

Abstract
A docking screen identified reversible, non-covalent inhibitors (e.g. 1) of the parasite cysteine protease cruzain. Chemical optimization of 1 led to a series of oxadiazoles possessing interpretable SAR and potencies as much as 500-fold greater than 1. Detailed investigation of the SAR series subsequently revealed that many members of the oxadiazole class (and surprisingly also 1) act via divergent modes of inhibition – competitive or via colloidal aggregation – depending on the assay conditions employed.
Small-molecule aggregation leading to promiscuous inhibition of enzymes is now well recognized as a common source of false positives in high-throughput screening.1–4 While these phenomena are not thoroughly understood at the molecular scale, common characteristics of aggregates can be useful in their identification, for example their capacity to inhibit unrelated enzymes5 and the ability of non-ionic detergents to disrupt them and reverse enzyme inhibition.6, 7 Aggregation is viewed by many medicinal chemists as an all-or-nothing phenomena of primary concern in high-throughput screening and hit validation. That non-specific modes of inhibition could emerge in the course of a standard hit-to-lead optimization campaign is not generally appreciated. Even less appreciated is the notion that promiscuous inhibition could be responsible for multiple logs of apparent (interpretable) SAR or that nanomolar-level inhibition can be conferred by small-molecule aggregates. Recently, we uncovered exactly these effects in the course of optimizing a novel class of reversible, non-electrophilic inhibitors of the trypanosome cysteine protease cruzain. Here we describe aspects of this work that bear consideration by any group engaged in chemical optimization guided by biochemical assay data.
Cruzain is the major cysteine protease of the protozoan parasite Trypanosoma cruzi, the causative agent of Chagas’ disease, which affects 16–18 million people in Latin America. The two currently available drugs, benznidazole and nifurtimox, are efficacious only in the acute stage of the disease and present severe side-effects, motivating the search for new agents. The importance of cruzain as a target has been demonstrated through the efficacy of inhibitors of this enzyme in mouse8 and dog9 models of the disease. Several efforts have been conducted to find cruzain inhibitors and diverse classes of compounds have been reported, but mostly covalent warhead-based chemotypes.10, 11
Motivated to find novel inhibitors with a reversible, non-covalent mode of inhibition, we turned to virtual screening of commercially available lead-like12, 13 compounds that could subsequently be optimized by medicinal chemistry. Publically available structures of cruzain14,15 were interrogated computationally with half a million compounds from the lead-like subset of the ZINC database16 using DOCK 3.5.54 (see Supporting Information for details). Following visual inspection of the 500 top-ranked docked molecules, 17 were selected for purchase and testing in an enzyme assay. Among these, glycolamide ester 1 inhibited cruzain with an IC50 of 77 μM (Figure 1, see Supporting Information for a docking pose of 1 bound in the cruzain active site).
Figure 1.

Structures and cruzain inhibition data for original screening hit 1 and structurally-related analogs obtained from commercial sources.
Inhibition of cruzain by 1, though relatively modest, was independent of incubation time and was competitive, with a Ki of 32 μM (Figure 2). Both observations were consistent with a reversible, non-covalent mechanism of inhibition. The assays were conducted in the presence of 0.01% of the detergent Triton X-100—a control for colloidal aggregation2,17 —and close congeners of 1 such as 2 and 3 were also competitive inhibitors of cruzain under similar conditions. Thus, the initial compounds in this series (1–3) were deemed “clean” as far as aggregation and reversibility was concerned, and chemistry was initiated on the series.
Figure 2.
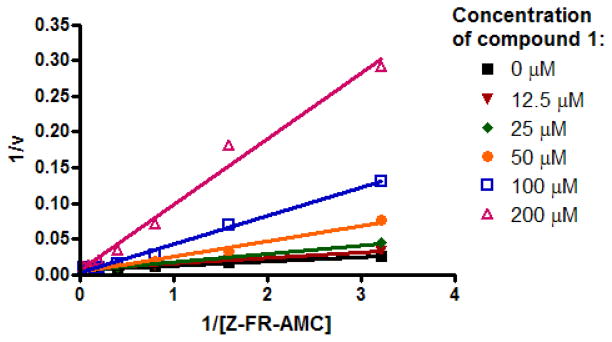
Lineaweaver-Burk plot indicating competitive inhibition of cruzain by 1 (Ki = 32 μM).
