

Abstract
Poorly soluble drugs often encounter low bioavailability and erratic absorption patterns in the clinical setting. Due to the rising number of compounds having solubility issues, finding ways to enhance the solubility of drugs is one of the major challenges in the pharmaceutical industry today. Polymeric micelles, which form upon self-assembly of amphiphilic macromolecules, can act as solubilizing agents for delivery of poorly soluble drugs. This manuscript examines the fundamentals of polymeric micelles through reviews of representative literature and demonstrates possible applications through recent examples of clinical trial developments. In particular, the potential of polymeric micelles for delivery of poorly water-soluble drugs, especially in the areas of oral delivery and in cancer therapy, is discussed. Key considerations in utilizing polymeric micelles’ advantages and overcoming potential disadvantages have been highlighted. Lastly, other possible strategies related to particle size reduction for enhancing solubilization of poorly water-soluble drugs are introduced.
Keywords: Polymeric micelle, oral delivery, anticancer, drug solubilisation, clinical trial, micelle stability, nanocrystal, nanoemulsion
1. INTRODUCTION
Over the past ten years, the number of drug candidates with solubility problems has steadily increasedas a result of using combinatory chemistry and high-throughput screening in drug discovery. At present it is estimated that approximately 70% of new chemical entities are poorly soluble in aqueous and many even in organic media, and approximately 40% of currently marketed immediate-release oral drugs are considered practically insoluble (solubility less than 100 μg/ml) in water1, 2. Poor solubility leads to a variety of issues. Low solubility limits the drug dissolution rate, which frequently results in low bioavailability of the orally administered drug 3. In such a case the therapeutic drug concentration in the blood can be achieved by dose escalation. However, dose escalation is often undesirable for the following reasons: 1) possibility of increased toxicity and therefore decreased patient compliance; 2) difficulty in designing formulations for drug product with high drug load; and 3) increase in manufacturing costs associated with higher consumption of active pharmaceutical ingredients (API). Low solubility also may result in erratic absorption patterns, which detract from the clinical efficacy of the drugs. Consequently one of the major challenges of the pharmaceutical industry is developing strategies to enhance the aqueous solubility of drugs. This is particularly pertinent to drugs within class II and IV of the biopharmaceutical classification system (BCS), where dissolution velocity is a rate limiting step for absorption.
Various methods to overcome the poor aqueous solubility of drug candidates have been investigated in the research and development of oral formulations. These methods include changing the chemical structure of drug candidate in lead optimization phase and utilizing pro-drug approaches whereby a polar functional group is introduced into the structure of the drug molecule 4. The most often used approach is to enhance the dissolution of these poorly water-soluble drugs, especially in the case of BCS class II and IV drugs. According to the Noyes – Whitney equation, the rate of dissolution is affected by the effective surface area, diffusion coefficient, diffusion layer thickness, saturation solubility, the amount of dissolved drug as well as volume of dissolution media 3. Among them, effective surface area, diffusion layer thickness and saturation solubility are factors that can be modified by formulation efforts. Ways to modify these factors include crystal modification (e.g., metastable polymorphs, cocrystal and salt formation), particle size reduction (e.g. micronization, nanocrystals), amorphization, pH modification and self-emulsification.
Particle size reduction to the nanometer range is one of the most widely investigated approaches to enhance dissolution. Nanonization, i.e., production of drug nanocrystals, reduces the drug particle size to the sub-micron range via either bottom-up methods such as precipitation and self-assembly or top-down technologies such as milling and high pressure homogenization 5-9. Nanocrystals dramatically increase the drug particle surface area, thereby enhancing the dissolution rate of poorly soluble drugs. In addition, an increase in saturation solubility is also expected as described by Ostwald-Freundlich’s equation 10. To stabilize nanocrystal formulations, hydrophilic polymers and/or surfactants are usually added to the nanocrystal suspensions. These formulations have been found to demonstrate 1.7-60folds and 2-30 folds enhancements in Cmax and AUC, respectively, when compared with crystalline formulations with particle sizes in the micrometer range 11-13.
In addition to the nanocrystal approach, other types of nanonization strategies have emerged as new nanoplatforms for the delivery of poorly soluble drugs. Typical examples of these nanoplatforms include nanoemulsions and polymeric micelles. A common feature of these nanoplatforms is the ability to solubilize poorly water-soluble drugs in a hydrophobic reservoir or core. Poorly water-soluble drugs are encapsulated within the reservoir or core in a dissolved state, and the reservoir or core is often stabilized by surfactants or polymeric shell to prevent rapid diffusion of encapsulated drug from the reservoir or core. More specifically, for nanoemulsionsnanoscopic oil droplets (typical size 20 – 200 nm) are suspended in aqueous phase. The oil droplets are the reservoirs for hydrophobic drugs. Widely used oil molecules include saturated and unsaturated fatty acids, fatty acid esters and soybean oils. Although nanoemulsions have tendency to phase separate and flocculate, kinetically stable nanoemulsions with sufficient shelf life stability can be achieved by using common surfactants such as poloxamers, lecithin and Tween 80. Combinations of various surfactants have also been explored for controlling particle size and improving nanoemulsion stability. Commercially available nanoemulsion-based formulations include Estrasorb® (estradiol, Novavax/Graceway), Flexogan® (camphor, menthol and methyl salicylate, AlphaRX, Canada) and Restasis® (cyclosporine, Allergan).
Polymeric micelles have gained considerable attention in the last two decades as a multifunctional nanotechnology-based delivery system for poorly water soluble drugs. Typically polymeric micelles are formed from self-aggregation of amphiphilic polymers with the hydrophobic part of the polymer on the inside (core) and hydrophilic on the outside (shell). As a result of this characteristic, the advantages of polymeric micelles as delivery vehicles are two-fold: first, the hydrophobic core serves as a solubilisation depot for drugs with poor aqueous solubility; second, the hydrophilic shell provides some protection in limiting opsonin adsorption, which contributes towards a longer blood circulation time or better blood stability. The small size of polymeric micelles also contributes towards longer blood circulation time by evading scavenging by the mononuclear phagocytic system in the liver and bypassing the filtration of inter-endothelial cells in the spleen. Ultimately longer circulation time leads to improved accumulation at tissue sites with vascular abnormalities. This last particular characteristic provides one of the strongest arguments for using polymeric micelles for delivering anti-cancer drugs, most of which also have very low aqueous solubility.
Despite increasing attention in polymer micelles as drug delivery vehicles for poorly soluble drugs, so far much of the work has been conducted in a laboratory setting with very few in clinical trial studies. Those currently in clinical trials include phase II and phase IV studies of paclitaxel-loaded polymer micelles for non-small cell lung cancer (Samyang Biopharmaceuticals Corporation) and recurrent breast cancer (Korean Breast Cancer Study Group), respectively. While polymeric micelle delivery systems have remained promising, thus far significant problems have impeded their progress and limited their applications. The fundamentals of polymer micelle drug delivery systems (PMDDS) is reviewed with a focus on application of PMDDS in oral delivery and anti-cancer therapy, two of the most widely investigated applications for PMDDS. The scope is limited to polymeric micelles that encapsulate poorly soluble drugs by purely physical interactions rather than via chemical linkages, which describe a group of micelles formed by self-assembly of hydrophilic polymer-drug conjugates. A priority is placed on PMDDS that form by physical drug loading because of the preference for intact drug molecules during drug development. Alternative nanotechnology-based delivery methods for poorly water soluble compounds are proposed at the end.
2. STRUCTURE AND COMPOSITION
Amphiphilic polymers self-assemble in aqueous environment to form supramolecular core-shell structures, either with a solid core or a more fluid structure. In the former case, nanospheres are formed and in the latter structures called polymeric micelles are formed (Fig.1). The core of polymeric micelle is a dense region consisting of the hydrophobic part of the amphiphilic polymer. In PMDDS the core serves as a reservoir for drugs with low aqueous solubility due to the tendency of these drugs to partition into the core as a result of hydrophobic interactions. Due to the non-covalent nature of interaction, it is unlikely that drugs will encounter chemical stability issues as a result of encapsulation inside micelle cores. The shell of polymeric micelles is composed of the hydrophilic part of the amphiphilic polymer. The shell is a physical shield that stretches away from the core and limits micelle-micelle or micelle-protein (opsonin) interactions. Typically, average hydrodynamic diameter of polymer micelles is within the 20-80 nm range 14. Primary methods used to study micelle dimensions are dynamic light scattering (DLS), static light scattering (SLS), transmission electron microscopy (TEM) and atomic force microscopy (AFM). TEM and AFM also provide direct images of micelles and insight into shape, which is generally spherical in nature.
Figure 1.
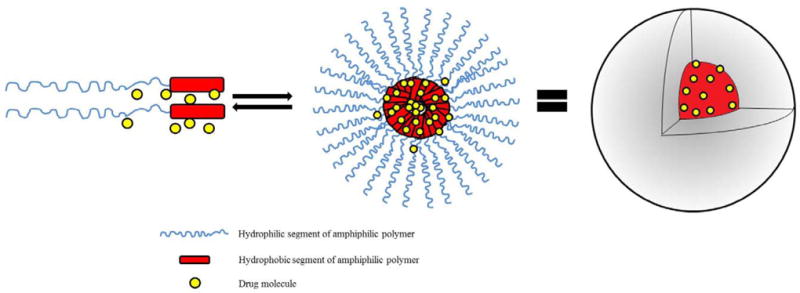
Schematic representation of supramolecular structure of polymeric micelles.
2.1. Core of Polymeric Micelles
The hydrophobic core is a key component in determining the micelle’s capacity to solubilize a poorly water-soluble compound. The ability of the core to encapsulate drug is largely dependent upon the compatibility between the hydrophobic core and the drug molecule 15. Generally, a good indication of compatibility is structural similarity between drug molecule and the hydrophobic part or hydrophobic side chain of core-forming amphiphilic polymer. Compatibility can also be estimated by comparing the polarity of the poorly water-soluble drug compound and the hydrophobic segment of polymer. Somewhat similar to the ‘like dissolves like’ rule (though in the case of polymer micelles the core does not really dissolve the drug), a general rule of thumb is drug and core-forming block with similar polaritiesare more compatible than a combination with larger differences in polarity. To quantify this interaction, a commonly used parameter to estimate compatibility using polarity is the Flory-Huggins interaction parameter, χsp. The equation to calculate χsp is given below:
where δs and δp are solubility parameters for drug and core-forming polymer segment respectively, νs is the molar volume of the drug, k is the Boltzmann constant, and T is the temperature in Kelvins. The solubility parameters of drug (small molecule) and polymer can be calculated using group contribution method, and the solubility parameter of drug can also be determined experimentally by measuring mole fraction solubility of drug in different solvent systems. Theoretically, minimization of χsp leads to better compatibility and therefore better core encapsulation of the poorly soluble drug, though more studies involving wide range of compounds and polymers are needed to strongly validate this conclusion.
Although polymer-drug miscibility is apparently one of the most important parameters to govern drug encapsulation within the micelle core, factors such as hydrophilic-lipophilic balance (HLB) of block copolymers and polymer – drug ratio (P/D ratio) are also worth noting. For example, block copolymer PEO-b-PCL with longer PCL hydrophobic segment demonstrated better drug loading than one shorter PCL segment16. Furthermore, the encapsulation capacity was shown to increase with increasing initial concentration of drug in the preparation (decrease in P/D ratio). In this case, the drug displayed good compatibility with the core polymer, which was confirmed by dilution and drug content determination studies. In such situations, relatively low P/D ratio enables drug solubilisation. However, when compatibility between drug and core polymer is low, P/D ratio generally should be increased to achieve sufficient solubilisation. It should be noted that changes in P/D ratio not only affects drug encapsulation but also other properties of the formulation such as absorption and elimination. In the example given here, lowering P/D ratio to achieve higher encapsulation efficiency also resulted in increased hemolytic activity of the formulation compared with the commercial formulation.
In terms of the composition of the hydrophobic core, biocompatibility and non-toxicity are key prerequisites in selecting the appropriate hydrophobic segment. Commonly used core-forming hydrophobic polymers for drug delivery can be classified into the following groups: poly(propylene oxide) (PPO) as in Pluronics®17; poly(esters) such as poly(lactic acid) (PLA)18 and poly(ε-caprolactone) (PCL) 19, 20; poly(L-amino acids) such as poly(L-lysine) 21; and phospholipids and lipid-derivatives such as phosphatidyl ethanoloamine 22. In addition, core-forming polymers such as polystyrene have been used in both in drug delivery systems 23 as well as fundamental research regarding polymer micelles 24. These core-forming constituents cover a wide range of structural diversity and polarity for solubilizing a wide range of poorly water-soluble drugs. The encapsulation of drug within hydrophobic cores constructed from these polymers occurs via hydrophobic interactions that are thermodynamically driven. Besides hydrophobic interactions, micelles can also take up bioactive compounds by electrostatic interactions such as in the case of PEGylated gene nanocarriers based on block catiomers with ethylenediamine repeating units 25, but such polyion complex micelles and interactions are not within the scope of this article. Polymeric micelle core can also take up drug through metal complexation, though this approach is less commonly employed than the previous two approaches.
2.2 Shell of Polymeric Micelles
As previously mentioned, the shell of polymeric micelles is composed of hydrophilic portion of amphiphilic polymer. In almost all cases studied, poly(ethylene glycol) (PEG) is invariably the shell-forming polymer of choice. There are several reasons for using PEG in PMDDS. First, it’s non-toxic and one of the few synthetic polymers already approved by FDA for use in the drug products. Second, in aqueous environment, PEG is highly hydrated and can move rapidly to sweep out a large exclusion volume. In micelles, PEG forms a dense, brush-like shell that stretches away from the core. These characteristics act to limit micelle interaction with other micelles (leading to aggregation) and proteins (opsonin), which promote uptake and removal by the mononuclear phagocytic system. Third, PEG can be easily functionalized to tether ligands for targeted drug delivery. This particular property has generated a lot of excitement in delivery of highly potent compounds such as anti-cancer agents, which would benefit immensely both in terms of efficacy and safety profiles. The above mentioned reasons all contribute to the large number of studies on polymer micelles involving PEG.
