

Abstract
Background:
Serum uric acid (UA) has been shown to be an independent predictor of outcome in the general population and in patients with heart failure. There are, however, limited data regarding the prognostic value of UA in the context of acute coronary syndromes (ACS) particularly in medium-term follow up and the available results are contradictory.
Materials and methods:
Study of consecutive patients admitted with an ACS (with and without ST-segment elevation) at a single-centre coronary care unit. Primary endpoint was all-cause mortality at 1-year follow up. We evaluated if serum UA is an independent predictor of outcome and if it has any added value on top of GRACE risk score for risk prediction.
Results:
We included 683 patients, mean age 64±13 years, 69% males. In-hospital and 1-year mortality were 4.5 and 7.6% respectively. The best cut-off of UA to predict 1-year mortality was 6.25 mg/dl (sensitivity 59%, specificity 72%) and 30.2% of the patients had an increased UA according to this cut off. Independent predictors of UA were male gender (β= 0.078), body mass index (β=0.163), diuretics before admission (β=0.142), and admission serum creatinine (β=0.403). One-year mortality was significantly higher in patients with increased UA (15.5 vs. 4.2%, p<0.001; log rank, p<0.001). After adjustment, both increased UA as a categorical variable (HR 2.25, 95% CI 1.23–4.13, p=0.008) and as a continuous variable (HR 1.26, 95% CI 1.13–1.41, p<0.001) are independent predictors of mortality. The AUC increases only slightly after inclusion of UA in the model with GRACE risk score (from 0.78 to 0.79, p=0.350). Both models had a good fit; however, model fit worsened after inclusion of UA. Overall, the inclusion of UA in the original was associated with an improvement in both the net reclassification improvement (continuous NRI=44%), and the integrated discrimination improvement (IDI=0.052) suggesting effective reclassification.
Conclusions:
Serum UA is an independent predictor of all-cause mortality in medium-term after the whole spectrum of ACS and has an added value for risk stratification.
Keywords: Acute coronary syndromes, prognosis, serum uric acid
Introduction
Uric acid (UA) is a byproduct of purine metabolism and an increase in its concentration may reflect increased xanthine oxidase (XO) pathway activity, as well as decreased elimination by the kidneys. The XO pathway is an important source of oxygen-free radicals, with several detrimental processes.1 Therefore, hyperuricaemia has been implicated as a marker of poor outcome, both in the general population and in patients with stroke and heart failure.2–5 There is limited data in the context of acute coronary syndromes (ACS) with contradictory results.
The aim of this study was to investigate whether serum UA after admission, has an independent and added prognostic value to conventional risk factors in the context of the whole spectrum of acute coronary syndromes in a real-life contemporary population.
Materials and methods
This is an observational study of consecutive patients admitted to our intensive care unit (ICU) with ACS (with and without ST-segment elevation) during the years 2007–10. Data was collected prospectively and recorded on a computer database of ACS patients admitted to our institution’s ICU (single-centre registry of ACS). Inclusion criteria were a history of chest pain at rest or other symptoms suggestive of an ACS, with the most recent episode occurring within 24 hours of admission. This could be associated with new or presumed new significant ST-segment or T wave changes, new left bundle branch block, or elevated levels of biomarkers of myocardial damage (cardiac troponin I and creatine kinase). Myocardial infarction was defined by a rise and/or fall of cardiac troponin I with at least one value above 0.10 ng/ml. We evaluated demographic characteristics of the patients, risk factors for coronary artery disease, previous cardiac history, laboratory data, and vital signs on admission as well as in-hospital treatment. Serum UA levels were measured in the first 48 hours after admission from peripheral venous blood samples. Since this blood test is not available off hours, we selected the 48 hours timeline. We used a modification of the Fossati method (uricase-based method) (Beckman Coulter AU270). The within-run precision for serum samples is less than 2% CV and total precision is less than 3% CV. Serum UA information was collected retrospectively because this data was not originally entered in our database. Patients with increased serum UA level were defined by a cut off obtained with the statistical analysis described below. Hypertension, diabetes, and hyperlipidaemia were defined as either previously known or on specific therapy. Patients that had smoked during the previous 6 months were classified as smokers and were self-reported. Body weight was measured to the nearest kilogram using a digital scale, and height to the nearest centimeter in the standing position. Body mass index was calculated as weight in kilograms divided by the height in metres, squared. Estimated glomerular filtration rate was calculated according to the Cockcroft–Gault formula.6 For each patient, a numerical classification according to the previously described Global Registry of Acute Coronary Events (GRACE) risk score was calculated from the initial clinical history, electrocardiogram, and laboratory values collected on admission.7
Follow up was obtained in every patient that survived to discharge by reviewing the medical records and/or by telephone interview with the patient or family members at 30 days and 1 year after admission. Follow-up information was obtained in 99.8% of the patients. The study primary endpoint was all-cause mortality at 1-year follow up. In a secondary analysis we also analysed the in-hospital and 30-day all-cause mortality.
