

Abstract
The native environment for membrane proteins is the highly asymmetric phospholipid bilayer, and this has a large effect on both their structure and dynamics. Reproducing this environment in samples containing suitable for spectroscopic and diffraction experiments is a key issue, and flexibility in sample preparation is essential in order to accommodate the diverse size, shape, and other physical properties of membrane proteins. In most cases, in order to assure that the biological activities are maintained, this means reconstituting the proteins in fully hydrated planar phospholipid bilayers. The asymmetric character of protein-containing bilayers means that it is possible to prepare either oriented or unoriented (powder) samples. Here we demonstrate the equivalence of mechanical, magnetic, and what we refer to as “rotational alignment” of membrane proteins in phospholipid bilayer samples for solid-state NMR spectroscopy. The trans-membrane domain of virus protein “u” (Vpu) from human immunodeficiency virus (HIV-1) and the full-length membrane-bound form of fd bacteriophage coat protein in phospholipid bilayers are used as examples. The equivalence of structural constraints from oriented and unoriented (powder) samples of membrane proteins is based on two concepts: (1) their alignment is defined by the direction of the bilayer normal relative to the magnetic field and (2) they undergo rapid rotational diffusion about the same bilayer normal in liquid crystalline membranes. The measurement of angular constraints relative to a common external axis system defined by the bilayer normal for all sites in the protein is an essential element of oriented sample (OS) solid-state NMR.
Keywords: OS solid-state NMR, protein structure, MAS solid-state NMR, Vpu, fd
Introduction
There are two broad categories of methods for determining the structures of proteins by NMR spectroscopy. In oriented sample OS (oriented sample) solid-state NMR the orientation dependence of the chemical shift and heteronuclear dipole-dipole interactions associated with 1H, 13C, and 15N nuclei in labeled sites provide angular constraints for structure calculations.1 In contrast, isotropic approaches have their roots in solution NMR of liquids where the anisotropic properties of the spin-interactions are averaged out in proteins undergoing rapid isotropic reorientation; however, variants can be applied to immobile, unoriented proteins in MAS (magic angle spinning) solid-state NMR.2–6 The isotropic approach works well for densely packed globular proteins and there are now examples of structures that have been determined by both solution NMR and MAS solid-state NMR with similar results;6 although, notably, the structural resolution is improved by the inclusion of anisotropic constraints in the calculations in both cases.
OS solid-state NMR can be performed on samples where there is uniaxial alignment of the molecules relative to the direction of the applied magnetic field. It has its roots in solid-state NMR of single crystals,7 and has been extended to aligned synthetic polymers,8 magnetically aligned virus particles,1 and both mechanically9,10 and magnetically11,12 aligned membrane proteins in phospholipid bilayers. OS solid-state NMR is particularly well suited for proteins whose structures are not compact, are dominated by regular secondary structure elements, and are generally not well suited for isotropic approaches. Solid-state NMR spectra obtained on stationary oriented samples rely on the orientation dependence of the anisotropic nuclear spin-interactions both for spectral resolution and as the source of angular constraints as input for calculation of three-dimensional structures.
With the increase in attention to the structures of membrane proteins, there is a heightened concern about the influence of the surrounding environment that may be provided by detergents, unusual lipids, organic solvents, crystal packing, etc. Nonetheless, considerable progress has been made in describing the structures of membrane proteins by a number of methods despite these limitations and concerns. These include the early applications of electron microscopy of tilted, unstained samples to identify the seven trans-membrane helices of bacteriorhodopsin in its native purple membrane environment.13 As shown by a variety of experimental measurements, the bacteriorhodopsin protein molecules are constrained in this highly ordered environment containing di-phytanyl lipids.14, 15 However, when the protein is purified and reconstituted into bilayers consisting primarily of phosphatidyl choline lipids, it undergoes rapid rotational diffusion about the bilayer normal at temperatures above that of the gel to liquid crystalline phase transition temperature of the lipids.16,17 Taken together, these early studies of bacteriorhodopsin demonstrated that fast rotational diffusion of membrane proteins about the bilayer normal depends on lipid composition and temperature, although these factors may be mainly affecting the aggregation state of the protein rather than directly the energetics of the rotational diffusion. Hong and coworkers18 have shown that the rate of rotational diffusion of a small membrane protein can be significantly modulated by lipid composition. Similarly, we have shown that the rate of rotational diffusion of filamentous bacteriophage particles is dramatically affected by both the concentration19 and pH of the solution20. More typically for membrane proteins in phospholipid bilayers, the rate of rotational diffusion about the bilayer normal can be switched between frequencies that are “slow” or “fast” on the relevant NMR timescales by changing the temperature between values that are below or above, respectively, the gel to liquid crystalline phase transition temperature of the dominant lipid species. And this is the principal comparison made in the examples presented here, as in the initial example of bacteriorhodopsin in DMPC bilayers.15
Because of the difficulty of applying nearly all methods of structure determination to membrane proteins in phospholipid bilayer environments where they exhibit their natural mobility, including rapid rotational diffusion about the bilayer normal, compromises in the sample conditions have had to be made in order for the field to move forward. Although the initial 7 Å resolution map of bacteriorhodopsin was obtained in the natural environment of the purple membrane using electron microscopy on unstained specimens, 13 by operating under cryo-electron microscopy conditions and using chemical derivatives the resolution was improved to 3.5 Å.22 Notably early atomic-resolution structures of membrane proteins were obtained using purified proteins crystallized from detergents with the x-ray diffraction performed at cryogenic temperatures.23–24 The crystal structure of bacteriorhodopsin ultimately refined to 1.55 Å resolution with this approach.25 Recently, there has been a great deal of interest in using lipid cubic phases (LCP) for crystallography of membrane proteins,26 although these too are limited by the lack of a planar bilayer environment and the cryogenic conditions of the data collection. Although it is possible to crystallize membrane proteins in bicelles at room temperature, 27 their organization and bilayer characteristics are highly temperature dependent, and the data are collected under cryogenic conditions.
Complementary methods of the structure determination are being developed and applied to membrane proteins, some of which have the potential to approach atomic resolution, including electron crystallography of two-dimensional crystals, 28 single particle image reconstruction,29 and atomic force microscopy (AFM).30 While these techniques vary in the details of the samples and their resolution, none of them provide experimental conditions compatible with fast rotational diffusion about the bilayer normal that has been observed in liquid crystalline bilayer samples by a variety of methods for a membrane proteins with a range of sizes.14–17 Although fast rotational diffusion is not demonstrated to be a universal property of all membrane proteins, it is clearly present in many of the examples examined and is likely to be fundamental to their adopting their native, functional conformations. Several theoretical approaches are being developed and applied in order to understand and predict the rates of diffusion of membrane proteins about the bilayer normal in liquid crystalline phospholipid bilayers31–34 by taking into account the viscosity of the bilayer environment, the size, and shape of the proteins. In general, these calculations are complicated by the presence of both lateral and rotational diffusion of the membrane proteins35–36 and interactions among the same and different proteins and other cellular components present in membranes. With the immediate goal of determining the structures of purified proteins in lipid bilayers, their rotational diffusion plays an integral role in the experimental studies and their interpretation.