Despite the obvious advantages outlined above for using PEG, it is important to note that there are several major drawbacks in the use of PEG, especially in the clinical setting. The potentially unfavorable effects of using PEG can be attributed to several causes: (i) immunological response due to the polymer itself or side products during synthesis; (ii) unexpectedchanges in the pharmacokinetic profile of PEGylated nanocarriers and (iii) non-biodegradability of PEG and relatively easy degradation upon exposure to oxygen. Adverse reactions of intravenously administered PEG occur through complement activation, which causes hypersensitivity reactions that can lead to anaphylactic shock 26, 27. In addition, accelerated blood clearance phenomenon was seen with the use of PEG 28, 29. This phenomenon not only affects the drug bioavailability, but also the blood circulation and extravasation process 29, 30. The third drawback of PEG is its non-biodegradability. Therefore, lower molecular weight PEG would be preferable. In drug formulation lower molecular weight PEG is generally used as a solvent, and higher molecular weight PEG is used as component of micelles, possibly because oxidative degradation significantly decreases with increasing molar mass. However, care should be taken not to exceed the renal clearance threshold molar mass to allow complete excretion of the polymer. These considerations have important impact on polymeric micelle design and development.
Besides PEG, several other hydrophilic shell-forming polymers have been used in polymer micelle formation. Poly(N-vinyl-2-pyrrolidone) (PVP) is a frequently used PEG alternative31. Another alternative is the hydrophilic, non-immunogenic and biocompatible polymer poly[N-(2-hydroxypropyl) methacrylamide] (pHPMA)32. pHPMA has been investigated for use as the building block for hydrophilic shell. An advantage of pHPMA over PEG is greater multi-functionality, which allows multiple drugs or targeting ligands to be conjugated to the same polymer chain. Examples of pHPMA as the shell-forming block include A-B-A triblock copolymers of pHPMA (A block) with PCL (B block) 33as well as star-shaped PCL-b-pHPMA34. These pHPMA-based copolymers self-assemble at concentrations above CMC in aqueous solutions to form micelles with pHPMA shell and PCL core. Poly(N-isopropylacrylamide) (pNIPAAm) is a temperature sensitive polymer that has been investigated to prepare thermo-sensitive polymeric micelles 35. pNIPAAm exhibits a lower critical solution temperature (LCST) of approximately 33 °C in aqueous solution, above which it is water insoluble and below which it becomes water soluble. This unique property allows pNIPAAm to be used either as the hydrophilic shell-forming segment at temperatures below LCST, or the core-forming segment at temperatures above LCST. Examples of block-copolymers used in micelle formation with pNIPAAm as the shell segment include pNIPAAm-b-PLA, p(NIPAAm-co-methacrylic acid-co-octadecyl acrylate), p(NIPAAm-co-N,N-dimethylacrylamide-co-10-undecenoic acid) among many other examples36-38.
2.3. Thermodynamic and Kinetic Stability
The major driving force behind self-assembling of amphiphilic polymers is hydrophobic interactions that lower the free energy of the system by removing the hydrophobic segments from the aqueous environment. The threshold at which unimers (non-assembled amphiphilic polymer molecule) start to assemble into polymeric micelles is called the critical micelle concentration (CMC). Below the CMC in aqueous environment, amphiphilic molecules exist separately; above the CMC unimers exist in equilibrium with polymer micelles. One of the best models to describemicellar colloidal solutions is the closed association model. In this model, we assume that each micelle is composed of n amphiphilic unimers (M) and that each micelle is formed in a single step. That is:
The equilibrium constant for this pathway is therefore:
From the above equation it is easy to see that the rate of micellation is heavily dependent upon the concentration of unimer M. Perhaps less obvious from the equation but more intuitive is the dependence of K on temperature. The effect of temperature on micellation can be derived from the following equations, which describe the standard free energy ΔG0 associated with micelle formation:
where CMC is expressed in mole fraction, R is the gas constant and T is the temperature in kelvins. Since:
where ΔH0 and ΔS0 represent standard enthalpy and entropy changes, respectively, substituting into the first equation and solving for ln(CMC) would give the following expression:
If we plot ln(CMC) versus 1/T we would obtain a straight line with slope equals ΔH0/R and intercept of - ΔS0/R, assuming ΔH0 is independent of temperature. The above equation implies that for certain polymers such as Pluronics®, where ΔH0 is positive, the value of ln(CMC), and therefore CMC, would decrease with increasing T. The practical implication is that the increase in temperature lowers CMC, which allows CMC to form at a lower concentration of unimers. As applied in drug delivery, a lower CMC means a greater resistance to dissociation by dilution when the PMDDS is introduced into the physiological environment. The opposite effect of temperature on CMC can be said for polymers with negative ΔH0, where CMC increases with increasing T.
Besides thermodynamic stability, kinetic stability also has several important implications for drug delivery. At equilibrium, polymeric micelles exhibit inordinate kinetic stability with regards to the dissociation and exchange of unimers between different polymeric micelles. Numerous studies involving polymeric micelles with a poly(styrene) core show that exchange of unimers between micelles in water at ambient temperature is imperceptibly low to none 24, 39, but the exchange rate can be modified by changes in temperature 39 and presence of co-solvents or co-surfactants 40. The implications for polymeric micelle-based drug delivery are: (i) preparation of polymeric micelles may not be possible for some polymersby direct dissolution in water at ambient temperature; and (ii) blood components may modify the extent of exchange between micelles and promote dissociation of polymeric micelles 41, even when they are administered far beyond the CMC. In regards to the first implication, it should be noted that for some moderately hydrophobic copolymers such as poloxamers with low PPO content the direct dissolution approach can be employed to prepare drug-loaded polymeric micelles 42. Direct dissolution was used to prepare PLA / PEG micelles containing paclitaxel 43. In addition, liquid copolymers such as low molecular weight PEG-b-poly(CL-co-trimethylenecarbonate) can be easily mix with hydrophobic drug in the absence of organic solvents to prepare micelles by direct dissolution 44.
3. POLYMERIC MICELLES FOR ORAL DELIVERY
The oral route of drug delivery remains the most preferred route of drug administration. From the drug developer’s point of view, the oral route of drug administration is widely accepted by the authorities, is well studied and understood. From the patient’s point of view, it is easy and painless to administer, and allows for self-medication, which is especially convenient for chronic therapy. However, even though it is a widely utilized approach and well-understood, the formulation of drugs for oral delivery remains an intricate process, especially for the poorly water-soluble drugs. In order for absorption of orally administered drug to take place, it must first dissolve into its molecular form. For a poorly soluble drug, the rate of dissolution may be so slow or the saturation solubility so low that there is incomplete or inadequate release of drug, which ultimately leads to poor bioavailability and low drug efficacy. In this regard, polymeric micelles can positively impact bioavailability by solubilizing the poorly water-soluble drug which otherwise would precipitate in the aqueous fluids of the GI tract. In addition, encapsulation of drug inside the core of polymeric micelle may protect against rapid clearance from circulation, which can lead to reduced amount of drug available for absorption. The following sections provide some practical considerations in the formulation of polymeric micelles for oral delivery.
3.1. Maintaining Micelle Stability
All orally administered drugs must pass through the gastrointestinal (GI) environment, and conditions in the GI vary depending on location. The most obvious example is the pH value, which ranges from 1-2 in the stomach to 5-7 in the small intestine 45. The fluid volumes inside the GI tract also vary depending on location as well as fasted or fed state. In the fasted state, total fluid volume in stomach and small intestine is approximately 130 ml whereas in the fed state, the total volume increases to 740 ml 46. The implication for PMDDS development is that the micelle carriers must be able to resist rapid and premature dissociation upon dilution and exposure to the harsh and changing conditions of the GI tract.
Generally speaking, lower CMC values denote more resistance to effects of dilution and therefore greater stability 47. As an indicative guide, a CMC value of less than 135 mg/ml should be resistant to rapid dissociation by dilution in orally administered PMDDS. A relatively low CMC value is usually conferred by the presence of highly hydrophobic regions within the micelle core. In order to achieve a lower CMC, we can keep the shell-forming polymer at the same chain length but increase the chain length of the core-forming polymer. For example, Peng, Liu and Tong demonstrated that increasing hydrophobic polystyrene (PS) or poly(methyl methacrylate) (PMMA) block length decreased CMC of micelles formed from triblock co-polymers of PS-b-PEG-b-PS or PMMA-b-PEG-b-PMMA 48. Conversely we can alsokeep the core-forming polymer at the same chain length but decrease chain length of hydrophilic shell-forming polymer block, though the effect of this change on CMC is less dramatic than the previous approach. For example, Ashok et al. examined the effect of various PEG chain lengths (2000, 3000 and 5000) on CMC of PEGylated phospholipid micelles and found that the CMC was higher for micelles made from longer PEG chain lengths 49.
An examination of CMC alone is not sufficient to ensure polymeric micelle stability within the GI tract. As previously mentioned, from the point of administration to absorption polymeric micelles may encounter a range of pH values. In addition, the effect of various digestive enzymes and bile salts must also be taken into consideration. The most straight-forward in vitro studies involve investigating drug release from micelles upon exposure to both simulated gastric fluid (SGF) and simulated intestinal fluid (SIF). Francis et al. examined cyclosporine A (cyA) release from micelles in SGF (pH 1.2) and SIF (pH 6.8) and found that in both cases, the drug release reached a plateau within 4 hours with less than 12% cyA release, indicating good micelle integrity under these conditions 50. Elsewhere in another study, less than 50% of griseofulvin was released from PEG-b-PLA micelles in phosphate buffered saline (PBS, pH 7.4), SGF and SIF, though such a release may be too slow for oral drug delivery purposes 51. Although these studies seem to indicate good micelle stability in GI, the results should be interpreted with caution. The major limitation of the above studies and indeed many other in vitro studies carried out to predict polymeric micelle stability in GI in vivo is that no enzymes or bile salts added in SIF, which is different from the actual situation where these components abound in the small intestinal environment. In a study that did include bile salts in SIF a decrease in micelle size was detected compared with micelles in SIF without bile salts, indicating partial destabilization 52. The effect may be more pronounced in vivo since the presence of enzymes must also be accounted for. Drug retention within polymeric micelles is a prerequisite to successful delivery of poorly soluble drugs to the absorption site, but the retention should not so extended that it hinders the absorption of drug molecules through the GI mucosa.
Interestingly, several studies have been carried out to investigate the effect of using pH sensitive polymeric micelles on drug release and oral bioavailability of poorly water-soluble drugs. Satturwar et al. constructed pH-sensitive polymeric micelles using PEG-b-poly(alkyl(meth)acrylate-co-methacrylic acid) and the poorly water-soluble drug candesartan cilexetil was encapsulated within the micelles in amorphous form 53. The release of candesartan cilexetil was monitored in vitro as a function of pH. Results show release of drug from micelles was triggered when pH increased from 1.2 to 7.2. In another study, Sant et al. synthesized the pH sensitive block copolymer PEG-b-poly(alkyl acrylate-co-methacrylic acid) by atom transfer radical polymerization. This ionizable block copolymer formed self-assembled micelles at pH below 4.7 and dissociated partially or completely above this pH 54. It was hypothesized that these polymeric micelles can enhance bioavailability of orally administered poorly water-soluble compounds by preventing drug release and subsequent phase separation in the low pH environment of the stomach, but releases the drug in molecularly dispersed form upon the more basic environment of the small intestine. The poorly water-soluble model drugs fenofibrate and progesterone were encapsulated by oil-in-water emulsion or film-casting methods. One important result of the study demonstrated that relative bioavailability of fenofibrate incorporated in pH-sensitive micelles increased by 156% and 15% compared withfenofibrate coarse dispersion and commercial formulation. The increase in relative bioavailability is attributed to enhanced solubility of drug in GI track as well as reduction of leakage and precipitation in stomach. These studies show that use of pH-sensitive block copolymers to construct polymer micelles can alter the stability profile of micelles under different pH environment, a fact that can be utilized for controlled drug release and as a possible way to increase bioavailability of poorly water-soluble drugs.
3.2. Interactions with Intestinal Mucosa
Different experimental methods have been employed to study the interaction of polymeric micelles with intestinal membrane, mostly using Caco-2 monolayer as the model membrane. In general, polymeric micelles are not known to interact extensively with cell membranes, probably due to steric hindrance from shell-forming polymer segments. Therefore, indicators of paracellular permeability such as the often used trans-epithelial electrical resistance (TEER) usually remain unaltered in the presence of polymeric micelles. Instead most of the in vitro studies carried out assess the effect of micelle encapsulation on drug permeability compared with un-encapsulated drug permeability. Theoretically the encapsulation of BCS class II drugs in polymeric micelles should bring about an increase in absorption, but cell permeability studies have been known to give contradicting results. For example, cyA loaded in hydrophobically-modified dextran or hydroxypropyl cellulose micelles demonstrated increases of 1.5- and 3-fold respectively in permeability 50, but risperidone loaded in PEG-b-P(CL-co-TMC) micelles did not show any improvement in permeability 55. However, caution should be used in the interpretation of these results, because the in vitro cell permeability studies do not accurately represent the conditions that lead to micelle dissociation and drug release in vivo. In fact, the correlation between cell permeability studies and in vivo pharmacokinetics is still a subject of much discussion in current literature.
As mentioned previously, the intestinal mucosa is normally relatively impermeable to polymeric micelles. However, there are other pathways that allow the transport of micellar carrier systems across the membrane (Fig. 2). First, in the absence of targeting moieties on its surface, polymeric micelle can be absorbed in its intact form by enterocytes or M cells through an endocytotic pathway triggered by non-specific interactions such as hydrogen bonding or van der Waal interactions between the micelle surface and the cell 56. Second, the micelles can be absorbed through the process of pinocytosis, in which the cell surface forms invagination that engulfs the micelle carrier. Third, polymeric micelles can be absorbed through receptor-mediated pathway, which is an approach widely investigated in parenteral drug delivery but rarely researched on in oral drug delivery. Still, the ability to enhance oral absorption through increased receptor-mediated endocytotic pathways remains an attractive prospect. Francis et al. 57 studied the permeability of cyA encapsulated in dextran-g-PEO-C16 micelles decorated with vitamin B12 (VB12) on the surface. VB12 promotes receptor-mediated endocytosis by binding to intrinsic factor, and together the complex is transported across the mucosa. The permeation coefficient of cyA transported by VB12-decorated micelles was 3.3 cm/sec compared to 1.4 cm/sec for undecorated micelles.
Figure 2.