The study complies with the Declaration of Helsinki and informed consent was obtained from all the subjects.
Statistical analysis
Continuous variables are expressed as mean±standard deviation and were compared with the two-tailed Student’s t-test. Normality was tested with the Kolmogorov–Smirnov test. Non-normally distributed variables are presented as median and interquartile range and were compared with the Mann–Whitney test. Categorical variables are expressed as percentage and were compared with Pearson’s chi-squared test (with Yates correction) or Fisher’s Exact test, when appropriate.
Liner regression analysis was used to select independent predictors of serum UA levels. Receiver operator characteristics (ROC) curve analysis and the area under the curve (AUC), or c-statistic, was used as a measure of the predictive accuracy of UA on the considered mortality endpoint. The best cut off of UA was selected by maximizing the sum of sensitivity and specificity. Estimates of the event-free survival were calculated by the Kaplan–Meier method and curves were compared with the log-rank test. Predictors of all-cause mortality were assessed by univariate and stepwise multivariate Cox proportional-hazards regression models, with the p level for entry into and removal from the model set at 0.05 and 0.10, respectively.
We performed tests to discriminate and calibrate different prediction models. To test discrimination, we used ROC curve analysis. The AUC is presented with 95% bias-corrected bootstrap confidence intervals (CI). Calibration of the models was tested with Hosmer–Lemeshow goodness of fit test. A model with good fit will have lower chi-square statistic value and a non-significant p-value, suggesting that there is no difference between the events expected (based on the model) and the events observed. To compare the two models (with and without UA), continuous net reclassification improvement (NRI) and integrated discrimination improvement (IDI) with corresponding bootstrap 95% confidence intervals were also calculated. NRI is a measure which quantifies the degree of correct upward and downward reclassification or movement of predicted probabilities as a result of adding a new marker and IDI may be viewed as a difference between improvement in average sensitivity and any potential increase in average in 1 – specificity.8
Two-tailed tests of significance are reported. For all comparisons, a p-value <0.05 was considered statistically significant. Statistical analysis was performed with SPSS version 12.0 (SPSS, Chicago, Illinois, USA). For model comparison, we used STATA version 12.0 (Stata, College Station, Texas, USA).
Results
A total of 792 consecutive patients were included in the registry. From these, 683 had information on serum UA levels in the first 48 hours after admission, with a mean age of 64±13 years, 69% males, and were included in the present analysis. In this population, 67% had hypertension, 39% were smokers, 25% had diabetes, and 51% had known hyperlipidaemia. As for previous cardiac history, 18% had myocardial infarction, 11% had percutaneous coronary intervention (PCI), and 5% had coronary artery bypass grafting. The majority of the patients were admitted with an ST-elevation acute myocardial infarction (62%). In our population, 21% presented with signs of heart failure on admission, mean GRACE risk score was 141±35 and 6% had a left ventricular ejection fraction <35%. In respect to treatment, 85% received a beta-blocker, 87% angiotensin-converting enzyme inhibitor, 93% a statin, and 79% underwent PCI. The in-hospital, 30-day, and 1-year mortality rates were 4.5, 5.4 and 7.6%, respectively.
Serum UA showed a normal distribution (mean±SD 5.7±1.8 mg/dl; range 0.8–19.6 mg/dl; Figure 1). UA levels were similar between ST-elevation myocardial infarction and non-ST-elevation ACS patients (5.65±1.77 vs. 5.88±1.81 mg/dl, p=0.102). Serum UA correlated positively with age (β=0.15, p<0.001), body mass index (β=0.18, p<0.001), renal function (β=0.46, p<0.001), previous use of diuretics (β=0.21, p<0.001), and male gender (β=0.08, p=0.032) (Figure 2). It showed also a positive correlation with GRACE risk score (β=0.15, p<0.001). However, after multivariate linear regression analysis, age was no longer an independent predictor of serum UA (Table 1).
Figure 1.
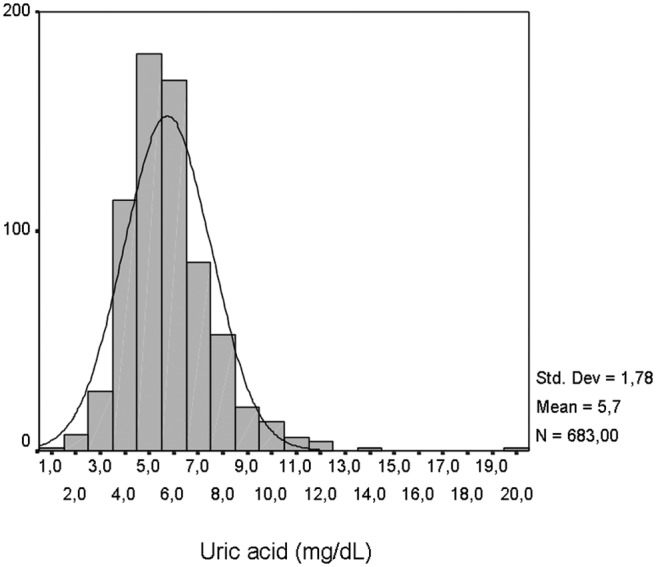
Distribution of serum uric acid levels in the studied population.
