

Abstract
Inappropriate Na+ reabsorption by thick ascending limbs (THALs) induces hypertension. Nitric oxide (NO) produced by NO synthase type 3 (NOS3 or eNOS) inhibits NaCl reabsorption by THALs. Tumor necrosis factor alpha (TNF-α) decreases NOS3 expression in endothelial cells and contributes to increases in blood pressure. However, the effects of TNF-α on THAL NOS3 and the signaling cascade are unknown. TNF-α activates several signaling pathways including Rho/Rho kinase (ROCK) which is known to reduce NOS3 expression in endothelial cells. Therefore, we hypothesized that TNF-α decreases NOS3 expression via Rho/ROCK in rat THAL primary cultures. THAL cells were incubated with either vehicle or 1 nmol/L TNF-α for 24 hrs and NOS3 expression was measured by Western blot. TNF-α decreased NOS3 expression by 51±6% (p<0.002) and blunted stimulus-induced NO production. A 10-minutes treatment with TNF-α stimulated RhoA activity by 60±23% (p<0.04). Inhibition of Rho GTPase with 0.05 μg/mL C3 exoenzyme blocked TNF-α-induced reductions in NOS3 expression by 30±8% (p<0.02). Inhibition of ROCK with 10 μmol/L H-1152 blocked TNF-α-induced decreases in NOS3 expression by 66±15 % (p<0.001). Simultaneous inhibition of Rho and ROCK had no additive effect. Myosin light chain kinase, NO, protein kinase C, mitogen-activated kinase kinase, c-Jun amino terminal kinases and Rac-1 were also not involved in TNF-α-induced decreases in NOS3 expression. We conclude that TNF-α decreases NOS3 expression primarily via Rho/ROCK in rat THALs. These data suggest that some of the beneficial effects of ROCK inhibitors in hypertension could be due to the mitigation of TNF-α-induced reduction in NOS3 expression.
Keywords: eNOS, hypertension, kidney, cytokines
Introduction
Thick ascending limbs (THALs) reabsorb 20 to 30% of the filtered NaCl load and contribute to the maintenance of the renal corticomedullary osmotic gradient1. Increased NaCl reabsorption by this nephron segment induces salt-sensitive hypertension2,3. Conversely, NO produced by NO synthase type 3 (NOS3, endothelial NOS or eNOS) inhibits Cl- transport by THALs4 and thus leads to natriuresis. Activation of NOS3 by physiological stimulators such as luminal flow5, endothelin-16 and angiotensin II7 is mediated by phosphatidylinositol triphosphate kinase (PI3K) and phosphatidylinositol 3,4,5-triphosphate (PIP3). NOS3 activity is also modulated by hormones, cytokines, mechanical stress and oxygen tension8. Changes in NOS3 activity can be caused by changes in NOS3 expression6,9, phosphorylation10 or both11. Because reduced NOS3 levels results in enhanced Na+ retention by the kidney and elevated blood pressure12, studying the factors that decrease NOS3 expression is of physiological relevance.
Tumor necrosis factor alpha (TNF-α) is a pro-inflammatory cytokine elevated in hypertension13-15 and heart failure16. Angiotensin II stimulates TNF-α release by THALs17 and TNF-α mediates part of the increase in blood pressure in angiotensin II-dependent hypertension18,19. TNF-α also plays a role in the elevation of blood pressure caused by insulin resistance20 and systemic lupus erythematosus21.
Part of the pro-hypertensive actions of chronic elevations of TNF-α appears to be due to reductions in NO produced by NOS320. TNF-α decreases NOS3 expression in adipocytes, myocytes22 and endothelial cells23,24. However, whether TNF-α reduces NOS3 expression in the THAL has not been studied.
TNF-α activates several intracellular signaling cascades. These include protein kinase C (PKC)25, myosin light chain kinase (MLCK)26, mitogen-activated protein kinase kinase (MAPKK)27, c-Jun amino terminal kinases (JNK)28 and Rac-129. Contrary to the chronic effects, acute treatment with TNF-α increases NO production30 and enhances the activity of Rho GTPase and Rho kinase (ROCK)27. The latter two pathways can reduce NOS3 expression31-34. On one hand we have shown that angiotensin II decreases NOS3 expression in THALs via peroxynitrite31 and thus its inhibition depends on NO. On the other hand, hypoxia and thrombin reduce NOS3 expression via Rho/ROCK in endothelial cells33,34. Reductions in NOS3 expression by high glucose were prevented by blocking both peroxynitrite and ROCK activity in endothelial cells.35 Therefore, we hypothesized that TNF-α decreases NOS3 expression via Rho/ROCK in THALs.
Methods
Primary cultures of medullary THALs (mTHALs)
All protocols involving animals were approved by the Institutional Animal Care and Use Committee (IACUC) of Henry Ford Hospital. The composition of physiological saline used was (in mmol/L) 130 NaCl, 2.5 NaH2PO4, 4 KCl, 1.2 MgSO4, 6 D/L-alanine, 1 trisodium citrate, 5.5 glucose, 2 calcium dilactate, and 10 HEPES. The solution was adjusted to 320 ± 3 mosmol/kgH2O with mannitol and was pH 7.4 at room temperature. Rat mTHALs primary culture were generated as previously described31. In the first protocol, forty hours after cells were seeded they were treated either with vehicle (DMEM/F12 medium) or 1 nmol/L TNF-α (Sigma) for 24 hrs. In subsequent experiments mTHALs were seeded in 4 wells; 1) vehicle, 2) TNF-α, 3) inhibitor alone and 4) inhibitor plus TNF-α. Cells were pre-incubated with the desired inhibitor or vehicle 1 hour before adding vehicle or TNF-α. When Rho was inhibited, cells were pre-treated with exoenzyme C3 transferase or vehicle for 12 hours before stimulating them with TNF-α.
Drugs concentration and source
please see online supplement material at http://hyper.ahajournals.org.
Western blot analysis
NOS3 expression was measured as previously described31 with some modifications (please see online supplement material http://hyper.ahajournals.org). One set of samples (i.e. vehicle, TNF-α, inhibitor, inhibitor plus TNF-α) were loaded using a single gel so each experiment had its own control.