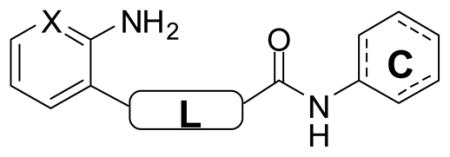
Preliminary SAR profiling around the glycolamide scaffold was carried out with commercially available and synthesized analogs (Figure 1, Table 1, and Supporting Information). The dichloroaniline ring in 1 could be replaced by aromatic, benzylic, or aliphatic ring systems without substantially impacting potency (e.g., 2 and 3). In contrast, the SAR of the aminopyridine ring was much less forgiving. For example, an aniline ring could be substituted for aminopyridine (compare 1 and 6, Table 1), but only if the amine was positioned in the ortho position. Analogs lacking an ortho amino group or having the amino group in another position on the ring were essentially inactive (e.g., 4 and 5, Figure 1).
Table 1.
SAR for cruzain inhibitors bearing ester, amide, or heterocyclic linkers.
| ||||
|---|---|---|---|---|
| Compd | Linker (L) | X | C-Ring | cruzain IC50 (μM) |
| 1 |
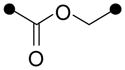
|
N | DiCl-Pha | 77 |
| 6 | CH | DiCl-Pha | 58 | |
| 2 | N | cycloheptyl | 87 | |
|
| ||||
| 7 |
|
N | cyclohexyl | 16% at 200 μM |
| 8 | CH | cyclohexyl | 15% at 200 μM | |
|
| ||||
| 9 |
|
CH | cyclohexyl | 78 |
| 10 | CH | cycloheptyl | 25 | |
| 11 | CH | DiMeOPhb | 27 | |
DiCl-Ph = 2,5-dichlorophenyl,
DiMeOPh = 2,4-dimethoxyphenyl
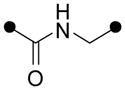
With an aminopyridine (or aniline) ring established as a putative pharmacophore, we set about replacing the labile ester function in 1 with more drug-like surrogates. The most straightforward modification –ester to amide – uniformly produced less potent analogs (e.g., analogs 7 and 8, Table 1). The application of 1,2,4-oxadiazole and related heterocycles as ester bioisosteres is well established18 and so we next walked down this well trod path. Gratifyingly, oxadiazole analogs (e.g., 9–11) were found to be at least as potent as the esters while offering expected advantages with respect to metabolic stability in vivo. As in the ester series, both aliphatic and aromatic “C-rings” were tolerated in the oxadiazole series (e.g., 10 and 11).
With more lead-like scaffolds in hand, we sought to expand our survey of C-ring variants in the oxadiazole series. An established method18 for preparing the oxadiazole ring system was well suited for library generation since diversification of the C-ring would occur in the final step (see Supporting Information). The end result of the C-ring survey was ~50 new analogs, prepared over three cycles of synthesis, with potencies ranging from >100 μM to 200 nM and with clear and interpretable SAR (Table 2, and Supporting Information for the complete data set). Hence, analogs bearing basic, amine containing rings (17, 18) and analogs bearing heterocycles linked by aliphatic chains of more than one carbon atom (19) were generally without measurable activity (IC50 >100 μM). Consistent with the preliminary SAR profile, analogs bearing aliphatic, benzylic, or aryl/heteroaryl C-rings generally possessed mid-low micromolar potencies against cruzain. Most exciting was the finding that substituted pyrazole C-ring analogs inhibited cruzain with sub-micromolar potencies (e.g., cruzain IC50 ~ 200 nM for analog 20). Further exploration of the pyrazole series produced additional interpretable SAR. Hence, aryl (20, 21, 22), heteroaryl (23) or bulky alkyl (24) substitution on the pyrazole ring conferred superior potency than did small alkyl (e.g. Me, 25) substituents. Strikingly, methylation of the pyrazole N-H function completely abrogated activity (analog 26; IC50 > 100 μM), implying that the enhanced potency of the pyrazole series might result in part from a hydrogen bonding interaction not present in the other aryl and benzylic C-rings analogs studied.