Figure 2.

Significant correlations between serum uric acid and continuous variables.
Table 1.
Multivariate linear regression analysis for predictors of serum uric acid.
| Variable | β | p-value |
|---|---|---|
| Age | 0.050 | 0.178 |
| Male gender | 0.078 | 0.031 |
| Body mass index | 0.163 | <0.001 |
| Previous use of diuretics | 0.142 | <0.001 |
| Admission creatinine | 0.403 | <0.001 |
By ROC curve analysis, the AUC of serum UA for the prediction of 1-year mortality was 0.67 (95% CI 0.58–0.76, p<0.001), and the best cut off was 6.25 mg/dl, with a sensitivity of 59% and specificity of 72%. In our population, 30.2% of the patients had increased serum UA levels according to this cut off. Comparing the two groups, patients with an increase serum UA level were older, had higher body mass index, had more hypertension and diabetes, were less often smokers, and had more previous history of myocardial infarction and coronary artery bypass grafting. On the initial presentation, they had higher heart rate and worse Killip class, higher blood glucose (trend), and higher creatinine as well as higher GRACE risk score (Table 2). No differences were found on admission diagnosis. Prior to admission, patients with increased UA were more frequently on ACEI, beta-blockers, diuretics, and statins (Table 3). In-hospital treatment showed higher use ACEI and diuretics in patients with increased UA, as well as after discharge. Patients with increased UA were more often treated with PCI. As for mortality endpoints, patients with increased serum UA levels had significantly higher in-hospital, 30-day, and 1-year mortality (Table 2). By Kaplan–Meier analysis, patients with increased serum UA level had a higher event rate (mortality) in the follow up (log-rank, p<0,001) (Figure 3).
Table 2.
Clinical characteristics by serum uric acid group (cut off 6.25 mg/dl).
| Increased uric acid (n=206) | Normal uric acid (n=477) | p-value | |
|---|---|---|---|
| Age (years) | 67±13 | 63±13 | <0.001 |
| Male gender | 70.4 | 68.6 | 0.699 |
| BMI (kg/m2) | 28±5 | 26±4 | <0.001 |
| Risk factors (%) | |||
| Hypertension | 73.8 | 63.7 | 0.013 |
| Hyperlipidaemia | 46.6 | 53.2 | 0.131 |
| Diabetes | 32.0 | 21.8 | 0.006 |
| Smoking | 31.6 | 41.5 | 0.018 |
| Previous history | |||
| Myocardial infarction | 25.7 | 15.3 | 0.002 |
| PCI | 12.6 | 10.9 | 0.605 |
| CABG | 8.7 | 3.8 | 0.013 |
| Stroke/TIA | 8.7 | 6.5 | 0.379 |
| Initial presentation | |||
| STEMI | 57.8 | 63.3 | 0.208 |
| Killip class ≥2 | 33.5 | 15.1 | <0.001 |
| Heart rate (bpm) | 84±21 | 76±18 | <0.001 |
| Systolic blood pressure (mmHg) | 138±32 | 136±29 | 0.399 |
| Laboratory data | |||
| Admission blood glucose (mg/dl) | 141 (114–199) | 133 (110–177) | 0.087a |
| Peak CK (IU/l) | 960 (318–2472) | 1167 (365–2792) | 0.221a |
| Admission creatinine (mg/dl) | 1.26±0.48 | 0.91±0.28 | <0.001 |
| eGFR <60 ml/min/1.73 m2 | 40.3 | 29.6 | 0.008 |
| GRACE risk score | 153±37 | 136±32 | <0.001 |
| LVEF <35% | 11.2 | 3.8 | <0.001 |
| PCI at index hospitalization | 83.2 | 68.0 | <0.001 |
| All-cause mortality | |||
| In-hospital mortality | 9.2 | 2.5 | <0.001 |
| 30-day mortality | 9.7 | 3.6 | 0.002 |
| 1-year mortality | 15.5 | 4.2 | <0.001 |
Values are mean±SD, %, or median (IQR).
Mann–Whitney test.
