

Abstract
Many bacterial and archaeal lineages have a history of extensive and ongoing horizontal gene transfer and loss, as evidenced by the large differences in genome content even among otherwise closely related isolates. How ecologically cohesive populations might evolve and be maintained under such conditions of rapid gene turnover has remained controversial. Here we synthesize recent literature demonstrating the importance of habitat and niche in structuring horizontal gene transfer. This leads to a model of ecological speciation via gradual genetic isolation triggered by differential habitat association of nascent populations. Further, we hypothesize that subpopulations can evolve through local gene exchange networks by tapping into a gene pool that is adaptive towards local, continuously changing organismic interactions and is, to a large degree, responsible for the observed rapid gene turnover. Overall, these insights help explain how bacteria and archaea form populations that display both ecological cohesion and high genomic diversity.
Keywords: ecological speciation, frequency dependent selection, environmental niche, genotypic clusters
“Though this be madness, yet there is method in’t.” {Shakespeare: Hamlet, Act 2, scene 2]
The madness we refer to is rapid gene turnover due to horizontal gene transfer (HGT) and gene loss, which can lead to extensive genome differences among closely related bacteria and archaea. Even isolates that are nearly identical across most of the genome can have hundreds of unique genes [1], and recent comparison of gene transfer networks among hundreds of sequenced genomes has suggested that on average 20% of genes have recently been acquired [2]. Because of this vast diversity, it is difficult to estimate the total gene pool even for groups of closely related microbes [3, 4](BOX 1). Observations like these have led to critiques of the species concept for microbes and call into question to what extent closely related microbes could possibly maintain ecologically cohesive populations considering the fast pace of gene additions and losses [5, 6].
Box 1. The ‘gene pool’ of a species: core, flexible and pan genomes – an ecological critique.
It is common for microbiologists to speak of the ‘core’ genome of a species. This is a response to the observation that in many cases when multiple genomes of the same named species are compared with one another, we observe extensive variation in gene content. A classic paper demonstrated this in three isolates of Escherichia coli that were found to have less than 40% of their genes in common [50]. Those loci not found in all genomes in a sample are often termed the ‘flexible’ genome (alongside many other names – see glossary) and the total set of genes found in a sample, including the core and flexible genomes, is termed the ‘pan’ genome. While the designation of core and flexible genomes is a natural response to varying gene content, it may also be misleading and should not be over-interpreted as a straightforward correlate for ecological differentiation.
The core genome is usually interpreted as encoding those basic functions necessary for the common niche of a sample of isolates onto which more specialized properties are grafted through the flexible genome. The pan genome is then taken to represent the genes that are necessary to colonize the totality of habitats within which the isolates making up the sample are found. While this overall picture is generally correct, it must be remembered that the contents of the core, flexible or pan genome are artifacts of the sampling criteria. Most importantly, the gene content depends on how one defines the phylogenetic bounds of the sample of isolates. For example, most taxonomically defined species have not been tested for ecological cohesion in environmental samples. Moreover we cannot assume a simple relationship between gene content and ecology, because these concepts take no account of frequency: a gene found in a single isolate is weighted the same as one found in all but a few isolates in the sample.
The definition of the core vs. flexible genome also relies on the ability to determine whether two sequences represent different genes, or different alleles of the same gene. This is not necessarily straightforward. A further complication is the increasing importance of draft genomes (e.g., [10, 51]). Where draft genomes show a gene that is apparently missing from one genome in a sample, this may simply reflect poor assembly in that region rather than a genuine absence. Finally, we must also recall the presence of numerous prophage and other mobile elements in bacterial genomes. The genes within elements that encode only their own mobility make little if any contribution to niche adaptation, and yet these will be included in any analysis of gene content.
In short, microbiologists should practice extreme caution in defining and interpreting the contents of the core, flexible, and pan genomes of a sample. Although the terms are a useful conceptual shorthand, they fall far short of being an adequate description of the relationship between gene content and ecology.
Recent data, however, suggest the method in the madness of gene transfer arises from strong constraints on both sequence similarity and environmental niche. This insight provides a mechanistic explanation for why, in spite of the potential for free flowing genes, microbes remain geno- and phenotypically clustered. Here we first review the evidence for and significance of organization into genotypic clusters in the core genome (Glossary) and then outline how recent observations of ecological differentiation among closely related genomes support a new evolutionary model of cluster formation. Finally, we put the observed high gene transfer rates in the flexible genome into an ecological context and show its importance in the evolution of local population structure.
