

Abstract
A novel coronavirus, the Middle East respiratory syndrome coronavirus, recently emerged through zoonotic transmission, causing a severe lower respiratory tract infection in humans. In two recent papers, one published in Cell Research, the crystal structure of the viral receptor-binding domain in complex with the host CD26/dipeptidyl peptidase 4 receptor has now been characterized.
In mid 2012, a novel coronavirus (CoV) was isolated form the sputum of a patient with acute pneumonia and renal failure1. As of July 10th 2013, this virus, named Middle East respiratory syndrome (MERS)-CoV, has caused 80 laboratory-confirmed infections of which 44 were fatal2. The limited data available suggest that the virus is introduced into the human population through multiple independent, zoonotic transmission events from a — so far unknown — animal source with subsequent limited human-to-human spread. However, scenarios in which a single zoonotic transmission event has led to sustained, largely asymptomatic and non-detected human-to-human transmission cannot be excluded yet. Genetically, MERS-CoV is related to SARS-CoV, which killed nearly 10% of approximately 8 000 persons that were infected in the 2003 outbreak. It is therefore of utmost importance to better understand the biology and pathogenesis of this virus.
Coronaviruses infect mammals and birds, and occasionally cross the species barrier. The primary determinant of coronavirus host and cell tropism is the viral spike (S) entry protein that functions by binding to a cell surface receptor. The MERS-CoV S protein is a type I membrane glycoprotein, assembled as trimers that constitute the typical crown-like peplomers on the surface of the enveloped coronavirus. Functionally, two regions, S1 and S2, can be defined in the S protein, which are involved in binding and fusion with host cells, respectively. Recent studies have mapped the receptor-binding domain (RBD) to a ∼231-amino acid long region within the S1 region of MERS-CoV3.
MERS-CoV uses a cell surface amino peptidase, dipeptidyl peptidase 4 (DPP4), also known as CD26, as a functional receptor4. The multifunctional DPP4 — highly conserved among mammals — plays a major role in glucose metabolism by its degradation of incretins. It has been further implicated in T-cell activation, chemotaxis modulation, cell adhesion, and apoptosis5. In humans, it is primarily expressed on the epithelial cells in the lungs, kidney, small intestine, liver and prostate, and on activated leukocytes, while it also occurs in a soluble form in the circulation4,5.
The spike-receptor binding interface can be seen as a lock-and-key interaction where minor mutations within the interacting domain of the S protein or the receptor can abrogate infection, placing a barrier for cross-species transmission. Zoonotic potential of coronaviruses has been attributed to the adaptability of the S protein to human receptor orthologs. Intriguingly, the MERS-CoV S protein seems promiscuous in binding to orthologous receptors. Whereas coronaviruses generally tend to have a narrow host tropism, MERS-CoV can infect cells of a wide variety of species, at least in vitro4,6. The broad cell species tropism suggests that MERS-CoV has acquired facile cross-species transmissibility by binding to an evolutionarily conserved receptor.
Just four months after the discovery of the receptor, two Chinese research teams have now independently described the MERS-CoV spike-receptor interface. The study by Wang et al.7 recently published in Cell Research, and a recent study by Lu et al.8 published in Nature, both reveal the crystal structure of the RBD of the MERS-CoV S protein bound to its receptor, human DPP4. DPP4, of which the structure was published before9, consists of an N-terminal eight-bladed β-propeller domain and a C-terminal α/β-hydrolase domain. The RBD of the MERS-CoV S protein contains two subdomains: a conserved core subdomain and a receptor-binding subdomain, with the latter contacting blades 4 and 5 of the DPP4 β-propeller domain. Structural comparison with the RBD of the related betacoronavirus SARS-CoV (using the ACE2 peptidase as a receptor) reveals a conserved core domain and highly variable — both in length and in residues — receptor-binding region, explaining the differential receptor usage.
Both teams have scrutinized the RBD-receptor interface and identified critical residues within the interacting domain of the S protein or receptor, which allow MERS-CoV to bind to its receptor. Structural analysis and mutational analysis have identified several key residues in the RBD of the S protein shown to be critical for DPP4 binding and viral entry. This information is crucial to understand the adaptation of MERS-CoV to humans. Studies with SARS-CoV isolated from humans and civet cats (which function as the intermediate host) revealed 2 amino acids in the RBD that caused an > 1 000-fold difference in binding affinity to human receptor ACE210. Analysis of the MERS-CoV RBD sequences of the isolates characterized thus far shows no sequence variation except that 2 virus samples isolated from patients in the UK (GenBank: KC667074 and KC164505) had a leucine-to-phenylalanine substitution at position 506 of the S protein (L506F). As shown by Wang et al.7, residue L506 contacts DPP4 and its substitution to alanine reduced MERS-CoV S-mediated infectivity by over 50%.
