

Abstract
Increased remissions in multiple sclerosis (MS) during late pregnancy may result from high levels of sex steroids such as estrogen and estriol. Estrogen (E2=17β-estradiol) protects against experimental autoimmune encephalomyelitis (EAE), but the cellular basis for E2-induced protection remains unclear. Treatment with relatively low doses of E2 can protect against clinical and histological signs of MOG-35-55 induced EAE through mechanisms involving the PD-1 coinhibitory pathway and B-cells. The current study evaluated the contribution of PD-1 ligands, PD-L1 and PD-L2, on B-cells in E2-mediated protection against EAE in WT, PD-L1−/− and PD-L2−/− mice. Unlike PD-L2−/− mice that were fully protected against EAE after E2 treatment, E2-implanted PD-L1−/− mice were fully susceptible to EAE, with increased numbers of proliferating Th1/Th17 cells in the periphery and severe cellular infiltration and demyelination in the CNS. Moreover, transfer of B-cells from MOG-immunized PD-L1−/− or PD-L2−/− donors into E2-preconditioned B-cell deficient μMT−/− recipient mice revealed significantly reduced E2-mediated protection against EAE in recipients of PD-L1−/− B-cells, but near-complete protection in recipients of PD-L2−/− B-cells. We conclude that PD-1 interaction with PD-L1 but not PD-L2 on B-cells is crucial for E2-mediated protection in EAE and that strategies that enhance PD-1/PD-L1 interactions might potentiate E2 treatment effects in MS.
Keywords: EAE, Multiple Sclerosis, Estrogen, PD-L, Regulatory B cells
Introduction
Multiple sclerosis (MS) is a chronic autoimmune disease of the central nervous system (CNS) that mainly affects young adults and may lead to significant disability over time [1]. MS comprises of a heterogeneous string of inflammatory, demyelinating and degenerative events of presumed Th1 origin [2]. The exact pathogenic mechanisms have not been completely unraveled but peripheral activation of autoreactive CD4+ T cells targeting proteins of the myelin sheath of neurons has been hypothesized as a key process in the development of the disease [3]. Although the incidence of MS is higher in women [4], its relapse rates are decreased during late pregnancy, [5–7] and treatment with pregnancy levels of estriol reduces central nervous system lesions [8,9]. Pregnancy is characterized by an array of biological changes that could mediate both immunomodulatory and neuroprotective effects and one such effect is a pronounced systemic shift from Th1-type cellular immunity towards Th2-type humoral immunity [10]. Experimental autoimmune encephalomyelitis (EAE) is an induced autoimmune disease in mice that mimics MS in disease susceptibility and histopathology. EAE is specifically mediated by IFN-γ and IL-17-producing T cells while IL-10 is critical in negative regulation [11–14]. Our group has previously demonstrated that relatively low doses of 17β-estradiol (E2) and estriol confer potent protection against clinical and histological signs of EAE [15,16]. Studies from our lab have also demonstrated that though E2 is a potent regulator of autoimmunity, it does not directly act on autoreactive T cells [17]. However, the molecular and cellular mechanisms by which estrogens regulate MS and EAE have not yet been well characterized.
The activation of T cells via the various antigen-presenting cells (APCs) forms an integral part of the inflammatory process and this process gains importance especially since the inflammatory cells in MS include CD4 and CD8 T lymphocytes, microglia and macrophages [18]. T cell activation is a complex and multistep phenomenon and the B7 family of costimulatory molecules plays a pivotal role in optimal activation of T cells [19]. Programmed cell death-1 (PD-1), a member of the B7-CD28 family, is a co-inhibitory receptor expressed by a variety of activated immune cells, including CD4+ and CD8+ T cells, natural killer T (NKT) cells, B cells, monocytes and some dendritic cell subsets [20]. PD-L1 and PD-L2 are the 2 known ligands for PD-1 [21] and they have different expression patterns. While PD-L1 is found constitutively expressed on murine lymphoid cells like T cells, B cells, macrophages, dendritic cells (DCs), mesenchymal stem cells, and bone marrow-derived mast cells [22], it is also expressed by non-hematopoietic cells [21, 23–27]. Expression of PD-L2 on the other hand is restricted to macrophages, DCs, bone marrow-derived mast cells, and peritoneal B1 cells [21,22,28–30]. Various studies performed in EAE have underlined the contribution of PD-1 and its ligands to dampening disease susceptibility or severity [25,31–33].
With respect to the role of the PD-1/PD-L pathway in estrogen treatment of EAE, our previous studies have demonstrated that E2 treatment of WT mice selectively upregulated the level of PD-1 in the CD4+FoxP3+ Treg compartment in MOG-immunized mice, with the level of PD-1 in Tregs being linked closely to the reduction in disease severity induced by E2. Down-regulation of PD-1 expression further reduced the frequency and suppressive activities of Treg cells, increased IL-17 production and thus permitted myelin-reactive T cells to launch an immune attack [34]. A more recent study demonstrated that E2 did not inhibit EAE in PD-L1−/− mice [35], and that PD-L1 expression was upregulated on B cells in the EAE-protected WT E2-implanted mice. These findings indicated that PD-1 and PD-L1 are critical mediators of the immunoregulatory effects of E2 and suggested that upregulation of PD-L1 on B cells is important for E2-mediated protection against EAE.
In lieu of the importance of PD-1 and PD-L1 in E2-mediated protection against EAE, we sought to extend our findings to the second ligand of PD-1, PD-L2, and also determine the nature of immune responses generated in E2-implanted mice in the absence of PD-L1 and PD-L2. Lastly, since we now know that the B cells may act as regulators [36] in an E2-rich milieu, the current study investigated the contribution of the PD-L1 and PD-L2 on the B cells. Results from our present study demonstrate that interactions between PD-1/PD-L1, and not PD-1/PD-L2, play an important role in attenuating autoimmune responses in the presence of E2. Furthermore, our results demonstrate that in the absence of PD-L1, the protective effects of E2 against EAE are completely lost, resulting in high levels of Th1/Th17 responses and also high proliferative capacities of T cells in the periphery. EAE disease severity in the E2-treated PD-L1−/− mice was accompanied by a severe cellular infiltration and active demyelination of the CNS. In contrast, the absence of PD-L2 was not permissive for development of any of these inflammatory immune responses. E2-treated PD-L2−/− mice were completely protected from EAE, with minimal damage to the CNS and the immune responses similar to those demonstrated by the protected E2-implanted WT mice. Transfer of B cells from MOG-immunized PD-L1−/− & PD-L2−/− donors into E2-preconditioned B cell deficient μMT−/− recipient mice resulted in partial protection against EAE in recipients of PD-L1−/− B cells, whereas full protection against EAE occurred in recipients of PD-L2−/− B cells. This study is a first report to define a central role of PD-L1 but not PD-L2 in E2-mediated protection against EAE, and as well demonstrated that PD-1 interaction with PD-L1 but not PD-L2 on B cells is crucial for the protective mechanism. Thus, we conclude that strategies that enhance PD-1/PD-L1 interactions might potentiate E2 treatment effects in EAE and possibly MS.
Materials and Methods
Ethics statement
The study was conducted in accordance with National Institutes of Health guidelines for the use of experimental animals, and the protocols were approved by the Portland Veteran Affairs Medical Center’s Institutional Animal Care and Use Committee, protocol #2415-10, local database ID #2415. Subsequent to onset of EAE, soft mix was placed in the cages of any animals that are not able to obtain water and nutrients through the normal route, monitored by weighing the animals daily after immunization for the induction of EAE. If an animal lost >20% of its body weight, the animal was treated with 1 ml/day isotonic sterile supplemental nutrients (5% dextrose in water/normal saline) administered i.p. until the weight returned to normal. The criteria for euthanasia to alleviate pain and distress were: if the animal did not respond to the above treatment after 48 hours (assessed by daily body weight measurement), the animal was euthanized with CO2 for humane reasons. Other than weight loss, loss of sensation and mobility, mice with EAE typically did not show other signs that warranted euthanasia. None of the mice used for the present study reached a condition that required euthanasia.