RhoA GTPase activation
RhoA GTPase was measured using the colorimetric G-LISA RhoA activation assay biochemical kit from Cytoskeleton Inc (Denver, CO). Briefly, aliquots of mTHALs suspensions were seeded in 24 wells plate (150 μg/well) in DMEM/F12 media. Tubules were incubated at 37°C and 95/5% O2/CO2 for 4 hours. Tubules were then treated for 0 or 10 min with 1 nmol/L TNF-α and RhoA activity was measured as described by the manufacturer protocol and detailed in online supplement material (please see http://hyper.ahajournals.org).
Measurement of NO Production by Fluorescence Microscopy
NO was measured before and after treatment with PIP3 in mTHAL cells cultured on glass coverslips and previously treated with either vehicle of TNF-α for 24 hours (please see online supplement material at http://hyper.ahajournals.org).
In experiments where the acute effect of TNF-α on NO production was measured, mTHALs were isolated from 100 to 150 g male Sprague Dawley rats as previously described36. Tubules were held between glass pipettes at 4 °C in a chamber designed for live cell imaging on the stage of an inverted microscope as done routinely in our laboratory36 and detailed in supplement material (please see http://hyper.ahajournals.org).
Statistical analysis
Results are expressed as percentage of control ± standard error. Data was analyzed by the Biostatistics and Research Epidemiology Department from Henry Ford Hospital. In some experiments ANOVA was used with post hoc testing. When multiple pair-wise comparisons were done, a procedure for multiple tests of significance was applied using Hochberg’s significance limits37.
Results
To begin testing our hypothesis that TNF-α decreases NOS3 expression in mTHALs we first treated rat mTHAL primary cultures with either vehicle or 1 nmol/L TNF-α for 24 hours. TNF-α reduced NOS3 expression by 51 ± 6% (Figure 1; n=5, p<0.002) compared to controls. When corrected by β-tubulin, the effect of TNF-α was 51 ± 8% (n=5 p<0.003). These data indicate that TNF-α decreases NOS3 expression in mTHALs.
Figure 1. Effect of TNF-α in NOS3 expression in mTHALs.
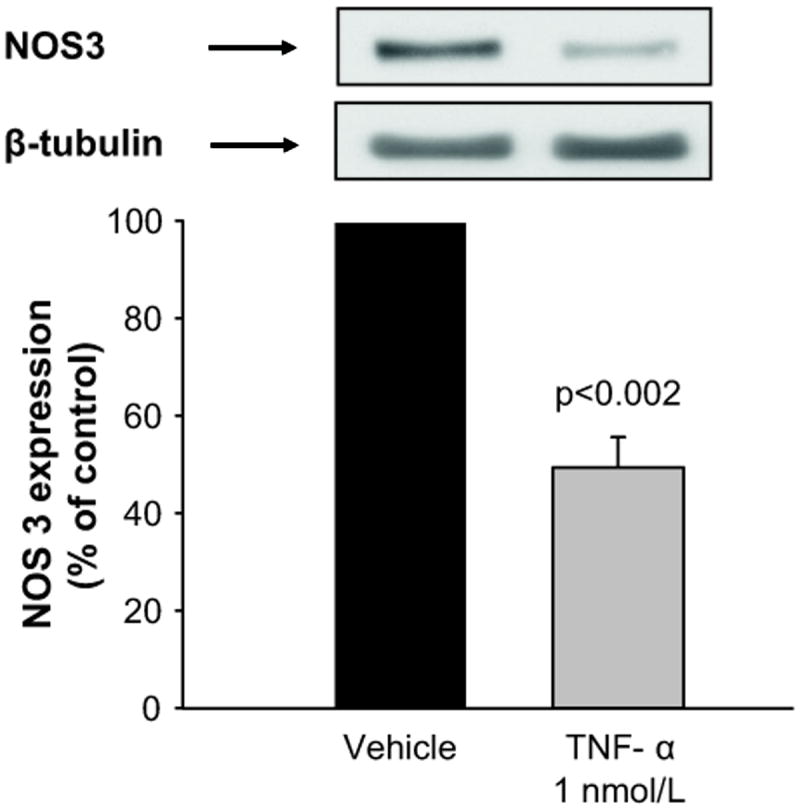
Top: representative Western blot for NOS3 and the loading control β-tubulin. Bottom: cumulative data (n=5).
Next, we tested whether TNF-induced decreases in NOS3 expression resulted in impaired NO production. PIP3 was used to stimulate NOS35-7. In vehicle treated cells, NO production increased from 4.83 ± 0.56 fluorescence units (FU)/min to 7.01 ± 0.68 FU/min in response to PIP3 (p<0.02, n=6, Figure 2A). In contrast, in cells treated for 24 hrs with TNF-α, PIP3 did not significantly increase NO production (basal: 5.24 ± 0.75 vs PIP3: 5.69 ± 0.86, n=6 Figure 2B). These data indicate that chronic exposure to TNF-α reduces stimulus-induced NO production by mTHALs.
Figure 2.