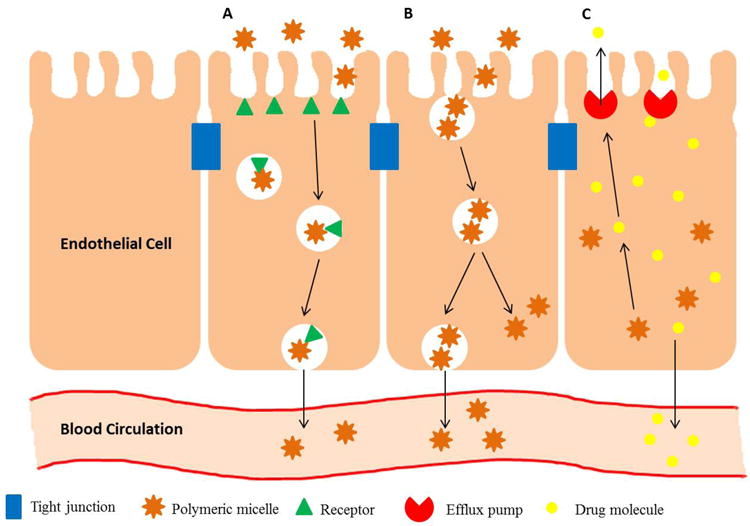
Schematic summary of the pathways by which polymeric micelles may interact with the intestinal mucosa. A) receptor-mediated endocytosis; B) pinocytosis; C) Efflux of drug molecules
Besides uptake, drugs can beand often are pumped out of enterocytes by efflux transporters on the surface of intestinal mucosa. The extent of absorption for poorly water-soluble drugs (and indeed all orally administered drugs in general) is affected by these efflux pathways. Among the efflux transporters, the most well-known and widely studied is the P-glycoprotein (Pgp) efflux transporters. Pgp is thought to be one of the most significant causes of decreased permeability and therefore oral bioavailability. Consequently modulation of its activity is seen as a way to improve absorption for orally administered drugs. Inhibition of Pgp has been demonstrated most extensively with the use of d-alpha-tocopheryl polyethylene glycol (TPGS) and poloxamers, though exact mechanisms are still unclear. In studies involving TPGS and poloxamers, improvement in permeability was seen at polymer concentrations below CMC and maximal at concentrations just below CMC 58-60. These findings seem to suggest that while amphiphilic unimers are able to influence Pgp activity, the formation of micelles will likely negatively impact drug permeability by rendering the encapsulated drug impermeable to the intestinal mucosa. Rather interestingly, when Zastre et al. 61 carried out a comprehensive study of the inhibition of Pgp activity by PEG-b-PCL micelles of various compositions (different hydrophobic and hydrophilic block lengths), they found that maximal permeability rhodamine-123 (R-123) was seen at concentrations 8 – 100 times over CMC of PEG-b-PCL. However, even at these polymer concentrations, less than 20% of the dye was encapsulated inside the micelles, so the increased permeability could be ascribed to either or both the Pgp inhibitory action of PEG-b-PCL or the high concentration of free dye in the system. It’s important to point out that not all polymers used for polymeric micelles have an effect on Pgp, despite some being able to alter membrane fluidity 62. The criteria that make a polymer good inhibitor of efflux transporters remain unclear.
3.3. In vivo Investigation of Polymeric Micelles
Very limited studies have been carried out to investigate the pharmacokinetics of orally administered polymeric micelles for delivery of poorly water-soluble drugs. A polymeric micelle formulation of paclitaxel (aqueous solubility < 0.1 μg/ml)63was administered intravenously or orally to canulated rats64. Data from oral administration indicated an estimated bioavailability of 12.4%, which is significantly higher than reported 6.5% 65for Cremophor EL micelle formulation of paclitaxel (Taxol®) given orally. Interestingly, the polymeric micelle formulation administered via the portal vein showed a 50% reduction in AUC compared to the i.v. infusion, indicating high metabolism of paclitaxel despite encapsulation in polymeric micelle. In an in vivo study involving risperidone (aqueous solubility ~ 103 μg/ml)66 bioavailability was not improved by formulating into polymeric micelles 55. Another study of polymeric micelles containing itraconazole (aqueous solubility 1.8 μg/ml)67 showed similar performance between micelle formulation and commercial formulation using cyclodextrin as solubilizing agent 68. Ould-Ouali et al. studied oral delivery of risperidone encapsulated in self-assembling PEG-p(CL-co-trimethylene carbonate) structures in male Wistar rats69. The micellar solution was compared to an aqueous solution of risperidone in tartaric acid. Results show no statistically significant differences between the plasma concentration time profiles of the two formulations, though Cmax of risperidone was lower in micelle formulation (162 ± 12 ng/ml vs. 256 ± 56 ng/ml for aqueous risperidone solution, p=0.16), which was thought to indicate a more sustained drug absorption, but the authors cautioned that additional data will be required to validate this observation. The apparent lack of statistically significant enhancement in bioavailability in some of these studies between micelle formulation and non-micelle formulation suggest that although micelles may enhance solubilisation of poorly water-soluble drugs, absorption is not necessarily increased, possibly due to the low availability of drug in its readily absorbable form within its absorption window in the gastrointestinal tract.
This issue can be potentially overcome using pH-sensitive micelles as delivery vehicles instead of non-sensitive micelles. As previously mentioned, Sant et al. incorporated fenofibrate into pH-responsive micelles and administered these micelles to male Sprague-Dawley rats that were fasted overnight by oral gavage 54. Relative bioavailability was enhancedcompared to coarse drug dispersion and commercial formulation. While these studies have greatly advanced our understanding of polymeric micelle delivery systems for poorly water-soluble drugs, there remain many unanswered questions. For one, in almost all such studies the micelle formulations were administered after fasting or infused directly into the duodenum, which do not provide sufficient information regarding the effect of stomach conditions or release of bile salts and enzymes (as occurs during digestion) on micelle performance, especially non-pH responsive micelles. It suffices to say that designing PMDDS for poorly water-soluble compounds is a complex procedure, and many more fundamental studies need to be carried out in order to fully understand these processes.
4. POLYMERIC MICELLES IN ONCOLOGY
Polymeric micelles are perhaps most extensively explored for use in anti-cancer treatmentsfor several reasons. First, the hydrophobic cores of polymeric micelles help to solubilize anti-cancer drugs, which are often poorly water-soluble. Second, encapsulation of anti-cancer drugs inside polymer micelles may minimize drug degradation and loss. As previously mentioned in this manuscript, the hydrophilic shell provides some protection in limiting opsonin adsorption. The small size of polymeric micelles also enables avoidance of scavenging by the mononuclear phagocytic system in the liver and filtration of inter-endothelial cells in the spleen. Both factors contribute towards a longer blood circulation time, which allows drug-loaded PMDDS sufficient time to travel to tumor site. Third, once the micelles are in the tumor vicinity, their small size allows them to escape into the affected tissue area via the leaky vasculature found at tumor sites, and because of the lack of lymphatic drainage in these areas, the micelles can be retained there for an extended period of time. Such an effect is known as the enhanced permeability and retention effect (EPR). Last but not least, we can modify the shell of polymer micelles by attaching specific ligands to promote PMDDS-cell specific interactions, which is especially useful for preventing harmful side-effects stemming from highly potent anti-cancer agents acting on normal cells. Because of the above reasons, the potential benefit of using polymeric micelles in cancer therapy is great. This section will focus on research in using PMDDS for oncology.
4.1. Improvements in Solubility
Solubility enhancement of several commonly studied anti-cancer drugs by incorporation into polymeric micelles will be discussed in this section. Paclitaxel, an anti-cancer agent with an aqueous solubility of approximately 0.3 μg/ml, was loaded into 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy(PEG)/TPGS (PEG-DSPE/TPGS) micelles 52. The aqueous solubility of paclitaxel was enhanced by up to 5000 times to achieve an aqueous solubility of approximately 5 mg/ml. The most impressive enhancement in paclitaxel solubility was achieved by the work of Kim et al 70, in which it was reported that the aqueous solubility of paclitaxel was as high as 38.9 mg/ml through encapsulation in micelles. In this study, nicotinamide derivatives, i.e. N,N-diethylnicotinamide and N-picolylnicotinamide were shown to be powerful hydrotropes for paclitaxel. Copolymers with a segment containing such nicotinamide derivatives could be used to produce polymeric micelles with hydrotropic properties toward paclitaxel. In addition, micelles composed of PEG-b-poly(vinylbenzyloxy)-N,N-diethylnicotinamide (PEG-b-PVBODENA) could achieve a remarkably high drug loading (37.4% w/w) for micelle-based delivery systems. The drug loading increased proportionally to the length of the hydrotropic DENA segment. As a comparison, PEG-b-PLA micelles could only load up to 27.6 % w/w of paclitaxel under similar conditions. The difference in drug loading capacity is very likely due to the extent of polymer-drug compatibility, as explained in more detail in section 2.1 ‘core of polymeric micelles’.
Camptothecin is an inhibitor of topoisomerase I – an enzyme involved in the replication of DNA. As such it is a widely investigatedpossible anti-cancer agent for several forms of cancer 71, 72with an aqueous solubility of approximately 1.3 μg/ml 73. Camptothecin was loaded into polymeric micelles consisting of Pluronics® (PEO-PPO-PEO) covalently conjugated to poly(acrylic acid) (Pluronic-PAA) 74. This micellar formulation demonstrated an approximately 3- to 4-fold enhancement in the aqueous solubility of camptothecin at pH5, a pH at which the lactone form (or the therapeutically active form) of camptothecin is stable but still insoluble. Rather interestingly, the amount of camptothecin solubilized per unit PPO was considerably greater in Pluronic-PAA formulation than in the Pluronic-alone formulation, which seems to suggest solubilisation not only by the hydrophobic core but also by the hydrophilic PEO-PAA micelle shells of the micelles.
Tamoxifen is another anticancer hydrophobic drug with extremely low aqueous solubility (~ 0.24 μg/ml)75. Tamoxifen was incorporated into micelles consisting of a new self-assembling polyaspartylhydrazide co-polymer, which is synthesized by grafting both PEG(2000) chains and hydrophobic palmitic acid (C(16)) moieties on the the hydrosoluble polyaspartylhydrazide (PAHy) backbone 76. The PAHy-PEG2000-C16 micelles were able to achieve 4% w/w drug loading with tamoxifen, which was three times more efficient than previously studied systems containing similar polyaspartic copolymers 77. The solubility enhancement of tamoxifen after incorporation into PAHy-PEG2000-C16 micelles was approximately 500-fold, reaching an aqueous solubility of 0.12 mg/ml. The enhancement in solubility is substantial and rather remarkable. Using the polymeric micelle approach, other anticancer agents besides the three examples given above have also achieved increases in solubility.
4.2. Improvements in Stability
Polymeric micelles improve drug stability by inhibiting drug degradation. For example, the therapeutically active lactone form of camptothecin was physically incorporatedinto hydrophobic core of N-phthaloylchitosan-grafter PEG methyl ether (PLC-g-MPEG) micelles by the dialysis method 78 and analyzed for in vitro release behaviors as well as stability. The in vitro release profile of camptothecin-loaded PLC-g-MPEG micelles showed sustained release of over 96 hours when the drug loading was around 10% w/w for the micelles. More importantly, when compared to the unprotected camptothecin, camptothecin loaded PLC-g-MPEG micelles were able to protect the lactone form of the drug from being hydrolyzed. The prevention of lactone hydrolysis is crucial for camptothecin formulation development to prevent severe systemic toxicity and poor tumor response efficacy associated with lactone form hydrolysis. Furthermore, camptothecin-loaded PLC-g-MPEG micelles showed an increased half-life in the presence of human serum albumin (HSA) and fetal bovine serum (FBS) from 94 minutes to 76.15 hours compared to un-encapsulated drug. When camptothecin was loaded into micelles composed from Pluronic-PAA 74 hydrolysis of the lactone form of the drug was prevented for up to 2 hours at pH 8 in water. When comparison was made between the encapsulated formulation and unprotected drug, it was found that drug hydrolysis in human serum was approximately 10-fold slower for the Pluronic-PAA formulation. The half-lives of unprotected camptothecin versus micelle-encapsulated camptothecin were 0.16 and 1.1 – 1.7 hours respectively.
Another substantial advantage of encapsulation in polymeric micelles is that non-specific interactions, including RES, may be reduced due to steric repulsion by the hydrophilic polymers surrounding the drug-encapsulated hydrophobic core. The steric hindrance effect provides a possibility for designing PMDDS with prolonged circulation time in the blood, which is ultimately beneficial for accumulation at tumor site. So far, experimental results have been promising. Doxorubicin is one of the most powerful and widely investigated anticancer drugs in the clinical field. However, dose-limiting toxicity often occurs with doxorubicin therapy because of the drug lacks sufficient selectivity against tumor cells. Protection from pre-mature drug release and prolonged circulation until reaching tumor site would improve clinical usefulness of doxorubicin immensely. Yu et al. 79 incorporated doxorubicin into self-assembling aggregates consisting of cholesterol-modified glycol chitosan (CHGC) using a dialysis method. In rat pharmacokinetic studies, doxorubicin-CHGC formulation demonstrated significant increase in mean residence time (2.477 ± 0.297 hours) compared with free doxorubicin (0.125 ± 0.016 hours), indicating a remarkably delayed blood clearance. The area under the plasma concentration-time curve (AUC) of the encapsulated formulation was approximately 6.61 times higher than free doxorubicin. A previous paper reported the detection of self-assembled glycol chitosan in the blood for three days 80; thus it was postulated that the glycol chitosan offers steric hindrance to plasma opsonin that contributed to the delayed blood clearance. When doxorubicin was physically loaded into PEG-poly(beta-benzyl-L-aspartate) (PEG-PBLA) block copolymer micelles by an o/w emulsion method, approximately 77.5% of PEG-PBLA dose was cleared from blood circulation after 1 hour compared to almost 100% clearance of un-encapsulated doxorubicin 81.
One particular aspect that is important to note is the effect of specific surface area on the ability of the micelles to stabilize encapsulated drug. Elsabahy et al. synthesized PEO-b-poly(styrene oxide) (EO-SO) and PEO-b-poly(butylene oxide) (EO-BO) of different chain lengths and studied their self-assembling properties in water as well as the resulting polymeric micelles’ ability to solubilize and protect docetaxel from degradation in vitro82. The size and shape of micelles are controlled by various factors such as length and nature of core and shell-forming segments. In the present study, micelles composed of EO-BO with number-average block lengths of 45 and 15 for PEO and poly(butylene oxide) units, respectively, (denoted EO45-BO15) had the smallest diameter measured by dynamic light scattering compared with EO45-BO24, EO45-SO15 and EO45-SO25 but EO45-BO24 micelles yielded the lowest specific surface area based on calculations. When chemical stability of docetaxel was investigated in water over a period of 24 hours at 50 °C, only EO45-BO24 was able to preserve most of the docetaxel chemical integrity. The vastly different protective effect was partially attributed to the lower specific surface area of EO45-BO24 micelles, which decreases interaction of drug and aqueous medium at the water-micelle interface.