Animals
Female wild-type (WT) C57BL/6 mice were purchased from Harlan Laboratories (Livermore, CA). Female PD-L1−/− mice were a gift from Dr. Indira Guleria, Ph.D. (Transplantation Research Center, Brigham and Women’s Hospital and Children’s Hospital Boston, Harvard Medical School, Boston, MA, USA). Female PD-L2−/− mice were a gift from Arlene Sharpe, Ph.D. (Department of Pathology, Harvard Medical School, Boston, MA, USA). μMT−/− mice containing no B cells because of targeted disruption of the membrane exon of the immunoglobulin mu chain gene, originally obtained from Jackson Laboratories (Bar Harbor, ME), were bred at the Animal Resource Facility at the Portland Veteran Affairs Medical Center. All mice (on a C57BL/6 background) were used at 7–8 weeks of age and were housed in the Animal Resource Facility at the Portland Veterans Affairs Medical Center in accordance with institutional guidelines.
Hormone treatment and induction of EAE
Female WT, PD-L1−/− and PD-L2−/− mice were implanted with 2.5 mg/60-day release 17β-estradiol pellets (Innovative Research of America, Sarasota, FL) or sham-treated (control) one week prior to immunization with 200 μg mouse (m) MOG35-55 peptide (PolyPeptide Laboratories, San Diego, CA) in 400 μg Complete Freund’s adjuvant (CFA, H37Ra, Difco, Detroit, MI). Mice received pertussis toxin (Ptx, List Biologicals, Campbell, CA) on the day of immunization (75 ng) and 2 days later (200 ng). All mice were monitored daily for clinical signs of disease and scored using the following scale: 0=normal; 1=limp tail or mild hind limb weakness; 2=moderate hind limb weakness or mild ataxia; 3=moderately severe hind limb weakness; 4=severe hind limb weakness or mild forelimb weakness or moderate ataxia; 5=paraplegia with no more than moderate forelimb weakness; and 6=paraplegia with severe forelimb weakness or severe ataxia or moribund condition. Mice were scored daily and were evaluated for incidence, day of onset, day of maximal clinical signs (peak) and for total disease score over the course of the experiment (Cumulative Disease Index). Mean ± SD were calculated for these parameters for each of experimental group.
Histopathology
Intact spinal columns removed from mice at the end of the study (i.e. Day 24–26 post-immunization) were fixed in 4% paraformaldehyde. Dissected spinal cords were embedded in paraffin before sectioning. Sections were stained with Hematoxylin and Eosin to assess inflammatory lesions or modified eriochrome cyanine (EC) protocol to assess the sparing of the white and gray matter (demyelination) [37]. Slides were analyzed by light microscopy.
Cytokine detection by Luminex bead array
Single-cell suspensions of spleen cells obtained from control and E2-treated WT, PD-L1−/− and PD-L2−/− mice on day 24 post-immunization (p.i.) were cultured in the presence of 25 μg/ml MOG35-55 peptide for 48 h. Culture supernatants were assessed for cytokine levels using a Luminex Bio-Plex cytokine assay kit according the instructions by the manufacturer (Bio-Rad, Richmond, CA).
Proliferation assay
Splenocytes, collected from control and E2-treated WT, PD-L1−/− and PD-L2−/− mice on Day 24 post-immunization, were plated onto 96-well flat bottom plates (400,000 cells per well). Cells were stimulated with 10 or 50 μg/ml of MOG35-55 peptide for 72 h and the last 18 h in the presence of 3H-Thymidine. Cells harvested onto filter mats were read on a Perkin Elmer micro-beta scintillation counter. Stimulation indices were determined by calculating the ratio of antigen specific cpm to medium alone cpm.
Flow cytometry
Spleens and brains from control and E2-treated WT, PD-L1−/− and PD-L2−/− mice were processed for lymphocyte isolation. Briefly, the single cell suspension was prepared by dissociating tissue through a 100 μm nylon mesh screen. Red blood cells were lysed using 1X red cell lysis buffer (eBioscience, San Diego, CA). Cells were washed and resuspended in staining medium (1X PBS, 0.5% BSA, 0.02% sodium azide) and then stained with a combination of the following antibodies obtained from BD Bioscience: CD4 (L3T4), CD44 (1M7), CD62L (MEL-14), CD19 (1D3), CD1d (1B1), CD5 (53–7.3), PD-L1 (MIH5), PD-L2 (TY25), CD11b (M1/70), CD11c (HL-3), CD45 (30-F11). The intracellular staining of Foxp3 (MF23) and PD-1 (J43) was completed following overnight incubation in fixation/permeabilization buffer (eBiosciences). Dead cells were gated out using propidium iodide discrimination. Cells were gated on CD19 to determine expression of the CD1dhighCD5+, PD-L1 and PD-L2 populations. Data were collected using CellQuest (BD Biosciences) and FCS Express (De Novo) software on a FACSCalibur (BD Biosciences). Absolute numbers were calculated from live-gated cells.
Adoptive transfer of B cells
Female PD-L1−/− and PD-L2−/− mice that served as donors of B cells were immunized with 200 μg MOG35-55 peptide (PolyPeptide Laboratories, San Diego, CA) in 200 μg Complete Freund’s adjuvant (CFA, H37Ra, Difco). Pertussis toxin was not given to ensure the retention of MOG-primed cells in the spleens. Spleens from MOG-immunized PD-L1−/− and PD-L2−/− mice were harvested on day 14 p.i. and processed for B cell isolation. Splenic CD19+ B cells were purified using paramagnetic bead-conjugated antibodies (Abs) from the CD19 cell isolation kit and subsequently separated by AutoMACS (Miltenyi Biotec, Auburn, CA). The positive fraction of the cells thus separated were CD19+ B cells with a purity of ≥ 95%. Approximately 10×106 purified splenic B cells from the donor mice were transferred i.v. into μMT−/− mice, on the same day as EAE induction. The B cell-deficient (μMT−/−) mice were either sham-treated or implanted with estrogen pellet 7 days before the B cell transfer and immunization.
Statistical analysis
Data are reported using GraphPad Prism (v 4.0, San Diego, CA) and expressed as the mean ± SEM. Statistical significance for the disease course between control and E2-treated WT, PD-L1−/− and PD-L2−/− mice and also for the recipient μMT−/− control and E2-implanted mice was calculated using the Mann-Whitney U test. The Student’s t-test and the Kruskal–Wallis Test (non-parametric analysis of variance) with Dunn’s multiple comparison of means post-test were used for analysis of the cumulative disease index. Statistical significance for the Luminex and flow cytometry data was calculated using One-way ANOVA (one way analysis of variance and Newman-Keuls Multiple Comparison post test). The Student’s t-test was used to compare stimulation indices between control and E2-treated WT, PD-L1−/− and PD-L2−/− mice, within the same strain. P-values ≤ 0.05 were considered to be significant (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
Results
The co-inhibitory ligand, PD-L2, but not PD-L1, is dispensable in E2-mediated protection against EAE
Our previous work [35] demonstrated that E2-treatment did not protect PD-L1−/− mice, which developed EAE disease scores equivalent to those of control (sham-treated) WT and PD-L1−/− mice. These results indicated that PD-L1 is a critical co-inhibitory molecule in E2-mediated protection against EAE. The current study further explores whether or not the second ligand of the PD-1/PD-L co-inhibitory family, PD-L2, is redundant in mediating E2-related protective effects in EAE. Age-matched female WT, PD-L1−/− and PD-L2−/− mice were either sham-treated (control) or implanted with E2 pellets 7 days prior to immunization with mMOG35-55 peptide with CFA/Ptx and the EAE disease course was followed for 24 days post-immunization (p.i.).