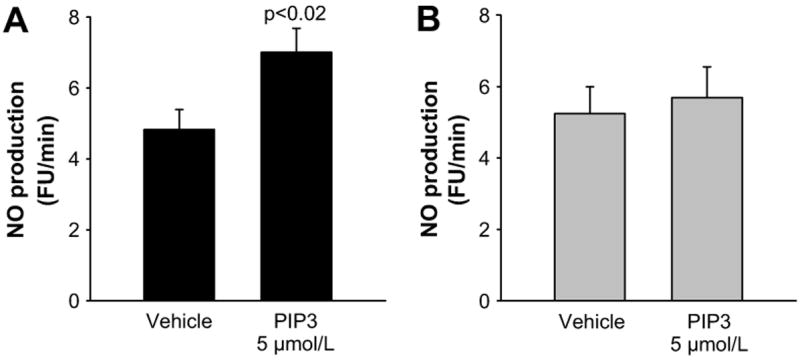
A. Basal and PIP3-induced NO production in mTHALs cells treated with A) vehicle or B) TNF-α for 24 hrs (n=6).
TNF-α has been shown to activate RhoA GTPase in tubular27 and endothelial cells 38. Therefore, we next tested whether TNF-α increased RhoA activity in mTHALs. Basal RhoA activity was 0.520 ± 0.038 OD, acute treatment with TNF-α (10 min) increased RhoA activity to 0.816 ± 0.090 OD (Δ= 60 ± 23%, n=4 p<0.04, Figure 3). These data indicate that TNF-α stimulates RhoA GTPase activity in mTHALs. Therefore, we tested whether TNF-α decreased NOS3 expression via Rho by incubating cells with 0.05 μg/mL exoenzyme C3 transferase. Figure 4 shows that Rho inhibition blocked TNF-α-induced decreases in NOS3 expression by 30 ± 8% (n=7 p<0.02 vs TNF-α) whereas treatment with C3 exoenzyme did not significantly affect basal NOS3 expression (Δ= 14 ± 15% vs vehicle n=7). These data suggest that TNF-α reduces NOS3 expression via Rho GTPase.
Figure 3.
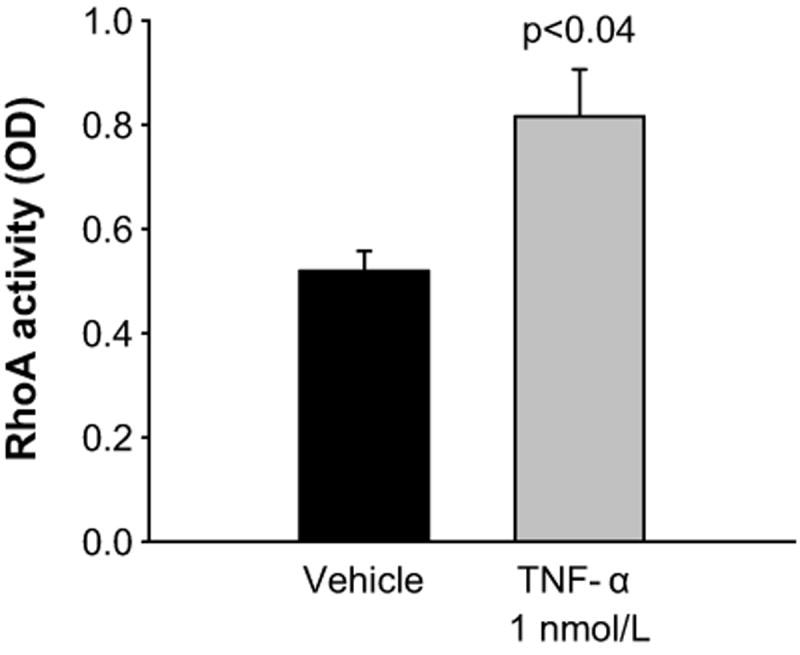
Effect of TNF-α on RhoA GTPase activity in mTHALs (n=4)
Figure 4. Effect of Rho GTPase blockade on TNF-α-induced decreases in NOS3 expression in mTHALs.
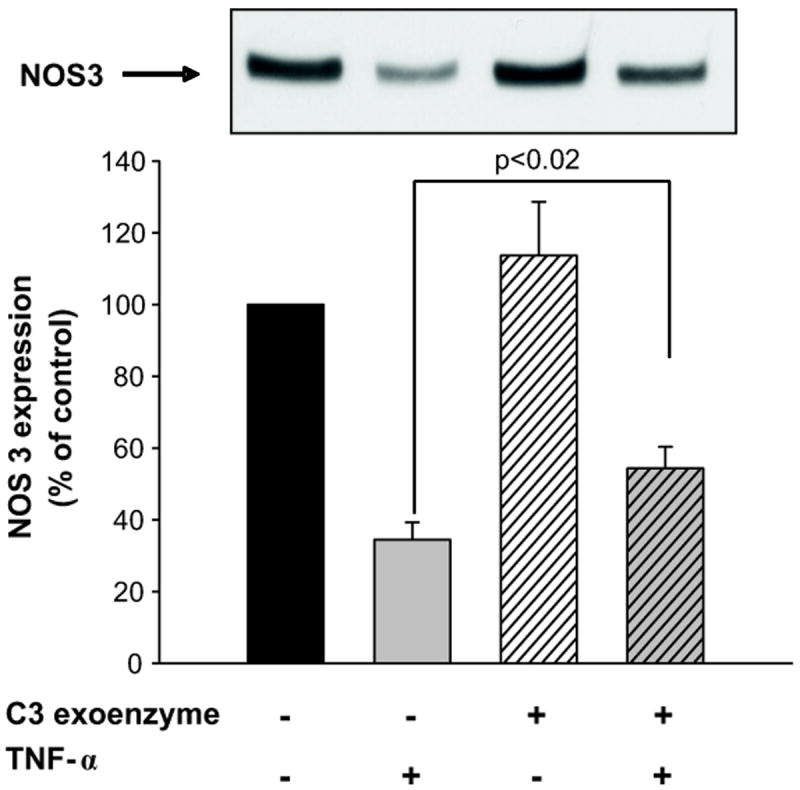
Top: representative Western blot for NOS3. Bottom: cumulative data (TNF-α vs vehicle p<0.001; TNF-α vs C3+ TNF-α p<0.02; vehicle vs C3 not significant, n=7).
Active Rho binds and activate ROCK, thus we tested whether ROCK mediated TNF-α’s effect on NOS3 expression. Figure 5 shows that TNF-α alone decreased NOS3 expression by 56 ± 7% (n=6, p<0.001). Inhibition of ROCK with 10 μmol/L H-1152 blocked TNF-α-induced reductions in NOS3 expression by 66 ± 15 % (n=6, p<0.01 vs TNF-α alone). ROCK inhibitor alone did not significantly affect basal NOS3 expression (Δ= 19 ± 11% vs vehicle n=6). Lower and higher concentrations of the inhibitor were tested; however, the ability to blunt TNF-α-induced inhibition was the same. These data indicate that TNF-α reduces NOS3 expression in part via activation of ROCK. In addition, concomitant inhibition of RhoA and ROCK did not have an additive effect, indicating that RhoA and ROCK are part of the same signaling cascade (please see online supplement material at http://hyper.ahajournals.org).