Genotypic clustering
Plants and animals are organized into phenotypic and genotypic clusters and this forms the vernacular notion of a species. For bacteria and archaea, the identification of natural clusters was difficult before sequencing became widely available because phenotypic traits used for traditional taxonomy were arbitrarily defined. Although traditional classification allowed reliable identification [7], it is clear that many taxonomic species do not necessarily represent natural units. Hence it was an important step forward when widespread application of multilocus sequencing and (meta) genomic approaches demonstrated that organization into genotypic clusters is also pervasive among microbes [1, 8-13].
These clusters are evident from comparison of multiple genes representing the core genome and can take different shapes and forms, possibly reflecting varying recombination rates within and between clusters as well as evolutionary age and/or demography (reviewed in [14]). Clusters can be sharply delineated or fuzzy, and while some appear relatively monomorphic, others contain considerable sequence diversity. Similarly, sequence diversity between clusters can differ considerably and range from nearly indistinguishable to deeply divergent. Although the significance of such differences is not fully understood, new data suggest that there may be a unifying process of cluster formation across both bacteria and archaea [1, 11].
Formation of genotypic clusters
Recent population genomic data allow a synthesis of past theories and observations that were seemingly at odds in explaining cluster formation. In presenting this new evidence, we first pose the important question of whether genotypic clusters can originate among sympatric microbes in the absence of selection. The focus on sympatric differentiation is because geographic isolation, although thought to be an important factor in animals and plants, appears to be the exception in microbes having primarily been invoked for microbes from isolated hotsprings [15].
A neutral model of mutation and drift has previously been used to identify conditions under which populations might separate into permanently differentiated clusters, [16]. Taking into account different propensity for recombination and the exponential drop in recombination rate vs. sequence divergence typically observed for bacteria, the main conclusion of the model is that strong and permanent clustering is unlikely in the absence of some selective pressure. Although highly clonal populations (those with extremely low recombination rates) may differentiate into transient and unstable clusters, microbes with commonly observed recombination to mutation rates only form clusters if an unrealistically steep drop in recombination vs. sequence diversity is assumed [8]. Hence theory suggests that selection is required for microbes to differentiate into genotypic clusters.
The second important question is whether genotypic clusters can form as a consequence of ecological specialization, i.e., can the spread of adaptive genes trigger the process of clustering? The answer to this fundamental question must lie in the balance between recombination and selection and can be illustrated by a simple comparison of rates. Typically observed recombination rates are not high enough to unlink a gene under selection from the rest of the genome [17, 18], unless we assume extremely small fitness differences, which are hard to reconcile with the role of selection in generating clusters. Importantly, this suggests a mechanism for cluster formation as expressed by the ‘ecotype’ theory [19]. As the gene under selection increases in frequency within the population, so will the genome it is contained in until it has outcompeted all other genomes within its niche. Subsequently, the winning genome may diversify by neutral processes, eventually forming a genotypic cluster in which all loci should show a similar pattern, except for discordant alleles introduced by homologous recombination from other populations [20].
The ecotype theory provides an appealing explanation for the widespread observation of genotypic clusters, but it is inconsistent with several observations. First, populations of microbes have been observed in which individuals from distinct geographic locations carry environment-specific genes [21] or alleles [22], sometimes with obvious adaptive significance. For example, the globally distributed ocean cyanobacteria Prochlorococcus have differentiated into local populations in the Atlantic and Pacific where horizontally acquired genes that match differing nutrient regimes have reached high frequency, although the bulk of the genome remains indistinguishable across populations [21]. Second, in a few cases, reduced diversity at single loci in the genome has been observed, providing further evidence of gene-specific sweeps independent of their genomic background [23-25]. Taken together, these observations suggest that the process of genotypic cluster formation in natural populations subject to homologous and non- homologous recombination is more complex.
Two studies targeting recently diverged sympatric populations of bacterial and archaeal genomes may help reconcile observations of genotypic clustering and gene/allele-specific sweeps within populations. One study of hotspring archaea provided evidence for sympatric differentiation in the absence of genome-wide selective sweeps [11]. Genomic analysis indicated that over time the contribution of homologous recombination to diversification decreased, consistent with ongoing divergence among populations; however, it was less clear what specific adaptive genes or genome regions might be driving this divergence.