As reported earlier, mice lacking PD-L1 not only developed more extensive EAE (higher CDI) than WT control mice, but also were significantly less protected after E2 treatment (Figure 1A and Table 1), resulting in EAE disease scores equivalent to those of sham-treated WT mice. In contrast to the PD-L1−/− mice, control PD-L2−/− mice developed EAE similar to the control WT mice and E2-implanted PD-L2−/− mice were completely protected against EAE through day 24 post-immunization similar to E2-treated WT mice (Table 1). Consistent with the clinical results, spinal cord sections from E2-implanted PD-L1−/− mice demonstrated massive leukocyte infiltration with several foci of inflammation (Figure 1B) along with severe demyelination similar to control PD-L1−/− mice, whereas spinal cord sections from the PDL2−/− E2-treated mice showed no obvious signs of inflammation and demyelination (Figure 1C). These results demonstrate conclusively that PD-L2 is redundant and that PDL1 plays the major role in mediating E2-dependent protection against EAE.
Figure 1. The co-inhibitory ligand, PD-L2, but not PD-L1, is dispensable in E2 mediated protection against EAE.

Seven to 8 week old WT, PD-L1−/− and PD-L2−/− mice were implanted with E2 pellets one week prior to immunization with mouse (m) MOG35-55 peptide in CFA/Ptx. Mice were monitored for signs of clinical EAE. A) mean disease scores from 2 independent experiments with 3–4 mice/group/experiment. *significant difference (p ≤ 0.05) as compared to the respective E2-implanted mice (Mann-Whitney U test). B and C) Histopathological evaluation of spinal cords from PD-L1−/− (control and E2-implanted) and PD-L2−/− (control and E2-implanted) mice. Spinal cords from each group, collected on day 24 post-immunization, were fixed in PFA and embedded in paraffin. Ten μm transverse sections from different regions of the spinal cord from each of the groups were stained with Hematoxylin & Eosin to enumerate infiltrating leukocytes and with Eriochrome cyanine to visualize extent of demyelination. Magnification 50 times and 200 times (inset). Sections are representative of 2 experiments (n=3–4/group/experiment).
Table 1.
Clinical course of EAE in WT, PD-L1−/− and PD-L2−/− mice*.
| Mice | Incidence | Onset | Peak | CDI | Mortality |
|---|---|---|---|---|---|
| WT Ctrl | 8/8 | 15.3 ± 1.3b | 4.6 ± 0.1 | 38.5 ± 5.1b | 0 |
| WT E2 | 1/8 | 20.0 ± 0a,c | 2.0 ± 0a,c | 0.9 ± 0.8a,c | 0 |
| PD-L1−/− Ctrl | 6/6 | 11.1 ± 0.4 | 4.6 ± 0.1 | 55.2 ± 2.8 | 0 |
| PD-L1−/− E2 | 5/6 | 14.9 ± 0 | 3.6 ± 0.3a | 33.3 ± 5.4a | 1 |
| PD-L2−/− Ctrl | 7/8 | 11.7 ± 0.6 | 4.4 ± 0.4 | 33.9 ± 6.7b | 0 |
| PD-L2−/− E2 | 0/8 | 0 ± 0a,c | 0 ± 0a,c | 0 ± 0a,c | 0 |
significant (p ≤ 0.05) as compared to respective Ctrl group
significant (p ≤ 0.05) as compared to PD-L1−/− Ctrl group
significant (p ≤ 0.05) as compared to PD-L1−/− E2 group
Statistical evaluation of EAE disease course for WT and PDL1−/− and PD-L2−/− Control and E2-implanted mice, including the Cumulative Disease Index (CDI), through Day 24 post-immunization for the 2 experiments (n=3–4 mice/group/experiment). The daily disease scores from pooled data and corresponding CDI data from each of the three independent experiments (mean ± SEM) are presented. Additional information for the experiments is shown in Figure 1.
Absence of PD-L1 but not PD-L2 elicits a high pro-inflammatory environment in spleens
Pathogenic T cells are known to be induced in the periphery (spleens and draining lymph nodes) after which they migrate to the CNS to initiate an inflammatory process responsible for clinical signs of EAE [3]. Therefore, the cytokine milieu was evaluated in the spleens of the control and E2-implanted PD-L knockout (KO) mice. MOG-specific responses generated in spleens, 24 days p.i., were determined for cytokine expression by culturing mononuclear cells from sham treated and E2-implanted WT, PD-L1−/− and PD-L2−/− mice and collecting supernatants 48 hours later. As reported earlier [35], sham-treated WT mice produced higher levels of IL-17, TNF-α and IFN-γ as compared to their E2-implanted counterparts (Figure 2A). The splenocytes of both the sham-treated and E2-implanted PD-L2−/− mice produced almost equivalent levels of IL-17, TNF-α and IFN-γ as the WT sham-treated and E2-implanted mice, respectively. However, splenocytes from not only the sham-treated but also the E2-implanted PD-L1−/− mice produced significantly higher levels of proinflammatory cytokines as compared to both the E2-implanted WT and PD-L2−/− mice (Figure 2A). Moreover, proliferation responses of splenic T cells were inhibited by E2 treatment in control and PD-L2−/− mice, but no inhibition was observed in PD-L1−/− mice (Figure 2B).
Figure 2. Absence of PD-L1 but not PD-L2 elicits a high pro-inflammatory environment in spleens.

Splenocytes were isolated from control and E2-implanted WT, PDL1−/− and PD-L2−/− mice, on Day 24 post-immunization and: A) cultured in the presence of 25 μg/ml mMOG35-55 peptide for 48h. Culture supernatants were assayed for secreted levels of cytokines by Luminex Bead array. Data are representative of 2 independent experiments (mean ± SEM). B) Plated in a 96-well flat bottom microtiter plate and cultured with 10 μg/ml of mMOG35-55 peptide for 72 h, the last 18 h in the presence of 3H-thymidine. Stimulation indices (S.I.) were determined by calculating the ratio of cpm from antigen-induced vs. unstimulated cultures. Equal number of cells from each mouse in a group were pooled and detection was done in pooled cells from n=3–4 mice/group from each of two separate experiments (mean ±SEM). Significant differences between the groups (*p ≤ 0.05) were determined by One-way ANOVA with one way analysis of variance and Newman-Keuls Multiple Comparison post test.
Higher infiltration and loss of E2-mediated reduction of proinflammatory immune cells in the CNS of PD-L1−/− vs. PD-L2−/− mice
Since PD-L1−/− mice demonstrated more severe clinical and histological signs of EAE, we evaluated the immune cell trafficking into the CNS of these mice compared to sham-treated and E2-implanted WT and PD-L2−/− mice. Mononuclear cells isolated from brains of control and E2-implanted WT, PD-L1−/− and PD-L2−/− mice were stained for several cell surface markers. Brains of control and E2-implanted PD-L1−/− mice contained significantly higher numbers of total mononuclear as compared to E2-implanted WT and PD-L2−/− mice (Figure 3A). Upon further characterizing infiltrating mononuclear cells in the brains, it was revealed that brains of E2-implanted PD-L1−/− mice had a significant increase in percentages of the CD11b+CD45high cell population represented by resident activated microglia and/or infiltrating monocytes/macrophages as well as dendritic cells (CD11c+CD45+) (Figure 3B). Both of these populations have been known to participate in disease pathogenesis through effects on blood brain barrier permeability, antigen presentation and immune regulation [17]. The PD-L2−/− E2-implanted mice on the other hand demonstrated significantly less infiltration of activated microglia/monocytes and dendritic cells (Figure 3B). Thus, the inhibitory effects of E2 on infiltration of inflammatory cells into the CNS were totally abrogated in the absence of PD-L1 whereas they were retained in the absence of PD-L2.