4.3. Clinical Trials
Clinical trials of polymeric micelles containing anti-cancer agents are few in comparison to the large number of research conducted in laboratory settings. A summary of PMDDS-based formulations in clinical trials can be found in Table 1. In this section, we will examine each of these formulations, with a concentration on formulation and important results of clinical trials.
Table 1.
Summary of polymeric micelle-based formulations containing anticancer agents in clinical trials
| Formulation | Drug | Polymer | Particle Size (nm) | Drug loading (%) | PK parameters (fold change over free drug) | Phase | Company | ||
|---|---|---|---|---|---|---|---|---|---|
| t1/2 | AUCblood | AUCtumor | |||||||
| Genexol-PM83, 84, 118, 119 | Paclitaxel | mPEG-PDLLA | < 50 | 16.7 | 0.62 | 0.74 | 1.74 | III, IV | Samyang, Korea |
| NK105120 | Paclitaxel | PEG-P(Asp) | 85 | 23.0 | 6.11a, 3.71b | 86.11a, 50.40b | 24.00a, 24.06b | II, III | Nanocarrier/Nippon Kayaku, Japan |
| SP1049C93, 94 | Doxorubicin | Pluronic L61, F127 | 30 | 8.2 | 1.38c, 1.05d | 2.06c, 1.20d | 1.69 | III | Suprateck, Canada |
| DTXL-TNP96 | Docetaxel | PLA-PEG, PLA-PEG-ACUPA | 100 | 10 | n.a | n.a | n.a | I | BIND Biosciences |
| NC6004 97, 121, 122 | Cisplatin | PEG-P(Glu)-Cisplatin | 30 | 39.0 | 0.19 | 64.77 | 3.59 | I, II | Nanocarrier, Japan |
| NK012 99, 100 | SN-38 | PEG-P(Glu)-SN38 | 20 | 20.0 | 16.41e | 14.09e | 9.53e | II | Nippon Kayaku, Japan |
| NK911 91, 103 | Doxorubicin | PEG-P(Asp)-Dox | 40 | N/A | 2.62 | 28.88 | 3.46 | II | Nippon Kayaku, Japan |
Dose: 50 mg/kg
Dose: 100 mg/kg
Data gathered in normal mice
Data gathered in tumor-bearing mice
Marketed in South Korea in 2007
4.3.1. Genexol®-PM
Genexol®-PM is a polymeric micelle-based formulation of paclitaxel encapsulated in monomethoxy-PEG-b-poly(D,L-lactide) (MPEG-PDLLA). The amphiphilic polymer was synthesized by a ring-opening polymerization reaction with MPEG molecular weight of 2000 g/mol 83. Physical encapsulation of paclitaxel was carried by using a solid dispersion technique. The final formulation contained PMDDS less than 50 nm in diameter with a drug loading of approximately 16.7% 84.
MPEG-PDLLA was shown to be non-toxic and biocompatible in both in vitro and in vivo studies 85. In comparison to Taxol® (paclitaxel solubilized by Cremophore EL), Genexol® PM displayed similar cytotoxicity against various human cancer cells, including breast, colon, ovarian and non-small cell lung cancer (NSCLC) cells 86. However, unlike usual studies involving micelle-based formulations, Genexol® PM showed an 82% decrease in AUC after IV administration when compared with Taxol® given in equivalent dose86. It was postulated that such dramatic decrease was likely due to rapid dissociation of MPEG-PDLLA micelles in presence of α- and β-globulin in the blood, resulting in the rather rapid release of paclitaxel from the micelles 87. Despite the decrease in AUC, the formulation was deemed superior to Taxol® for its higher efficacies in subsequent clinical studies. In Phase II and III trials comparing Genexol®, Abraxane® (an albumin nanoparticle formulation of paclitaxel) and Taxol® for metastatic breast cancer, the response rate to Genexol® (administrated over 3 hours every 3 weeks at 300 mg/m2) was higher than both Abraxane® and Taxol®88, 89. For NSCLC, Genexol® PM combined with cisplatin was more effective than Abraxane® alone 90. Due to its superior efficacies and lower adverse reactions, Genexol® PM is currently available commercially for treatment of NSCLC, ovarian cancer, breast cancer and gastric cancer in some countries. Current phase III and IV clinical trials are ongoing.
4.3.2. NK105
NK105 is a formulation consisting of paclitaxel physically incorporated into polymeric micelles self-assembled from PEG-poly(aspartic acid) (PEG-P(Asp)) modified with 4-phenyl-1-butanol to increase the hydrophobicity 91. The drug loading obtained for this formulation was approximately 23% w/w. Prior to clinical injections, the lyophilized powder was dissolved in 5% glucose solution, and average particle size was approximately 85 nm, with a rather wide size distribution ranging from 20 nm to 430 nm.
Phase I clinical trials of NK105 began in 2004 in 19 patients with pancreatic, bile duct, gastric or colonic cancers 92. In the typical dose escalation study (10 mg/m2 to 180 mg/m2), NK105 demonstrated reduced toxicity and lowered adverse reactions compared to Taxol® treatments. Neutropenia was the only grade-4 toxicity observed, and neuropathy, the most common adverse reaction associated with Taxol® treatments, was only grade 1 or 2. Allergic reactions were not observed except for one patient who had grade-2 hypersensitivity at dose of 180 mg/m2. Amongst the 19 patients, partial response was observed for 1 (out of 11) patient with metastatic pancreatic cancer; colon (1 patient) and gastric (2 patients) patients experienced stable disease state lasting for ten and seven courses of treatments respectively. In pharmacokinetics studies, AUC and total clearance of NK105 administered at 150 mg/m2 were 32- fold higher and 72-fold lower respectively compared with that of Genexol® PMat 300 mg/m2, indicating higher blood stability of NK105 84. Phase II clinical studies were conducted in Japan in 2007 and completed in 2010, and Phase III trials are in preparation.
4.3.3. SP1049C
SP1049C is a Pluronic® based polymeric micelle formulation of doxorubicin. This formulation is prepared by reconstituting doxorubicin with a 0.9% sodium chloride solution containing 0.25% w/v Pluronic® L61 and 2% w/v of F127 to a final concentration of 2 mg/ml 93. The rationale for using Pluronics® is the discovery that such non-ionic surfactants can reduce drug resistance considerably, which is expected to improve effectiveness of clinical treatments by decreasing multidrug resistance (MDR) in cancer cells. L61 was found to be the most effective Pluronic® modulator of doxorubicin activity against various MDR cell lines, but L61 with doxorubicin alone would not produce stable formulation due to liquid phase separation 94. Therefore, F127 was added as a stabilizer. The average particle size in SP1049C is approximately 30 nm with 8.2% drug loading.
Phase I clinical trials of SP1049C was conducted in Canada in 1999 93. In initial dose escalation studies (5 mg/m2 to 90 mg/m2), SP1049C showed similar spectrum of toxicities as conventional Doxil treatment at doses of 35 mg/m2 and above. Neutropenia was the primary toxicity observed. Unlike Doxil treatment, hand-foot syndrome was not observed for SP1049C. Amongst the patients enrolled in the clinical trial, 3 (corresponding to 11.5%) had a complete or partial response during treatment, and 8 (30.8%) had stable disease with time to progression ranging from 9 to 24 weeks with median of 17.5 weeks. Phase II clinical trials began in 2002 in patients with advanced adenocarcinoma of the esophagus and gastro esophageal junction95. The dosing regimen consisted of 30 minute IV infusion at dose of 75 mg/m2 given once every 3 weeks for up to 6 cycles. The results showed some unexpected toxicities associated with this treatment. 61.9% of patients experienced grade 3/4 neutropenia, with 1 patient requiring granulocyte colony-stimulating factor treatment for grade 4 neutropenia and fever. 1 patient experienced grade 3/4 mucositis. Gradual absolute decrements in left ventricular ejection fraction (LVEF, a measure of how much blood is pumped out of the left ventricle with each contraction) were also observed with cumulative treatment. These adverse responses were likely due to micelle instability and subsequent degradation issues. However, the encouraging results are that out of 19 patients evaluated, 9 had partial response and 8 had stable disease. The median overall survival and progression-free survival were longer than for formulation consisting of free doxorubicin combined with cisplatin and 5-FU, reaching 10 and 6.6 months respectively. The drug is currently in Phase III clinical studies and was designated an orphan drug by the FDA in 2008.
4.3.4 Docetaxel-loaded targeted polymeric nanoparticle (DTXL-TNP)
A drug delivery system that allows targeted delivery of therapeutic agents to the disease location is a particularly desirable strategy in cancer treatments, because the therapeutic agents are often cytotoxic and cause damage to normal cells and tissues. Hrkach et al. developed targeted polymeric micelles containing the chemotherapeutic agent docetaxel (DTXL) for the treatment of patients with advanced and metastatic solid tumors 96. The micelles were targeted to the extracellular domain of prostate-specific membrane antigen (PSMA) using a PMSA substrate analog inhibitor S,S-2-[3-[5-amino-1-carboxylpentyl]-ureido]-pentanedioic acid (ACUPA). A combinatory library of DTXL-TNPs was prepared by self-assembly of particles from varied proportions of PLA-PEG polymer conjugated to ACUPA, DTXL, and PLA, PLGA, PLA-PEG, and PLGA-PEG copolymers of varying PLA, PLGA and PEG block lengths and PLGA ratio of glycolic to lactic acid units. The micelles formed thus consisted of a hydrophobic core with encapsulated DTXL, and a hydrophilic shell with PEG and PEG-ACUPA. The suspension was diluted with aqueous polysorbate 80 solution. After being purified and concentrated, the final formulation was stored as a frozen suspension in a 10% aqueous sucrose solution. The formulations were screened to optimize drug loading, consistency of particle size distribution across different batches, stability and drug release properties. The most promising formulations were evaluated in healthy Sprague Dawley rats. On the basis of their findings, the final formulation selected for clinical evaluation consisted of 10 wt% DTXL encapsulated in 100 nm particles composed of PLA-PEG (16-kD PLA, 5-kD-PEG), and PLA-PEG-ACUPA (also 16-kD PLA, 5-kD PEG), with PLA-PEG and PLA-PEG-ACUPA representing 97.5% and 2.5% of polymer mass respectively.
In Phase I clinical trial (NCT01300533), DTXL-TNP was given intravenously every 3 weeks in patients with advanced or metastatic cancer. The study is currently ongoing and full results have yet to be published. In interim analysis of patients receiving doses up to 75 mg/m2 DTXL-TNP displayed pharmacological properties different from commercially available, solvent-based DTXL formulation sb-DTXL that is consistent with results from the animal models. In the PK profile for patients receiving dose of 30 mg/m2, DTXL-NP showed higher plasma levels for all time points greater than 1 hour post-administration. The plasma levels were at least two orders of magnitude higher compared to equivalent dose of sb-DTXL. Furthermore, the high plasma concentration was maintained for at least 48 hours. In dose-ranging studies of 3.5 to 75 mg/m2, PK for DTXL-TNP was essentially dose proportional, with correlation coefficients of 0.87 and 0.79 for Cmax and AUC versus dose respectively (Figure 3). In terms of efficacy, 2 patients exhibit stable disease at dose below 30 mg/m2. The computed tomography images of a 51-year-old male patient with metastatic cholangiocarcinoma showed disappearance or shrinkage of multiple lung metastases after 2 treatment cycles of 15 mg/m2 DTXL-TNP. A 63-year-old patient with tonsillar cancer showed 25% shrinkage of a target tonsillar lesion after 2 dosing cycles at 30 mg/m2. These results are highly promising at this early stage of clinical development.
Figure 3.
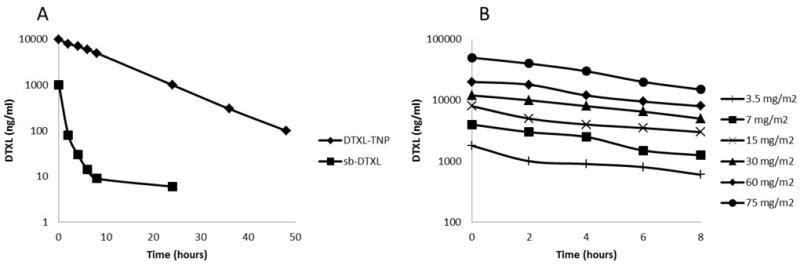
(A) Pharmacokinetic profile of DTXL-TNP in humans: A) comparison of plasma concentration time profile of DTXL-TNP at a dose of 30 mg/m2 compared to published sb-DTXL data at the same dose in patients with advanced solid tumors (n=3). (B) Plasma concentration time profile in dose-ranging studies over the first 8 hours after single dose administration of DTXL-TNP. (Figures adapted from Reference 96).
4.3.5. Non-physically Incorporated Formulations
The above mentioned formulations all incorporate drugs into the micelle through physical interaction. There are also a few formulations in clinical trials that load cancer drugs into polymeric micelles via chemical conjugation or metal complexation. Since the focus of this manuscript is on PMDDS that form by physical drug loading, we will only briefly examine the non-physically loaded formulations in clinical trials.
NC-6004 is a cis-dichlorodiammineplatinum(II) (CDDP) loaded polymer micelle formulation consisting of PEG and polymer-metal complex between poly(glutamic acid) (P(Glu)) and CDDP. The average particle size was approximately 30 nm with a drug loading of 39% 97. Clinical trials of NC-6004 began in the UK in 2006, with a dosing regimen of IV administration over 1 hour every 3 week at a dosing range of 10 mg/m2 to 120 mg/m2. Minor nephrotoxicity was observed with no significant myelosuppresion, ototoxicity or neurotoxicity. However, unexpectedly hypersensitivity reactions occurred more frequently than CDDP alone regardless of dosing level. In terms of anti-tumor efficacy, the best response was stable disease in 7 out of total of 17 patients. Currently, clinical Phase I/II studies are taking place in Singapore and Taiwan in patients with advanced or metastatic pancreatic cancer.