Figure 5. Effect of ROCK blockade on TNF-α-induced decreases in NOS3 expression in mTHALs.
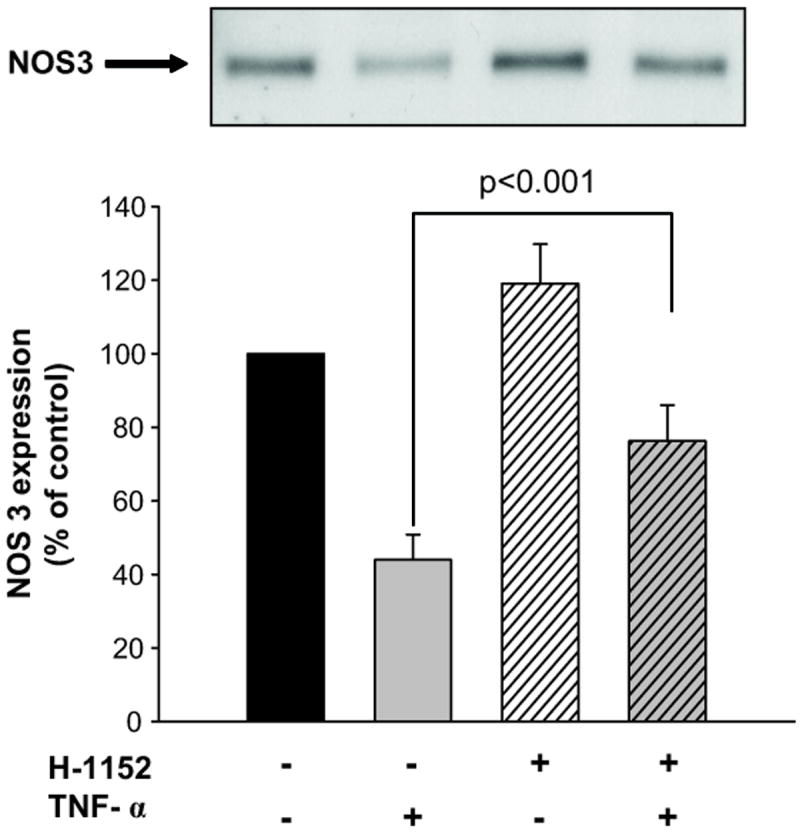
Top: representative Western blot for NOS3. Bottom: cumulative data (TNF-α vs vehicle p<0.001; TNF-α vs H-1152+ TNF-α p<0.001; vehicle vs H-1152, not significant, n=6).
NO can decrease NOS3 expression via peroxynitrite formation 31. Therefore, we next tested whether TNF-α acutely increased NO production by isolated mTHALs. Treatment with TNF-α increased NO production from -0.153 ± 0.116 FU/min to 0.408 ± 0.071 FU/min (Figure 6; n=5; p<0.02) whereas vehicle treatment did not significantly affect NO production (basal NO production: -0.020 ± 0.103 FU/min; vehicle treatment 0.065 ± 0.040 FU/min; n=4). These data indicate that similar to angiotensin II, TNF-α acutely stimulates NO production by mTHALs.
Figure 6.
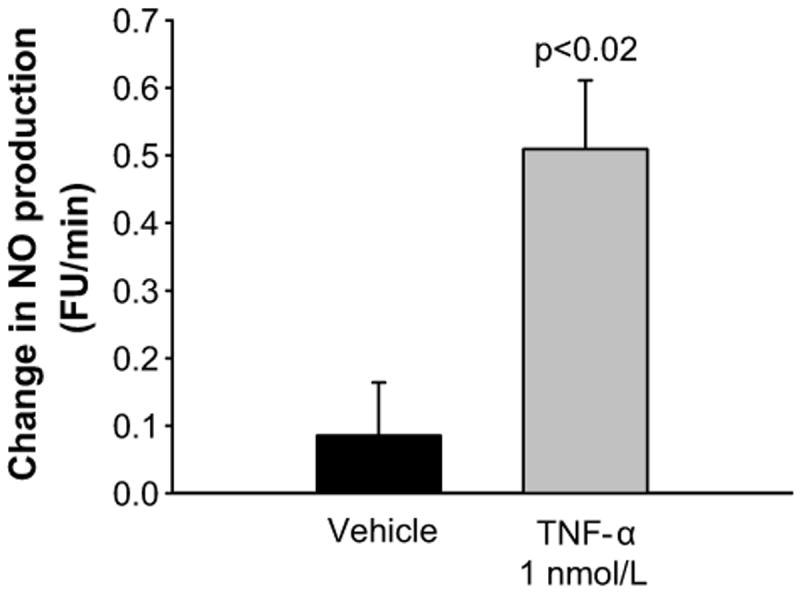
Acute effect of TNF-α on NO production by mTHAL (vehicle n=4, TNF n=6)
Next, we tested whether the TNF-α-induced reductions in NOS3 expression was mediated by NO by inhibiting NO production with L-NAME. TNF-α alone decreased NOS3 expression by 48 ± 6% (n=4, p<0.001 vs vehicle); however, L-NAME (4 mmol/L) did not block the effect of TNF-α on NOS3 expression (Δ= -43 ± 5% vs vehicle, Figure 7). L-NAME treatment did not affect basal NOS3 expression. Thus, NO, and therefore peroxynitrite, did not appear to mediate TNF-α-induced reductions in NOS3 expression.
Figure 7. Effect of NOS inhibition on TNF-α-induced decreases in NOS3 expression in mTHALs.