Table 2.
Results of enzyme inhibition assays and secondary assays of aggregation for selected oxadiazole analogs and glycolamide 1.
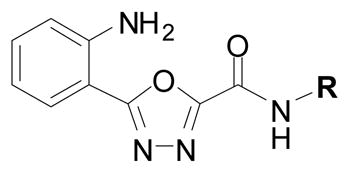
|
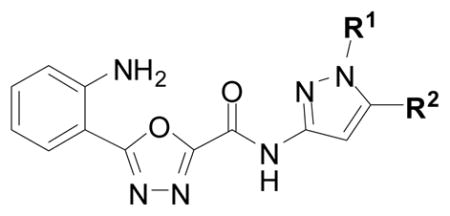
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| cpd | R = | cruzain IC50 (μM)a
|
inhibitsAmpC ? b | particles? (μM)c | cpd | R1 = R2 = |
cruzain IC50 (μM)a
|
inhibitsAmpC ? b | particles? (μM)c | ||||
| 0.001 % triton | 0.01% triton | 0.001 % + BSA | 0.001 % triton | 0.01% triton | 0.001 % + BSA | ||||||||
| 12 |
|
2.0 | >100 | --- | yes 50 μM | >100 | 20 | H 4-MePh |
0.50 | >100 | --- | --- | --- |
| 13 |
|
6.0 | >100 | --- | --- | --- | 21 | H 4-MeOPh |
0.20 | >100 | --- | yes 13 μM | 3.1 |
| 14 |
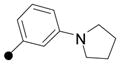
|
7.0 | >100 | 99 | yes 50 μM | 50 | 22 | H Ph |
0.8 | >100 | --- | --- | --- |
| 15 |
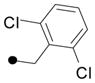
|
8.0 | >100 | --- | --- | --- | 23 | H 2-thioph. |
1.0 | 82.4 | 102 | --- | --- |
| 16 |
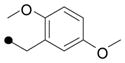
|
8.0 | 44 | 45 | --- | --- | 24 | H t-Bu |
2.0 | >50 | --- | yes 13 μM | 3.1 |
| 17 |
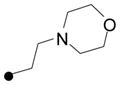
|
>100 | >100 | --- | --- | >200 | 25 | H Me |
11 | >100 | --- | no 50 μM | 50 |
| 18 |
|
>100 | >100 | --- | --- | >200 | 26 | Me H |
>100 | >100 | --- | --- | 200 |
| 19 |
|
>100 | >100 | --- | --- | >200 | 1 | --- | 27 | 77 | 116 | yes 100 μM | 25 |
cruzain IC50s were determined in either 0.01% or 0.001% Triton, and in the later case either with or without pre-incubation with BSA;
compounds were tested against β-lactamase either in the absence or in the presence of 0.01% Triton, at the concentrations noted, and classified as aggregators if inhibition was detergent-sensitive;
particle formation was evaluated by flow cytometry and in some cases examined and confirmed by DLS. The concentrations noted are the lowest evaluated concentration at which particle formation was observed.
With around 50 oxadiazoles synthesized, we were in possession of three logs of consistent, intepretable SAR and had identified analogs that were nearly 500-fold more potent than compound 1. We were troubled, however, that few of the oxadiazole analogs exhibited measurable activity against cultured T. cruzi or Trypanosoma brucei parasites and moreover, the handful of analogs that did were not among the most potent enzyme inhibitors. This discrepancy was initially rationalized as arising from poor cell permeability or active efflux from parasite, but a close inspection of the enzyme dose-response curves, many of which turned out to have unusually high Hill slopes, suggested a second possibility: that the inhibitors were acting by super-stoichiometric mechanisms.1, 19 At this juncture we also determined that the analogs from the C-ring survey had inadvertently been assayed at a 10-fold lower detergent concentration (0.001% Triton X-100) than was employed in the original profiling of analogs 1–11 (0.01%).