NK012 contains 7-ethyl-10-hydroxy-camptothecin (SN38) (aqueous solubility < 5 μg/ml)98, which is an active metabolite of irinotecan hydrochloride (CPT-11) with powerful cytotoxic effects against various cancerous cell lines in vitro. NK012 is formed by covalently conjugating SN38 with the P(Glu) segment of PEG-P(Glu) copolymer followed by self-assembling of the amphiphilic copolymer PEG-P(Glu)(SN38) in aqueous media 99. Average particle size in NK012 formulation is approximately 20 nm with a drug loading of 20% w/w. Phase I clinical trials were conducted in both Japan 100 and the US 101 with different dosing regimens. NK012 was administered IV for 30 minutes every 3 weeks with an SN38 equivalent dose range of 2 – 28 mg/m2 and 9 – 28 mg/m2 in Japan and US respectively. Prior to infusion, NK012 was diluted to total volume of 250 ml with 5% glucose solution. No dose limiting toxicity (DLT) was observed for both until 28 mg/m2 with the exception of one elevated γ-glutamyl transpeptidase at 20 mg/m2 in the Japanese trial. Non-hematologic toxicities were minimal. In comparison to CPT-11 trials, cholinergic reactions appeared less frequently. NK012 also exhibited a higher systemic exposure and slower elimination than CPT-11. 8 total partial responses were reported in Japanese and US trials. Phase II clinical trials are currently underway in Japan and US 102.
NK911 is yet another polymer micelle-based formulation in clinical trials involving chemical conjugation of drug to hydrophobic segment of amphiphilic block polymer. NK911 contains doxorubicin (DOX) conjugated to P(Asp) with PEG (molecular weight 5000 g/mol) as the hydrophilic segment. Upon reconstitution, the product contains particles with average size of 40 nm. Drug loading was dictated by the extent of DOX substitution, which for NK911 was approximately 45% substituted 103. Clinical trials began in 2001 in Japan 23 patients. Dose escalation studies showed similar toxicity spectrum as that of free DOX. Grade 3/4 hematological toxicity was observed until dose reached 50 mg/m2, at which 3 patients had grade 3 leucocytopenia, 5 had grade 3 neutropenia and 2 had grade 4 neutropenia. No DLT was observed for non-hematological toxicity. Common side effects associated with Doxil administration were rare and mild. However, AUC of NK911 was more than 1429-fold lower than Doxil at the same dose of 50 mg/m2, indicating the lower stability of NK911 than Doxil. Nevertheless, 1 patient had a partial response and 8 had stable disease. Phase II clinical trials are underway for NK911.
5. EVALUATION OF POLYMERIC MICELLES AS DELIVERY VEHICLES
Early clinical trials and abundant laboratory research have revealed several advantages of using polymeric micelles as solubilizing vehicles for delivery of poorly water-soluble drugs. These advantages include their small size, lower toxicity and advent of adverse reactions, and potentially long blood circulation times. However, several aspects of polymeric micelle-based delivery systems remain to be elucidated. Without a clearer understanding of these issues the potential of polymeric micelles as solubilizing vehicles may not be fully realized.
One of the key issues of polymeric micelles is their stability in the physiological environment. From the clinical studies mentioned previously, we have seen how insufficient stability may lead to unexpected side effects and adverse reactions. Therefore, it is critical that polymeric micelles should have enough structural integrity to remain stable in the body after administration or until they have arrived at their destination site in the case of cancer therapy. However, polymeric micelles are liable to dissociate, especially upon administration when they are diluted to a concentration below the CMC. The kinetic stability of polymeric micelles is also important to consider when evaluating PMDDS stability, because blood components can alter the kinetic stability of micelles and cause dissociation. Usefulness of PMDDS would be drastically reduced if the micelle carrier system cannot maintain the poorly water-soluble drug for the desired period of time. Chemical crosslinking of micelles is one way to prevent micelle dissociation and preserve drug inside the hydrophobic core 104, 105. However, a concern is that these crosslinked micelles are too stable and may not release sufficient amounts of drug to achieve therapeutic efficacy, or that their prolonged circulation may result in unpredictable physiological disruption 44. Biodegradable or physical crosslinking may be more suitable for drug delivery purposes by introducing reversible crosslink bonds, but so far only limited data have been made available on studies in vivo. Undoubtedly, the issue of micelle stability has to be resolved before PMDDS can achieve more clinical significance.
Biocompatibility and cytotoxicity are highly important in the development of effective PMDDS. Consequently, it is critical to gain a more comprehensive understanding of the fate of amphiphilic copolymers after administration into the body. However, this is rather challenging to do, so most of the studies on PMDDS choose to use amphiphilic copolymers with well characterized biocompatibility and minimal side effects, for example PEG-PLA, PEG-PCL or PEG-poly(Asp). As a result, the application of PMDDS becomes severely limited by the few copolymers already in use. On the other hand, studies that report novel copolymer strands for PMDDS are often focused on drug loading efficiency, drug release behavior, targeting capabilities and stability, with little attention (if any at all) devoted to biocompatibility or cytotoxicity. This approach has stunted the development of more suitable copolymers for PMDDS application. More research in this area may lead to increasing number of PMDDS with clinical significance.
6. ALTERNATIVES TO POLYMERIC MICELLES
As briefly mentioned in the beginning of this manuscript, there are currently many ways to overcome the poor aqueous solubility of BCS Class II and IV drugs, including crystal modification, amorphization, pH modification and self-emulsification. Particle size reduction to the micro- and often nanometer range is a widely explored option to solubilize poorly soluble drugs. The use of polymeric micelles is one possible approach to achieve particle size reduction. Alternatively, we can also achieve particle size reduction through the nanocrystal approach or nanoemulsions approach. In this section, we will examine these two alternative approaches separately and as a comparison to polymeric micelles.
6.1. Drug Nanocrystals
Drug nanocrystals are essentially nanoscopic crystals of the parent compound. By definition, the dimensions of nanocrystals are less than 1 μm but for practical purposes they are often less than 500 nm in dimensions. Preparation of nanocrystals takes place either via top-down or bottom-up techniques. Top-down techniques generally involve physically breaking up larger particles of the parent compound into smaller particles via high shear, high pressure or a combination of both. Typically used methods are milling and high pressure homogenization. Disadvantages associated with these methods include contamination issues from beads and equipment, and long processing times required to physically grind particles down to the nanometer range. On the other hand, bottom-up techniques such as nano-precipitation form nanocrystals by nucleation events followed by growth of drug crystals. The major problem with this approach is difficulty in controlling crystal growth and preventing further growth beyond target size. Nevertheless, a number of nanocrystal-based drug products have made it to the market and this information is summarized in Table 2.
Table 2.
Representative nanocrystal or nanoemulsion based formulations of poorly soluble drugs that are currently available commercially
| Formulation approach | Commercial name | Drug | Free drug solubility (μg/ml) | Major indication | Dosage form |
|---|---|---|---|---|---|
| Nanocrystal | Triglide® | Fenofibrate | 0.3 123 | Hypercholesterolemia | Tablet |
| Rapamune® | Sirolimus | 2.6 124 | Immunosuppresion | Tablet | |
| Emend® | Aprepitant | 3 - 7125 | Antiemetic | Capsule | |
| Tricor® | Fenofibrate | 0.3 | Hypercholesterolemia | Tablet | |
| Megace ES® | Megestrol | Antianorexia | Suspension (oral) | ||
| Invenga™ | Paliperidonepalmitate | 11 – 30 126 | Schizophrenia | Suspension (intramuscular) | |
|
| |||||
| Nanoemulsion | Estrasorb® | Estradiol | 2 127 | Menopausal vasomotor symptoms | Emulsion (topical) |
| Restasis® | Cyclosporine | 20 128 | Chronic dry eye | Emulsion (ophthalmic) | |
| SandimmunNeoral® | Cyclosporine | 20 128 | Prophylaxis of organ rejection following organ transplant | SMEDDS | |
| Norvir® | Ritonavir | 1 129 | HIV infection | SMEDDS | |
| Fortovase®* | Saquinavir | 29 130 | HIV infection | SMEDDS | |
SMEDDS: self-microemulsifying drug delivery system
Currently discontinued in the US
The basis for using nanocrystals (and indeed other particle size reduction techniques) as a solubilisation strategy for poorly soluble drugs can be explained by the Noyes-Whitney and Ostwald-Freundlich equation. According to the Noyes-Whitney equation dissolution velocity dC / dt is proportional to the concentration gradient A(Cs–Cx)/h, where A is the surface area of the solid, Cs is the concentration of the solid in the diffusion layer, Cx is the bulk concentration of solid and h is the diffusion layer thickness 106. The nanonization of drug particles leads to great enhancements in solid surface area, which the equation predicts will lead to increased dissolution velocity. An additional effect of nanonization is reduced diffusion layer thickness, which also contributes towards the increase in dissolution rate. The higher saturation solubility of nanocrystals can be explained by Ostwald-Freundlich equation, which states that log (Cs/C∞) is proportional to the inverse of r, where Cs is the saturation solubility, C∞ the solubility of bulk and r is the radius of drug particles 107. Thus smaller particles are also expected to demonstrate higher saturation solubility than parent compound.
After nanonization, drug nanocrystals are often formulated into conventional dosage forms such as tablets, capsules, pellets and suspensions for IV administration. Prior to formulation, extra steps should be taken to remove any residual organic solvents below maximum acceptable concentrations and concentrate the drug nanocrystals without compromising the physical and chemical properties of these crystals108. Techniques often used to achieve such results include freeze drying, spray drying, centrifugation and ultrafiltration. For solid dosage forms, the final product may also contain excipients such as fillers, binders, humectants, disintegration agents and lubricants to ensure that the drug nanocrystals maintain their physical, chemical and pharmaceutical properties both during storage and when administered into the body.
6.2. Nanoemulsions
Nanoemulsions consist of two immiscible liquids (usually an oil phase and an aqueous phase) where one liquid is dispersed as droplets in the other liquid. The nanoscopic droplets typically have dimensions ranging from 20 – 200 nm 109. In a broad sense, the term ‘nanoemulsions’ consist of two closely related systems termed ‘microemulsions’ and ‘submicron emulsions’ 110. The defining hallmark of microemulsions from submicron emulsions is thermodynamic stability 111. Microemulsions are described as thermodynamically stable, whereas submicron emulsions are described as approaching thermodynamic stability 112. Nanoemulsions used for solubilisation of poorly water-soluble drugs usually consist of oil phase as the dispersed phase and aqueous phase the dispersing medium, because the oil droplets can serve as reservoirs for hydrophobic drugs113. The most widely used components of oil phase are saturated and unsaturated fatty acids, fatty acid esters and soybean oils 109. In order to stabilize these drug-loaded oil droplets, nonionic or amphoteric surfactants such as poloxamers, lecithin and Tween 80 are commonly used. Nanoemulsion-based products that are commercially available are listed in Table 2.
Theoretically, the arrangement of emulsifier molecules occurs spontaneously, possibly with the aid of co-surfactants. This low energy emulsification method or so-called self-emulsifying method enables formation of nanoemulsions spontaneously when an oil/surfactant mixture is added to water or when a water/surfactant mixture is diluted with oil, and mixing of all the components occur in the final composition. Low energy emulsification method is mainly adopted for preparation of microemulsions, and hence microemulsions typically exist as microemulsion preconcentrate or so-called self-microemulsifying drug delivery systems (SMEDDS). The major disadvantage associated with this method is the lack of control over droplet size, and quite often a large size distribution is seen. In contrast to this low energy method, in some cases energy is input into the system to accelerate the re-arrangement of the surfactant molecules or to overcome a small kinetic energy barrier. Methods such as high pressure homogenization, microfluidization and ultrasonication are included in this category114. High pressure homogenization, microfluidization and ultrasonication are similar in that they all form nanoemulsions by high disruptive forces that essentially break apart the oil droplets into smaller ones. In high pressure homogenization, the disruptive forces are created by high pressure (as the name implies); in microfluidization the parent emulsion is forced through many microchannels in the central chamber of the microfluidizer; in ultrasonication, the disruptive force is supplied by ultrasonic energy. Generally, microfluidization produces nanoemulsions with the most narrow size distribution 115, 116.
Nanoemulsions have been shown to increase bioavailability compared with conventional drug suspensions. Ezetimibe, a BCS class II molecule with lipid-lowering effects, was formulated into nanoemulsions form with various surfactants and subjected to in vitro and in vivo testing 117. Plasma concentration profile of ezetimibe nanoemulsions formulation in rats showed greater improvement in drug absorption than the marketed formulation and simple drug suspension, with approximate Cmax and AUC values of 69.53 ng/ml and 948 ng hr/ml respectively for nanoemulsions formulation, 43.74 ng/ml and 222 ng hr/ml for marketed tablet formulation and 47.42 ng/ml and 294 ng hr/ml for simple drug suspension. The relative bioavailability of nanoemulsions formulation with respect to marketed tablet formulation was 477.09%, whereas with respect to simple drug suspension relative bioavailability was found to be 323.02%. The remarkable improvement in bioavailability of nanoemulsions-based formulation was attributed to increase in ezetimibe solubility and immediate dispersion in the GI tract.
6.3. Comparison of Nano-formulation Strategies
Table 3 summarizes and compares the three major nano-formulation strategies discussed in this manuscript: polymeric micelles, nanocrystals and nanoemulsions. Currently, the most established and widely used technique (especially in the industry setting) is the nanocrystal approach. This approach has also resulted in more clinically approved pharmaceutical products compared with the other two strategies. The major reasons for its popularity in industry are its excellent reproducibility and applicability to wide range of drugs with various solubility profiles, including those drugs that are poorly soluble in both water and oils. However, nanocrystal approach sometimes requires high energy input which drives up the cost of production. Moreover, the nanocrystals formed usually require extra steps to ensure stability. Unmodified nanocrystals are not suitable for cytotoxic drugs with small therapeutic indices such as anticancer agents due to rapid dissolution kinetics and lack of controlled release mechanism.
Table 3.
A comparison of nanocrystal-, nanoemulsion- and polymeric micelle-based nanonization approaches for delivery of poorly water-soluble compounds
| Approach | Advantages | Disadvantages |
|---|---|---|
| Polymeric micelles |
|
|
| Nanocrystal |
|
|
| Nanoemulsion |
|
|
Nanoemulsions offer several advantages over nanocrystals. First of all, nanoemulsions can be administered via various routes of administration with several clinically approved products for topical and ophthalmic use already on the market. High drug loading is easily achievable using this approach. Moreover, there is the potential to enhance bioavailability further by increasing permeability in GI tract (in the case of oral delivery) or mucous layer (in the case of sublingual or intranasal delivery), which would be particularly beneficial to BCS class IV compounds. The major disadvantage of nanoemulsions is lack of stability, with flocculation and coalescence often taking place during storage. The lack of controlled release mechanism is also a limitation if delivering cytotoxic compounds such as anticancer drugs.