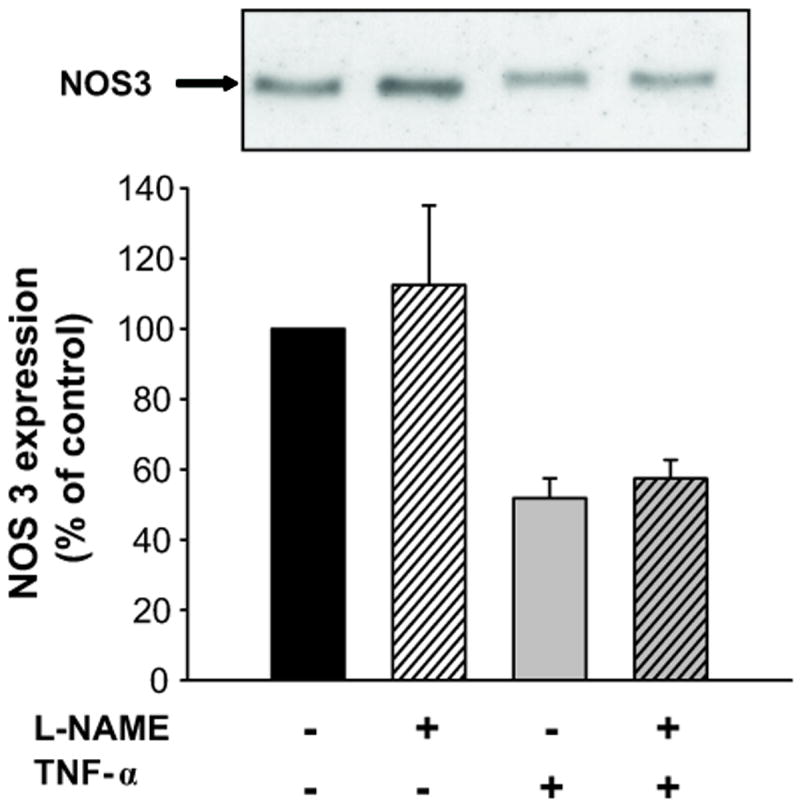
Top: representative Western blot for NOS3. Bottom: cumulative data (TNF-α vs vehicle p<0.003; TNF-α vs L-NAME + TNF-α not significant; vehicle vs L-NAME not significant; n=4).
Finally, we tested a number of other signaling cascades. These pathways have been shown to: 1) be activated by TNF-α; or 2) play a role in NOS3 expression. Inhibition of MLCK, PKC, MAPKK, JNK, JAK or Rac-1 did not prevent TNF-α from decreasing NOS3 expression (please see online supplement material at http://hyper.ahajournals.org).
Discussion
We hypothesized that TNF-α decreases NOS3 expression via RhoA GTPase and its associated ROCK in mTHALs. We found that: 1) chronically, TNF-α decreases NOS3 expression and NO production in rat mTHAL cells; 2) acutely, TNF-α activates RhoA GTPase; 3) two thirds of the TNF-α-induced decrease in NOS3 expression was mediated by the Rho/ROCK pathway; and 4) none of the several other signaling molecules we tested could account for the remaining third including, NO, MLCK, PKC, MAPKK, JAK, JNK and Rac-1.
We used primary cultures of mTHALs to avoid the systemic effects of TNF-α that could confound our interpretation of the data. The concentration of TNF-α in the interstitium of the outer medulla under physiological and pathophysiological conditions is unknown. Thus, we used 1 nmol/L TNF-α, which is approximately the concentration produced by THALs stimulated with angiotensin II in vitro17. All the experiments using inhibitors had their own paired controls to account for anticipated variability in the effect of TNF-α on NOS3 expression in primary cultures generated from different rats at different times.
The effect of TNF-α on NOS3 expression in mTHAL cells is similar to those shown by other investigators studying different tissues. Valerio et al.22 reported that obesity caused a reduction in NOS3 expression in white and brown fat tissue as well as in skeletal muscle due to TNF-α. Agnoletti et al.16 demonstrated that serum from patients with severe heart failure had elevated levels of TNF-α compared to controls. Incubation of endothelial cells with serum from those heart failure patients decreased NOS3 expression. This effect was reversed by a TNF-α neutralizing antibody confirming the role of TNF-α. Finally, addition of TNF-α to the media of cultured endothelial cells reduces NOS3 expression.23,24. However, to our knowledge, the effect of TNF-α on NOS3 expression in renal epithelial cells had not been previously reported.
MTHALs express all three NOS isoforms: NOS1, 2 and 339. NOS2 is constitutively active and thus its activity is mainly dependent on its expression level40. TNF-α has been shown to increase NOS2 expression and thus activity in human umbilical vein endothelial cells 41. However, we found that basal NO production was not elevated in mTHAL cells treated with TNF-α for 24 hrs, indicating that NOS2 was not likely to be elevated. On the other hand, NOS1 and NOS3 activity can be regulated independently of their expression11,40. In mTHALs, increases in NO production by luminal flow, endothelin-1 and angiotensin II are mediated by NOS3 and depend upon PI3K, PIP3 and Akt5-7. In line with the reduced NOS3 expression, PIP3-induced increases in NO production were blunted in mTHAL cells treated with TNF-α for 24 hrs. These data indicate that chronic exposure to elevated levels of TNF-α decreases NOS3 expression and blunts the ability of physiological stimuli to increase NO production. These results are in agreement with those found by Valerio22 and Goodwin 42 in which TNF-α decreased NOS3 expression and NO production in fat, muscle and endothelial cells.
Although the effect of TNF-α on NOS3 expression in non-renal cells has been widely tested, data addressing the signaling pathway by which TNF-α impairs NOS3 expression are scarce. Here we show for the first time that Rho/ROCK mediates most of the inhibitory effect of TNF-α on NOS3 expression in mTHALs. This conclusion was supported by four lines of evidence. First, TNF-α increased RhoA GTPase activity, as shown by the increase in GTP-bound RhoA levels. Second, exoenzyme C3 transferase, an inhibitor of Rho, blunted the inhibitory effect of TNF-α. Third, a ROCK inhibitor reduced the effect of TNF-α by almost 70%. Finally concomitant inhibition of RhoA and ROCK had no additive effect.
Our finding that ROCK mediates the effects of TNF-α on NOS3 expression is unique. Although ROCK mediates the decrease in NOS3 expression induced by high glucose 35 hypoxia33 and thrombin34 in endothelial cells, there have been no other reports of this kinase mediating TNF-α-induced reductions in NOS3 expression. Thus, our data suggest that TNF-α, high glucose, hypoxia and thrombin utilize ROCK as a common pathway to decrease NOS3 expression.