Our second example provides a model of how ecological differentiation may drive genotypic differentiation [1]. Two genetically nearly indistinguishable but ecologically divergent populations of Vibrio cyclotrophicus were targeted for whole genome sequencing to examine how they had diversified. These populations co-occur in the coastal ocean but display differential propensity for being free-living or attached to zoo-and phytoplankton [26, 27]. Analysis of whole genomes provided evidence that specific genome regions but not whole genomes have swept each of the two populations [1]. Moreover, several of these regions are consistent with differentiation into attached vs. free-living lifestyles providing evidence for differential acquisition of adaptive genes/alleles. Finally, analysis of homologous recombination within and across populations demonstrated that both populations had been actively recombining in the past but that the most recent events had already become population specific, suggesting an independent evolutionary trajectory of these nascent populations [1].
These observations suggest a new model of genotypic cluster formation in which an ancestral, ecologically uniform and recombining population differentiates into novel, ecologically distinct populations, which gradually develop into genotypic clusters [1](Figure 1). The first step in this process is evolution of an adaptive allele or gene, either via mutation or recombination (both homologous and non-homologous) in at least one member of the ancestral population. Such adaptive genes may subsequently spread by recombination to other genomes within the ancestral populations. Importantly, if this process triggers differential environmental association, new subpopulations are formed with decreased gene flow between them due to spatial separation (Figure 1). Finally, if the new population structure remains stable over time, accumulation of population-specific mutations will start to genetically differentiate genomes. In bacteria, this may strongly and quickly enhance cluster formation by inhibiting homologous recombination whose rate decreases exponentially with sequence divergence [28]. This is thought to be primarily due to (i) a requirement for a 20 bp stretch of identical nucleotides that lines up at one or both ends of the recombining stretches of DNA for initiation of the process [28], and (ii) the ability of the methyl-directed mismatch repair (MMR) system to strongly inhibit recombination among even slightly dissimilar sequences [29]. In contrast to bacteria, in the archaeon Sulfolobus only a few identical nucleotides are sufficient to initiate homologous recombination (incidentally, Sulfolobus also lacks a MMR system) [30] leading to a less rapid decrease in rates with sequence divergence [31]. Because genotypic clustering is nonetheless observed in archaea this suggests that the influence of environmental separation in inhibiting gene flow must be a very strong factor, which is also supported by the very recently diverged Vibrio populations discussed above [1].
Figure 1.
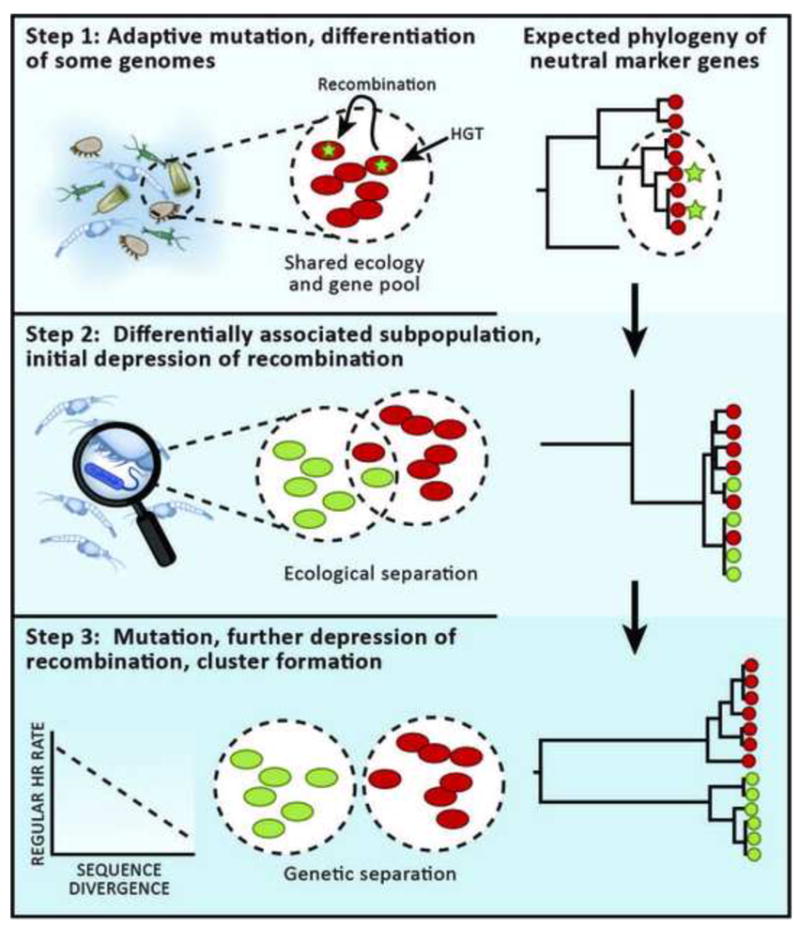
Model of the gradual process of genotypic cluster formation via sympatric ecological specialization using the example of an ancestral population associated with zooplankton giving rise to a free-living population in ocean water. In Step 1, at least one genome receives at least one mutation (denoted by green star) via recombination, point mutation or gene loss that adapts it for an alternate habitat. This mutation can spread to other members of the population by recombination. In a phylogenetic tree of a neutral marker gene, the population remains homogeneous. In Step 2, ecological separation is beginning to depress gene flow between the two populations (novel population denoted by green color, which is starting to occupy a niche separate from the zooplankton). The phylogeny still appears mixed because of a history of recombination; only the most recent recombinations show high population-specificity but may not yet be evident in neutral marker genes. Also note that different genes will give different phylogenetic trees due to different degrees of recombination and hitchhiking with the adaptive gene(s). In Step 3, mutations accumulate within each population. Because in bacteria there is an exponential decrease of homologous recombination rate with sequence divergence, this process quickly depresses gene flow between nascent populations. At this point, the phylogenetic tree shows genotypic clusters, which are consistent with ecological differentiation.