We have discussed in depth the advantages and disadvantages of the third nano-formulation approach, polymeric micelles, in the rest of the manuscript; here we will focus on those improvements or lack thereof as compared to nanocrystals or nanoemulsions. The major advantage of polymeric micelles is its improved controlled release properties, which makes possible to deliver a variety of poorly water-soluble cytotoxic drugs for chemotherapy. In addition, it is relatively easy to design a multifunctional particle for polymeric micelles, incorporating imaging, targeting and therapeutic agents all into one vehicle, whereas this multifunctional design is rather difficult to implement for nanocrystal or nanoemulsions approach. However, major disadvantages of polymeric micelles include lack of stability, limited polymers for use and lack of suitable methods for large-scale production.
7. CONCLUSION
This manuscript has attempted to explain the fundamentals of polymeric micelles through reviews of representative literature and recent clinical trial developments. The primary purpose of this manuscript is to illustrate the potential of polymeric micelles for delivery of poorly water-soluble drugs, especially in the areas of oral delivery and in cancer therapy, which would benefit most from using polymeric micelles. Key considerations to utilize polymeric micelles’ advantages and overcome potential disadvantages have been highlighted. Lastly, other possible particle size reduction related strategies to solubilize poorly water-soluble drugs were introduced.
When designing an appropriate PMDDS for solubilisation, it is crucial to consider compatibility between core and drug. The Flory-Huggins interaction parameter is one possible way to gauge core polymer-drug compatibility. The shell of polymeric micelle acts as a physical barrier to protect the drug-loaded core. At present the most commonly used component is PEG and its various conjugates. However, use of PEG may be limited by immunological responses, unpredictable pharmacokinetics profile and lack of biodegradability. Therefore, in order to choose the appropriate amphiphilic copolymer, biocompatibility and cytotoxicity issues must be taken into consideration. Currently understanding of non-PEG based amphiphilic copolymers is limited, which prevents development of clinically significant PMDDS. Another important issue in designing PMDDS is the stability of the polymeric micelle, which is dependent on both thermodynamic and kinetic factors.
Polymeric micelles have been investigated for both oral and IV administration of poorly soluble compounds. Although oral delivery of drugs using polymeric micelles is an attractive approach, few studies have been carried out in vivo. One possible reason is the availability of other well-understood methods such as drug nanocrystals to formulate poorly soluble drugs into orally delivered formulations, while polymeric micelles are still not yet well-understood for applications in this field. As a result, most recent progress in polymeric micelles for drug delivery has been almost exclusively in the field of intravenous delivery of anticancer agents. Several polymeric micelle-based formulations for anticancer agents have made it to clinical trials and a few are commercially available. Still, in order to fully realize the potential of polymeric micelles as a solubilisation strategy for poorly water-soluble drugs, more fundamental research promoting deeper understanding of amphiphilic copolymer degradation mechanisms and micelle stability characterization in vivo is needed.
Acknowledgments
This study was supported in part by NIH through grant CA129287 and GM095879 and Ralph W. and Grace M. Showalter Research Trust Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lipinski C. Drug-like properties and the causes of poor solubility and poor permeability. Journal of Pharmacological and Toxicological Methods. 2000;44(1):235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 2.Merisko-Liversidge EM, Liversidge GG. Drug Nanoparticles: Formulating Poorly Water-Soluble Compounds. Toxicologic Pathology. 2008;36(1):43–48. doi: 10.1177/0192623307310946. [DOI] [PubMed] [Google Scholar]
- 3.Horter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Advanced Drug Delivery Reviews. 2001;46(1-3):75–87. doi: 10.1016/s0169-409x(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 4.Kawabata Y, Wada K, Nakatani M, Yamada S, Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. International Journal of Pharmaceutics. 2011;420(1):1–10. doi: 10.1016/j.ijpharm.2011.08.032. [DOI] [PubMed] [Google Scholar]
- 5.Kawakami K, Yoshikawa T, Moroto Y, Kanaoka E, Takahashi K, Nishihara Y, Masuda K. Microemulsion formulation for enhanced absorption of poorly soluble drugs: I. Prescription design. Journal of Controlled Release. 2002;81(1-2):65–74. doi: 10.1016/s0168-3659(02)00049-4. [DOI] [PubMed] [Google Scholar]
- 6.Liversidge GG, Cundy KC. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs. International Journal of Pharmaceutics. 1995;125(1):91–97. [Google Scholar]
- 7.Colin WP. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and self-microemulsifying drug delivery systems. European Journal of Pharmaceutical Sciences. 2000;11(Supplement 2 (0)):S93–S98. doi: 10.1016/s0928-0987(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 8.Keck CM, Muller RH. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. European Journal of Pharmaceutics and Biopharmaceutics. 2006;62(1):3–16. doi: 10.1016/j.ejpb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 9.Horn D, Rieger J. Organic Nanoparticles in the Aqueous Phase—Theory, Experiment, and Use. Angewandte Chemie International Edition. 2001;40(23):4330–4361. doi: 10.1002/1521-3773(20011203)40:23<4330::aid-anie4330>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 10.Muller RH, Peters K. Nanosuspensions for the formulation of poorly soluble drugs: I. Preparation by a size-reduction technique. International Journal of Pharmaceutics. 1998;160(2):229–237. [Google Scholar]
- 11.Xia D, Cui F, Piao H, Cun D, Piao H, Jiang Y, Ouyang M, Quan P. Effect of Crystal Size on the Dissolution and Oral Absorption of Nitrendipine in Rats. Pharmaceutical Research. 2010;27(9):1965–1976. doi: 10.1007/s11095-010-0200-0. [DOI] [PubMed] [Google Scholar]
- 12.Sylvestre J-P, Tang M-C, Furtos A, Leclair G, Meunier M, Leroux J-C. Nanonization of megestrol acetate by laser fragmentation in aqueous milieu. Journal of Controlled Release. 2011;149(3):273–280. doi: 10.1016/j.jconrel.2010.10.034. [DOI] [PubMed] [Google Scholar]
- 13.Jinno J-i, Kamada N, Miyake M, Yamada K, Mukai T, Odomi M, Toguchi H, Liversidge GG, Higaki K, Kimura T. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. Journal of Controlled Release. 2006;111(1-2):56–64. doi: 10.1016/j.jconrel.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 14.Kwon GS. Polymeric micelles for delivery of poorly water-soluble compounds. Critical Reviews in Therapeutic Drug Carrier Systems. 2003;20(5):357–403. doi: 10.1615/critrevtherdrugcarriersyst.v20.i5.20. [DOI] [PubMed] [Google Scholar]
- 15.Nagarajan R, Barry M. Unusual selectivity in solubilization by block copolymer micelles. Abstracts of Papers of the American Chemical Society. 1986;191:287. COLL. [Google Scholar]
- 16.Elhasi S, Astaneh R, Lavasanifar A. Solubilization of an amphiphilic drug by poly(ethylene oxide)-block-poly(ester) micelles. European Journal of Pharmaceutics and Biopharmaceutics. 2007;65(3):406–413. doi: 10.1016/j.ejpb.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 17.Rapoport N. Combined cancer therapy by micellar-encapsulated drug and ultrasound. International Journal of Pharmaceutics. 2004;277(1-2):155–162. doi: 10.1016/j.ijpharm.2003.09.048. [DOI] [PubMed] [Google Scholar]
- 18.Ruan G, Feng SS. Preparation and characterization of poly(lactic acid)-poly(ethylene glycol)-poly(lactic acid) (PLA-PEG-PLA) microspheres for controlled release of paclitaxel. Biomaterials. 2003;24(27):5037–5044. doi: 10.1016/s0142-9612(03)00419-8. [DOI] [PubMed] [Google Scholar]
- 19.Vangeyte P, Gautier S, Jerome R. About the methods of preparation of poly(ethylene oxide)-b-poly(epsilon-caprolactone) nanoparticles in water analysis by dynamic light scattering. Colloids and Surfaces Physicochemical and Engineering Aspects. 2004;242(1-3):203–211. [Google Scholar]
- 20.Meier MAR, Aerts SNH, Staal BBP, Rasa M, Schubert US. PEO-b-PCL block copolymers: Synthesis, detailed characterization, and selected micellar drug encapsulation behavior. Macromolecular Rapid Communications. 2005;26(24):1918–1924. [Google Scholar]
- 21.Stapert HR, Nishiyama N, Jiang DL, Aida T, Kataoka K. Polyion complex micelles encapsulating light-harvesting ionic dendrimer zinc porphyrins. Langmuir. 2000;16(21):8182–8188. [Google Scholar]
- 22.Woodle MC, Engbers CM, Zalipsky S. New amphipatic polymer lipid conjugates forming long-circulating reticuloendothelial system-evading liposomes. Bioconjugate Chemistry. 1994;5(6):493–496. doi: 10.1021/bc00030a001. [DOI] [PubMed] [Google Scholar]
- 23.Jiang GH, Wang Y, Zhang R, Wang RJ, Wang XH, Zhang M, Sun XK, Bao SY, Wang T, Wang S. Preparation of Redox-Sensitive Shell Cross-Linked Nanoparticles for Controlled Release of Bioactive Agents. ACS Macro Letters. 2012;1(4):489–493. doi: 10.1021/mz300063g. [DOI] [PubMed] [Google Scholar]
- 24.Tian M, Qin A, Ramireddy C, Tuzar Z, Munk P. Hybridization of block-copolymer micelles. Abstracts of Papers of the American Chemical Society. 1993;206:89. PMSE. [Google Scholar]
- 25.Arnida, Nishiyama N, Kanayama N, Jang WD, Yamasaki Y, Kataoka K. PEGylated gene nanocarriers based on block catiomers bearing ethylenediamine repeating units directed to remarkable enhancement of photochemical transfection. Journal of Controlled Release. 2006;115(2):208–215. doi: 10.1016/j.jconrel.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 26.Chanan-Khan A, Szebeni J, Savay S, Liebes L, Rafique NM, Alving CR, Muggia FM. Complement activation following first exposure to pegylated liposomal doxorubicin (Doxil): possible role in hypersensitivity reactions. Annals of Oncology. 2003;14(9):1430–1437. doi: 10.1093/annonc/mdg374. [DOI] [PubMed] [Google Scholar]
- 27.Szebeni J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology. 2005;216(2-3):106–121. doi: 10.1016/j.tox.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 28.Laverman P, Brouwers AH, Dams ETM, Oyen WJG, Storm G, Van Rooijen N, Corstens FHM, Boerman OC. Preclinical and clinical evidence for disappearance of long-circulating characteristics of polyethylene glycol liposomes at low lipid dose. Journal of Pharmacology and Experimental Therapeutics. 2000;293(3):996–1001. [PubMed] [Google Scholar]
- 29.Ishida T, Maeda R, Ichihara M, Irimura K, Kiwada H. Accelerated clearance of PEGylated liposomes in rats after repeated injections. Journal of Controlled Release. 2003;88(1):35–42. doi: 10.1016/s0168-3659(02)00462-5. [DOI] [PubMed] [Google Scholar]
- 30.Ishida T, Masuda K, Ichikawa T, Ichihara M, Irimura K, Kiwada H. Accelerated clearance of a second injection of PEGylated liposomes in mice. International Journal of Pharmaceutics. 2003;255(1-2):167–174. doi: 10.1016/s0378-5173(03)00085-1. [DOI] [PubMed] [Google Scholar]
- 31.Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Advanced Drug Delivery Reviews. 2004;56(9):1273–1289. doi: 10.1016/j.addr.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Talelli M, Rijcken CJF, van Nostrum CF, Storm G, Hennink WE. Micelles based on HPMA copolymers. Advanced Drug Delivery Reviews. 2010;62(2):231–239. doi: 10.1016/j.addr.2009.11.029. [DOI] [PubMed] [Google Scholar]
- 33.Kang N, Leroux J-C. Triblock and star-block copolymers of N-(2-hydroxypropyl)methacrylamide or N-vinyl-2-pyrrolidone and d,l-lactide: synthesis and self-assembling properties in water. Polymer. 2004;45(26):8967–8980. [Google Scholar]
- 34.Lele BS, Leroux JC. Synthesis of novel amphiphilic star-shaped poly(e-caprolactone)-block-poly(N-(2-hydroxypropyl)methacrylamide) by combination of ring-opening and chain transfer polymerization. Polymer. 2002;43(21):5595–5606. [Google Scholar]
- 35.Wei H, Cheng SX, Zhang XZ, Zhuo RX. Thermo-sensitive polymeric micelles based on poly(N-isopropylacrylamide) as drug carriers. Prog Polym Sci. 2009;34(9):893–910. [Google Scholar]
- 36.Taillefer J, Jones MC, Brasseur N, van Lier JE, Leroux JC. Preparation and characterization of ph-responsive polymeric micelles for the delivery of photosensitizing anticancer drugs. Journal of Pharmaceutical Sciences. 2000;89(1):52–62. doi: 10.1002/(SICI)1520-6017(200001)89:1<52::AID-JPS6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 37.Kohori F, Sakai K, Aoyagi T, Yokoyama M, Sakurai Y, Okano T. Preparation and characterization of thermally responsive block copolymer micelles comprising poly(N-isopropylacrylamide-b-dl-lactide) Journal of Controlled Release. 1998;55(1):87–98. doi: 10.1016/s0168-3659(98)00023-6. [DOI] [PubMed] [Google Scholar]