At present, solid-state NMR spectroscopy is the only method capable of high-resolution studies of membrane proteins in liquid crystalline phospholipid bilayer environments that are compatible with the global rotational diffusion and local motions.38,39 The term “high-resolution” refers to three distinct aspects of the method. The most conventional meaning is that signals from individual atomic sites are resolved. The second is the measurement of spectral parameters, typically the frequencies of resonances or the splittings between doublets that reflect the angular dependence of a single nuclear spin-interaction with respect to a common external axis, defined by the direction of the applied magnetic field or of fast rotational diffusion. And the third is the precision and accuracy of the structures that are calculated from the angular constraints gleaned from these measurements, largely because the measurements are made relative to the same external axis and errors do not accumulate.
In NMR spectroscopy the experimental data reflect both time-average (structure) and time-dependent (dynamic) properties; thus the timescales and directions of the global and local motions are intrinsic to the approach. Most previous studies have focused on the effects of backbone and side chain motions of individual residues on the spectra, their possible influence on the structure calculations, and their implications for the functions of the proteins. In this article, we focus on the influence of global rotational diffusion of membrane proteins about the bilayer normal, not only to understand its effects on the structure and functions of the proteins, but also with the goal of exploiting this fundamental property of membrane proteins to expand the repertoire of solid-state NMR spectroscopy.
The basic features of mechanical and “rotational alignment” of bilayers were first demonstrated by McLaughlin and coworkers40 with 31P NMR spectra of oriented and unoriented samples of phospholipids, which themselves undergo rapid rotational diffusion about the bilayer normal. Griffin and coworkers15 showed that the 13C NMR spectra of unoriented bacteriorhodopsin labeled with 13C in some backbone carbonyl sites could be used to monitor the global rotational diffusion of the protein in unoriented bilayers; at temperatures below the gel to liquid crystalline transition of the lipids the protein is immobile on the relevant NMR timescales and the full frequency span and asymmetry of the carbonyl 13C chemical shift tensor are observed; at higher temperatures the protein undergoes rotational diffusion that is rapid enough to yield a motionally averaged powder pattern with dramatically reduced frequency span. By itself, the observation of narrow single-line resonances in the spectra from protein-containing bilayers oriented perpendicular to the magnetic field (Figure 1B and E) demonstrates that both of these proteins undergo rapid rotational diffusion about their bilayer normals. Similar observations have been shown in G-protein coupled receptors. Rhodopsin37 and CXCR141, which like bacteriorhodopsin have seven trans-membrane helices, undergo rapid rotational diffusion about the bilayer normal under similar conditions. Even more convincing are ‘tilted coil’ experiments where single-line resonances are observed over the full range of orientations of the bilayer normals with respect to the magnetic field.42
Figure 1.

A. Structure of the transmembrane domain of Vpu in lipid bilayers. B. and C. 15N chemical shift spectra of uniformly 15N labeled transmembrane domain of Vpu aligned with its bilayer normal perpendicular (B) or parallel (C) to the magnetic field. D. Structure of the membrane bound form of the major coat protein of fd bacteriophage in lipid bilayers. E. and F. 15N chemical shift spectra of uniformly 15N labeled fd coat protein aligned with its bilayer normal perpendicular (E) or parallel (F) to the magnetic field. The spectra were recorded at 40°C in magnetically aligned DHPC:DMPC q=3.2 bilayers. The spectra in C. and F. were from samples with 3 mM Yb added to ‘flip’ the direction of the bilayer normal to parallel to the field. The spectrum in B. was taken from reference 30.
Other solid-state NMR studies of powder patterns averaged by the axial diffusion of membrane proteins have mainly focused on determining the orientations of their secondary structure in unoriented samples.43–45 In contrast, we focus on demonstrating the equivalence of angular constraints measured from resolved resonances from single sites in mechanically or magnetically aligned samples and from the frequency associated with the parallel edge of the powder pattern from the same sites in unoriented samples where the proteins undergo rapid rotational diffusion. The goal is to complete structure determination based on measurements from individual backbone and side chain sites.
The rotational diffusion of membrane proteins has the potential to merge OS solid-state NMR and MAS solid-state NMR experimental methods, enabling the complementary use of aligned and unoriented samples, and increasing the potential for synergy between the isotropic and anisotropic nuclear spin interaction approaches to protein structure determination. This is feasible because the same rotationally averaged powder patterns observed in stationary unoriented samples can be measured in MAS solid-state NMR spectra of unoriented samples. Here we use the spectral parameters associated with the 15N chemical shift and 1H-15N dipolar couplings in specifically labeled backbone and side chain sites of two different membrane proteins to illustrate the equivalence of mechanical and magnetic uniaxial alignment of protein-containing bilayers and “rotational alignment” of the membrane proteins resulting from their rapid rotational diffusion about the bilayer normal in unoriented samples when the temperature is higher than that of gel to liquid crystalline transition for the phospholipids. Because the resonance frequencies are determined by the orientations of functional groups (chemical shift) or chemical bonds (dipole-dipole coupling) relative to a single axis defined by the direction of the magnetic field or by rotational diffusion about the bilayer normal (in practice, these are the same axis), the protein structure is mapped onto the resulting spectra by the anisotropy of the spin interactions. 46–50 It is therefore feasible to determine the three-dimensional structure of a protein by relying only on the angular dependence of these two spin-interactions.12
The three-dimensional backbone structures of the membrane proteins shown in Figure 1 were determined in aligned phospholipid bilayers by OS solid-state NMR spectroscopy. The samples were obtained by expression in E. coli, enabling selective (by residue type) and uniform labeling with 15N. The trans-membrane channel-forming domain of the protein Vpu from HIV-1 (Figure 1A) has 36 residues. Its structure has been determined in several different mechanically and magnetically aligned lipid bilayer environments,51,52 and the tilt angle of the trans-membrane helix displays an exquisite sensitivity to the hydrophobic thickness of the bilayers.53 The structure of the membrane-bound form of fd coat protein shown in Figure 1D has been determined in mechanically aligned phospholipid bilayers,54 as well as the assembled virus particles.55 This protein has 50 residues and includes a hydrophobic trans-membrane helix and an in-plane amphipathic helix connected by a very short linker. These proteins serve as convenient systems to illustrate the spectroscopic principles of OS solid-state NMR, especially since it is possible to prepare samples with only one or two sites labeled with 15N. The spectroscopy is equally applicable to larger membrane proteins with multiple trans-membrane helices because they also undergo rapid rotational diffusion about the bilayer normal, although multidimensional experiments at high magnetic fields are essential to resolve individual resonances in uniformly labeled proteins.