BMI, body mass index; CABG, coronary artery bypass grafting; CK, creatine kinase; eGFR, estimated glomerular filtration rate; GRACE, Global Registry of Acute Coronary Events; LVEF, left ventricular ejection fraction; PCI, percutaneous coronary intervention; STEMI, ST-elevation myocardial infarction; TIA, transient ischaemic attack.
Table 3.
Medication prior to and during admission and at discharge.
| Increased uric acid | Normal uric acid | p-value | |
|---|---|---|---|
| Prior to admission | |||
| Diuretic | 29.6 | 11.5 | <0.001 |
| ACEI | 28.2 | 17.6 | 0.003 |
| Beta-blocker | 21.8 | 13.2 | 0.006 |
| Statin | 33.0 | 24.7 | 0.033 |
| In hospital | |||
| Diuretic | 44.2 | 23.5 | <0.001 |
| ACEI | 81.1 | 89.1 | 0.007 |
| Beta-blocker | 86.4 | 83.9 | 0.464 |
| Statin | 93.2 | 93.5 | 1.000 |
| Dischargea | |||
| Diuretic | 46.5 | 22.4 | <0.001 |
| ACEI | 67.9 | 76.1 | 0.040 |
| Beta-blocker | 81.8 | 77.8 | 0.308 |
| Statin | 89.3 | 93.8 | 0.074 |
Values are %.
In patients that survived to discharge (n=652: 187 with increased uric acid; 465 with normal uric acid).
ACEI, angiotensin-converting enzyme inhibitor.
Figure 3.
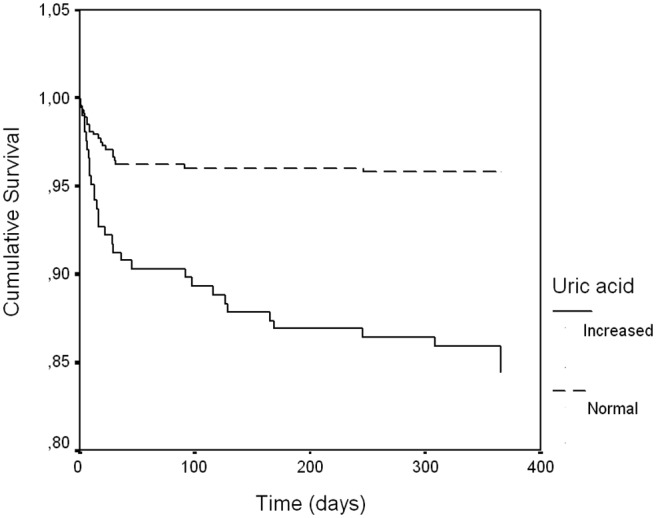
Kaplan–Meier survival curve according to serum uric acid.
In the univariate analysis, age, male gender, diabetes, heart rate, systolic blood pressure, ACEI and beta-blockers use, Killip class ≥2, estimated glomerular filtration rate <60 ml/min/1.73 m2, PCI, and serum UA (as a continuous and categorical variable) were significant predictors of outcome. In the adjusted Cox proportional-hazard regression analysis, an increased serum UA level remained an independent predictor of all-cause mortality (Table 4). Since the discriminative capacity of serum UA to predict mortality is somewhat low (AUC <0.70), we also performed multivariate analysis with serum UA as a continuous variable and it remained as an independent predictor of mortality (Table 4). Age, heart rate, SBP, beta-blocker use, and estimated glomerular filtration rate <60 ml/min/1.73 m2 were the other independent predictors of mortality.
Table 4.
Multivariate Cox proportional-hazard regression analysis (independent predictors of outcome).
| Variable | HR (95% CI) | p-value | HR (95% CI) | p-value |
|---|---|---|---|---|
| Age (per year) | 1.060 (1.032–1.090) | <0.001 | 1.062 (1.033–1.092) | <0.001 |
| Heart rate (per bpm) | 1.022 (1.008–1.036) | 0.002 | 1.020 (1.006–1.034) | 0.005 |
| SBP (per mmHg) | 0.981 (0.971–0.991) | <0.001 | 0.983 (0.973–0.993) | 0.001 |
| Beta-blocker | 0.423 (0.223–0.800) | 0.008 | 0.436 (0.230–0.825) | 0.011 |
| eGFR <60 | 1.999 (1.087–3.677) | 0.026 | 1.890 (1.017–3.512) | 0.044 |
| Uric acid (categorical) | 2.254 (1.231–4.128) | 0.008 | – | – |
| Uric acid (continuous) (per mg/dl) | – | – | 1.260 (1.125–1.412) | <0.001 |
Adjusted for age, male gender, diabetes, heart rate, SBP, angiotensin-converting enzyme inhibitor, and beta-blocker use, Killip class ≥2, eGFR <60 ml/min/1.73 m2 and PCI.
eGFR, estimated glomerular filtration rate; SBP, systolic blood pressure.