A habitat specific gene pool?
The above model shows how genotypic clusters in the core genome might arise but has yet to fully consider the intricacies of the flexible genome, which has very high turnover, can contribute a large portion of the total genes and makes up the bulk of the vast pan-genome. In fact, one of the puzzles of microbial biology is that genomes can be highly optimized energetically and functionally while tolerating the disruptive effect of horizontally acquired genes that are both phylogenetically and functionally diverse. However, new data that explicitly incorporate ecology in the study of gene flow led us to hypothesize that horizontal acquisition of genes is highly structured by function in an explicit local ecological context [2, 32, 33]. We propose that there exist local (i.e., habitat-specific) gene pools with high adaptive potential for continuously changing organismal interactions such as viral predation and interference competition. Rather than barriers to gene flow, which undoubtedly create structure in gene flow [34], we point to the additional importance of environmental selection in enriching and rapidly transferring specific types of genes in the flexible genome, thus creating a niche-specific horizontal gene pool. In the following, we summarize three observations that support this hypothesis.
The first observation, already made over a decade ago, is that flexible genes are not randomly dispersed across the genome but are highly clustered into islands and islets [35, 36]. This has the obvious advantage that incorporation of novel genes is relegated to specific genome regions without disruption of vital functions. The underlying mechanisms of island/islet formation are varied and include preferential insertion sites for temperate phages and integrative conjugative elements as well as gene capture machines, such as integrons. The latter represent large and highly variable islands in many bacteria and facilitate the acquisition and expression of gene cassettes in a conveyor belt like fashion [37]. That is, genes are continuously added downstream of an integrase and promoter and are subsequently lost randomly along the length of the integron. Hence while the gene content of the integron changes continuously, the integron position in the genome remains stable. A further, perhaps underappreciated factor, which can contribute to maintenance of highly variable islands within core regions, is that even divergent genes can be shared by homologous recombination of conserved flanking regions (Figure 2). The best example of this is the O-antigen, which is highly variable even among the closest of relatives (e.g., [38-40]) and is, in spite of its considerable variation in sequence composition and gene content, most often replaced via homologous recombination between specific conserved flanking genes [41].
Figure 2.
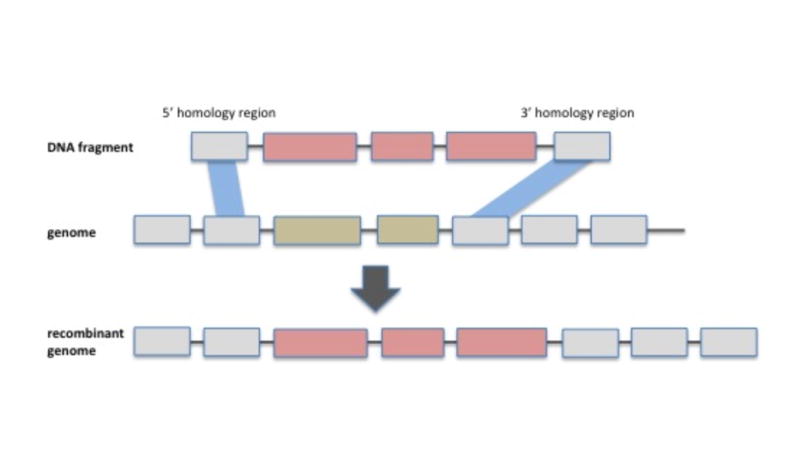
Schematic representation of homologous recombination spreading flexible genes among genomes. Comparative genomics increasingly reveals the importance of homologous recombination rather than non-homologous mechanisms in spreading genes among close relatives. This suggests a population-specific gene pool, which can rapidly add or delete genes that are under frequency dependent selection such as the O-antigen or various surface receptors.