Experimental Section
Protein preparation
The trans-membrane domain of Vpu used in the experiments has the sequence QPIQIAIVALVVAIIIAIVVWSIVIIEGRGGKKKK. Except for the six additional residues GGKKKK added to the C-terminus to facilitate purification and sample handling residues, the sequence corresponds to the trans-membrane domain from native Vpu of BH10 isolate. The protein was expressed as a fusion with Keto Steroid Isomerase and purified as previously described by RP-HPLC,32 after which the fractions containing pure polypeptide were pooled. The organic solvents and trifluoroacetic acid were removed by flowing nitrogen gas, and then the sample was lyophilized repeatedly from water, and stored as a powder at −20°C before being reconstituted into the various lipid preparations.
The Y21M mutant of the major coat protein of fd bacteriophage used in the experiments has the sequence AEGDDPAKAA FDSLQASATE MIGYAWAMVV VIVGATIGIK LFKKF TSKAS. The coat protein was purified by RP-HPLC after dissolution of the bacteriophage particles in a solvent (50% trifluoroethanol, 49.9% H2O, 0.1% trifluoroacetic acid). The purified protein was lyophilized and stored as a powder at − 20°C before being reconstituted into the various lipid preparations.
Unoriented proteoliposomes
The trans-membrane domain of Vpu and fd coat protein powders were dissolved in trifluoroethanol and the appropriate lipids in chloroform, which were then mixed. The lipids were purchased from Avanti Polar Lipids (Alabaster, AL). The solvents were removed and the protein-containing bilayers were lyophilized, redissolved in excess water, adjusted pH, and then spun down by ultracentrifugation at 300,000 g for 2 hrs at 15°C. The resulting, fully hydrated pellet was used as the sample for the unoriented NMR experiments.
Mechanically aligned bilayers
Oriented samples of 15N-labeled peptides in hydrated lipid bilayers between glass plates were prepared as previously described.,56 For trans-membrane domain of Vpu, 75 mg of lipid mixture (9:1, DOPC:DOPG, w/w) was dissolved in 300 μl chloroform and mixed with 3 mg of peptide dissolved in 300 μl trifluoroethanol. The peptide/lipid mixture was spread onto 15 glass slides. For fd coat protein, proteoliposome in cholate buffer was prepared at a protein/lipid molar ratio of 1.2:100 and contained 16 mg of coat protein and 170 mg of lipid mixture (POPC:POPG) in a molar ratio of 8:2. The proteoliposome was spread onto 40 glass slides. The samples in stacked glass slides dried completely by using a vacuum pump, and then hydrated to 95% of relative humidity. The samples were sealed with a polymer wrap and placed into the coil of the NMR probe.
Magnetically aligned bilayers
The methods for the preparation of magnetically aligned protein-containing phospholipid bilayers have been previously described in detail.57 Briefly, both Vpu trans-membrane domain and fd coat protein were solubilized in short chain phospholipid DHPC (1,2-dihexanoyl-sn-glycero-3-phosphocholine) or its ether-linked equivalent 6-O-PC and then mixed with an aqueous solution containing DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) or its ether-linked equivalent 14-O-PC. The DMPC:DHPC ratio was 3.2 (q=3.2), and the samples were all maintained at 40°C for the NMR experiments.
Solid-State NMR spectroscopy
The stationary sample experiments were performed on a Bruker 700 MHz spectrometer with a homebuilt probe with a solenoid coil double-tuned to the 1H and 15N resonance frequencies. The magic angle spinning experiments were performed on a Bruker 750 MHz spectrometer with a Bruker triple-resonance MAS probe.
The one-dimensional 15N chemical shift spectra were obtained by cross-polarization with a mix time of 1 ms, recycle delay of six seconds, and 50 kHz RF field strength on both the 1H and 15N channels. Conditions for two-dimensional PISEMA58 (polarization inversion spin exchange at the magic angle) experiments were as described.53,54 The number of t1 increments varied between 32 and 64. The signal was acquired with 256 transients for each t1 increments. The data were zero filled in both the t2 and t1 dimensions, yielding a 1024x1024 real matrix. A 30° phase shifted sine bell multiplication followed by 100 Hz of exponential multiplication was applied in t1.
The 15N chemical shifts in the Figures are externally referenced to 15N labeled ammonium sulfate at 26.8 ppm. The NMR data were processed using the program NMRPipe/NMRDraw.59
Results
The atomic-resolution structures of the 36-residue oligomeric trans-membrane domain of Vpu51,52 (Figure 1A) and that of the 50-residue full-length membrane-bound form of fd coat protein54 (Figure 1D) have been determined by OS solid-state NMR spectroscopy. The one-dimensional 15N solid-state NMR spectra shown in Figure 1 were obtained from samples of uniformly 15N labeled proteins in magnetically aligned DMPC:DHPC (q=3.2) bilayers. All four spectra consist primarily of resolved or partially overlapped single-line resonances from backbone amide sites, although a few distinctive signals from side chain nitrogen sites are also present, such as from the amino groups in lysine residues and the N-terminus near 40 ppm. The backbone amide sites that contribute the vast majority of resonances in these spectra have substantial 1H-15N heteronuclear dipole-dipole couplings. Irradiation at the 1H resonance frequency during data acquisition provides the heteronuclear decoupling necessary to observe narrow 15N resonances, and the 15N signals themselves result from Hartmann-Hahn matched spin-lock cross-polarization60 that relies on the presence of these same dipole-dipole couplings to effect transfer of magnetization from 1H to 15N.
Aligned vs. unoriented samples
The spectra in Figures 1B and 1E were obtained from samples of protein-containing bilayers with their normals aligned perpendicular to the direction of the applied magnetic field. In contrast, the spectra in Figures 1C and 1F were obtained from the same samples except that their bilayer normals were aligned parallel to the direction of the field as a result of ‘flipping’ the bilayers by the addition of lanthanide ions.61 Parallel alignment can also be obtained by using different long chain phospholipids.62 Notably, the frequency spans of the spin-interactions, in this case the 15N chemical shift, are twice as large in the parallel alignment compared to that observed in the perpendicular alignment; they are also reversed relative to their isotropic chemical shifts.
The structures of membrane proteins are affected by their environment; most obviously, the tilt angles of trans-membrane helices reflect the hydrophobic thickness of the phospholipids that constitute the planar bilayers.53 The experimental results presented in the Figures were obtained on samples with bilayers consisting of several different long chain phospholipids; consequently, there are minor discrepancies in some of the experimental comparisons that are explained by the expected differences in protein structure due to the different hydrophobic thicknesses and/or order parameters of the phospholipids in the bilayers.
