway CJ, Chartron JW, Zaslaver M, Clemons WM., Jr. Model for eukaryotic tail-anchored protein binding based on the structure of Get3. Proc Natl Acad Sci U S A. 2009;106:14849–14854. doi: 10.1073/pnas.0907522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamagata A, et al. Structural insight into the membrane insertion of tail-anchored proteins by Get3. Genes Cells. 2010;15:29–41. doi: 10.1111/j.1365-2443.2009.01362.x. [DOI] [PubMed] [Google Scholar]
- 20.Hu J, Li J, Qian X, Denic V, Sha B. The crystal structures of yeast Get3 suggest a mechanism for tail-anchored protein membrane insertion. PLoS ONE. 2009;4:e8061. doi: 10.1371/journal.pone.0008061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auld KL, et al. The conserved ATPase Get3/Arr4 modulates the activity of membrane-associated proteins in Saccharomyces cerevisiae. Genetics. 2006;174:215–227. doi: 10.1534/genetics.106.058362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leznicki P, Clancy A, Schwappach B, High S. Bat3 promotes the membrane integration of tail-anchored proteins. J Cell Sci. 2010;123:2170–2178. doi: 10.1242/jcs.066738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuldiner M, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 24.Costanzo M, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chartron JW, Suloway CJM, Zaslaver M. a., Clemons WM. Structural characterization of the Get4/Get5 complex and its interaction with Get3. Proc Natl Acad Sci USA. 2010;107:12127–12132. doi: 10.1073/pnas.1006036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renthal R. Helix insertion into bilayers and the evolution of membrane proteins. Cell Mol Life Sci. 2010;67:1077–1088. doi: 10.1007/s00018-009-0234-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brambillasca S, et al. Transmembrane topogenesis of a tail-anchored protein is modulated by membrane lipid composition. EMBO J. 2005;24:2533–2542. doi: 10.1038/sj.emboj.7600730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma A, Mariappan M, Appathurai S, Hegde RS. In vitro dissection of protein translocation into the mammalian endoplasmic reticulum. Methods Mol Biol. 2010;619:339–363. doi: 10.1007/978-1-60327-412-8_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brambillasca S, Yabal M, Makarow M, Borgese N. Unassisted translocation of large polypeptide domains across phospholipid bilayers. J Cell Biol. 2006;175:767–777. doi: 10.1083/jcb.200608101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panzner S, Dreier L, Hartmann E, Kostka S, Rapoport TA. Posttranslational protein transport in yeast reconstituted with a purified complex of Sec proteins and Kar2p. Cell. 1995;81:561–570. doi: 10.1016/0092-8674(95)90077-2. [DOI] [PubMed] [Google Scholar]
- 31.Rothblatt JA, Meyer DI. Secretion in yeast: reconstitution of the translocation and glycosylation of alpha-factor and invertase in a homologous cell-free system. Cell. 1986;44:619–628. doi: 10.1016/0092-8674(86)90271-0. [DOI] [PubMed] [Google Scholar]
- 32.Hansen W, Garcia PD, Walter P. In vitro protein translocation across the yeast endoplasmic reticulum: ATP-dependent posttranslational translocation of the prepro-alpha-factor. Cell. 1986;45:397–406. doi: 10.1016/0092-8674(86)90325-9. [DOI] [PubMed] [Google Scholar]
- 33.Gorlich D, Rapoport TA. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell. 1993;75:615–630. doi: 10.1016/0092-8674(93)90483-7. [DOI] [PubMed] [Google Scholar]
- 34.Fons RD, Bogert BA, Hegde RS. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J Cell Biol. 2003;160:529–539. doi: 10.1083/jcb.200210095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Deshaies RJ, Schekman R. SEC62 encodes a putative membrane protein required for protein translocation into the yeast endoplasmic reticulum. J Cell Biol. 1989;109:2653–2664. doi: 10.1083/jcb.109.6.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.