- 38.Soppimath KS, Tan DCW, Yang YY. pH-triggered thermally responsive polymer core-shell nanoparticles for drug delivery. Advanced Materials. 2005;17(3):318. [Google Scholar]
- 39.Wang YM, Balaji R, Quirk RP, Mattice WL. Detection of rate of the rate of exchange of chains between micelles formed by diblock copolymers in aqueous solution. Polymer Bulletin. 1992;28(3):333–338. [Google Scholar]
- 40.van Stam J, Creutz S, De Schryver FC, Jerome R. Tuning of the exchange dynamics of unimers between block copolymer micelles with temperature, cosolvents, and cosurfactants. Macromolecules. 2000;33(17):6388–6395. [Google Scholar]
- 41.Kabanov AV, Batrakova EV, Alakhov VY. Pluronic (R) block copolymers as novel polymer therapeutics for drug and gene delivery. Journal of Controlled Release. 2002;82(2-3):189–212. doi: 10.1016/s0168-3659(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 42.Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. Block copolymer micelles: preparation, characterization and application in drug delivery. Journal of Controlled Release. 2005;109(1-3):169–188. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 43.Yang L, Wu XH, Liu F, Duan YR, Li SM. Novel Biodegradable Polylactide/poly(ethylene glycol) Micelles Prepared by Direct Dissolution Method for Controlled Delivery of Anticancer Drugs. Pharmaceutical Research. 2009;26(10):2332–2342. doi: 10.1007/s11095-009-9949-4. [DOI] [PubMed] [Google Scholar]
- 44.Qiu LY, Zheng C, Jin Y, Zhu KJE. Polymeric micelles as nanocarriers for drug delivery. Expert Opinion on Therapeutic Patents. 2007;17(7):819–830. [Google Scholar]
- 45.Daugherty AL, Mrsny RJ. Transcellular uptake mechanisms of the intestinal epithelial barrier - Part one. Pharmaceutical Science & Technology Today. 1999;2(4):144–151. doi: 10.1016/s1461-5347(99)00142-x. [DOI] [PubMed] [Google Scholar]
- 46.Schiller C, Frohlich CP, Giessmann T, Siegmund W, Monnikes H, Hosten N, Weitschies W. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Alimentary Pharmacology & Therapeutics. 2005;22(10):971–979. doi: 10.1111/j.1365-2036.2005.02683.x. [DOI] [PubMed] [Google Scholar]
- 47.Francis MF, Cristea M, Winnik FM. Polymeric micelles for oral drug delivery: Why and how. Pure and Applied Chemistry. 2004;76(7-8):1321–1335. [Google Scholar]
- 48.Peng ZP, Liu XX, Tong Z. Effect of hydrophobic blocks on the aggregrate behavior of amphiphilic triblock copolymers in aqueous solution. Acta Polymerica Sinica. 2009;(9):936–941. [Google Scholar]
- 49.Ashok B, Arleth L, Hjelm RP, Rubinstein I, Önyüksel H. In vitro characterization of PEGylated phospholipid micelles for improved drug solubilization: Effects of PEG chain length and PC incorporation. Journal of Pharmaceutical Sciences. 2004;93(10):2476–2487. doi: 10.1002/jps.20150. [DOI] [PubMed] [Google Scholar]
- 50.Francis MF, Cristea M, Yang YL, Winnik FM. Engineering polysaccharide-based polymeric micelles to enhance permeability of cyclosporin a across Caco-2 cells. Pharmaceutical Research. 2005;22(2):209–219. doi: 10.1007/s11095-004-1188-0. [DOI] [PubMed] [Google Scholar]
- 51.Pierri E, Avgoustakis K. Poly(lactide)-poly(ethylene glycol) micelles as a carrier for griseofulvin. Journal of Biomedical Materials Research Part A. 2005;75A(3):639–647. doi: 10.1002/jbm.a.30490. [DOI] [PubMed] [Google Scholar]
- 52.Dabholkar RD, Sawant RM, Mongayt DA, Devarajan PV, Torchilin VP. Polyethylene glycol-phosphatidylethanolamine conjugate (PEG-PE)-based mixed micelles: Some properties, loading with paclitaxel, and modulation of P-glycoprotein-mediated efflux. International Journal of Pharmaceutics. 2006;315(1-2):148–157. doi: 10.1016/j.ijpharm.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 53.Satturwar P, Eddine MN, Ravenelle F, Leroux JC. pH-responsive polymeric micelles of poly(ethylene glycol)-b-poly(alkyl(meth)acrylate-co-methacrylic acid): Influence of the copolymer composition on self-assembling properties and release of candesartan cilexetil. European Journal of Pharmaceutics and Biopharmaceutics. 2007;65(3):379–387. doi: 10.1016/j.ejpb.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 54.Sant VP, Smith D, Leroux JC. Enhancement of oral bioavailability of poorly water-soluble drugs by poly(ethylene glycol)-block-poly(alkyl acrylate-co-methacrylic acid) self-assemblies. Journal of Controlled Release. 2005;104(2):289–300. doi: 10.1016/j.jconrel.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 55.Ould-Ouali L, Noppe M, Langlois X, Willems B, Riele PT, Timmerman P, Brewster ME, Arien A, Preat V. Self-assembling PEG-p(CL-co-TMC) copolymers for oral delivery of poorly water-soluble drugs: a case study with risperidone. Journal of Controlled Release. 2005;102(3):657–668. doi: 10.1016/j.jconrel.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 56.Norris DA, Puri N, Sinko PJ. The effect of physical barriers and properties on the oral absorption of particulates. Advanced Drug Delivery Reviews. 1998;34(2-3):135–154. doi: 10.1016/s0169-409x(98)00037-4. [DOI] [PubMed] [Google Scholar]
- 57.Francis MF, Cristea M, Winnik FM. Exploiting the vitamin B-12 pathway to enhance oral drug delivery via polymeric micelles. Biomacromolecules. 2005;6(5):2462–2467. doi: 10.1021/bm0503165. [DOI] [PubMed] [Google Scholar]
- 58.Chang T, Benet LZ, Hebert MF. The effect of water-soluble vitamin E On cyclosporine pharmacokinetics in healthy volunteers. Clinical Pharmacology & Therapeutics. 1996;59(3):297–303. doi: 10.1016/S0009-9236(96)80007-5. [DOI] [PubMed] [Google Scholar]
- 59.Batrakova EV, Li S, Elmquist WF, Miller DW, Alakhov VY, Kabanov AV. Mechanism of sensitization of MDR cancer cells by Pluronic block copolymers: Selective energy depletion. British Journal of Cancer. 2001;85(12):1987–1997. doi: 10.1054/bjoc.2001.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Batrakova EV, Han HY, Alakhov VY, Miller DW, Kabanov AV. Effects of pluronic block copolymers on drug absorption in Caco-2 cell monolayers. Pharmaceutical Research. 1998;15(6):850–855. doi: 10.1023/a:1011964213024. [DOI] [PubMed] [Google Scholar]
- 61.Zastre J, Jackson J, Bajwa M, Liggins R, Iqbal F, Burt H. Enhanced cellular accumulation of a P-glycoprotein substrate, rhodamine-123, by caco-2 cells using low molecular weight methoxypolyethylene glycol-block-polycaprolactone diblock copolymers. European Journal of Pharmaceutics and Biopharmaceutics. 2002;54(3):299–309. doi: 10.1016/s0939-6411(02)00119-4. [DOI] [PubMed] [Google Scholar]
- 62.Mathot F, Rieux AD, Arien A, Schneider YJ, Brewster M, Preat V. Transport mechanisms of mmePEG(750)P(CL-co-TMC) polymeric micelles across the intestinal barrier. Journal of Controlled Release. 2007;124(3):134–143. doi: 10.1016/j.jconrel.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 63.Konno T, Watanabe J, Ishihara K. Enhanced solubility of paclitaxel using water-soluble and biocompatible 2-methacryloyloxyethyl phosphorylcholine polymers. Journal of Biomedical Materials Research Part A. 2003;65A(2):209–214. doi: 10.1002/jbm.a.10481. [DOI] [PubMed] [Google Scholar]
- 64.Lee SC, Huh KM, Lee J, Cho YW, Galinsky RE, Park K. Hydrotropic polymeric micelles for enhanced paclitaxel solubility: In vitro and in vivo characterization. Biomacromolecules. 2007;8(1):202–208. doi: 10.1021/bm060307b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peltier S, Oger JM, Lagarce F, Couet W, Benoit JP. Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsules. Pharmaceutical Research. 2006;23(6):1243–1250. doi: 10.1007/s11095-006-0022-2. [DOI] [PubMed] [Google Scholar]
- 66.Patel RB, Patel BG, Patel MR, Bhatt KK. HPTLC Method development and validation for analysis of risperidone in formulations, and in-vitro release study. Acta Chromatographica. 2010;22(4):549–567. [Google Scholar]
- 67.Jung J-Y, Yoo SD, Lee S-H, Kim K-H, Yoon D-S, Lee K-H. Enhanced solubility and dissolution rate of itraconazole by a solid dispersion technique. International Journal of Pharmaceutics. 1999;187(2):209–218. doi: 10.1016/s0378-5173(99)00191-x. [DOI] [PubMed] [Google Scholar]
- 68.Yi Y, Yoon HJ, Kim BO, Shim M, Kim SO, Hwang SJ, Seo MH. A mixed polymeric micellar formulation of itraconazole: Characteristics, toxicity and pharmacokinetics. Journal of Controlled Release. 2007;117(1):59–67. doi: 10.1016/j.jconrel.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 69.Ould-Ouali L, Noppe M, Langlois X, Willems B, Te Riele P, Timmerman P, Brewster ME, Arien A, Preat V. Self-assembling PEG-p(CL-co-TMC) copolymers for oral delivery of poorly water-soluble drugs: a case study with risperidone. Journal of Controlled Release. 2005;102(3):657–668. doi: 10.1016/j.jconrel.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 70.Kim JY, Kim S, Papp M, Park K, Pinal R. Hydrotropic solubilization of poorly water-soluble drugs. Journal of Pharmaceutical Sciences. 2011;99(9):3953–3965. doi: 10.1002/jps.22241. [DOI] [PubMed] [Google Scholar]
- 71.Dey J, Warner IM. Spectroscopic and photophysical studies of the anticancer drug: Camptothecin. Journal of Luminescence. 1997;71(2):105–114. [Google Scholar]
- 72.O’Leary J, Muggia FM. Camptothecins: a review of their development and schedules of administration. European Journal of Cancer. 1998;34(10):1500–1508. doi: 10.1016/s0959-8049(98)00229-9. [DOI] [PubMed] [Google Scholar]
- 73.Kang J, Kumar V, Yang D, Chowdhury PR, Hohl RJ. Cyclodextrin complexation: influence on the solubility, stability, and cytotoxicity of camptothecin, an antineoplastic agent. European Journal of Pharmaceutical Sciences. 2002;15(2):163–170. doi: 10.1016/s0928-0987(01)00214-7. [DOI] [PubMed] [Google Scholar]
- 74.Barreiro-Iglesias R, Bromberg L, Temchenko M, Hatton TA, Concheiro A, Alvarez-Lorenzo C. Solubilization and stabilization of camptothecin in micellar solutions of pluronic-g-poly(acrylic acid) copolymers. Journal of Controlled Release. 2004;97(3):537–549. doi: 10.1016/j.jconrel.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 75.Buchanan CM, Buchanan NL, Edgar KJ, Lambert JL, Posey-Dowty JD, Ramsey MG, Wempe MF. Solubilization and dissolution of tamoxifen-hydroxybutenyl cyclodextrin complexes. Journal of Pharmaceutical Sciences. 2006;95(10):2246–2255. doi: 10.1002/jps.20710. [DOI] [PubMed] [Google Scholar]
- 76.Licciardi M, Cavallaro G, Di Stefano M, Pitarresi G, Fiorica C, Giammona G. New self-assembling polyaspartylhydrazide copolymer micelles for anticancer drug delivery. International Journal of Pharmaceutics. 2010;396(1-2):219–228. doi: 10.1016/j.ijpharm.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 77.Cavallaro G, Maniscalco L, Licciardi M, Giammona G. Tamoxifen-Loaded Polymeric Micelles: Preparation, Physico-Chemical Characterization and In Vitro Evaluation Studies. Macromolecular Bioscience. 2004;4(11):1028–1038. doi: 10.1002/mabi.200400089. [DOI] [PubMed] [Google Scholar]
- 78.Opanasopit P, Ngawhirunpat T, Chaidedgumjorn A, Rojanarata T, Apirakaramwong A, Phongying S, Choochottiros C, Chirachanchai S. Incorporation of camptothecin into N-phthaloyl chitosan-g-mPEG self-assembly micellar system. European Journal of Pharmaceutics and Biopharmaceutics. 2006;64(3):269–276. doi: 10.1016/j.ejpb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 79.Yu JM, Li YJ, Qiu LY, Jin Y. Polymeric nanoparticles of cholesterol-modified glycol chitosan for doxorubicin delivery: preparation and in-vitro and in-vivo characterization. Journal of Pharmacy and Pharmacology. 2009;61(6):713–719. doi: 10.1211/jpp.61.06.0003. [DOI] [PubMed] [Google Scholar]
- 80.Park K, Kim JH, Nam YS, Lee S, Nam HY, Kim K, Park JH, Kim IS, Choi K, Kim SY, Kwon IC. Effect of polymer molecular weight on the tumor targeting characteristics of self-assembled glycol chitosan nanoparticles. Journal of Controlled Release. 2007;122(3):305–314. doi: 10.1016/j.jconrel.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 81.Kataoka K, Matsumoto T, Yokoyama M, Okano T, Sakurai Y, Fukushima S, Okamoto K, Kwon GS. Doxorubicin-loaded poly(ethylene glycol)-poly(beta-benzyl-l-aspartate) copolymer micelles: their pharmaceutical characteristics and biological significance. Journal of Controlled Release. 2000;64(1-3):143–153. doi: 10.1016/s0168-3659(99)00133-9. [DOI] [PubMed] [Google Scholar]
- 82.Elsabahy M, Perron ME, Bertrand N, Yu GE, Leroux JC. Solubilization of docetaxel in poly(ethylene oxide)-block-poly(butylene/styrene oxide) micelles. Biomacromolecules. 2007;8(7):2250–2257. doi: 10.1021/bm070226v. [DOI] [PubMed] [Google Scholar]
- 83.Kim SC, Kim DW, Shim YH, Bang JS, Oh HS, Kim SW, Seo MH. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. Journal of Controlled Release. 2001;72(1-3):191–202. doi: 10.1016/s0168-3659(01)00275-9. [DOI] [PubMed] [Google Scholar]
- 84.Kim TY, Kim DW, Chung JY, Shin SG, Kim SC, Heo DS, Kim NK, Bang YJ. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clinical Cancer Research. 2004;10(11):3708–3716. doi: 10.1158/1078-0432.CCR-03-0655. [DOI] [PubMed] [Google Scholar]
- 85.Burt HM, Zhang XC, Toleikis P, Embree L, Hunter WL. Development of copolymers of poly(D,L-lactide) and methoxypolyethylene glycol as micellar carriers of paclitaxel. Colloids and Surfaces B-Biointerfaces. 1999;16(1-4):161–171. [Google Scholar]