Although the spectra of the uniformly 15N labeled samples have been fully resolved and assigned using multidimensional experiments, by using samples where the proteins are isotopically labeled in only one or two specific sites, the essential features of the NMR spectra can be elucidated by direct comparisons between spectra obtained from oriented and unoriented samples. The trans-membrane domain of Vpu is well-characterized. 51–53 This 36-residue polypeptide contains one leucine residue at position 11 that is specifically labeled through biosynthetic incorporation of 15N-leucine from the growth media. Leu11 is located within the hydrophobic trans-membrane helix, and is fully representative of the spectroscopic, structural, and motional features of this helix. The 15N NMR spectrum in Figure 2A was obtained from an unoriented sample of 15N - Leu11 labeled Vpu TM domain in DMPC bilayers in their gel phase. The spectrum is a powder pattern with a span of about 170 ppm, which is typical of an immobile 15N backbone amide site in a protein. The spectrum of the same sample in the liquid crystalline phase of the phosphopholipids (Figure 2B) is also a powder pattern, but it is axially symmetric with a reduced span due to motional averaging by fast rotational diffusion about the bilayer normal.
Figure 2.
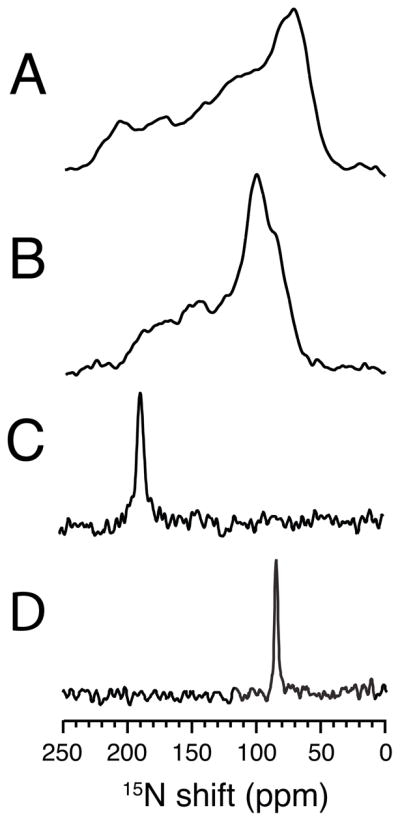
15N NMR spectra of the single site 15N-leucine-11 labeled transmembrane domain of Vpu in lipid bilayers. A. and B. Spectra of an unoriented sample in DMPC bilayers. A. Low temperature 5°C. B. High temperature 30°C. C. and D. Spectra of a magnetically oriented sample in 14-O-PC:6-O-PC q=3.2 bilayers at 40°C. C. The sample is aligned so that the bilayer normal is parallel to the field. D. The sample is aligned so that the bilayer normal is perpendicular to the field.
The aligned sample with its bilayer normal parallel to the field (Figure 2C) is a single-line resonance with the same frequency as the parallel edge of the motionally averaged powder pattern in Figure 2B. Similarly, the aligned sample with its bilayer normal perpendicular to the field (Figure 2D) has the same resonance frequency as the perpendicular edge of the motionally averaged powder pattern. The spectra in Figure 2 demonstrate that the orientationally-dependent 15N chemical shift frequency from the backbone amide of Leu11 can be determined from measuring the edges of the powder pattern in Figure 2B or from the resonance frequencies from the aligned samples in Figure 2C and D. Notably, the frequency edges of the powder pattern are measured from an unoriented bilayer sample, and the single resonance frequencies are measured from magnetically aligned bilayer samples. The slight difference in the chemical shift frequency between the parallel edge of the powder pattern for the unoriented sample and the single-line resonance of the oriented sample is due to the slight decrease in order parameter of the oriented sample compared to the unoriented sample due to the addition of DHPC to the lipids.
The results shown in Figure 3 were obtained on a sample of the membrane-bound form of fd coat protein labeled in its two leucine sites with 15N; Leu 41 is in the hydrophobic trans-membrane helix and Leu 14 in the amphipathic in-plane surface helix. The spectra in Figure 3 demonstrate the equivalence of results obtained from proteins in mechanically aligned bilayer samples and in unoriented samples that undergo rapid rotational diffusion about the bilayer normal. The structure of this protein in Figure 1D shows that the backbone amide sides of these two leucine residues are approximately orthogonal to each other because of the arrangement of the protein in the bilayer. The spectrum in Figure 3A is of the 15N Leu labeled coat protein in DMPC bilayers in their gel phase at a relatively low temperature, and corresponds to a powder pattern for an immobile 15N amide site in a protein. Both of the 15N Leu amide sites appear to have similar chemical shift tensors as expected for residues in α-helices, and can not be differentiated in the powder pattern spectrum in Figure 3A. However, they can be differentiated in the motionally averaged powder pattern in Figure 3B, which was obtained in the liquid crystalline phase of the phosphopholipids at a higher temperature where the protein undergoes rapid rotational diffusional motion about its bilayer normal. The difference between the static and rotationally averaged powder patterns in Figure 3A and B is striking. Not only is the span of the powder pattern from Leu 41 significantly reduced, but also the powder pattern from Leu 14 is changed in sign and even further reduced in span. The experimental motionally averaged overlapping powder patterns in Figure 3B are simulated in Figure 3D. The results are in complete agreement with the previously measured frequencies of the single line resonances observed from a mechanically aligned sample.56 As shown in Figure 3C, the chemical shifts of their 15N resonances are separated by 76 ppm in a mechanically aligned sample with its bilayer normal parallel to the direction of the magnetic field. The spectra in Figure 3B and 3C of the membrane-bound form of fd coat protein, demonstrate that these two amide sites have very different orientations relative to the direction of the applied magnetic field and to the bilayer normal. The parallel edge of the rotationally averaged axially symmetric 15N chemical shift powder patterns corresponds to the single frequency of the aligned sample with the bilayer parallel to the direction of the magnetic field for both 15N sites.
Figure 3.
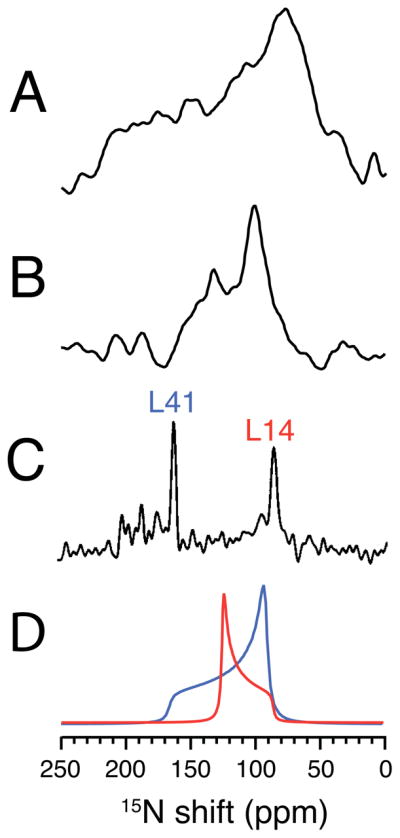
15N NMR spectra of 15N-leucine-14, and 41 labeled fd coat protein in lipid bilayers. A. and B. Spectra of an unoriented sample in DMPC bilayers. A. Low temperature 5°C. B. High temperature 30°C. C. Spectrum of a mechanically aligned sample at 22°C on glass plates aligned with the bilayer normal parallel to the magnetic field. D. Simulated powder line shapes for two leucine sites that are consistent with the spectra in B. and C. and the structure of the protein in bilayers. The spectrum in C. was previously published in reference 35.