Concerned, we repeated the IC50 determinations for selected oxadiazole and glycolamide analogs at both low and high detergent concentration (Table 2, Figure 3).7, 20 Significantly, each of the oxadiazoles examined showed either no measurable dose-response or a significantly higher IC50 when tested at the higher Triton concentration. Only one analog (16) exhibited potency comparable to the early oxadiazole leads 9–11 under high Triton conditions. In contrast, the IC50 values of glycolamide analogs 1–3 were unchanged or only modestly altered (~3-fold in the case of 1, essentially unaltered for 2 and 3) at the different detergent concentrations. Also consistent with aggregation, some oxadiazoles were sensitive to pre-incubation with Bovine Serum Albumin (BSA), which at high-concentration competes with enzyme for colloid particles (aggregates), preventing or reducing inhibition of the target enzyme.21 Thus, oxadiazoles 14 and 23 showed, respectively, a 10- and 100-fold increases in IC50 value in the presence of 1mg/ml BSA, while analog 16 was unaffected by BSA pre-treatment.
Figure 3.
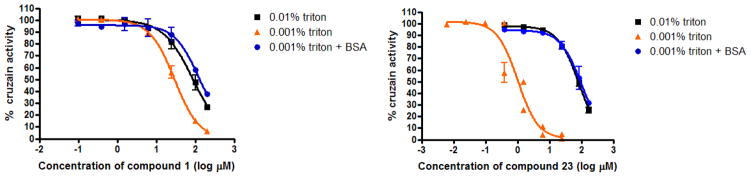
IC50 curves for original lead compound 1 (left panel) and oxadiazole 23 (right panel) at various concentrations of Triton X-100 and with or without pre-incubation with BSA. The oxadiazole shows a much more significant dose-response shift at low detergent concentrations, likely owing to the inadvertent optimization of this series under low Trion X-100 conditions.
Detergent-reversible inhibition of AmpC β-lactamase is another marker of promiscuous aggregation and so this was examined next. Four of five oxadiazoles tested did indeed inhibit β-lactamase in a detergent-reversible fashion at relevant compound concentrations, the exception being analog 25. Quite surprisingly, the original glycolamide lead 1 (but not 2 or 3) was also found to inhibit AmpC, and its inhibition was reversed by 0.01% Triton. This result suggested that glycolamide 1 might also act as an aggregator under certain assay conditions, although clearly this was not true of 1 in its inhibition of cruzain where competitive inhibition (at 0.01% Triton) had already been established (Figure 2).
Next, we sought direct evidence of particle (aggregate) formation by dynamic light scattering (DLS)22 and flow cytometry.23 We studied both suspected aggregators as well as analogs that fail to inhibit enzyme under any conditions, as the latter compounds presumably do not aggregate (an thus do not inhibit). At relevant inhibitory concentrations for low detergent conditions (0.001% Triton X-100), suspected oxadiazole aggregators and also compounds 1–3 were above their critical aggregation concentrations (CACs)16 and formed colloidal aggregates (the exception being 12) of size 250 to 400 nm. In contrast, inactive analogs (e.g. 17–19) did not show particle formation even at high concentration.
These studies provided strong evidence for a non-specific mode of inhibition by oxadiazoles under low Triton (0.001%) assay conditions. Not all the secondary assay data were in perfect agreement however, and the DLS and AmpC inhibition data were puzzling as they suggested that at least some of the original glycolamide analogs could also form aggregates under low Triton conditions. We therefore re-evaluated the nature of cruzain inhibition at both high and low Triton concentrations for glycolamide 1 and a representative oxadiazole (23). Under low Triton conditions, oxadiazole 23 (at 5 μM) was a non-competitive inhibitor of cruzain, consistent with aggregation-based inhibition. In contrast, under high Triton conditions, 23 (at 50 μM) was a competitive inhibitor of cruzain. Similarly, glycolamide 1 was found to be non-competitive under low Triton conditions and at a relevant inhibitor concentration (25 μM). Hence, in both glycolamide 1 and oxadiazole 23, divergent modes of enzyme inhibition are operating under the two different assay conditions. Even so, there are subtle differences between these two chemotypes. Members of the oxadiazole class, having been unwittingly optimized for aggregation (driven by an assay condition favoring aggregation) can be >100-fold more potent as aggregators than as competitive inhibitors (e.g. cmpds 20–23 Table 2, 1Figure 3). In contrast, glycolamides –3 confer similar potencies by the two modes of inhibition. Thus, the differences in detergent effects (Figure 3) and initially surprising results concerning AmpC inhibition by 1 can be reconciled.