Figure 3. Higher infiltration of proinflammatory immune cells in the CNS of PD-L1−/− vs. PD-L2−/− mice.
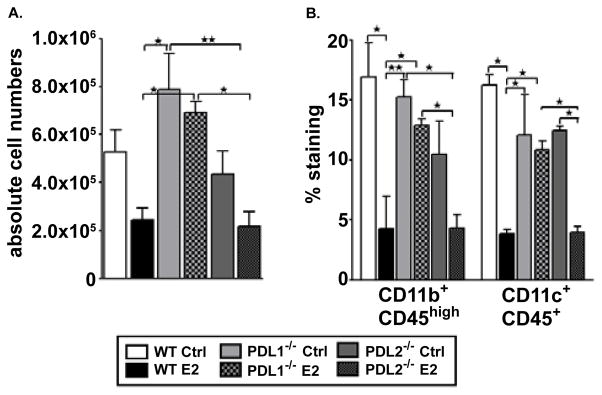
On Day 24 post-immunization, mononuclear cells isolated from brains of control and E2-implanted WT, PD-L1−/− and PD-L2−/− mice were analyzed for: A) Total live cell numbers counted on a hemocytometer; and B) CD11b+CD45high infiltrating monocytes/activated microglia and CD11c+CD45+ dendritic cells (DCs). Frequencies of monocytes/microglia and DCs were determined in individual brains processed and indicate the percentages of total gated live leukocytes (n=6–8). Data are representative of 2 independent experiments (mean ± SEM). Significant differences between the groups (*p ≤ 0.05 and **p ≤ 0.01) were determined using One-way ANOVA with one way analysis of variance and Newman-Keuls Multiple Comparison post test and are indicated by brackets.
Absence of PD-L1 but not PD-L2 leads to a higher number of activated CD4+ T cells in the CNS of E2-implanted mice
The control and E2-implanted PD-L1−/− mice also had a significant increase in percentages of brain-infiltrating CD4+ and CD8+ T cells as compared to E2-implanted WT and PD-L2−/− mice (Figure 4A). CD44hiCD4+ T-cells are increased in the spleen and CNS of mice with EAE [38], and it thus was of interest to further characterize the CD4+ T cells in the brains of WT, PD-L1−/− and PD-L2−/− mice. The percentage of CD44−CD62L+ (naive), CD44+CD62L+ (activated) and CD44+CD62L− (memory) were evaluated amongst the CD4+ T population. The percentage of CD4+ T cells expressing CD44hiCD62L+ was significantly increased in PD-L1−/− control and E2-implanted mice (Figure 4B), indicating that the infiltrating CD4+ T cells were significantly more activated than those found in the brains of E2-protected WT and PD-L2−/− mice (Figure 4B). The PD-L2−/− E2-implanted mice on the other hand demonstrated significantly less infiltration of CD8+ T cells and activated CD4+ T cells (Figures 4A and 4B).
Figure 4. Absence of PD-L1 results in higher activated CD4+ T cells in CNS of E2-implanted mice.
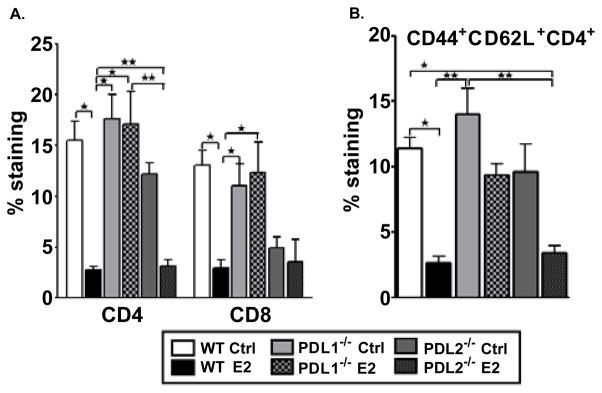
Mononuclear cells isolated from brains of control and E2-implanted WT, PDL1−/− and PD-L2−/− mice were analyzed on Day 24 post-immunization. A) Frequencies of CD4+ and CD8+ T cells were determined in individual brains and indicate the percentages of total gated live leukocytes (n=3–4). Data are pooled from 2 independent experiments (mean ± SEM). B) Flow antibodies for CD44 and CD62L were used to differentiate between naïve, memory and activated (CD44+CD62L+) CD4+ T cells. Cells were gated on live CD4+ cells for the analysis of activated CD4+ T cells. Data are pooled from 2 independent experiments (mean ±SEM). Significant differences between the groups (*p ≤ 0.05 and **p ≤ 0.01) were determined using One-way ANOVA with one way analysis of variance and Newman-Keuls Multiple Comparison post test and are indicated by brackets.
Absence of PD-L1 but not PD-L2 leads to a significant decrease in the percentage of CD1dhiCD5+ (regulatory) B cells
As demonstrated earlier [35], the E2-protected WT mice exhibited a significant increase in the percentage of CD1dhiCD5+ B cells in the spleens. This regulatory B cell sub-population is a strong candidate for mediating the protective properties of E2 against the development of EAE. The complete absence of EAE signs in the E2-implanted PD-L2−/− mice led us to investigate the contribution of the regulatory B cell-subset in these mice. The CD1dhiCD5+ Breg cell subpopulation increased significantly in the spleens of PD-L2−/− E2-implanted mice not only as compared to its sham-treated counterparts, but also compared to the sham-treated and E2-implanted PD-L1−/− mice (Figure 5A). Moreover, at day 24, splenocytes from PD-L2−/− E2-implanted mice demonstrated a remarkable increase in production of IL-10 comparable to that produced by the E2-implanted WT mice but in contrast to the minimal levels of IL-10 produced by splenocytes from control or E2-treated PD-L1−/− mice (Figure 5B). Further characterization of the B cells in the different strains of mice and in the sham-treated and E2-implanted groups led to demonstration of a similar increase in the PD-L1 on B cells in the E2-implanted PD-L2−/− mice as the E2-implanted WT mice (Figure 5C). However, the splenocytes of E2-implanted PD-L1−/− mice failed to demonstrate any change in the expression of the PD-L2 by their B cells (Figure 5D). These results indicate that the expression of PD-L1 and CD1dhiCD5+ by the B cells endows a protective function to the B cells in an E2-rich milieu.
Figure 5. Absence of PD-L1 but not PD-L2 leads to a significant decrease in Breg cells.

Splenocytes from control and E2-implanted WT, PD-L1−/− and PD-L2−/− mice were harvested on day 24 post-immunization and assessed for expression of: A) CD1dhiCD5+ B cells, C) PD-L1-expressing B cells and D) PD-L2-expressing B cells Data represent CD1dhiCD5+, PD-L1+ and PD-L2+ expression on gated CD19+ cells of total gated and live cells. Data are representative of 2 independent experiments with spleens processed from 6–8 individual mice (mean ± SEM). B) Splenocytes from control and E2-implanted WT, PD-L1−/− and PD-L2−/− mice, harvested on Day 24 post-immunization, were cultured in the presence of 25 μg/ml mMOG35-55 peptide for 48 h. Cells were pooled from mouse groups (6–8 mice total; 2 individual experiments) (mean ± SEM) and cytokines were detected using the Luminex bead array. Significantdifferences between the groups (*p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001) were determined using One-way ANOVA with one way analysis of variance and Newman-Keuls Multiple Comparison post test and are indicated by brackets.