With the structure available, the promiscuous binding of MERS-CoV to DPP4 orthologs can now be analyzed at the molecular level. Relevant to functional usage of orthologous receptors by MERS-CoV is the degree of conservation of the amino acid residues in DPP4 that were identified to contact the viral RBD7,8. DPP4 sequence comparison reveals that mammalian DPP4 orthologs (e.g., of macaque, horse, rabbit and pig) have no or little variation for residues contacting MERS-CoV RBD in human DPP4 (Table 1). The level of identity of the RBD-contacting residues in human DPP4 and DPP4 orthologs is generally lower than the homology between full-size DPP4 proteins. Notably, the DPP4 proteins of ferret and pig have an overall sequence-identity of 88% relative to human DPP4, but the identity of the RBD-contacting residues differs substantially, being 91% for pig DPP4 and only 45% for ferret DPP4 (Table 1). Consistently, MERS-CoV can infect porcine cells, but cannot use the ferret DPP4 as a functional receptor [6, unpublished results from BLH]. The conservation degree of RBD-contacting residues in DPP4 orthologs has predictive value for functional receptor usage and may hence help to track the animal source for MERS-CoV. Mutational analysis of the RBD-contacting residues in DPP4 in combination with biochemical and functional assays is needed to better understand the critical molecular determinants for the unusual broad cell species tropism of MERS-CoV.
Table 1. Conservation of DPP4 residues that contact the RBD of MERS-CoV.
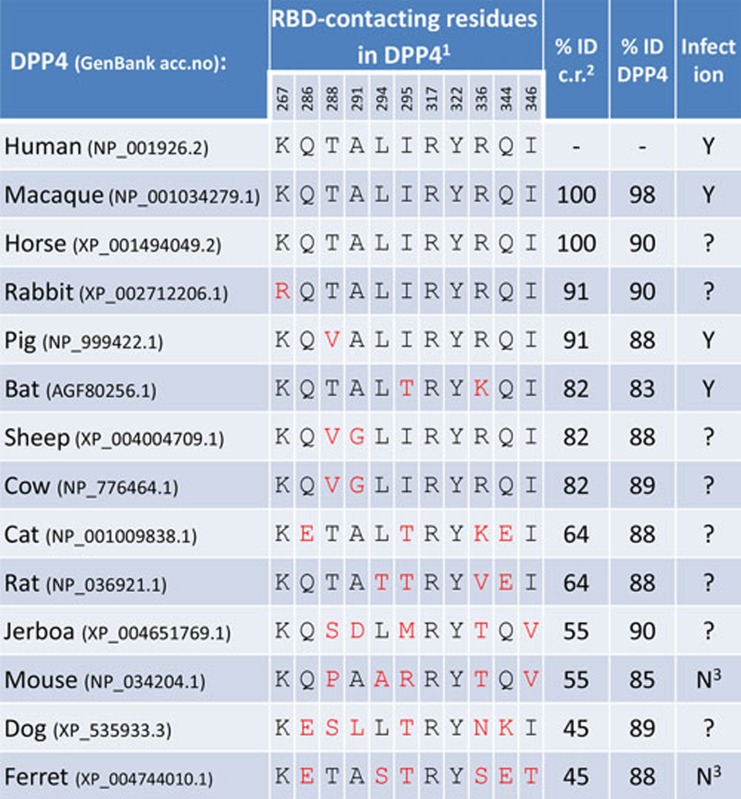 |
1 Critical residues in DPP4, which contact the MERS-CoV RBD, identified by Wang et al.7 and Lu et al.8. Position (human DPP4 numbering) and single-letter identity of RBD-contacting residues are indicated;
2% identity of RBD-contacting residues in relative to those in human DPP4;
3 unpublished results from BLH.
In conclusion, knowing the molecular details of the coronavirus-receptor interface will be highly instrumental in predicting interactions between MERS-CoV and orthologous receptors and mutation-driven host range expansion, and may help to identify susceptible host species and hence the host reservoir(s) for MERS-CoV. In addition, the structural information of the interface between the S protein and the receptor may provide novel strategies for developing effective antibodies or drugs that target the spike-receptor interface.
References
- Zaki AM, van Boheemen S, Bestebroer TM, et al. N Engl J Med. 2013. pp. 1814–1820. [DOI] [PubMed]
- Source WHO ( ( http://www.who.int/csr/don/2013_07_07/en/index.html ).
- Mou H, Raj VS, van Kuppeveld FJ, et al. J Virol. 2013. pp. 9379–9383. [DOI] [PMC free article] [PubMed]
- Raj VS, Mou H, Smits SL, et al. Nature. 2013. pp. 251–254. [DOI] [PMC free article] [PubMed]
- Boonacker E, Van Noorden CJ. Eur J Cell Biol. 2003. pp. 53–73. [DOI] [PubMed]
- Müller M, Raj SV, Muth D, et al. MBio 20123e00515–e00512.23232719
- Wang N, Shi X, Jiang L, et al. Cell Res. 2013. pp. 986–993. [DOI] [PMC free article] [PubMed]
- Lu G, Hu Y, Wang Q, et al. Nature. 2013. pp. 227–231. [DOI] [PMC free article] [PubMed]
- Rasmussen HB, Branner S, Wiberg FC, et al. Nat Struct Biol. 2003. pp. 19–25. [DOI] [PubMed]
- Li W, Zhang C, Sui J, et al. EMBO J. 2005. pp. 1634–1643. [DOI] [PMC free article] [PubMed]