The second observation made more recently is that the flexible genome is highly biased in the functions it encodes [36]. Although a very high proportion of genes in the flexible genome annotate poorly, those that do are enriched in certain functional categories. These include many surface structures, such as the above-mentioned O-antigen or membrane-spanning transporters, as well as secondary metabolite production/modification [32, 36]. These biased functions suggest a gene pool adapted for organismal interactions. Rapidly changing surface antigens can ensure resistance to rapidly changing biological factors, like immune defenses or viral and protozoan predation [36, 42], all of which are initiated by molecular recognition among specific surface structures. Similarly, secondary metabolite production and modification genes suggest a role in organismal interactions either via communication within and between populations, interference competition or predation resistance.
Finally, several studies that systematically analyzed the relative frequency of very recent HGT among genomes revealed strong structuring by habitat and ecology [2, 32, 33]. A comprehensive analysis of recent gene transfers among 657 bacterial and archaeal genomes revealed that the large majority of highly connected donors and recipients reside in the same general habitat [2]. Similarly, comparison of 2,235 sequenced genomes suggested that recent transfers, recognized as genes with near identity between donor and recipient, is more common among microbes that share similar ecology even after controlling for phylogenetic divergence [33]. This trend was evident across all environments but particularly so in the human microbiome where recent sampling of body sites has established a high-resolution map of associated microbes. The preferential transfer of genes with apparent relevance for the specific habitat suggested that transferred genes are under local, niche-specific selection [33]. In contrast, genes for antibiotic resistance spread more freely among habitats in accordance with the widespread application of antibiotics in humans and animals [33].
A third study compared Vibrio cholerae from two locations across the globe and concluded that, while the core genome appeared globally well mixed, the integron represented a locally differentiated, rapidly transferred gene pool [32]. Importantly, integron genes were more similar between locally co-occurring V. cholerae and V. metecus than between the globally dispersed V. cholerae populations indicating that habitat supersedes phylogeny in structuring of this gene pool. On the other hand, the core genomes of V. cholerae and V. metecus were distinct and showed little evidence of gene flow crossing the species boundaries [32]. The analysis further showed that the integron represents a gene pool enriched in the types of interaction genes outlined above and discrete from the remainder of the genome, that is, although the genes can originate from the core genome of other, unrelated microbes, there seems to be little exchange between the core and integron genes [32].
Taken together, we propose the existence of ecologically determined gene transfer networks that enable rapid sharing of niche-adaptive genes in the flexible genome. Far from being a random sampling of genes, the flexible genome plays roles in adaptation to rapidly changing environmental conditions of which frequency dependent interactions with predators and competitors are prime candidates [36]. The enrichment of surface structure determinants and secondary metabolites in the flexible gene pool is consistent with this view. Novel surface receptors may provide immunity against phage or protozoa and hence an immediate strong fitness advantage, at least until recombination equips these predators with the matching docking proteins. Similarly, antibiotic production genes may provide populations with weapons against competitors until resistance spreads and the antibiotic loses effectiveness [43, 44]. At the population level, diversification by constant exchange of genes with adaptive value represents a type of bet hedging similar to the random phenotype switching observed in experimental evolution studies [45].
Concluding remarks
Although we are only beginning to understand the intricacies of gene flow in the wild, it is becoming clear that gene transfer networks need to be analyzed in the context of ecology. The recent results reviewed here add an evolutionary dimension to this ecological structure by showing how clusters may arise as a consequence of ecological specialization. The gradual mechanism by which this happens (Figure 1) is unexpected since theoretical considerations suggested genome-wide selective sweeps as a mechanism of cluster formation based on weighing the effect of realistic recombination and selection rates [17, 18]. This discrepancy of observation and theory shows that additional forces depressing the effect of positive selection for ecological adaptation are at work. Prime candidates for such forces are frequency dependent interactions such as predation and competition, which appear to have a disproportionate impact on the flexible genome [36]. It is thus an important observation that the flexible genome, and the transfer among lineages of the genes that comprise it, appears highly structured by habitat and ecology. Together with the observation that frequently transferred genes are compositionally highly biased, this gives rise to the hypothesis that there exists a horizontal gene pool with high adaptive value for local interactions. These genes may thus be considered ‘habitat’ associated [6], and an intriguing possibility is that genes have different habitat ranges dependent on environmental selection. More limited distribution and low frequency of occurrence might be expected for genes mediating frequency dependent organismal interactions; however, genes under positive selection, e.g., for adaptation to regional nutrient regimes [21], may spread much more widely. In short, in bacteria it may be useful to think of genes as having niches in a fashion akin to whole organisms. The dimensions of that niche can be considered to include other genes in the same genome.