MAS solid-state NMR spectra
Identifying the parallel and perpendicular edges of the motionally averaged powder pattern in a stationary unoriented sample is generally feasible when only one site is labeled, as shown in Figure 2. In a very favorable case, such as that shown in Figure 3, it is possible to do this with two labeled sites. However, this approach is clearly limited to very few sites. With the goal of studying proteins isotopically labeled in many sites, including uniformly labeled proteins, it is essential to disentangle the many overlapping powder patterns using MAS solid-state NMR, as has been demonstrated for uniformly labeled polycrystalline proteins6. Here we utilize proteins with one or two labeled sites to demonstrate the feasibility of observing both static and rotationally averaged powder patterns in a membrane protein in phospholipid bilayers.
The experimental MAS solid-state NMR spectra in Figure 4 are from the same unoriented sample of 15N-Leu labeled Vpu TM domain used to generate the data in Figure 2. The spectra were obtained under conditions of relatively slow magic angle spinning so that a sufficient number of spinning sidebands would have measurable intensities in order to reconstruct the underlying powder patterns. This was done under static conditions (low temperature) in Panels A and B and while the protein is undergoing rapid rotational diffusion (high temperature) in Panels C and D. The data in Figure 4 were obtained at two different spinning frequencies. The spectra in panels A and C were obtained at a spinning rate of 2.2 kHz, and those in panels B and D at a spinning rate of 3.0 kHz. In each panel of the Figure, there is from top to bottom, a reconstructed powder pattern generated from the experimental sideband intensities at the bottom of the panel. The experiments performed at two different spinning rates give essentially identical results. The middle spectrum is back calculated from the powder pattern at the top that was derived from the experimental side band intensities observed in the bottom spectrum; the comparison of the sideband intensities in the middle and bottom spectra in each panel provides a measure of the accuracy of the method. The results shown in Figure 4 demonstrate that MAS solid-state NMR of unoriented samples can measure the parallel and perpendicular edges of motionally averaged powder patterns. And comparison to the data in Figure 2 demonstrates that these frequencies are the same as those measured from stationary aligned and unoriented samples.
Figure 4.

MAS solid-state 15N NMR spectra of 15N-leucine-11 labeled transmembrane domain of Vpu in unoriented DMPC lipid bilayers (vesicles). A. and B. were obtained at high temperature 30°C. C. and D. were obtained at low temperature 5°C. In each panel the three spectra are: Top: Powder pattern calculated from the experimental sideband intensities. Middle: Sideband intensities ‘back calculated’ from the powder pattern on top. Bottom: Experimental NMR spectrum. The calculated spectra were simulated with δ11=65 ppm, δ22=75 ppm, δ33=217 ppm and δiso=119 ppm and η=0.1 for the low temperature and δ⊥ =82 ppm, δ||=194 ppm, δiso=119 ppm and η=0 for high temperature. The experimental spectra resulted from signal averaging 3000 transients and 6 second recycle delay. The MAS spinning rates were 2.2 kHz for A and C, and 3.0 kHz for B and D.
Two-dimensional solid-state NMR spectra
In order to characterize protein secondary structure through the observation of regular spectral patterns, such as PISA wheels and Dipolar Waves, and ultimately to calculate the three-dimensional structures of proteins, at least two orientationally dependent frequencies are needed for each backbone site. In most applications to proteins, these have been from the 15N chemical shift and the 1H-15N heteronuclear dipolar coupling,12 but they can also come from 13C labeled sites in 1H/13C/15N triple-resonance experiments.63 In general, two-dimensional experiments are required in order to measure two frequencies from two different spin-interactions in both MAS and stationary samples.
Under fast MAS conditions, the center band at approximately 120 ppm, which corresponds to the isotropic chemical shift frequency, is the only observable signal from the single 15N-Leu labeled site in the Vpu trans-membrane domain. The associated 1H-15N heteronuclear dipolar coupling can be measured using the cross-polarization build-up under Lee-Goldburg conditions to provide selectivity.64,65 These data are shown in Figure 5 for a static sample measured at 5°C (Figure 5A) and a rotationally averaged sample measured at 30°C (Figure 5B). The large center band results from the initial part of the buildup being linear, however, there are sufficient oscillations in the build up curve to provide the doublet used to measure the magnitude of the dipolar coupling following Fourier transformation. In a simple way, the reduction of the 1H-15N heteronuclear dipolar coupling from a static protein at low temperature to one undergoing fast rotational diffusion at a higher temperature can be used to measure the angle between the N-H bond and the bilayer normal in an unoriented sample.
Figure 5.

Two-dimensional MAS SLF spectra of an unoriented 15N-leucine-11 labeled sample of the transmembrane domain of Vpu in unoriented DMPC lipid bilayers (vesicles) obtained with a spinning rate of 7 kHz. A. Low temperature 0°C. B. High temperature 30°C. A. Top: Simulated spectrum for 10 kHz dipolar coupling to match the experimental data. Middle: Slice from experimental spectrum. Bottom: Experimental two-dimensional spectrum obtained from the frequencies observed in the cross-polarization build up curve under Lee-Goldburg irradiation. B. Same as A. except that the simulated spectrum is for 7 kHz dipolar coupling to match the experimental data. 2000 scans and 16 t1 real data points were collected for each two-dimensional spectrum.
Two-dimensional SLF (separated local field spectroscopy)66 is a powerful approach to structure determination of uniaxially aligned samples.8 High resolution versions of the experiment, such as PISEMA,58 enable the resolution and assignment of signals from essentially all of the backbone sites in uniformly 15N labeled proteins,56 and the simultaneous measurement of the orientationally-dependent 15N chemical shift and 1H-15N heteronuclear dipole-dipole coupling frequencies associated with each resonance provides sufficient angular constraints to calculate the three dimensional structures of proteins.54 In the spectra in Figure 6 obtained from mechanically aligned samples of 15N-Leu labeled trans-membrane domain of Vpu, the long chain lipid is DOPC:DOPG (1,2-dioleoyl-sn-glycero-3-phosphocholine: 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol), hence the trans-membrane helix has a smaller tilt angle than in the thinner DMPC bilayers used for the magnetically aligned samples in Figure 2. Nonetheless, the close similarity, and completely predictable differences, of the chemical shift and heteronuclear dipolar coupling frequencies demonstrate the equivalence of the data that can be obtained with mechanically and magnetically aligned protein-containing bilayer samples. In addition the comparison between Figure 6A where the bilayer normal is parallel to the field and Figure 6B where it is perpendicular, show that there is a factor of two difference in the magnitude of the dipolar couplings and an equivalent different in the chemical shifts. However, the same angular measurements come from both spectra.