Two important conclusions can be drawn from the studies described here. First, divergent modes of inhibition can exist in a homologous SAR series, and these divergent mechanisms are only discernable by careful consideration of assay conditions. We would argue that such a possibility should be considered as the default position, since the migration from competitive to non-competitive regimes will not usually be obvious from structure alone. The ester bioisostere approach described here is a standard one, and could not have been predicted a priori to have led to a series of aggregators with nanomolar potencies. That this in fact occurred owes to the use of an assay condition under which the non-competitive mode of inhibition was predominant.
The second important finding is that interpretable SAR spanning a range of potencies can originate from a wholly non-specific mode of inhibition. Thus, the presence of SAR does not necessarily imply a specific binding interaction. Unfortunately, the medicinal chemist’s natural aptitude for pattern recognition and hypothesis generation works against them here. The poor activity of analogs such as 17, 18, and 26 was easily rationalized in structural terms (e.g., poor tolerance of a charged substituent in 17, entropic effects in 18, loss of a hydrogen bond in 26) when, in fact, the failure of these analogs to inhibit cruzain owes to a failure to aggregate. The danger for the medicinal chemist lies in interpreting SAR in terms of a specific binding interaction when, as shown here, such patterns of varying potency (SAR) may derive from a relative likelihood of aggregation (i.e., an SAR of aggregation). This is a humbling realization and emphasizes the peril of interpreting SAR derived from biochemical assays without considering the possibility of promiscuous inhibition, and more generally emphasizes the importance of understanding molecular mechanism throughout a series.24 Fortunately, experiments to exclude aggregation-based inhibition are straight-forward.25 Docking methods may also be helpful in spotting problematic scaffolds; a retrospective DOCK ranking of oxadiazole analogs revealed no correlation with enzyme inhibition data obtained under low Triton conditions (see Supporting Information). The cautionary contribution of this study is to point out that even within a clear SAR series, one is never entirely free from the concern that non-stoichiometric, artifactual mechanisms are contributing to the inhibition one observes.
Supplementary Material
Acknowledgments
This work was generously supported by funding from the Sandler Foundation (to ARR), the QB3-Malaysia Program (KKHA) and by GM71630 (to BKS). We thank BD Biosciences for a long-term loan of a BD Solubility Scanner for flow cytometry of aggregates and Ana Lazic for providing the cruzain construct. We acknowledge Priyadarshini Jaishankar and DongMei Zhou for experimental contributions in the early stages of this work. We thank Drs. Michelle Arkin, and Jim Wells for helpful suggestions and Allison Doak and Oliv Eidam for reading the manuscript.
Abbreviations
- DLS
dynamic light scattering
- BSA
bovine serum albumin
Footnotes
Supporting Information Available: Experimental details for the DOCK screen (including docking pose for 1) and for all biochemical assays and secondary assays of aggregation. Synthetic procedures and spectroscopic characterization data for all new compounds. Table of cruzain inhibition data at 0.001% triton for the full set of ~50 oxadiazole analogs synthesized. Retrospective comparison of DOCK scores and biochemical inhibition data for oxadiazole analogs. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Feng BY, SA, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J Med Chem. 2007;50(10):2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 2.Feng BY, SA, Doman TN, Guy RK, Shoichet BK. High-throughput assays for promiscuous inhibitors. Nat Chem Biol. 2005;1(3):146–8. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- 3.Liu HY, Wang Z, Regni C, Zou X, Tipton PA. Detailed Kinetic Studies of an Aggregating Inhibitor; Inhibition of Phosphomannomutase/Phosphoglucomutase by Disperse Blue 56†. Biochemistry. 2004;43(27):8662–8669. doi: 10.1021/bi0491907. [DOI] [PubMed] [Google Scholar]