The fact that ROCK inhibition only accounted for 66% of TNF-α’s effect on NOS3 expression is unlikely to be due to using too low of a concentration of the inhibitor. We used a concentration that is several times the Ki and higher concentrations induced no further blockade. To achieve maximal ROCK inhibition we used the compound H-1152. H-1152 has been shown to be more selective and potent than fausudil and Y-2763243. The lack of complete blockade of TNF-α’s effect on NOS3 expression by ROCK inhibition is not unique to our study. El-Remessy et al. showed that in bovine endothelial cells high glucose reduced NOS3 expression by 39% and that treatment with a ROCK inhibitor blocked this effect by about 70%35. Based on these findings, we sought other signaling pathways to account for the remaining 34%.
We previously demonstrated that angiotensin II decreases NOS3 expression in mTHALs via peroxynitrite31 and TNF-α induces protein-tyrosine nitration in endothelial cells 44. Our data shows for the first time that TNF-α acutely increased NO production mTHALs. However, contrary to angiotensin II, inhibition of NO production did not prevent TNF-α from reducing NOS3 expression in mTHALs. These results are similar to those found in endothelial cells, where NO inhibition did not block TNF-α-induced destabilization of NOS3 mRNA 45 nor TNF-α-induced reduction in NOS3 promoter activity46. However, El-Remessy et al. showed that high glucose and diabetes reduced NOS3 expression via peroxynitrite and ROCK35 and both conditions have been shown to increase TNF-α levels 47-49. Therefore, it is possible that high glucose and angiotensin II enhance peroxynitrite formation leading to an increase in TNF-α release which in turn reduces NOS3 expression primarily via Rho/ROCK.
TNF-α can activate a number of other signaling cascades. Consequently we tested several of them in a vain attempt to characterize the remaining 34% of TNF-α’s actions that were ROCK independent. Neither, MAPKK, PKC, Rac-1, JAK nor JNK appeared to play a role in TNF-α-induced reductions in NOS3 expression.
In conclusion, we found that TNF-α reduces NOS3 expression primarily via RhoA GTPase and ROCK in the mTHAL. MLCK, NO, PKC, MAPKK, JAK, JNK and Rac-1 were not involved.
Perspectives
A correlation between TNF-α levels and elevated blood pressure has been demonstrated in humans and in animals. In rodents, TNF-α blockade with pharmacologic inhibitors or genetic manipulations blocks or delays the progression of hypertension in angiotensin II-infused animals, systemic lupus erythematosus, insulin resistance and preeclampsia. TNF-α neutralizing drugs like etarnecept and infliximab are available for patients with chronic inflammatory diseases and thus the effect of TNF-α blockade on hypertension in humans could be studied. On the other hand, ROCK inhibitors like fasudil are currently used to treat pulmonary hypertension and cerebral vasospasm and have beneficial effects on patients with systemic hypertension and chronic heart failure. However, most of the studies employing ROCK inhibitors have been focused on the vascular effects of those compounds whereas the effects on the kidney remain largely unexplored. Our results indicate that these drugs would improve renal function and decrease blood pressure by mitigating the effect of TNF-α on NOS3 expression, elevating NO levels and thus enhancing natriuresis.
Supplementary Material
Novelty and Significance.
We have shown for the first time that chronic elevation of TNF-α decreases NOS3 expression and blunts stimulus-induced NO production by the mTHAL. In addition, this is the first report that identifies RhoA GTPase and ROCK as the main mediators of TNF-α-induced decreases on NOS3 expression. NO produced by the mTHAL not only inhibits mTHAL NaCl transport but also increases renal medullary blood flow. Elevated TNF-α has been found in hypertension, heart failure and metabolic syndrome and treatment with ROCK inhibitors reduces blood pressure and improves cardiac function in those settings. Our findings could be extrapolated to other tissues and they suggest that some of the beneficial effects of ROCK inhibitors in diseases where TNF-α is elevated could be due to the restoration of NOS3 expression and activity.
Acknowledgments
Sources of Funding
This work was supported by grants from National Institutes of Health (HL 028982, HL 070985, HL 090550) to J.L. Garvin and from American Heart Association (11PRE7510005) to V.D. Ramseyer.