Finally, relative rates of homologous and non-homologous recombination matter. As detailed in Figure 1, acquisition of ecologically differentiating genes can lead to independent evolutionary trajectories of populations if ecological separation depresses gene flow in both the core and flexible genome. This fits the examples of V. cyclotrophicus and hotspring archaea, which both appear to be in the process of speciating [1, 11]. On the other hand, homologous recombination rates may remain high enough for the core genome to remain mixed, while transfer of flexible genes by non-homologous mechanisms may be much faster such that local population structure can ensue. This might be the case in the globally distributed V. cholerae in which local differentiation is evident in the integron gene pool [32]. Overall, this demonstrates the complex interplay between recombination and ecology, and suggests a model for microbial speciation and evolution of local population structure in the highly structured microbial adaptive landscape.
Acknowledgments
Funding was provided by grants from the Moore Foundation (MFP), NSF DEB 0821391 (EJA and MFP), NIH/NIGMS GM088558-01 (WPH) and the MIDAS Center for Communicable Disease Dynamics at the Harvard School of Public Health (WPH). We also thank James McInerney for helpful discussions.
Glossary
- Core genome:
the genes shared by all members of a pre-defined group of bacteria or archaea (see also BOX 1).
- Flexible genome:
genome regions or genes present in only a subset of members of a predefined group of bacteria or archaea (see also BOX 1). May also be referred to as the ‘accessory’ (e.g., [46]), ‘dispensable’ (e.g., [47]), ‘auxiliary’ (e.g., [48]) or ‘distributed’ (e.g., [49]) genome.
- Frequency dependent interactions:
interactions whose outcomes depend on the frequency of the interacting genotypes. In negative frequency dependency, a particular genotype becomes more fit at low frequency; conversely, positive frequency dependency leads to higher fitness of more common genotypes.
- Fitness advantage:
expresses higher probability of survival by increased growth or decreased death rates.
- Homologous recombination:
mediates the change of a stretch of nucleotides in the genome by incorporation of a highly similar or identical stretch of nucleotides. In bacteria and archaea, it is unidirectional since it is decoupled from reproduction, and in bacteria but not archaea, an identical stretch of 20 bp at either one or both ends of the recombining DNA is required to initiate the process.
- Integron:
a gene capture system consisting of an integrase, which captures and integrates gene cassettes at a recombination site in a conveyor belt fashion. Genes are eventually lost as they are pushed along the integron by new additions.
- Interference competition:
occurs between two individuals by aggression rather than resource monopolization. In microbes, it is most often mediated by secondary metabolites, such as antibiotics.
- Metagenomics:
the direct study of genes and genomes present in environmental samples without culturing organisms.
- Multilocus sequencing:
comparison of multiple protein coding genes across genomes for reliable classification of isolates. The approach originated in epidemiology but is increasingly applied to microbial taxonomy.
- Non-homologous recombination:
denotes all recombination mediated by mechanisms other than homologous recombination and includes illegitimate recombination and integrase-mediated recombination. It generally requires no sequence similarity (illegitimate recombination) or very short stretches or recognition sequence (integrase-mediated recombination).
- Pan-genome:
the sum of core and flexible genes in a pre-defined group of bacteria or archaea (see also BOX 1).