Presence of PD-L1 but not PD-L2 on B cells is critical for E2-mediated protection against EAE
The splenic B cells in the E2-implanted PD-L2−/− mice demonstrate a significant increase in PD-L1 expression and our earlier studies demonstrated a steady increase in PD-L1-expressing B cells over the course of EAE disease in E2-implanted WT mice [35]. Thus, it was crucial to determine the contribution of these PD-ligands in the protective function of B cells in E2-dependent protection against EAE. Also, our recent study [36] demonstrated that B cells from WT donors are sufficient to restore E2-mediated protection in the μMT−/− mice and the E2-implanted B cell-recipients were significantly protected until day 25 p.i./after transfer as compared to the control recipients. In the current study, we first assessed the role of PD-L1 in B cells as a contributor in E2-mediated protection upon transfer into recipient μMT−/− mice on the day of EAE induction. B cells were obtained from MOG-immunized PD-L1−/− donors, day 14 p.i., transferred into sham-treated or E2-implanted recipient μMT−/− mice and EAE disease induced and scores followed through Day 26. μMT−/−-control and E2-treated mice receiving no cells were included as experimental controls. Upon transferring B cells from MOG-immunized PD-L1−/− donors, as expected the control recipient μMT−/− mice had an early onset (day 10–11 p.i.) similar to that observed in control mice receiving no B cells. However, both the PD-L1−/− B cell and the no cell E2-implanted recipient groups had an onset of disease around day 17 p.i. (Figure 6A), with slightly lower CDI in the E2-implanted PD-L1−/− B cell recipient group (Table 2). The E2-implanted recipients of PD-L1−/− B cells lost protection from day 20 p.i. as compared to the sham-treated recipients, thus indicating a partial protection for the E2-implanted PD-L1−/− B cell recipients. Spinal cord sections from E2-implanted PD-L1−/− B cell recipient μMT−/− mice also demonstrated leukocyte infiltration with several foci of inflammation (Figure 6B) along with somewhat less apparent demyelination than control recipient μMT−/− mice. These data indicate a crucial role of PD-L1 in B cells for E2-mediated protection in recipient μMT−/− mice.
Figure 6. PD-L1 but not PD-L2 on B cells is critical for E2-mediated protection against EAE.

Female PDL1−/− and PD-L2−/− mice that served as donors of B cells were immunized with mouse (m)MOG35-55 peptide in Complete Freund’s adjuvant (CFA) with no Ptx administration. Seven to 8 week old μMT−/− mice that served as recipients of B cells were either sham-treated (control) or implanted with E2 pellets one week prior to B cell transfer and immunization with MOG35-55 peptide in CFA/Ptx. Recipient mice were monitored for signs of clinical EAE. A) Mean disease scores from 2–3 independent experiments with 3–5 mice/group/experiment. *p ≤ 0.05, compared to the respective control mice (i.e. E2-implanted with no cells vs. controls with no B cells and E2-implanted recipients with B cells from the appropriate donors vs. control recipients with B cells) (Mann-Whitney U test). Histopathological evaluation of spinal cords from B) PD-L1−/−, C) PD-L2−/− B cell-recipient μMT−/− (control and E2-implanted) mice (day 26 post-immunization). Spinal cords from each group, collected on day 26 post-immunization, were fixed in PFA and embedded in paraffin. 10 μm thick transverse sections from different regions of the spinal cord from each of the groups were stained with Hematoxylin & Eosin to enumerate infiltrating leukocytes and with Eriochrome cyanine to visualize the extent of demyelination (magnification 50 times and 200 times (inset)). Sections are representative of 2 experiments (n=3–5/group/experiment).
Table 2.
Clinical course of EAE in B-cell-recipient μMT−/− mice*.
| μMT−/− recipient mice | Incidence | Onset | Peak | CDI | Mortality |
|---|---|---|---|---|---|
| no cells Ctrl | 6/6 | 12.2 ± 0.2 | 5.0 ± 0.0 | 64.8 ± 1.4 | 1 |
| no cells E2 | 7/7 | 17.1 ± 0.8a | 4.5 ± 0.4 | 40.9 ± 4.6a | 1 |
| +PD-L1−/− B cells Ctrl | 8/8 | 9.8 ± 0.3 | 4.8 ± 0.2 | 64.9 ± 1.9 | 0 |
| +PD-L1−/− B cells E2 | 8/8 | 17.3 ± 0.6a | 4.2 ± 0.2a | 32.6 ± 2.2a | 0 |
| +PD-L2−/− B cells Ctrl | 10/10 | 11.8 ± 0.5 | 5.0 ± 0.0 | 63.9 ± 2.8 | 0 |
| +PD-L2−/− B cells E2 | 6/11 | 22.8 ± 0.7a,b,c | 3.6 ± 0.7a | 6.8 ± 2.7a,b,c | 0 |
significant (p ≤ 0.05) as compared to respective Ctrl group
significant (p ≤ 0.05) as compared to E2-implanted PD-L1−/− B cell-recipient group
significant (p ≤ 0.05) as compared to E2-implanted no cell transfer μMT−/− recipient group
Statistical evaluation of EAE disease course for Control and E2-implanted recipient μMT−/− mice, including the Cumulative Disease Index (CDI), through day 26 post-immunization for the 2–3 experiments (n=3–5 mice/group/experiment). The daily disease scores from pooled data and corresponding CDI data from each of the independent experiments (mean ± SEM) are presented. Additional information for the experiments is shown in Figure 6.
Similarly, to verify the requirement of PD-L2 on the B cells in E2-mediated protection, B cells from MOG-immunized PD-L2−/− donors were transferred into sham-treated or E2-implanted μMT−/− mice on the day of induction of EAE. The control μMT−/− recipients of MOG-specific B cells from PD-L2−/− donors demonstrated an onset of EAE at day 11.8 p.i. similar to the no cell recipients (Figure 6A and Table 2). However, the E2-implanted μMT−/− recipients of MOG-specific PD-L2−/− B cells were significantly protected from EAE through day 26 p.i. (Figure 6A) with eventual loss of protection, there onwards, as compared to the control PDL2−/− B cell recipient counterparts (not shown). As shown in Table 2, the CDI and the peak of the disease of E2-implanted μMT−/− recipients of PD-L2−/− B cells were also strikingly and significantly lower than those of control recipients. Spinal cords sections were assessed for the extent of inflammation (H&E staining) and demyelination (Eriochrome staining) in CNS of the control and E2-implanted μMT−/− recipient mice. We demonstrated earlier [35] that spinal cord sections of E2-implanted μMT−/− mice (receiving no B cells) demonstrated massive leukocyte infiltration with several foci of inflammation along with severe demyelination similar to control μMT−/− mice. Here similarly, the control PD-L2−/− B cell transferred μMT−/− recipient mice demonstrated massive leukocyte infiltration along with demyelination. In contrast, spinal cord sections from the E2-implanted PD-L2−/− B cell transferred μMT−/− recipient mice showed no obvious signs of inflammation and demyelination (day 26 p.i.) (Figure 6C), mimicking the histological picture in WT E2-treated mice [15]. Taken together, the above results confirm that the presence of PD-L1 on B cells is critical for E2-mediated protection against EAE, whereas the protection imparted is independent of PD-L2.