Most of the mortality was accounted for in-hospital mortality, and for that reason we decided to perform a subgroup analysis of the patients that survived to discharge. In this group, 1-year mortality was still significantly higher in the group with increased UA (6.9 vs. 1.7%, p=0.002). After adjustment, increased UA remained as an independent predictor of outcome (HR 1.31, 95% CI 1.03–1.67, p=0.030).
We built prediction models with GRACE risk score alone and after inclusion of serum UA levels (Table 5). AUC increased slightly after inclusion of UA in the model, although it did not reach statistical significance. Both models had a good fit; however model fit worsened after inclusion of UA. Overall, the inclusion of UA in a model with GRACE risk score was associated with a NRI of 44%, suggesting effective reclassification. However, reclassification occurred more effectively in the non-events group. The new model reduced the calculated risk in 32.5% of those without events significantly. That is, the new model better identifies those who do not have events than those who do. The IDI again showed that the model diagnostic performance was significantly improved by adding UA to the GRACE risk score (IDI=0.052).
Table 5.
Statistics for model improvement with the addition of uric acid.
| Goodness of fit (GRACE risk score)a | 0.742 |
| Goodness of fit (GRACE + uric acid)a | 0.149 |
| Events (n) | 52 |
| Nonevents (n) | 631 |
| Continuous NRI | |
| cNRIevents | 11.5 (–8.7 to 32.0) |
| cNRInonevents | 32.5 (11.8 to 50.0) |
| cNRI | 44.0 (13.3 to 72.4) |
| IDI statistics | |
| IDIevents | 0.048 (0.004 to 0.111) |
| IDInonevents | 0.004 (0.000 to 0.009) |
| IDI | 0.052 (0.005 to 0.119) |
| AUC | |
| GRACE risk score | 0.78 (0.71 to 0.83) |
| GRACE + uric acid | 0.79 (0.73 to 0.85) |
| Difference (p-value) | 0.350 |
Values are % (95% CI) unless otherwise stated.
Hosmer–Lemeshow goodness of fit test (p-value).
AUC, area under the curve of the receiver operator characteristic curve; IDI, integrated discrimination improvement; NRI, net reclassification improvement.
In the 206 patients that presented with increased serum UA levels, 6.8% received allopurinol therapy after admission. In this group, none of the patients died at 1-year follow up, whereas 16.7% died in the group that did not receive allopurinol (p=0.133).
Discussion
Serum UA is produced by enzymatic activity of XO and is the main end product of metabolism of purins, which in turn are derived mostly from diet, de novo biosynthesis, and breakdown of nucleic acids. Serum UA levels reflect the degree of XO activation. During UA production, oxygen-free radicals are generated and therefore UA is a simple and useful clinical indicator of excess oxidative stress.1 In humans, one of the tissues with the highest activity of XO is the capillary endothelium and the endothelium of small arteries, an important source of oxygen-free radical production within the endothelium.9 One major factor responsible for the impaired regulation of vascular tone is the increase in oxidative stress.9 Synthesis of nitric oxide is disrupted and its degradation is accelerated by excessive free radical activity causing endothelial dysfunction. Thus, elevated levels of UA may be implicated in oxygen-free radical generation and indirectly cause endothelial dysfunction.
Patients with atherosclerosis have increased levels of both UA and total serum antioxidant capacity. UA is also one of the strongest determinants of plasma antioxidative capacity, with free-radical scavenging activity in human serum.9 For that reason, some authors suggest that increased levels of UA may be a compensatory mechanism trying to counteract oxidative stress.9 Other authors suggest that, in the early stages of the atherosclerotic process, serum UA acts as an antioxidant. However, later in the atherosclerotic process when serum UA levels are elevated, this previous antioxidant activity paradoxically becomes pro-oxidant (the so-called paradoxical antioxidant–prooxidant switch or the urate redox shuttle).10 In the accelerated atherosclerotic-vulnerable plaque, the intimae has been shown to be acidic, depleted of local antioxidants with an underlying increase in oxidative stress and reactive oxygen species, and associated with uncoupling of the endothelial nitric oxide synthase enzyme and a decrease in the locally produced naturally occurring antioxidant.10 It is possible that UA increase as an antioxidant response to the oxidative stress associated with risk factors and coronary disease. However, XO activation will increase oxidative stress even further. Then, either by insufficient antioxidant capacity of serum UA or by the proposed urate redox shuttle, the oxidative stress causes vascular injury.