Figure 6.
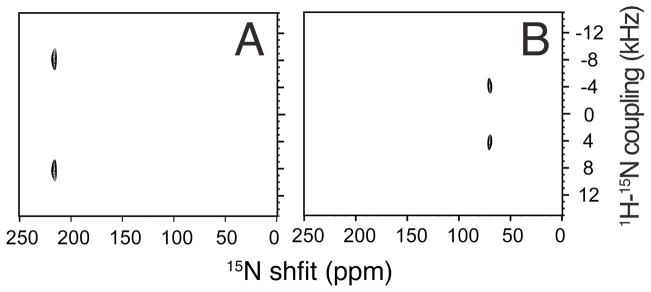
Two-dimensional SLF PISEMA spectra of 15N-leucine-11 labeled transmembrane domain of Vpu in DOPC:DOPG (9:1, w/w) bilayers oriented mechanically on glass plates at 22°C. A. The bilayer normal is parallel to the field. B. The bilayer normal is perpendicular to the field.
Example of Trp 26 of fd coat protein in phospholipid bilayers
The single tryptophan in fd coat protein (residue 26) has two nitrogens; one is an indole nitrogen in the side chain and the other is the backbone amide nitrogen. The spectral properties of these two sites provide an opportunity to demonstrate the equivalence among all of the solid-state NMR approaches to structure determination discussed individually with the data in Figures 1 – 6. The structure of the membrane-bound form of fd coat protein, including the backbone and side chain of Trp-26 has been determined in mechanically aligned phospholipid bilayers between glass plates.54
The resolution between the two nitrogen resonances in the fast MAS solid-state NMR spectrum in Figure 7A results from the difference in their isotropic chemical shifts, and they can be assigned on the basis of the well-established indole and amide nitrogen chemical shift data. The experimental spectrum in Figure 7E was obtained with slow magic angle spinning; it retains the resolution of the two lines as reiterated in the spinning sidebands whose intensities reflect the chemical shift anisotropy powder patterns of the indole and amide nitrogen sites. The sidebands for the two sites are distinguished by blue dots (amide backbone) and red dots (indole side chain). As demonstrated in Figure 4, the intensities of the sidebands enable the underlying chemical shift powder patterns to be reconstructed. The rotationally averaged powder patterns for the backbone amide nitrogen (Figure 7C (blue)) and the side chain indole nitrogen (Figure 7D (red)) are aligned above the corresponding experimental spectrum obtained from a uniformly 15N labeled oriented sample (Figure 7G).34
Figure 7.

Solid-state NMR spectra of 15N-tryptophan-26 labeled fd coat protein in lipid bilayers at 20°C. The tryptophan residue is labeled in both the sidechain indole nitrogen and the backbone amide nitrogen. A. and E. One-dimensional 15N MAS spectra of an unoriented sample of fd coat protein in DOPC:DOPG (9:1, w/w) bilayers. (A. 8.8 kHz spin rate and E. 2.5 kHz spin rate). B. Two-dimensional LG-CP spectrum showing the 1H-15N heteronuclear dipolar couplings obtained with a 8.6 kHz spin rate. C. Simulated 15N static power pattern for the backbone amide site. D. Simulated 15N static powder pattern for the side chain indole site. F. “Dot” representation of the resonances in G. corresponding to the sidechain indole nitrogen (red) and the backbone amide nitrogen (blue) assigned to Trp-26. G. =Two-dimensional PISEMA spectrum of uniformly 15N labeled fd coat protein in POPC:POPG (8:2, w/w) bilayers at 22°C oriented mechanically on glass plates with the bilayer normal parallel to the magnetic field, taken from reference 33.
The parallel edges of the rotationally averaged powder patterns calculated from the spinning sideband intensities are marked with vertical dotted lines. The same chemical shift frequencies are measured from the parallel edges of the motionally averaged powder patterns (Figure 7C and 7D) and the oriented sample spectrum in Figure 7G. To simplify the comparisons, the experimental resonances assigned to Trp 26 in the spectrum of the uniformly 15N labeled sample are simulated as two “dots” with the same frequencies as in the experimental spectrum in the two-dimensional spectral representation in Figure 7F. The vertical dotted blue and red lines demonstrate that the same frequencies can be measured from the parallel edge of the motionally averaged chemical shift powder patterns (Figure 7C and 7D) and from the frequencies of the single line resonances observed in the spectrum of an oriented sample (Figure 7F and 7G).
The fast MAS solid-state NMR spectrum in Figure 7A has two resonances that directly yield the isotropic chemical shift frequencies of the indole and amide nitrogen sites of Trp 26 in the protein. However, these resolved resonances also enable the measurement of the motionally averaged 1H-15N heteronuclear dipole-dipole couplings, as demonstrated in Figure 5. Here too we utilize the oscillations in the cross-polarization build up curves observed under Lee-Goldburg irradiation conditions. Fourier transformation of the build up curves for the two resolved isotropic resonances in Figure 7A results in the two-dimensional spectrum in Figure 7B. The dotted horizontal lines show that the frequencies correspond to those in Figure 7F. These results demonstrate that MAS experiments can be used to characterize the rotationally averaged frequencies that correspond to the mechanically aligned frequencies using unoriented samples where the sites are resolved by their isotropic chemical shift differences. The vertical lines show the measurement of the rotationally averaged chemical shift and horizontal lines show that for the associated heteronuclear dipolar coupling.
Discussion
The measurement of angular constraints for both backbone and side chain sites in a protein provides sufficient input to calculate the three-dimensional structure of a protein. These measurements are well suited to the study of non-globular proteins, such as membrane proteins in phospholipid bilayers. Moreover, these measurements are particularly forgiving with respect to experimental errors and uncertainties regarding the principal values and orientations of the tensors in the molecular frame because site-to-site errors do not accumulate. Each measurement is made independently relative to a common external axis, defined either by the direction of the magnetic field in magnetically or mechanically oriented samples or the bilayer normal for fast rotational diffusion of the protein. In the case of membrane proteins these axes are collinear.