- 86.Zhang XC, Burt HM, VonHoff D, Dexter D, Mangold G, Degen D, Oktaba AM, Hunter WL. An investigation of the antitumour activity and biodistribution of polymeric micellar paclitaxel. Cancer Chemotherapy and Pharmacology. 1997;40(1):81–86. doi: 10.1007/s002800050630. [DOI] [PubMed] [Google Scholar]
- 87.Chen H, Kim S, He W, Wang H, Low PS, Park K, Cheng JX. Fast release of lipophilic agents from circulating PEG-PDLLA micelles revealed by in vivo Forster resonance energy transfer imaging. Langmuir. 2008;24(10):5213–5217. doi: 10.1021/la703570m. [DOI] [PubMed] [Google Scholar]
- 88.Ibrahim NK, Samuels B, Page R, Doval D, Patel KM, Rao SC, Nair MK, Bhar P, Desai N, Hortobagyi GN. Multicenter phase II trial of ABI-007, an albumin-bound paclitaxel, in women with metastatic breast cancer. Journal of Clinical Oncology. 2005;23(25):6019–6026. doi: 10.1200/JCO.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 89.Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. Journal of Clinical Oncology. 2005;23(31):7794–7803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- 90.Green MR, Manikhas GM, Orlov S, Afanasyev B, Makhson AM, Bhar P, Hawkins MJ. Abraxane((R)), a novel Cremophor((R))-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Annals of Oncology. 2006;17(8):1263–1268. doi: 10.1093/annonc/mdl104. [DOI] [PubMed] [Google Scholar]
- 91.Matsumura Y, Hamaguchi T, Ura T, Muro K, Yamada Y, Shimada Y, Shirao K, Okusaka T, Ueno H, Ikeda M, Watanabe N. Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle-encapsulated doxorubicin. British Journal of Cancer. 2004;91(10):1775–1781. doi: 10.1038/sj.bjc.6602204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kato K, Hamaguchi T, Yasui H, Okusaka T, Ueno H, Ikeda M, Shirao K, Shimada Y, Nakahama H, Muro K, Matsumura Y. Phase I study of NK105, a paclitaxel-incorporating micellar nanoparticle, in patients with advanced cancer. Journal of Clinical Oncology. 2006;24(18):83S–83S. doi: 10.1038/sj.bjc.6603855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Danson S, Ferry D, Alakhov V, Margison J, Kerr D, Jowle D, Brampton M, Halbert G, Ranson M. Phase I dose escalation and pharmacokinetic study of pluronic polymer-bound doxorubicin (SP 1049C) in patients with advanced cancer. British Journal of Cancer. 2004;90(11):2085–2091. doi: 10.1038/sj.bjc.6601856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alakhov V, Klinski E, Li SM, Pietrzynski G, Venne A, Batrakova E, Bronitch T, Kabanov A. Block copolymer-based formulation of doxorubicin. From cell screen to clinical trials. Colloids and Surfaces B-Biointerfaces. 1999;16(1-4):113–134. [Google Scholar]
- 95.Valle JW, Armstrong A, Newman C, Alakhov V, Pietrzynski G, Brewer J, Campbell S, Corrie P, Rowinsky EK, Ranson M. A phase 2 study of SP1049C, doxorubicin in P-glycoprotein-targeting pluronics, in patients with advanced adenocarcinoma of the esophagus and gastroesophageal junction. Investigational New Drugs. 2011;29(5):1029–1037. doi: 10.1007/s10637-010-9399-1. [DOI] [PubMed] [Google Scholar]
- 96.Hrkach J, Von Hoff D, Ali MM, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, Low S, McDonnell K, Peeke E, Retnarajan B, Sabnis A, Schnipper E, Song JJ, Song YH, Summa J, Tompsett D, Troiano G, Hoven TV, Wright J, LoRusso P, Kantoff PW, Bander NH, Sweeney C, Farokhzad OC, Langer R, Zale S. Preclinical Development and Clinical Translation of a PSMA-Targeted Docetaxel Nanoparticle with a Differentiated Pharmacological Profile. Science Translational Medicine. 2012;4(128) doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 97.Uchino H, Matsumura Y, Negishi T, Koizumi F, Hayashi T, Honda T, Nishiyama N, Kataoka K, Naito S, Kakizoe T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. British Journal of Cancer. 2005;93(6):678–687. doi: 10.1038/sj.bjc.6602772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Meyer-Losic F, Nicolazzi C, Quinonero J, Ribes F, Michel M, Dubois V, de Coupade C, Boukaissi M, Chene AS, Tranchant I, Arranz V, Zoubaa I, Fruchart J-S, Ravel D, Kearsey J. DTS-108, A Novel Peptidic Prodrug of SN38: In vivo Efficacy and Toxicokinetic Studies. Clinical Cancer Research. 2008;14(7):2145–2153. doi: 10.1158/1078-0432.CCR-07-4580. [DOI] [PubMed] [Google Scholar]
- 99.Koizumi F, Kitagawa M, Negishi T, Onda T, Matsumoto S, Hamaguchi T, Matsumura Y. Novel SN-38-incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor-secreting bulky tumors. Cancer Research. 2006;66(20):10048–10056. doi: 10.1158/0008-5472.CAN-06-1605. [DOI] [PubMed] [Google Scholar]
- 100.Hamaguchi T, Doi T, Eguchi-Nakajima T, Kato K, Yamada Y, Shimada Y, Fuse N, Ohtsu A, Matsumoto S, Takanashi M, Matsumura Y. Phase I Study of NK012, a Novel SN-38-Incorporating Micellar Nanoparticle, in Adult Patients with Solid Tumors. Clinical Cancer Research. 2010;16(20):5058–5066. doi: 10.1158/1078-0432.CCR-10-0387. [DOI] [PubMed] [Google Scholar]
- 101.Burris HA. A phase I dose-escalation study of NK 012. J Clin Oncol. 2008;26:2538. [Google Scholar]
- 102.Nagano T, Yasunaga M, Goto K, Kenmotsu H, Koga Y, Kuroda J, Nishimura Y, Sugino T, Nishiwaki Y, Matsumura Y. Synergistic antitumor activity of the SN-38-incorporating polymeric micelles NK012 with S-1 in a mouse model of non-small cell lung cancer. International Journal of Cancer. 2010;127(11):2699–2706. doi: 10.1002/ijc.25282. [DOI] [PubMed] [Google Scholar]
- 103.Nakanishi T, Fukushima S, Okamoto K, Suzuki M, Matsumura Y, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Development of the polymer micelle carrier system for doxorubicin. Journal of Controlled Release. 2001;74(1-3):295–302. doi: 10.1016/s0168-3659(01)00341-8. [DOI] [PubMed] [Google Scholar]
- 104.Hu FQ, Ren GF, Yuan H, Du YZ, Zeng S. Shell cross-linked stearic acid grafted chitosan oligosaccharide self-aggregated micelles for controlled release of paclitaxel. Colloids and Surfaces B-Biointerfaces. 2006;50(2):97–103. doi: 10.1016/j.colsurfb.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 105.Zhang L, Katapodi K, Davis TP, Barner-Kowollik C, Stenzel MH. Using the reversible addition-fragmentation chain transfer process to synthesize core-crosslinked micelles. Journal of Polymer Science Part a-Polymer Chemistry. 2006;44(7):2177–2194. [Google Scholar]
- 106.Butcher JC, Garg S, Kim D, Sharma P. A modified approach to predict dissolution and absorption of polydisperse powders. Pharmaceutical Research. 2008;25(10):2309–2311. doi: 10.1007/s11095-008-9630-3. [DOI] [PubMed] [Google Scholar]
- 107.Muller RH, Peters K. Nanosuspensions for the formulation of poorly soluble drugs - I. Preparation by a size-reduction technique. International Journal of Pharmaceutics. 1998;160(2):229–237. [Google Scholar]
- 108.Van Eerdenbrugh B, Van den Mooter G, Augustijns P. Top-down production of drug nanocrystals: Nanosuspension stabilization, miniaturization and transformation into solid products. International Journal of Pharmaceutics. 2008;364(1):64–75. doi: 10.1016/j.ijpharm.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 109.Chen HB, Khemtong C, Yang XL, Chang XL, Gao JM. Nanonization strategies for poorly water-soluble drugs. Drug Discovery Today. 2011;16(7-8):354–360. doi: 10.1016/j.drudis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 110.Lu Y, Qi JP, Wu W. Absorption, Disposition and Pharmacokinetics of Nanoemulsions. Current Drug Metabolism. 2012;13(4):396–417. doi: 10.2174/138920012800166544. [DOI] [PubMed] [Google Scholar]
- 111.Jadhav KR, Shaikh IM, Ambade KW, Kadam VJ. Applications of microemulsion based drug delivery system. Current Drug Delivery. 2006;3(3):267–273. doi: 10.2174/156720106777731118. [DOI] [PubMed] [Google Scholar]
- 112.Tadros T, Izquierdo R, Esquena J, Solans C. Formation and stability of nano-emulsions. Advances in Colloid and Interface Science. 2004;108:303–318. doi: 10.1016/j.cis.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 113.Sonneville-Aubrun O, Simonnet JT, L’Alloret F. Nanoemulsions: a new vehicle for skincare products. Advances in Colloid and Interface Science. 2004;108:145–149. doi: 10.1016/j.cis.2003.10.026. [DOI] [PubMed] [Google Scholar]
- 114.Constantinides PP, Chaubal MV, Shorr R. Advances in lipid nanodispersions for parenteral drug delivery and targeting. Advanced Drug Delivery Reviews. 2008;60(6):757–767. doi: 10.1016/j.addr.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 115.Jahn A, Reiner JE, Vreeland WN, DeVoe DL, Locascio LE, Gaitan M. Preparation of nanoparticles by continuous-flow microfluidics. Journal of Nanoparticle Research. 2008;10(6):925–934. [Google Scholar]
- 116.Ganta S, Devalapally H, Baguley BC, Garg S, Amiji M. Microfluidic preparation of chlorambucil nanoemulsion formulations and evaluation of cytotoxicity and pro-apoptotic activity in tumor cells. Journal of Biomedical Nanotechnology. 2008;4(2):165–173. [Google Scholar]
- 117.Bali V, Ali M, Ali J. Study of surfactant combinations and development of a novel nanoemulsion for minimising variations in bioavailability of ezetimibe. Colloids and Surfaces B-Biointerfaces. 2010;76(2):410–420. doi: 10.1016/j.colsurfb.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 118.Lee KS, Chung HC, Im SA, Park YH, Kim CS, Kim SB, Rha SY, Lee MY, Ro J. Multicenter phase II trial of Genexol-PM, a Cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Research and Treatment. 2008;108(2):241–250. doi: 10.1007/s10549-007-9591-y. [DOI] [PubMed] [Google Scholar]
- 119.Kim DW, Kim SY, Kim HK, Kim SW, Shin SW, Kim JS, Park K, Lee MY, Heo DS. Multicenter phase II trial of Genexol-PM, a novel Cremophor-free, polymeric micelle formulation of paclitaxel, with cisplatin in patients with advanced non-small-cell lung cancer. Annals of Oncology. 2007;18(12):2009–2014. doi: 10.1093/annonc/mdm374. [DOI] [PubMed] [Google Scholar]
- 120.Hamaguchi T, Kato K, Yasui H, Morizane C, Ikeda M, Ueno H, Muro K, Yamada Y, Okusaka T, Shirao K, Shimada Y, Nakahama H, Matsumura Y. A phase I and pharmacokinetic study of NK105, a paclitaxel-incorporating micellar nanoparticle formulation. British Journal of Cancer. 2007;97(2):170–176. doi: 10.1038/sj.bjc.6603855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nishiyama N, Okazaki S, Cabral H, Miyamoto M, Kato Y, Sugiyama Y, Nishio K, Matsumura Y, Kataoka K. Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Research. 2003;63(24):8977–8983. [PubMed] [Google Scholar]
- 122.Plummer R, Wilson RH, Calvert H, Boddy AV, Griffin M, Sludden J, Tilby MJ, Eatock M, Pearson DG, Ottley CJ, Matsumura Y, Kataoka K, Nishiya T. A Phase I clinical study of cisplatin-incorporated polymeric micelles (NC-6004) in patients with solid tumours. British Journal of Cancer. 2011;104(4):593–598. doi: 10.1038/bjc.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li XM, Gu L, Xu YL, Wang YL. Preparation of fenofibrate nanosuspension and study of its pharmacokinetic behavior in rats. Drug Development and Industrial Pharmacy. 2009;35(7):827–833. doi: 10.1080/03639040802623941. [DOI] [PubMed] [Google Scholar]
- 124.Preetham AC, Satish CS. Formulation of a Poorly Water-Soluble Drug Sirolimus in Solid Dispersions to Improve Dissolution. Journal of Dispersion Science and Technology. 2011;32(6):778–783. [Google Scholar]
- 125.Shono Y, Jantratid E, Kesisoglou F, Reppas C, Dressman JB. Forecasting in vivo oral absorption and food effect of micronized and nanosized aprepitant formulations in humans. European Journal of Pharmaceutics and Biopharmaceutics. 2010;76(1):95–04. doi: 10.1016/j.ejpb.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 126.Patel RB, Patel MR, Bhatt KK, Patel BG. HPTLC method development and validation: Quantification of paliperidone in formulations and in vitro release study. Analytical Methods. 2010;2(5):525–531. [Google Scholar]
- 127.Sheu MT, Chen SY, Chen LC, Ho HO. Influence of micelle solubilization by tocopheryl polyethylene glycol succinate (TPGS) on solubility enhancement and percutaneous penetration of estradiol. Journal of Controlled Release. 2003;88(3):355–368. doi: 10.1016/s0168-3659(02)00492-3. [DOI] [PubMed] [Google Scholar]
- 128.Onoue S, Sato H, Ogawa K, Kawabata Y, Mizumoto T, Yuminoki K, Hashimoto N, Yamada S. Improved dissolution and pharmacokinetic behavior of cyclosporine A using high-energy amorphous solid dispersion approach. International Journal of Pharmaceutics. 2010;399(1-2):94–101. doi: 10.1016/j.ijpharm.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 129.Sinha S, Ali M, Baboota S, Ahuja A, Kumar A, Ali J. Solid Dispersion as an Approach for Bioavailability Enhancement of Poorly Water-Soluble Drug Ritonavir. AAPS PharmSciTech. 2010;11(2):518–527. doi: 10.1208/s12249-010-9404-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dodiya SS, Chavhan SS, Sawant KK, Korde AG. Solid lipid nanoparticles and nanosuspension formulation of Saquinavir: preparation, characterization, pharmacokinetics and biodistribution studies. Journal of Microencapsulation. 2011;28(6):515–527. doi: 10.3109/02652048.2011.590612. [DOI] [PubMed] [Google Scholar]