However, hyperuricaemia can also directly cause vascular injury. At high concentrations, UA promote vascular smooth muscle cell proliferation, platelet aggregation, cell apoptosis, and local inflammation.1,11–13 It is also associated with tubulointersticial inflammation, morphological and functional changes in the glomeruli and renal arteriole, and increased salt-sensitivity – hyperuricaemic or salt-sensitive kidney-dependent hypertension.14,15
All these mechanisms suggest that UA is potentially associated with cardiovascular disease. However, the Framingham Heart Study demonstrated that UA was not a risk factor for cardiovascular events, and therefore medical societies have not considered serum UA level as a cardiovascular risk factor.14,16 The role of UA as a prognostic factor in patients with myocardial infarction is also controversial. Whereas Homayounfar et al.17 concluded that UA was not an independent prognostic marker for in-hospital mortality after acute myocardial infarction, other studies18-22 showed that UA could be a marker of adverse prognosis. In an Italian study in the context of primary PCI, serum UA was an independent risk factor for in-hospital mortality.18 The same group showed later, in a larger series of patients with ST-elevation myocardial infarction that UA predicted the occurrence of complications in the ICU but not early mortality after adjustment for renal function and degree of myocardial necrosis.19 The Japanese Acute Coronary Syndrome Study and the Korea Acute Myocardial Infarction Registry, as well as other small studies, showed the prognostic value of serum UA in short-term mortality.20–22 Medium-term mortality studies are scarce, particularly in the whole spectrum of patients with ACS.
In the context of reperfusion injury, it is understood that XO-derived oxygen free radicals (reflected by elevated UA) are a major contributor to impaired flow and tissue damage and may form a vicious cycle. Some authors studied the pathophysiology related to the potential adverse prognosis in the context of ACS. Elevated levels of serum UA are reported to be an independent predictor for the presence of slow coronary flow as well as the no-reflow phenomenon during reperfusion therapy.13,23–25 Hyperuricaemia may also be related to impaired renal UA excretion caused by low cardiac output and tissue hypoxia.19
Our results in medium-term follow up in a population with the whole spectrum of ACS confirmed most of the previous results in short-term follow up in the context of PCI. The prognostic power of UA may be conveyed by cardiovascular risk accumulated in patients with increased levels of this biomarker. Elevated UA levels are associated with factors that connote a more adverse cardiovascular risk profile. However, the preservation of an independent association between UA and mortality after adjustment for cardiovascular risk and other relevant clinical variables shows that UA offers prognostic information beyond that mediated by risk factors that are clustered in patients with elevated UA levels. One can also argue that most of the mortality occurs in the short term (particularly during hospital stay). However, a subgroup analysis of the surviving population to discharge confirmed that patients with an increased serum UA fare worst at medium-term follow up.
Two very recent papers confirmed the independent prognostic value of serum UA in the context of PCI for ACS.25,26 Our study showed prognostic value in the whole spectrum of ACS.
Mere associations with incident all-cause mortality, although important, do not automatically mean that adding UA to traditional risk prediction models will improve outcome risk prediction. So, we performed tests to discriminate and calibrate different prediction models. Discriminative analysis of a model with GRACE risk score with and without serum UA showed that AUC increased only slightly after inclusion of UA in the model, although it did not reach statistical significance. However, in the presence of a fairly robust risk score, as in the case of GRACE risk score, the quantitative improvement in model performance introduced by adding new variables to the existing model is usually very small in magnitude with the AUC analysis. For that reason, the AUC is an insensitive measure of the ability of a new marker to add value to a pre-existing risk prediction model. Two new statistical metrics have been proposed to quantify the degree of correct reclassification: the NRI and the IDI.8 Both measures consider separately individuals who develop and who do not develop events. The NRI is interpreted as the proportion of patients reclassified to a more appropriate risk category. Overall, the inclusion of UA in a model with GRACE risk score was associated with a NRI of 44%, suggesting effective reclassification. However, the new model better identifies those who do not have events than those who do. The new model reduced the calculated risk in 32.5% of those without events, significantly. The IDI (which incorporates both the direction of change in calculated risk and the extent of change) again showed that the model diagnostic performance was significantly improved by adding UA to the GRACE risk score.
These results show that adding UA to a model with GRACE risk score could mainly better identify patients at low risk. This might not be ideal when we are evaluating a risk score to identify high-risk patients. However, recent cardiovascular disease guidelines are encouraging a practice shift toward greater focus on identification of ‘truly low-risk’ patients instead of focusing on identification of high-risk patients. This allows a better selection of patients avoiding unnecessary interventions that might increase costs as well as the risk of intervention-related adverse events. In the coming years economical issues will arise and this subject will probably be very relevant. In summary, the new score is better at identifying ‘truly low-risk’ patients and is as good as in identifying patients who develop events.