Footnotes
Disclosures
None
References
- 1.Greger R. Ion transport mechanisms in thick ascending limb of Henle’s loop of mammalian nephron. Physiol Rev. 1985;65:760–797. doi: 10.1152/physrev.1985.65.3.760. [DOI] [PubMed] [Google Scholar]
- 2.Hoagland KM, Flasch AK, Dahly-Vernon AJ, dos Santos EA, Knepper MA, Roman RJ. Elevated BSC-1 and ROMK expression in Dahl salt-sensitive rat kidneys. Hypertension. 2004;43:860–865. doi: 10.1161/01.HYP.0000120123.44945.47. [DOI] [PubMed] [Google Scholar]
- 3.Garcia NH, Plato CF, Stoos BA, Garvin JL. Nitric oxide-induced inhibition of transport by thick ascending limbs from Dahl salt-sensitive rats. Hypertension. 1999;34:508–513. doi: 10.1161/01.hyp.34.3.508. [DOI] [PubMed] [Google Scholar]
- 4.Plato CF, Shesely EG, Garvin JL. eNOS mediates L-arginine-induced inhibition of thick ascending limb chloride flux. Hypertension. 2000;35:319–323. doi: 10.1161/01.hyp.35.1.319. [DOI] [PubMed] [Google Scholar]
- 5.Ortiz PA, Hong NJ, Garvin JL. Luminal flow induces eNOS activation and translocation in the rat thick ascending limb. II. Role of PI3-kinase and Hsp90. Am J Physiol Renal Physiol. 2004;287:F281–F288. doi: 10.1152/ajprenal.00383.2003. [DOI] [PubMed] [Google Scholar]
- 6.Herrera M, Garvin JL. Endothelin stimulates endothelial nitric oxide synthase expression in the thick ascending limb. Am J Physiol Renal Physiol. 2004;287:F231–F235. doi: 10.1152/ajprenal.00413.2003. [DOI] [PubMed] [Google Scholar]
- 7.Herrera M, Garvin JL. Angiotensin II stimulates thick ascending limb NO production via AT(2) receptors and Akt1-dependent nitric-oxide synthase 3 (NOS3) activation. J Biol Chem. 2010;285:14932–14940. doi: 10.1074/jbc.M110.109041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1–12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- 9.Oemar BS, Tschudi MR, Godoy N, Brovkovich V, Malinski T, Luscher TF. Reduced endothelial nitric oxide synthase expression and production in human atherosclerosis. Circulation. 1998;97:2494–2498. doi: 10.1161/01.cir.97.25.2494. [DOI] [PubMed] [Google Scholar]
- 10.Herrera M, Hong NJ, Ortiz PA, Garvin JL. Endothelin-1 inhibits thick ascending limb transport via Akt-stimulated nitric oxide production. J Biol Chem. 2009;284:1454–1460. doi: 10.1074/jbc.M804322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herrera M, Silva GB, Garvin JL. A high-salt diet dissociates NO synthase-3 expression and NO production by the thick ascending limb. Hypertension. 2006;47:95–101. doi: 10.1161/01.HYP.0000196274.78603.85. [DOI] [PubMed] [Google Scholar]
- 12.Perez-Rojas JM, Kassem KM, Beierwaltes WH, Garvin JL, Herrera M. Nitric oxide produced by endothelial nitric oxide synthase promotes diuresis. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1050–R1055. doi: 10.1152/ajpregu.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexander BT, Cockrell KL, Massey MB, Bennett WA, Granger JP. Tumor necrosis factor-alpha-induced hypertension in pregnant rats results in decreased renal neuronal nitric oxide synthase expression. Am J Hypertens. 2002;15:170–175. doi: 10.1016/s0895-7061(01)02255-5. [DOI] [PubMed] [Google Scholar]
- 14.Furumoto T, Saito N, Dong J, Mikami T, Fujii S, Kitabatake A. Association of cardiovascular risk factors and endothelial dysfunction in japanese hypertensive patients: implications for early atherosclerosis. Hypertens Res. 2002;25:475–480. doi: 10.1291/hypres.25.475. [DOI] [PubMed] [Google Scholar]
- 15.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. 2005;19:149–154. doi: 10.1038/sj.jhh.1001785. [DOI] [PubMed] [Google Scholar]
- 16.Agnoletti L, Curello S, Bachetti T, Malacarne F, Gaia G, Comini L, Volterrani M, Bonetti P, Parrinello G, Cadei M, Grigolato PG, Ferrari R. Serum from patients with severe heart failure downregulates eNOS and is proapoptotic: role of tumor necrosis factor-alpha. Circulation. 1999;100:1983–1991. doi: 10.1161/01.cir.100.19.1983. [DOI] [PubMed] [Google Scholar]
- 17.Ferreri NR, Escalante BA, Zhao Y, An SJ, McGiff JC. Angiotensin II induces TNF production by the thick ascending limb: functional implications. Am J Physiol. 1998;274:F148–F155. doi: 10.1152/ajprenal.1998.274.1.F148. [DOI] [PubMed] [Google Scholar]
- 18.Elmarakby AA, Quigley JE, Pollock DM, Imig JD. Tumor necrosis factor alpha blockade increases renal Cyp2c23 expression and slows the progression of renal damage in salt-sensitive hypertension. Hypertension. 2006;47:557–562. doi: 10.1161/01.HYP.0000198545.01860.90. [DOI] [PubMed] [Google Scholar]
- 19.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–1351. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran LT, MacLeod KM, McNeill JH. Chronic etanercept treatment prevents the development of hypertension in fructose-fed rats. Mol Cell Biochem. 2009;330:219–228. doi: 10.1007/s11010-009-0136-z. [DOI] [PubMed] [Google Scholar]
- 21.Venegas-Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC, Glover PH, Jones AV, Drummond HA, Ryan MJ. Tumor necrosis factor-alpha antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension. 2010;56:643–649. doi: 10.1161/HYPERTENSIONAHA.110.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L, Cantoni O, Clementi E, Moncada S, Carruba MO, Nisoli E. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest. 2006;116:2791–2798. doi: 10.1172/JCI28570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alonso J, Sanchez dM, Monton M, Casado S, Lopez-Farre A. Endothelial cytosolic proteins bind to the 3’ untranslated region of endothelial nitric oxide synthase mRNA: regulation by tumor necrosis factor alpha. Mol Cell Biol. 1997;17:5719–5726. doi: 10.1128/mcb.17.10.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshizumi M, Perrella MA, Burnett JC, Jr, Lee ME. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ Res. 1993;73:205–209. doi: 10.1161/01.res.73.1.205. [DOI] [PubMed] [Google Scholar]