Uniaxial sample alignment where the direction of the molecular alignment is parallel to that of the magnetic field is the next most general type of sample after a single crystal. In this case, the sample has one direction of orientation and the other two directions are random or undefined. What is most notable is that while these samples are capable of yielding very high resolution spectra when they are aligned parallel to the field, the line shapes become broad and complex when they are tilted away from parallel. This is seen most dramatically when the axis of alignment is perpendicular to the direction of the field. A characteristic partial powder pattern is observed in these situations. However, the situation is greatly simplified when the molecule of interest undergoes fast rotational diffusion about a single axis; in the case of membrane proteins, this is the bilayer normal.
Membrane proteins in phosphopholipid bilayers can be aligned mechanically between glass plates. As with uniaxially aligned polymers, when the axis of alignment is parallel to the field, then single line resonances are observed regardless of whether any molecular motion is present. Magnetically aligned samples of membrane proteins were originally sought as direct replacements for mechanically aligned samples, and once it was discovered that the lanthanide ions added to the lipids could “flip” the alignment so that the bilayer normals were parallel to the field61 then that was the case.11 However, there is considerably more flexibility with magnetically aligned systems because the proteins undergo fast rotational diffusion about the bilayer normal.
Once stable magnetically aligned bilayer samples were prepared it was possible to compare the spectra obtained in parallel and perpendicular alignments. Under all conditions examined so far, the proteins yield single line spectra in parallel and perpendicular alignments, and at intermediate angles of alignment; this requires rapid axial diffusion of the protein about the bilayer normal.
The experimental results and their comparisons demonstrate that mechanically aligned, magnetically aligned, and rotationally aligned samples are all equivalent and complementary approaches that can be used to determine the three dimensional structures of membrane proteins. In practice, this provides much needed flexibility in sample preparation and in the spectroscopic experiments. The data in this article were obtained on polypeptides with a single trans-membrane helix because of the opportunities for direct comparisons among the various experiments. However, we have obtained similar results on proteins with two, three, and seven trans-membrane helices, demonstrating the generality of the approach.
Conclusions
OS solid-state NMR is based on the measurement of angular constraints as input for structure calculations in order to determine the structures of proteins. Sample preparation is a key component of experimental design, and often the limiting factor in the selection of protein samples for study. Experimental results on mechanically and magnetically aligned samples are directly compared to those from unoriented samples, demonstrating the equivalence of the angularly dependent frequencies from the chemical shift and dipole-dipole interactions as long as the membrane proteins undergo fast rotational diffusion about the bilayer normal and that this axis is collinear with the axes of alignment defined by the direction of the bilayer normal relative to the magnetic field. This expands the possibilities for applying the basic principles of OS solid-state NMR to unoriented samples, including those with many labeled sites with the use of MAS solid-state NMR methods that reveal or recouple the powder patterns from the motionally averaged chemical shift and dipole-dipole interactions. Angular constraints from these two sources for each peptide plane are sufficient to determine the backbone structure of a protein, and it can be extended to side chains for complete structure determination of membrane proteins in their native liquid crystalline phospholipid bilayer environment.
Acknowledgments
The research described in this article was supported by grants from the National Institutes of Health. It utilized the Biomedical Technology Resource for NMR Molecular Imaging of Proteins at the University of California, San Diego, which is supported by NIH grant P41EB002031.
References
- 1.Cross TA, Opella SJ. J Am Chem Soc. 1983;105:306. [Google Scholar]
- 2.Opella SJ, Frey MH, Cross TA. J Am Chem Soc. 1979;101:5856. [Google Scholar]
- 3.Baldus M. J Biomol NMR. 2007;39:73. doi: 10.1007/s10858-007-9177-3. [DOI] [PubMed] [Google Scholar]
- 4.Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Nature. 2002;420:98. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 5.McDermott A, Polenova T. Curr Opin Struct Biol. 2007;17:617. doi: 10.1016/j.sbi.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Wylie BJ, Rienstra CM. J Chem Phys. 2008;128:052207. doi: 10.1063/1.2834735. [DOI] [PubMed] [Google Scholar]
- 7.Hester RK, Ackerman JL, Neff BL, Waugh JS. Phys Rev Lett. 1976;36:1081. [Google Scholar]
- 8.Opella SJ, Waugh JS. J Chem Phys. 1977;66:4919. [Google Scholar]
- 9.Nicholson LK, Moll F, Mixon TE, LoGrasso PV, Lay JC, Cross TA. Biochemistry. 1987;26:6621. doi: 10.1021/bi00395a009. [DOI] [PubMed] [Google Scholar]
- 10.Bechinger B, Kim Y, Chirlian LE, Gesell J, Neumann JM, Montal M, Tomich J, Zasloff M, Opella SJ. J Biomol NMR. 1991;1:167. doi: 10.1007/BF01877228. [DOI] [PubMed] [Google Scholar]
- 11.Howard KP, Opella SJ. J Magn Reson B. 1996;112:91. doi: 10.1006/jmrb.1996.0116. [DOI] [PubMed] [Google Scholar]
- 12.De Angelis AA, Nevzorov AA, Park SH, Howell SC, Mrse AA, Opella SJ. J Am Chem Soc. 2004;126:15340. doi: 10.1021/ja045631y. [DOI] [PubMed] [Google Scholar]
- 13.Henderson R, Unwin PNT. Nature. 1975;257:28. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 14.Razi-Naqvi K, Gonzlez-Rodriguez J, Cherry RJ, Chapman D. Nature. 1973;345:249. doi: 10.1038/newbio245249a0. [DOI] [PubMed] [Google Scholar]
- 15.Lewis BA, Harbison GS, Herzfeld J, Griffin RG. Biochemistry. 1985;24:4671. doi: 10.1021/bi00338a029. [DOI] [PubMed] [Google Scholar]
- 16.Cherry RJ, Muller U, Schneider G. FEBS Lett. 1977;80:465. doi: 10.1016/0014-5793(77)80498-5. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann W, Restall CJ, Hyla R, Chapman D. Biochim Biophys Acta. 1980;602:531. doi: 10.1016/0005-2736(80)90332-6. [DOI] [PubMed] [Google Scholar]