- Sympatric differentiation:
the process of genotypic differentiation under potential for gene flow due to inhabiting the same geographic region.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shapiro BJ, et al. Population genomics of early events in the ecological differentiation of bacteria. Science. 2012;336:48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Popa O, et al. Directed networks reveal genomic barriers and DNA repair bypasses to lateral gene transfer among prokaryotes. Genome Research. 2011;21:599–609. doi: 10.1101/gr.115592.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tettelin H, et al. Comparative genomics: the bacterial pan-genome. Current Opinion in Microbiology. 2008;12:472–477. doi: 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Mira A, et al. The bacterial pan-genome:a new paradigm in microbiology. Int Microbiol. 2010;13:45–57. doi: 10.2436/20.1501.01.110. [DOI] [PubMed] [Google Scholar]
- 5.Doolittle WF, Zhaxybayeva O. On the origin of prokaryotic species. Genome Research. 2009;19:744–756. doi: 10.1101/gr.086645.108. [DOI] [PubMed] [Google Scholar]
- 6.Doolittle WF, Papke RT. Genomics and the bacterial species problem. Genome Biology. 2006;7:116. doi: 10.1186/gb-2006-7-9-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosselló-Mora R, Amann R. The species concept for prokaryotes. FEMS Microbiol Rev. 2001;25:39–67. doi: 10.1111/j.1574-6976.2001.tb00571.x. [DOI] [PubMed] [Google Scholar]
- 8.Hanage WP, et al. Sequences, sequence clusters and bacterial species. Phil Trans R Soc Lond B. 2006;361:1917–1927. doi: 10.1098/rstb.2006.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polz MF, et al. Patterns and mechanisms of genetic and phenotypic differentiation in marine microbes. Phil Trans R Soc Lond B. 2006;361:2009–2021. doi: 10.1098/rstb.2006.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Croucher NJ, et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331:430–434. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cadillo-Quiroz H, et al. Patterns of gene flow define species of thermophilic archaea. PLoS Biol. 2012;10:e1001265. doi: 10.1371/journal.pbio.1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denef VJ, et al. AMD biofilms: using model communities to study microbial evolution and ecological complexity in nature. The ISME Journal. 2010;4:599–610. doi: 10.1038/ismej.2009.158. [DOI] [PubMed] [Google Scholar]
- 13.Caro-Quintero A, Konstantinidis KT. Bacterial species may exist, metagenomics reveal. Environ Microbiol. 2012;14:347–355. doi: 10.1111/j.1462-2920.2011.02668.x. [DOI] [PubMed] [Google Scholar]
- 14.Polz MF, Hanage WP. Overview: Qunatitative and theoretical microbial population biology. In: Rosenberg E, et al., editors. The Prokaryotes. 4. Springer; 2012. [Google Scholar]
- 15.Whitaker RJ. Allopatric origins of microbial species. Philos Trans R Soc Lond B Biol Sci. 2006;361:1975–1984. doi: 10.1098/rstb.2006.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraser C, et al. Recombination and the nature of bacterial speciation. Science. 2007;315:476–480. doi: 10.1126/science.1127573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shapiro BJ, et al. Looking for Darwin’s footprints in the microbial world. Trends Microbiol. 2009;17:196–204. doi: 10.1016/j.tim.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Cohan FM. The effects of rare but promiscuous genetic exchange on evolutionary divergence in prokaryotes. Am Nat. 1994;143:965–986. doi: 10.1016/0169-5347(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 19.Cohan FM. What are bacterial species. Annu Rev Microbiol. 2002;56:457–487. doi: 10.1146/annurev.micro.56.012302.160634. [DOI] [PubMed] [Google Scholar]
- 20.Hanage WP, et al. Fuzzy species among recombinogenic bacteria. BMC Biology. 2005;3 doi: 10.1186/1741-7007-1183-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coleman ML, Chisholm SW. Ecosystem-specific selection pressures revealed through comparative population genomics. Proc Natl Acad Sci U S A. 2010;107:18634–18639. doi: 10.1073/pnas.1009480107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papke RT, et al. Searching for species in haloarchaea. Proc Natl Acad Sci U S A. 2007;104:14092–14097. doi: 10.1073/pnas.0706358104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guttman DS, Dykhuizen DE. Detecting selective sweeps in naturally occurring. Escherichia coli Genetics. 1994;138:993–1003. doi: 10.1093/genetics/138.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denef VJ, et al. Proteogenomic basis for ecological divergence of closely related bacteria in natural acidophilic microbial communities. Proc Natl Acad Sci U S A. 2010;107:2383–2390. doi: 10.1073/pnas.0907041107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eppley JM, et al. Genetic exchanges across a species boundary in the archaeal genus. Ferroplasma Genetics. 2007;177:407–416. doi: 10.1534/genetics.107.072892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunt DE, et al. Resource partitioning and sympatric differentiation among losely related bacterioplankton. Science. 2008;320:1081–1085. doi: 10.1126/science.1157890. [DOI] [PubMed] [Google Scholar]