Discussion
Over the past decade numerous studies using either monoclonal blocking antibodies or knockout mice on different strains have demonstrated varying roles for the ligands of the PD-1 co-inhibitory pathway in EAE. Initial studies by Salama et al. [25], wherein neutralizing antibodies specific for PD-1 (J43) or PD-L2 (TY25) were used, described PD-1 and PD-L2 to be critical in EAE. The blockade of PD-1 and PD-L2 led to the expansion of MOG-reactive T cells, increased lymphocytic infiltration of the CNS, and ultimately, accelerated disease onset and severity. However, studies in PD-1 and PD-L-deficient mice on the 129svEv background demonstrated that deficiency of PD-1 and PD-L1 led to exacerbated EAE with increased proliferation of inflammatory ex vivo recall responses. PD-L2 deficiency, however, led to EAE disease similar to WT mice and less dramatic ex vivo recall response [33,39]. With the literature pointing to the contradictory roles of PD-L1 and PD-L2 in EAE, it was essential to discern their role in E2-mediated protection against EAE, especially since our lab had already implicated PD-1 in the E2-protective pathway [34,40–42]. Our recent work [35] demonstrated that E2-implanted PD-L1−/− mice were not protected from EAE. However, the immune responses elicited in this strain of mice, which renders them susceptible to EAE even in the presence of E2, were not investigated. Also, the effect of E2 on the disease outcome of the PD-L2−/− mice had not been studied earlier. Therefore, to complete the picture, it was necessary to investigate the nature of interactions between PD-1 and both PD-Ligands for understanding the susceptibility, pathogenic mechanisms, and protection afforded in an E2-rich environment. Moreover, we sought to determine the contribution of both ligands on B cells transferring E2-mediated protection to B cell-deficient mice.
As demonstrated earlier, we were able to repeat our results in the E2-implanted PD-L1−/− mice, in that E2 treatment of the PD-L1−/− mice delayed but could not inhibit the disease severity, thereby leading to trafficking of immune cells into the CNS causing inflammatory foci and demyelination. Lack of PD-L1, as demonstrated by other EAE-related studies, led to a much more severe disease. Despite E2-treatment, the PD-L1−/− mice demonstrated EAE disease scores and CNS damage comparable to the sham-treated WT mice. On the other hand, even if the absence of PD-L2 led to a comparable EAE disease outcome as in sham-treated WT mice, the E2-implanted PD-L2−/− mice were completely protected from developing EAE like the E2-implanted WT mice, with no infiltrating immune cells and demyelination in the CNS.
EAE is known to be predominantly a T cell-mediated autoimmune disease with IFN-γ and IL-17-producing T cell subsets responsible for promoting EAE [11,12]. Studies have demonstrated that stimulated T cells from PD-1−/− and PD-L1−/− mice with active disease produced copious amounts of IL-17 and IFN-γ as compared to WT mice [33], suggesting that T cells with deficient PD-1/PD-L signaling may be preferentially polarized toward effector T-cell differentiation. Our lab has demonstrated that treatment with E2 is a powerful regulator of cytokines, decreasing the production of proinflammatory cytokines such as TNF-α and increasing the production of anti-inflammatory cytokines, such as IL-4 and IL-10 [16,43]. Hence, ex vivo recall responses by means of cytokine production by splenocytes of the control and E2-implanted PD-L1 and PD-L2-deficient mice on day 24 were analyzed. Our results confirmed that, in the absence of PD-L1, significantly higher MOG-specific Th1/Th17 responses were generated in the periphery, not only by the sham-treated PD-L1−/− mice but also but the E2-implanted PD-L1−/− mice. However, absence of PD-L2 led to a proinflammatory milieu similar to the sham-treated WT mice. The cytokine profile in the EAE-protected and E2-implanted PD-L2−/− mice was similar to that in the E2-implanted WT mice, demonstrating significantly lower expression of the proinflammatory cytokines IFN-γ, IL17 and TNF-α.
Estrogen is known to reduce Ag-specific T cell proliferative responses [35], albeit not by direct interaction with the T cells. Several studies have demonstrated that PD-Ls can inhibit T cell proliferation. However, there are a few studies which contradict these results and the reasons for these contradictory results remain unclear and controversial. Hence T cell proliferative responses were analyzed in the periphery for the each of the PD-L KO mice, with or without E2 treatment. Akin to the disease pattern in each of the control and E2-implanted PD-L knockout mice, T cell proliferative responses were significantly higher in the absence of PD-L1, but E2-implantation failed to reduce this increase. However, the splenocytes of the E2-implanted PD-L2−/− mice had significantly lower T cell proliferation capabilities.
The PD-1/PD-L pathway is recognized to control peripheral T-cell tolerance in several ways. This pathway can limit the initial phase of activation and expansion of self-reactive T cells, and restrict self-reactive T-cell effector function. The PD-1/PD-L interactions also play a role in inhibiting expansion of naive self-reactive T cells and/or their differentiation into effector T cells [20]. Thus, PD-L1 and PD-L2 can signal bidirectionally by engaging PD-1 on T cells and by delivering signals into PD-L-expressing cells. Similarly, our results demonstrate that increased severity of disease in the control and E2-implanted PD-L1−/− mice was accompanied by increased infiltration of immune cells into the CNS. Not only were the numbers of infiltrating cells increased but so were the percentages of activated microglia/monocytes and infiltrating dendritic cells. This phenomenon was, however, lacking in E2-treated protected PD-L2−/− mice. Loss of PD-L1 also led to a higher number of CD4+ T and CD8+ T cells in the brains. Further analysis of the activation states of the infiltrating CD4+ T cells demonstrated a significantly higher percentage of activated CD4+ T cells in the E2-implanted PD-L1−/− mice as compared to both the E2-treated WT and PD-L2−/− mice, indicating that the CNS-infiltrating CD4+ T cells have a pathogenic rather than a regulatory role. Thus, our results indicate that the PD-1/PD-L1 pathway in presence of E2 delivers inhibitory signals that regulate peripheral T-cell tolerance but that the PD-1/PD-L2 pathway is not redundant for E2-mediated protection against EAE.
Our earlier studies [35] demonstrated that the E2-rich milieu is able to induce a regulatory B cell subset (CD1dhiCD5+ B cells; Bregs) in WT mice which regulate EAE by virtue of their IL-10 production. The literature demonstrates that the interaction between PD-1 and PD-L1 results in the up-regulation of IL-10 production [44]. It is also known that in the presence of anti-CD3 and TGF-β, PD-L1-Ig can induce a profound increase in the de novo generation of CD4+Foxp3+ Tregs from naïve CD4+ T cells. PD-L1-Ig also can enhance Foxp3 expression and the suppressive function of established iTregs [45]. Furthermore, a role for the PD-1/PD-L pathway in generation of Tregs has been supported by a number of studies [46,47]. Thus, the CD1dhiCD5+ regulatory B cell subset along with IL-10 production and percentage of Foxp3+ Tregs were evaluated in the E2-implanted PD-L1−/− and PD-L2−/− mice and compared to WT mice in an attempt to decipher the regulatory/protective properties endowed in the E2-protected PD-L2−/− vs. PD-L1−/− mice. The spleens of E2-implanted PD-L2−/− mice could produce a significantly higher percentage of Breg cells and IL-10, thus confirming the critical role played by IL-10-producing regulatory B cells in E2-mediated protection against EAE. No difference in Treg percentages in the spleens of each of the PD-L deficient strains was observed. However, the absence of the PD-ligands had a trend of decreased percentage of Tregs even in presence of E2 in the spleens as compared to the E2-implanted WT mice (data not shown). Thus, while Tregs are important in controlling EAE-induced inflammatory responses, they are not solely responsible for E2-mediated protection, as observed previously [48].