The oxidative stress associated with reperfusion injury and XO activation is responsible for a vicious cycle of tissue damage. Adjunctive therapy designed to decrease XO activity and inhibit oxidative stress production is expected to sever this vicious cycle. Consistent with this is the notion that allopurinol, a XO inhibitor, not only reduces the serum UA levels but also improves vascular endothelial function. In the context of heart failure, some studies demonstrated a benefit in the outcome with allopurinol therapy.27,28 An experimental study showed that treatment with allopurinol improved endothelial function and increased stimulated post-ischaemia and flow-dependent blood flow in peripheral vascular beds.29 There was a direct relationship between the allopurinol-induced reduction of UA levels and improvement of flow-dependent flow. Thus, allopurinol may exert protective effects against the reperfusion injuries and possibly have a favourable effect on mortality. However, in the context of myocardial infarction, only a very small randomized study in 60 patients submitted to primary PCI is available.30 It showed a benefit with allopurinol therapy, with a reduction in myocardial necrosis extension as well as a 13% reduction in major acute coronary events at 30-day follow up. We tried to analyse this subject in the small group of patients with increased serum UA. The group that did not receive allopurinol had higher mortality, although it did not reach statistical significance because it was a small sample.
Limitations
This is a single-centre study, which might limit the conclusions of this study. It might not be applicable to other populations with different baseline characteristics. Our hospital is a tertiary centre and as such receives many patients for coronary angiography and angioplasty. A significant proportion of patients with non-ST-elevation ACS are admitted directly to the catheterization laboratory (more than 24 hours after the event) and return to the original hospital after intervention. These patients were not included in our registry. On the other hand, all ST-elevation myocardial infarction patients are admitted directly to the ICU before intervention and remain in our hospital until discharge. These patients are systematically included in our registry. This can explain the higher percentage of ST-elevation myocardial infarction patients compared to non-ST-elevation ACS in our population. However, this does not reflect the distribution in other cohort studies and some caution should be used when translating our results for other cohorts.
Since the previous use of allopurinol was not collected in our registry and since this information was lacking in several medical records, we could not assess the influence of the previous use of allopurinol in our results. These patients were not excluded from our analysis.
As for the influence of allopurinol use after admission, we studied a very small sample in our subgroup analysis, with inadequate statistical power to draw any definite conclusions. Patient compliance to allopurinol therapy in the follow up was also not assessed in the follow up. A larger randomized trial would be necessary to clarify the benefit of XO-inhibition/UA-lowering therapy on cardiovascular events in the follow up.
In conclusion, in a population of patients with the whole spectrum of ACS, elevated levels of serum UA level are an independent correlate of all-cause mortality in medium-term follow up and UA adds some predictive value on top of the GRACE risk score. For every 1 mg/dl increase in UA, the adjusted risk for 1-year mortality was increased by 26%.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest: The authors declare that there is no conflict of interest.
References
- 1. Leyva F, Anker S, Swan JW, et al. Serum uric acid as an index of impaired oxidative metabolism in chronic heart failure. Eur Heart J 1997; 18: 858–865 [DOI] [PubMed] [Google Scholar]
- 2. Fessel WJ. High uric acid as an indicator of cardiovascular disease: independence from obesity. Am J Med 1980; 68: 401–404 [DOI] [PubMed] [Google Scholar]
- 3. Fang J, Alderman MH. Serum uric acid and cardiovascular mortality: the NHANES I epidemiologic follow-up study, 1971–1992: National Health and Nutrition Examination Survey. JAMA 2000; 283: 2404–2410 [DOI] [PubMed] [Google Scholar]
- 4. Weir CJ, Muir SW, Walters MR, et al. Serum urate as an independent predictor of poor outcome and future vascular events after acute stroke. Stroke 2003; 34: 1951–1956 [DOI] [PubMed] [Google Scholar]
- 5. Levya F, Anker SD, Godsland IF, et al. Uric acid in chronic heart failure: a marker of chronic inflammation. Eur Heart J 1998; 19: 1814–1822 [DOI] [PubMed] [Google Scholar]
- 6. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976; 16: 31–41 [DOI] [PubMed] [Google Scholar]
- 7. Granger CB, Goldberg RJ, Dabbous O, et al. Predictors of hospital mortality in the Global Registry of Acute Coronary Events. Arch Intern Med 2003; 163: 2345–2353 [DOI] [PubMed] [Google Scholar]