- 27.Szabo G, et al. Reproducibility of Vibrionaceae population structure in coastal bacterioplankton. ISME Journal. 2012 doi: 10.1038/ismej.2012.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majewski J. Sexual isolation in bacteria. FEMS Microbiol Let. 2001;199:161–169. doi: 10.1111/j.1574-6968.2001.tb10668.x. [DOI] [PubMed] [Google Scholar]
- 29.Vulic M, et al. Mutation, recombination, and incipient speciation of bacteria in the laboratory. Proc Natl Acad Sci U S A. 1999;96:7348–7351. doi: 10.1073/pnas.96.13.7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grogan DW, Stengel KR. Recombination of synthetic oligonucleotides with prokaryotic chromosomes: substrate requirements of the Escherichia coli/lambdaRed and Sulfolobus acidocaldarius recombination systems. Molecular Microbiology. 2008;69:1255–1265. doi: 10.1111/j.1365-2958.2008.06356.x. [DOI] [PubMed] [Google Scholar]
- 31.Naor A, et al. Low species barriers in halophilic archaea and the formation of recombinant hybrids. Curr Biol . 2012;22:1444–1448. doi: 10.1016/j.cub.2012.05.056. [DOI] [PubMed] [Google Scholar]
- 32.Boucher Y, et al. Local mobile gene pools rapidly cross species boundaries to create endemicity within global Vibrio cholerae populations. mBio. 2011;2:e00335–00310. doi: 10.1128/mBio.00335-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smillie CS, et al. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- 34.Popa O, Dagan T. Trends and barriers to lateral gene transfer in prokaryotes. Current Opinion in Microbiology. 2011;14:615–623. doi: 10.1016/j.mib.2011.07.027. [DOI] [PubMed] [Google Scholar]
- 35.Hacker J, Carniel E. Ecological fitness, genomic islands and bacterial pathogenicity - a Darwinian view of the evolution of microbes. EMBO Reports. 2001;2:376–381. doi: 10.1093/embo-reports/kve097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Valera F, et al. Explaining microbial population genomics through phage predation. Nat Rev Microbiol. 2009;7:828–836. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- 37.Cambray G, et al. Integrons. Annu Rev Genet. 2010;44:141–166. doi: 10.1146/annurev-genet-102209-163504. [DOI] [PubMed] [Google Scholar]
- 38.Samuel G, Reeves P. Biosynthesis of O-antigens: genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr Res. 2003;338:2503–2519. doi: 10.1016/j.carres.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 39.Chatterjee SN, Chaudhuri K. Lipopolysaccharides of Vibrio cholerae II. Genetics of biosynthesis. Biochim Biophys Acta. 2004;1690:93–109. doi: 10.1016/j.bbadis.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 40.Wildschutte H, et al. O-antigen diversity and lateral transfer of the wbe region among Vibrio splendidus. Environ Microbiol. 2010;12:2977–2987. doi: 10.1111/j.1462-2920.2010.02274.x. [DOI] [PubMed] [Google Scholar]
- 41.Touchon M, et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. Plos Genetics. 2009;5:e1000344. doi: 10.1371/journal.pgen.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wildschutte H, et al. Protozoan predation, diversifying selection, and the evolution of antigenic diversity in Salmonella. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10644–10649. doi: 10.1073/pnas.0404028101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vetsigian K, et al. Structure and evolution of Streptomyces interaction networks in soil and in silico. Plos Biology. 2011;9:e1001184. doi: 10.1371/journal.pbio.1001184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cordero OX, et al. Ecological populations of bacteria act as socially cohesive units of antibiotic production and resistance. Science. 2012;337:1228–1231. doi: 10.1126/science.1219385. [DOI] [PubMed] [Google Scholar]
- 45.Beaumont HJ, et al. Experimental evolution of bet hedging. Nature. 2009;462:90–93. doi: 10.1038/nature08504. [DOI] [PubMed] [Google Scholar]
- 46.Duangsonk K, et al. Use of a variable amplicon typing scheme reveals considerable variation in the accessory genomes of isolates of Burkholderia pseudomallei. J Clin Microbiol. 2006;44:1323–1334. doi: 10.1128/JCM.44.4.1323-1334.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tettelin H, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial ‘pan-genome’ (vol102, pg 13950, 2005) Proceedings of the National Academy of Sciences of the United States of America. 2005;102:16530–16530. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lan R, Reeves PR. Gene transfer is a major factor in bacterial evolution. Mol Biol Evol. 1996;13:47–55. doi: 10.1093/oxfordjournals.molbev.a025569. [DOI] [PubMed] [Google Scholar]
- 49.Erdos G, et al. Construction and characterization of a highly redundant Pseudomonas aeruginosa genomic library prepared from 12 clinical isolates: application to studies of gene distribution among populations. Int J Pediatr Otorhinolaryngol. 2006;70:1891–1900. doi: 10.1016/j.ijporl.2006.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Welch RA, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harris SR, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327:469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]