Recent work indicates that PD-1/PD-L interactions can regulate B cell responses [49], but whether PD-1 or PD-L1 on the T or B cells or PD-L2 on the B cells is involved, is not yet clear. Also the regulatory contributions of B lymphocytes during EAE are linked to their ability to produce IL-10 [50–53]. Our studies have established that the upregulation of PD-L1 on B cells in MOG-immunized E2-implanted WT mice is associated with protection from EAE [35]. Our recent studies also demonstrated [36] that the B cells expressing estrogen receptor-α and GPR30 are sufficient for mediating E2-related protection in μMT−/− mice via upregualtion of PD-1+ Tregs. Hence, it was important to evaluate the importance of the presence of PD-L1 and PD-L2 on B cells for mediating E2-related protection. The transfer of B cells from PD-Ligand KO mice provided critical information regarding the direct effects of E2 on the transferred regulatory B cells. Restoration of μMT−/− mice with B cells lacking PD-L1 partially reduced EAE severity, unlike the complete protection rendered upon transfer of B cells lacking PD-L2. Thus, PD-L2 expression on B cells does not contribute to E2 protection, whereas PD-L1 expression forms a crucial arm of the regulatory property endowed on B cells. The partial rather than complete loss of protection upon transfer of PD-L1−/− B cells may be attributed to the presence of PD-L1 on other cells of lymphoid or non-hematopoietic origin in the μMT−/− recipients. In fact, determination of the expression of PD-L1 and PD-L2 in splenocytes from B cell-deficient mice indicated a reduction in PD-L1 expression from ~17% in WT to ~7% in μMT−/− mice. On the other hand, there was no difference in PD-L2-expressing splenocytes between WT and μMT−/− mice (~6% in WT and ~5% in μMT−/− mice; data not shown). Thus, acknowledging the fact that where the contribution of PD-L1 expressed by cells other than B cells can be crucial, PD-L1 expression on B cells forms an integral part of the regulatory B cell contribution, whereas PD-L2 expression on B cells is redundant for this regulatory property.
A recent study demonstrating the increased expression of PD-1 and PD-L1 and higher IL-10 production, lower proliferation, and increased apoptosis of MBP-specific cells was shown to be associated with remission of disease in MS patients, thus, confirming the importance of the PD-1/PD-L1 coinhibitory pathway also in human pathology [54]. In summary, our data in this study indicate an important role for the PD-1/PD-L1 coinhibitory pathway in modulating E2-mediated protection against EAE. We conclusively demonstrate that unlike PD-L2−/− mice that were fully protected against EAE after E2 treatment, E2-implanted PD-L1−/− mice were susceptible to EAE, with increased numbers of proliferating Th1/Th17 cells in the periphery and severe cellular infiltration and active demyelination in the CNS. Moreover, transfer of B cells from MOG-immunized PD-L1−/− or PD-L2−/− donors into E2-preconditioned B-cell deficient μMT−/− recipient mice revealed significantly reduced E2-mediated protection against EAE in recipients of PD-L1−/− B cells, but complete protection in recipients of PD-L2−/− B cells. Thus, our data indicate that PD-1and PD-L1 play important roles in determining the E2-mediated EAE susceptibility and in controlling chronic disease progression. We conclude that PD-1 interaction with PD-L1 but not PD-L2 on B cells is crucial for E2-mediated protection in EAE and that strategies that enhance PD-1/PD-L1 interactions might potentiate E2 treatment effects in EAE and MS.
Acknowledgments
The authors wish to thank Sandhya Subramanian and Lisa Miller for technical assistance, Xiaolin Yu for assistance in preparing the histology figures and Melissa Barber for assistance with manuscript preparation. This work was supported by National Multiple Sclerosis Society grant RG3405-C-6 and NIH/NINDS 1R01 NS075887. This material is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
Conflict of Interest
The authors declare no financial or commercial conflict of interest.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Whitacre CC. Sex differences in autoimmune disease. Nat Immunol. 2001;2:777–780. doi: 10.1038/ni0901-777. [DOI] [PubMed] [Google Scholar]
- 2.Hemmer B, Archelos JJ, Hartung HP. New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci. 2002;3:291–301. doi: 10.1038/nrn784. [DOI] [PubMed] [Google Scholar]
- 3.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 4.Sadovnick AD. European Charcot Foundation Lecture: the natural history of multiple sclerosis and gender. J Neurol Sci. 2009;286:1–5. doi: 10.1016/j.jns.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Abramsky O. Pregnancy and multiple sclerosis. Ann Neurol. 1994;36(Suppl):S38–S41. doi: 10.1002/ana.410360712. [DOI] [PubMed] [Google Scholar]
- 6.Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N Engl J Med. 1998;339:285–291. doi: 10.1056/NEJM199807303390501. [DOI] [PubMed] [Google Scholar]
- 7.Vukusic S, Hutchinson M, Hours M, Moreau T, Cortinovis-Tourniaire P, et al. Pregnancy and multiple sclerosis (the PRIMS study): clinical predictors of post-partum relapse. Brain. 2004;127:1353–1360. doi: 10.1093/brain/awh152. [DOI] [PubMed] [Google Scholar]
- 8.Sicotte NL, Liva SM, Klutch R, Pfeiffer P, Bouvier S, et al. Treatment of multiple sclerosis with the pregnancy hormone estriol. Ann Neurol. 2002;52:421–428. doi: 10.1002/ana.10301. [DOI] [PubMed] [Google Scholar]
- 9.Soldan SS, Alvarez Retuerto AI, Sicotte NL, Voskuhl RR. Immune modulation in multiple sclerosis patients treated with the pregnancy hormone estriol. J Immunol. 2003;171:6267–6274. doi: 10.4049/jimmunol.171.11.6267. [DOI] [PubMed] [Google Scholar]
- 10.Whitacre CC, Reingold SC, O’Looney PA. A gender gap in autoimmunity. Science. 1999;283:1277–1278. doi: 10.1126/science.283.5406.1277. [DOI] [PubMed] [Google Scholar]
- 11.Kuchroo VK, Martin CA, Greer JM, Ju ST, Sobel RA, et al. Cytokines and adhesion molecules contribute to the ability of myelin proteolipid protein-specific T cell clones to mediate experimental allergic encephalomyelitis. J Immunol. 1993;151:4371–4382. [PubMed] [Google Scholar]
- 12.Park H, Li Z, Yang XO, Chang SH, Nurieva R, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bettelli E, Das MP, Howard ED, Weiner HL, Sobel RA, et al. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J Immunol. 1998;161:3299–3306. [PubMed] [Google Scholar]
- 14.Kennedy MK, Torrance DS, Picha KS, Mohler KM. Analysis of cytokine mRNA expression in the central nervous system of mice with experimental autoimmune encephalomyelitis reveals that IL-10 mRNA expression correlates with recovery. J Immunol. 1992;149:2496–2505. [PubMed] [Google Scholar]
- 15.Bebo BF, Jr, Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, et al. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J Immunol. 2001;166:2080–2089. doi: 10.4049/jimmunol.166.3.2080. [DOI] [PubMed] [Google Scholar]
- 16.Ito A, Bebo BF, Jr, Matejuk A, Zamora A, Silverman M, et al. Estrogen treatment down-regulates TNF-alpha production and reduces the severity of experimental autoimmune encephalomyelitis in cytokine knockout mice. J Immunol. 2001;167:542–552. doi: 10.4049/jimmunol.167.1.542. [DOI] [PubMed] [Google Scholar]