- 8. Pencina MJ, D’Agostino RB, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Statis Med 2011; 30: 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nieto FJ, Iribarren C, Gross MD, et al. Uric acid and serum antioxidant capacity: a reaction to atherosclerosis? Atherosclerosis 2000; 148: 131–139 [DOI] [PubMed] [Google Scholar]
- 10. Hayden MR, Tyagi SC. Uric acid: a new look at an old risk marker for cardiovascular disease, metabolic syndrome, and type 2 diabetes mellitus: the urate redox shuttle. Nutr Metab 2004; 1: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hare JM, Johnson RJ. Uric acid predicts clinical outcomes in heart failure: insights regarding the role of xanthine oxidase and uric acid in disease pathophysiology. Circulation 2003; 107: 1951–1953 [DOI] [PubMed] [Google Scholar]
- 12. Anker SD, Doehner W, Rauchhaus M, et al. Uric acid and survival in chronic heart failure: validation and application in metabolic, functional, and hemodynamic staging. Circulation 2003; 107: 1991–1997 [DOI] [PubMed] [Google Scholar]
- 13. Basar N, Sen N, Ozcan F, et al. Elevated serum uric acid predicts angiographic impaired reperfusion and 1-year mortality in ST-segment elevation myocardial infarction patients undergoing percutaneous coronary intervention. J Invest Med 2011; 59: 931–937 [DOI] [PubMed] [Google Scholar]
- 14. Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med 2008; 359: 1811–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson RJ, Kang DH, Feig D, et al. Is there a pathogenic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 2003; 41: 1183–1890 [DOI] [PubMed] [Google Scholar]
- 16. Culleton BF, Lansom MG, Kannel WB, et al. Serum uric acid and risk for cardiovascular disease and death: The Framingham Heart Study. Ann Intern Med 1999; 131: 7–13 [DOI] [PubMed] [Google Scholar]
- 17. Homayounfar S, Ansari M, Kashani KM. Evaluation of independent prognostic importance of hyperuricemia in hospital death after acute myocardial infarction. Saudi Med J 2007; 28: 759–761 [PubMed] [Google Scholar]
- 18. Lazzeri C, Valente S, Chiostri M, et al. Uric acid in the acute phase of ST elevation myocardial infarction submitted to primary PCI: its prognostic role and relation with inflammatory markers. Int J Cardiol 2010; 21: 206–209 [DOI] [PubMed] [Google Scholar]
- 19. Lazzeri C, Valente S, Chiostri, et al. Uric acid in the early risk stratification of ST-elevation myocardial infarction. Intern Emerg Med 2012; 7: 33–39 [DOI] [PubMed] [Google Scholar]
- 20. Kojima S, Sakamoto T, Ishihara M, et al. Prognostic usefulness of serum uric acid after acute myocardial infarction (the Japanese Acute Coronary Syndrome Study). Am J Cardiol 2005; 96: 489–495 [DOI] [PubMed] [Google Scholar]
- 21. Bae MH, Lee JH, Lee SH, et al. Serum uric acid as an independent and incremental prognostic marker in addition to N-terminal Pro-B-type natriuretic peptide in patients with acute myocardial infarction. Circ J 2011; 75: 1440–1447 [DOI] [PubMed] [Google Scholar]
- 22. Car S, Trkulja V. Higher serum uric acid on admission is associated with higher short-term mortality and poorer long-term survival after myocardial infarction: retrospective prognostic study. Croat Med J 2009; 50: 559–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Akpek M, Kaya M, Uyarel H, et al. The association of serum uric acid levels on coronary flow in patients with STEMI undergoing primary PCI. Atherosclerosis 2011; 219: 334–341 [DOI] [PubMed] [Google Scholar]
- 24. Kato M, Hisatome I, Tomikura Y, et al. Status of endothelial dependent vasodilation in patients with hiperuricemia. Am J Cardiol 2005; 96: 1576–1578 [DOI] [PubMed] [Google Scholar]
- 25. Ndrepepa G, Braun S, Haase HU, et al. Prognostic value of uric acid in patients with acute coronary syndromes. Am J Cardiol 2012; 109: 1260–1265 [DOI] [PubMed] [Google Scholar]
- 26. Kaya M, Uyarel H, Akpek M, et al. Prognostic value of uric acid in patients with ST-elevated myocardial infarction undergoing primary coronary intervention. Am J Cardiol 2012; 109: 486–491 [DOI] [PubMed] [Google Scholar]
- 27. Gotsman I, Keren A, Lotan C. Changes in uric acid levels and allopurinol use in chronic heart failure: association with improved survival. J Card Fail 2012; 18: 694–701 [DOI] [PubMed] [Google Scholar]
- 28. Wei L, Fahey T, Struthers AD, et al. Association between allopurinol and mortality in heart failure patients: a long-term follow-up study. Int J Clin Pract 2009; 63: 1327–1333 [DOI] [PubMed] [Google Scholar]
- 29. Doehner W, Schoene N, Rauchhaus M, et al. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circulation 2002; 105: 2619–2624 [DOI] [PubMed] [Google Scholar]
- 30. Rentoukas E, Tsarouhas K, Tsitsimpikou C, et al. The prognostic impact of allopurinol in patients with acute myocardial infarction undergoing primary percutaneous coronary interventions. Int J Cardiol 2010; 145: 257–258 [DOI] [PubMed] [Google Scholar]