- 4.Reddie KG, Roberts DR, Dore TM. Inhibition of Kinesin Motor Proteins by Adociasulfate-2. Journal of Medicinal Chemistry. 2006;49(16):4857–4860. doi: 10.1021/jm060115z. [DOI] [PubMed] [Google Scholar]
- 5.Seidler J, MS, Doman TN, Shoichet BK. Identification prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem. 2003;46(21):4477–86. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 6.Feng BY, SB A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc. 2006;1(2):550–3. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryan AJ, Gray NM, Lowe PN, Chung C-w. Effect of Detergent on “Promiscuous” Inhibitors. Journal of Medicinal Chemistry. 2003;46(16):3448–3451. doi: 10.1021/jm0340896. [DOI] [PubMed] [Google Scholar]
- 8.Engel JC, DP, Hsieh I, McKerrow JH. Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J Exp Med. 1998;188(4):725–734. doi: 10.1084/jem.188.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barr SC, WK, Kornreic BG, Piscitelli J, Wolfe A, Benet L, McKerrow JH. A cysteine protease inhibitor protects dogs from cardiac damage during infection by Trypanosoma cruzi. Antimicrob Agents Chemother. 2005;49(12):5160–1. doi: 10.1128/AAC.49.12.5160-5161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katrien Brak PSD, McKerrow James H, Ellman Jonathan A. Identification of a New Class of Nonpeptidic Inhibitors of Cruzain. J Am Chem Soc. 2008;130:6404–6410. doi: 10.1021/ja710254m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaishankar P, HE, Zhao DM, Doyle PS, McKerrow JH, Renslo A. Potency selectivty of P2/P3-modified inhibitors of cysteine proteases from trypanosomes. Bioorg Med Chem Lett. 2008;18(2):624–8. doi: 10.1016/j.bmcl.2007.11.070. [DOI] [PubMed] [Google Scholar]
- 12.Teague SJ, DA, Leeson PD, Oprea T. The design of leadlike combinatorial libraries. Angew Chem Int Ed Engl. 1999;38(24):3743–3748. doi: 10.1002/(SICI)1521-3773(19991216)38:24<3743::AID-ANIE3743>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 13.Rishton GM. Nonleadlikeness and leadlikeness in biochemical screening. Drug Discov Today. 2003;8(2):86–96. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]
- 14.Huang L, BL, Ellman JA. Crystal structures of reversible ketone-based inhibitors of the cysteine protease cruzain. Bioorg Med Chem. 2003;11(1):21–9. doi: 10.1016/s0968-0896(02)00427-3. [DOI] [PubMed] [Google Scholar]
- 15.Gillmor SA, CC, Fletterick RJ. Structural determinants of specificity in the cysteine protease cruzain. Protein Sci. 1997;6(8):1603–1611. doi: 10.1002/pro.5560060801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irwin JJ, SB ZINC - a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–82. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Babaoglu K, Simeonov A, Irwin JJ, Nelson ME, Feng B, Thomas CJ, Cancian L, Costi MP, Maltby DA, Jadhav A, Inglese J, Austin CP, Shoichet BK. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against beta-lactamase. J Med Chem. 2008;51(8):2502–11. doi: 10.1021/jm701500e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borg S, Estenne-Bouhtou G, Luthman K, Csoeregh I, Hesselink W, Hacksell U. Synthesis of 1,2,4-Oxadiazole-, 1,3,4-Oxadiazole-, and 1,2,4-Triazole-Derived Dipeptidomimetics. J Org Chem. 1995;60(10):3112–3120. [Google Scholar]
- 19.Shoichet BK. Interpreting steep dose-response curves in early inhibitor discovery. J Med Chem. 2006;49(25):7274–7. doi: 10.1021/jm061103g. [DOI] [PubMed] [Google Scholar]
- 20.Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc. 2006;1(2):550–3. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coan KE, SB Stability and equilibria of promiscuous aggregates in high protein millieus. Mol Biosyst. 2007;3(3):208–13. doi: 10.1039/b616314a. [DOI] [PubMed] [Google Scholar]
- 22.McGovern SL, HB, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46(20):4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 23.Coan KE, SB Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J Am Chem Soc. 2008;130(29):9606–12. doi: 10.1021/ja802977h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardy JA, Wells JA. Searching for new allosteric sites in enzymes. Curr Opin Struct Biol. 2004;14(6):706–15. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Shoichet BK. Screening in a spirit haunted world. Drug Discovery Today. 2006;11(13–14):607–15. doi: 10.1016/j.drudis.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.