- 17.Polanczyk MJ, Jones RE, Subramanian S, Afentoulis M, Rich C, et al. T lymphocytes do not directly mediate the protective effect of estrogen on experimental autoimmune encephalomyelitis. Am J Pathol. 2004;165:2069–2077. doi: 10.1016/S0002-9440(10)63257-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goverman JM. Immune tolerance in multiple sclerosis. Immunol Rev. 2011;241:228–240. doi: 10.1111/j.1600-065X.2011.01016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bugeon L, Dallman MJ. Costimulation of T cells. Am J Respir Crit Care Med. 2000;162:S164–168. doi: 10.1164/ajrccm.162.supplement_3.15tac5. [DOI] [PubMed] [Google Scholar]
- 20.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002;169:5538–5545. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- 23.Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198:63–69. doi: 10.1084/jem.20022125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. PD-1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med. 2003;198:39–50. doi: 10.1084/jem.20022235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med. 2003;198:71–78. doi: 10.1084/jem.20022119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiendl H, Mitsdoerffer M, Schneider D, Chen L, Lochmüller H, et al. Human muscle cells express a B7-related molecule, B7-H1, with strong negative immune regulatory potential: a novel mechanism of counterbalancing the immune attack in idiopathic inflammatory myopathies. FASEB J. 2003;17:1892–1894. doi: 10.1096/fj.03-0039fje. [DOI] [PubMed] [Google Scholar]
- 27.Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33:2706–2716. doi: 10.1002/eji.200324228. [DOI] [PubMed] [Google Scholar]
- 28.Ishida M, Iwai Y, Tanaka Y, Okazaki T, Freeman GJ, et al. Differential expression of PD-L1 and PD-L2, ligands for an inhibitory receptor PD-1, in the cells of lymphohematopoietic tissues. Immunol Lett. 2002;84:57–62. doi: 10.1016/s0165-2478(02)00142-6. [DOI] [PubMed] [Google Scholar]
- 29.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong X, Tumang JR, Gao W, Bai C, Rothstein TL. PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for V(H)11/V(H)12 and phosphatidylcholine binding. Eur J Immunol. 2007;37:2405–2410. doi: 10.1002/eji.200737461. [DOI] [PubMed] [Google Scholar]
- 31.Kroner A, Schwab N, Ip CW, Ortler S, Göbel K, et al. Accelerated course of experimental autoimmune encephalomyelitis in PD-1-deficient central nervous system myelin mutants. Am J Pathol. 2009;174:2290–2299. doi: 10.2353/ajpath.2009.081012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu B, Guleria I, Khosroshahi A, Chitnis T, Imitola J, et al. Differential role of programmed death-ligand 1 [corrected] and programmed death-ligand 2 [corrected] in regulating the susceptibility and chronic progression of experimental autoimmune encephalomyelitis. J Immunol. 2006;176:3480–3489. doi: 10.4049/jimmunol.176.6.3480. [DOI] [PubMed] [Google Scholar]
- 33.Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, et al. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2007;182:124–134. doi: 10.1016/j.jneuroim.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 34.Wang C, Dehghani B, Li Y, Kaler LJ, Vandenbark AA, et al. Oestrogen modulates experimental autoimmune encephalomyelitis and interleukin-17 production via programmed death 1. Immunology. 2009;126:329–335. doi: 10.1111/j.1365-2567.2008.03051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bodhankar S, Wang C, Vandenbark AA, Offner H. Estrogen-induced protection against experimental autoimmune encephalomyelitis is abrogated in the absence of B cells. Eur J Immunol. 2011;41:1165–1175. doi: 10.1002/eji.201040992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bodhankar S, Vandenbark AA, Offner H. Oestrogen treatment of experimental autoimmune encephalomyelitis requires 17β-oestradiol-receptor-positive B cells that up-regulate PD-1 on CD4+ Foxp3+ regulatory T cells. Immunology. 2012;137:282–293. doi: 10.1111/imm.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rabchevsky AG, Fugaccia I, Sullivan PG, Scheff SW. Cyclosporin A treatment following spinal cord injury to the rat: behavioral effects and stereological assessment of tissue sparing. J Neurotrauma. 2001;18:513–522. doi: 10.1089/089771501300227314. [DOI] [PubMed] [Google Scholar]
- 38.Chu CQ, Wittmer S, Dalton DK. Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192:123–128. doi: 10.1084/jem.192.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J Neurosci Res. 2006;84:370–378. doi: 10.1002/jnr.20881. [DOI] [PubMed] [Google Scholar]
- 41.Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1) Int Immunol. 2007;19:337–343. doi: 10.1093/intimm/dxl151. [DOI] [PubMed] [Google Scholar]
- 42.Wang C, Li Y, Proctor TM, Vandenbark AA, Offner H. Down-modulation of programmed death 1 alters regulatory T cells and promotes experimental autoimmune encephalomyelitis. J Neurosci Res. 2010;88:7–15. doi: 10.1002/jnr.22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ito A, Buenafe AC, Matejuk A, Zamora A, Silverman M, et al. Estrogen inhibits systemic T cell expression of TNF-alpha and recruitment of TNF-alpha(+) T cells and macrophages into the CNS of mice developing experimental encephalomyelitis. Clin Immunol. 2002;102:275–282. doi: 10.1006/clim.2001.5175. [DOI] [PubMed] [Google Scholar]
- 44.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 45.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krupnick AS, Gelman AE, Barchet W, Richardson S, Kreisel FH, et al. Murine vascular endothelium activates and induces the generation of allogeneic CD4+25+Foxp3+ regulatory T cells. J Immunol. 2005;175:6265–6270. doi: 10.4049/jimmunol.175.10.6265. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, et al. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105:9331–9336. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Subramanian S, Yates M, Vandenbark AA, Offner H. Oestrogen-mediated protection of experimental autoimmune encephalomyelitis in the absence of Foxp3+ regulatory T cells implicates compensatory pathways including regulatory B cells. Immunology. 2011;132:340–347. doi: 10.1111/j.1365-2567.2010.03380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haas KM, Poe JC, Tedder TF. CD21/35 promotes protective immunity to Streptococcus pneumoniae through a complement-independent but CD19-dependent pathway that regulates PD-1 expression. J Immunol. 2009;183:3661–3671. doi: 10.4049/jimmunol.0901218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev. 2008;224:201–214. doi: 10.1111/j.1600-065X.2008.00661.x. [DOI] [PubMed] [Google Scholar]
- 51.Wolf SD, Dittel BN, Hardardottir F, Janeway CA., Jr Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med. 1996;184:2271–2278. doi: 10.1084/jem.184.6.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yanaba K, Bouaziz JD, Matsushita T, Magro CM, St Clair EW, et al. B-lymphocyte contributions to human autoimmune disease. Immunol Rev. 2008;223:284–299. doi: 10.1111/j.1600-065X.2008.00646.x. [DOI] [PubMed] [Google Scholar]
- 53.Yanaba K, Bouaziz JD, Matsushita T, Tsubata T, Tedder TF. The development and function of regulatory B cells expressing IL-10 (B10 cells) requires antigen receptor diversity and TLR signals. J Immunol. 2009;182:7459–7472. doi: 10.4049/jimmunol.0900270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trabattoni D, Saresella M, Pacei M, Marventano I, Mendozzi L, et al. Costimulatory pathways in multiple sclerosis: distinctive expression of PD-1 and PD-L1 in patients with different patterns of disease. J Immunol. 2009;183:4984–4993. doi: 10.4049/jimmunol.0901038. [DOI] [PubMed] [Google Scholar]