- 18.Luo W, Cady SD, Hong M. Biochemistry. 2009;48:6361. doi: 10.1021/bi900716s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cross TA, Opella SJ. J Mol Biol. 1985;182:367. doi: 10.1016/0022-2836(85)90197-4. [DOI] [PubMed] [Google Scholar]
- 20.Tsang P, Opella SJ. Biopolymers. 1986:1859. doi: 10.1002/bip.360251004. [DOI] [PubMed] [Google Scholar]
- 21.Henderson R, Unwin PNT. Nature. 1975;257:28. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 22.Grigorieff N, Cesska TA, Downing KH, Baldwin JM, Henderson R. J Mol Biol. 1996;259:393. doi: 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- 23.Deisenhober J, Epp O, Miki K, Huber R, Michel H. J Mol Biol. 1984;180:385. doi: 10.1016/s0022-2836(84)80011-x. [DOI] [PubMed] [Google Scholar]
- 24.Doyle DA, Cabral JM, Pfuetzner R, Kuo Anling A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. Science. 1998;280:69. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 25.Luecke H, Schobert B, Richter H-T, Cartailler J-P, Lanyi JK. J Mol Biol. 1999;291:899. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 26.Johansson LC, Wohri AB, Katona G, Engstrom S, Neutze R. Curr Opin Struct Biol. 2009;19:372. doi: 10.1016/j.sbi.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Faham S, Bowie JU. J Mol Biol. 2002;316:1. doi: 10.1006/jmbi.2001.5295. [DOI] [PubMed] [Google Scholar]
- 28.Raunser S, Walz T. Annu Rev Biophys. 2009;38:89. doi: 10.1146/annurev.biophys.050708.133649. [DOI] [PubMed] [Google Scholar]
- 29.Bartesaghi A, Subramaniam S. Curr Opinion Struct Biol. 2009;19:402. doi: 10.1016/j.sbi.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engel A, Gelb HE. Annu Rev Biochem. 2008;77:127. doi: 10.1146/annurev.biochem.77.062706.154450. [DOI] [PubMed] [Google Scholar]
- 31.Saffman PG, Delbruck M. Proc Natl Acad Sci USA. 1975;72:3111. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson GR, Mazo RM. Biopolymers. 1980;19:1597. [Google Scholar]
- 33.Jahnig F. Eur Biophys J. 1986;114:63. doi: 10.1007/BF00260404. [DOI] [PubMed] [Google Scholar]
- 34.Hughes BD, Pailthorpe BA, White LR, Sawyer WH. Biophys J. 1982;37:673. [PMC free article] [PubMed] [Google Scholar]
- 35.Ramadurai S, Holt A, Krasnikov V, van den Bogaart G, Killian JA, Poolman B. J Am Chem Soc. 2009;131:12650. doi: 10.1021/ja902853g. [DOI] [PubMed] [Google Scholar]
- 36.Watts A, Sternberg B, Ulrich AS, Whiteway CA, Seifert G, Sami M, Fisher P, Heyn MP, Wallat I. Biophys Chem. 1995;56:1–46. doi: 10.1016/0301-4622(95)00013-n. [DOI] [PubMed] [Google Scholar]
- 37.Goncalves JA, Ahuja S, Erfani S, Eilers M, Smith SO. Prog NMR Spectrosc. 2010;57:159. doi: 10.1016/j.pnmrs.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Opella SJ, Marassi FM. Chem Rev. 2004;104:3587. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDermott A. Annu Rev Biophys. 2009;38:385. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 40.McLaughlin AC, Cullis PR, Hemminga MA, Hoult DI, Radda GK, Ritchie GA, Seeley PJ, Richards RE. FEBS Lett. 1975;57:213. doi: 10.1016/0014-5793(75)80719-8. [DOI] [PubMed] [Google Scholar]
- 41.Park SH, Prytulla S, De Angelis AA, Brown JM, Kiefer H, Opella SJ. J Am Chem Soc. 2006;128:7402. doi: 10.1021/ja0606632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park SH, Mrse AA, Nevzorov AA, De Angelis AA, Opella SJ. J Magn Reson. 2006;178:162. doi: 10.1016/j.jmr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Song Z, Kovacs FA, Wang J, Denny JK, Shekar SC, Quine JR, Cross TA. Biophys J. 2000;79:767. doi: 10.1016/S0006-3495(00)76334-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong M, Doherty T. Chem Phys Lett. 2006;432:296. doi: 10.1016/j.cplett.2006.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cady SD, Goodman C, Tatko CD, DeGrado WF, Hong M. J Am Chem Soc. 2007;129:5719. doi: 10.1021/ja070305e. [DOI] [PubMed] [Google Scholar]
- 46.Nevzorov A, De Angelis AA, Park SH, Opella SJ. NMR Spectroscopy of Biological Solids. 2006;7:177. [Google Scholar]
- 47.Cady SD, Hong M. J Magn Reson. 2008;191:219. doi: 10.1016/j.jmr.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Marassi FM, Opella SJ. J Magn Reson. 2000;144:150. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R, Quine JR, Cross TA. J Magn Reson. 2000;144:162. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 50.Marassi FM. Biophys J. 2001;80:994. doi: 10.1016/S0006-3495(01)76078-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park SH, De Angelis AA, Nevzorov AA, Wu CH, Opella SJ. Biophys J. 2006;91:3032. doi: 10.1529/biophysj.106.087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park SH, Mrse AA, Nevzorov AA, Mesleh MF, Oblatt-Montal M, Montal M, Opella SJ. J Mol Biol. 2003;333:409. doi: 10.1016/j.jmb.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 53.Park SH, Opella SJ. J Mol Biol. 2005;350:310. doi: 10.1016/j.jmb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 54.Marassi FM, Opella SJ. Protein Sci. 2003;12:403. doi: 10.1110/ps.0211503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zeri AC, Mesleh MF, Nevzorov AA, Opella SJ. Proc Natl Acad Sci U S A. 2003;100:6458. doi: 10.1073/pnas.1132059100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marassi FM, Ramamoorthy A, Opella SJ. Proc Natl Acad Sci U S A. 1997;94:8551. doi: 10.1073/pnas.94.16.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Angelis AA, Opella SJ. Nat Protoc. 2007;2:2332. doi: 10.1038/nprot.2007.329. [DOI] [PubMed] [Google Scholar]
- 58.Wu CH, Ramamoorthy A, Opella SJ. J Magn Reson Ser A. 1994;109:270. [Google Scholar]
- 59.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 60.Pines A, Waugh JS, Gibby MG. J Chem Phys. 1972;56:1776. [Google Scholar]
- 61.Prosser RS, Hunt SA, DiNatale JA, Vold RR. J Am Chem Soc. 1996;118:269. [Google Scholar]
- 62.Park SH, Loudet C, Marassi FM, Dufourc EJ, Opella SJ. J Magn Reson. 2008;193:133. doi: 10.1016/j.jmr.2008.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sinha N, Grant CV, Park SH, Brown JM, Opella SJ. J Magn Reson. 2007;186:51. doi: 10.1016/j.jmr.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dvinskikh SV, Zimmermann H, Maliniak A, Sandstrom D. J Chem Phys. 2005:122. doi: 10.1063/1.1834569. [DOI] [PubMed] [Google Scholar]
- 65.Lorieau J, McDermott AE. Magn Reson Chem. 2006;44:334. doi: 10.1002/mrc.1773. [DOI] [PubMed] [Google Scholar]
- 66.Waugh JS. Proc Natl Acad Sci U S A. 1976;73:1394. doi: 10.1073/pnas.73.5.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]