- 25.Bao HF, Zhang ZR, Liang YY, Ma JJ, Eaton DC, Ma HP. Ceramide mediates inhibition of the renal epithelial sodium channel by tumor necrosis factor-alpha through protein kinase C. Am J Physiol Renal Physiol. 2007;293:F1178–F1186. doi: 10.1152/ajprenal.00153.2007. [DOI] [PubMed] [Google Scholar]
- 26.Wu X, Guo R, Chen P, Wang Q, Cunningham PN. TNF induces caspase-dependent inflammation in renal endothelial cells through a Rho- and myosin light chain kinase-dependent mechanism. Am J Physiol Renal Physiol. 2009;297:F316–F326. doi: 10.1152/ajprenal.00089.2009. [DOI] [PubMed] [Google Scholar]
- 27.Kakiashvili E, Speight P, Waheed F, Seth R, Lodyga M, Tanimura S, Kohno M, Rotstein OD, Kapus A, Szaszi K. GEF-H1 mediates tumor necrosis factor-alpha-induced Rho activation and myosin phosphorylation: role in the regulation of tubular paracellular permeability. J Biol Chem. 2009;284:11454–11466. doi: 10.1074/jbc.M805933200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 29.Papaharalambus C, Sajjad W, Syed A, Zhang C, Bergo MO, Alexander RW, Ahmad M. Tumor necrosis factor alpha stimulation of Rac1 activity. Role of isoprenylcysteine carboxylmethyltransferase. J Biol Chem. 2005;280:18790–18796. doi: 10.1074/jbc.M410081200. [DOI] [PubMed] [Google Scholar]
- 30.Barsacchi R, Perrotta C, Bulotta S, Moncada S, Borgese N, Clementi E. Activation of endothelial nitric-oxide synthase by tumor necrosis factor-alpha: a novel pathway involving sequential activation of neutral sphingomyelinase, phosphatidylinositol-3’ kinase, and Akt. Mol Pharmacol. 2003;63:886–895. doi: 10.1124/mol.63.4.886. [DOI] [PubMed] [Google Scholar]
- 31.Ramseyer V, Garvin JL. Angiotensin II decreases NOS3 expression via nitric oxide and superoxide in the thick ascending limb. Hypertension. 2008;53:313–318. doi: 10.1161/HYPERTENSIONAHA.108.124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 33.Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- 34.Eto M, Barandier C, Rathgeb L, Kozai T, Joch H, Yang Z, Luscher TF. Thrombin suppresses endothelial nitric oxide synthase and upregulates endothelin-converting enzyme-1 expression by distinct pathways: role of Rho/ROCK and mitogen-activated protein kinase. Circ Res. 2001;89:583–590. doi: 10.1161/hh1901.097084. [DOI] [PubMed] [Google Scholar]
- 35.El-Remessy AB, Tawfik HE, Matragoon S, Pillai B, Caldwell RB, Caldwell RW. Peroxynitrite mediates diabetes-induced endothelial dysfunction: possible role of Rho kinase activation. Exp Diabetes Res. 2010;2010:247861. doi: 10.1155/2010/247861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabral PD, Hong NJ, Garvin JL. Shear stress increases nitric oxide production in thick ascending limbs. Am J Physiol Renal Physiol. 2010;299:F1185–F1192. doi: 10.1152/ajprenal.00112.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hochberg Yosef. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800–802. [Google Scholar]
- 38.Peng J, He F, Zhang C, Deng X, Yin F. Protein kinase C-alpha signals P115RhoGEF phosphorylation and RhoA activation in TNF-alpha-induced mouse brain microvascular endothelial cell barrier dysfunction. J Neuroinflammation. 2011;8:28. doi: 10.1186/1742-2094-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shin SJ, Lai FJ, Wen JD, Lin SR, Hsieh MC, Hsiao PJ, Tsai JH. Increased nitric oxide synthase mRNA expression in the renal medulla of water-deprived rats. Kidney Int. 1999;56:2191–2202. doi: 10.1046/j.1523-1755.1999.00795.x. [DOI] [PubMed] [Google Scholar]
- 40.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia Z, Liu M, Wu Y, Sharma V, Luo T, Ouyang J, McNeill JH. N-acetylcysteine attenuates TNF-alpha-induced human vascular endothelial cell apoptosis and restores eNOS expression. Eur J Pharmacol. 2006;550:134–142. doi: 10.1016/j.ejphar.2006.08.044. [DOI] [PubMed] [Google Scholar]
- 42.Goodwin BL, Pendleton LC, Levy MM, Solomonson LP, Eichler DC. Tumor necrosis factor-alpha reduces argininosuccinate synthase expression and nitric oxide production in aortic endothelial cells. Am J Physiol Heart Circ Physiol. 2007;293:H1115–H1121. doi: 10.1152/ajpheart.01100.2006. [DOI] [PubMed] [Google Scholar]
- 43.Sasaki Y, Suzuki M, Hidaka H. The novel and specific Rho-kinase inhibitor (S)-(+)-2-methyl-1-[(4-methyl-5-isoquinoline)sulfonyl]-homopiperazine as a probing molecule for Rho-kinase-involved pathway. Pharmacol Ther. 2002;93:225–232. doi: 10.1016/s0163-7258(02)00191-2. [DOI] [PubMed] [Google Scholar]
- 44.Yang B, Rizzo V. TNF-alpha potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H954–H962. doi: 10.1152/ajpheart.00758.2006. [DOI] [PubMed] [Google Scholar]
- 45.Mohamed F, Monge JC, Gordon A, Cernacek P, Blais D, Stewart DJ. Lack of role for nitric oxide (NO) in the selective destabilization of endothelial NO synthase mRNA by tumor necrosis factor-alpha. Arteriosclerosis, Thrombosis & Vascular Biology. 1995;15:52–57. doi: 10.1161/01.atv.15.1.52. [DOI] [PubMed] [Google Scholar]
- 46.Anderson HD, Rahmutula D, Gardner DG. Tumor necrosis factor-alpha inhibits endothelial nitric-oxide synthase gene promoter activity in bovine aortic endothelial cells. J Biol Chem. 2004;279:963–969. doi: 10.1074/jbc.M309552200. [DOI] [PubMed] [Google Scholar]
- 47.Ramana KV, Tammali R, Reddy AB, Bhatnagar A, Srivastava SK. Aldose reductase-regulated tumor necrosis factor-alpha production is essential for high glucose-induced vascular smooth muscle cell growth. Endocrinology. 2007;148:4371–4384. doi: 10.1210/en.2007-0512. [DOI] [PubMed] [Google Scholar]
- 48.Coughlan MT, Oliva K, Georgiou HM, Permezel JM, Rice GE. Glucose-induced release of tumour necrosis factor-alpha from human placental and adipose tissues in gestational diabetes mellitus. Diabet Med. 2001;18:921–927. doi: 10.1046/j.1464-5491.2001.00614.x. [DOI] [PubMed] [Google Scholar]
- 49.Rothe H, Ongoren C, Martin S, Rosen P, Kolb H. Abnormal TNF-alpha production in diabetes-prone BB rats: enhanced TNF-alpha expression and defective PGE2 feedback inhibition. Immunology. 1994;81:407–413. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.