

Abstract
The mitochondrial enzyme 25-hydroxyvitamin D 1α-hydroxylase, which is encoded by the CYP27B1 gene, converts 25OHD to the biological active form of vitamin D, 1,25-dihydroxyvitamin D (1,25(OH)2D). Renal 1α-hydroxylase activity is the principal determinant of the circulating 1,25(OH)2D concentration and enzyme activity is tightly regulated by several factors. Fibroblast growth factor-23 (FGF-23) decreases serum 1,25(OH)2D concentrations by suppressing CYP27B1 mRNA abundance in mice. In extra-renal tissues, 1α-hydroxylase is responsible for local 1,25(OH)2D synthesis, which has important paracrine actions, but whether FGF-23 regulates CYP27B1 gene expression in extra-renal tissues is unknown. We sought to determine whether FGF-23 regulates CYP27B1 transcription in the kidney and whether extra-renal tissues are target sites for FGF-23-induced suppression of CYP27B1. In HEK293 cells transfected with the human CYP27B1 promoter, FGF-23 suppressed promoter activity by 70%, and the suppressive effect was blocked by CI-1040, a specific inhibitor of extracellular signal regulated kinase 1/2. To examine CYP27B1 transcriptional activity in vivo, we crossed fgf-23 null mice with mice bearing the CYP27B1 promoter-driven luciferase transgene (1α-Luc). In the kidney of FGF-23 null/1α-Luc mice, CYP27B1 promoter activity was increased by 3-fold compared to that in wild-type/1α-Luc mice. Intraperitoneal injection of FGF-23 suppressed renal CYP27B1 promoter activity and protein expression by 26% and 60% respectively, and the suppressive effect was blocked by PD0325901, an ERK1/2 inhibitor. These findings provide evidence that FGF-23 suppresses CYP27B1 transcription in the kidney. Furthermore, we demonstrate that in FGF-23 null/1α-Luc mice, CYP27B1 promoter activity and mRNA abundance are increased in several extra-renal sites. In the heart of FGF-23 null/1α-Luc mice, CYP27B1 promoter activity and mRNA were 2- and 5-fold higher, respectively, than in control mice. We also observed a 3- to 10-fold increase in CYP27B1 mRNA abundance in the lung, spleen, aorta and testis of FGF-23 null/1α-Luc mice. Thus, we have identified novel extra-renal target sites for FGF-23-mediated regulation of CYP27B1.
Introduction
Vitamin D is activated by sequential hydroxylations in the liver and kidney to produce the active form of vitamin D; 1,25 dihydroxyvitamin D (1,25(OH)2D), which plays a central role in calcium and phosphorus homeostasis, skeletal growth and mineralization, and tissue differentiation [1]. In the liver, the 25-hydroxylation of vitamin D is catalyzed by several enzymes including the mitochondrial enzyme CYP27A1 [2] and the microsomal enzyme CYP2R1 [3]. Hepatic synthesis of 25 hydroxyvitamin D (25OHD) is dependent on substrate concentration, which is determined by dietary intake of vitamin D and exposure to sunlight. In the kidney, 1,25(OH)2D is produced by the mitochondrial enzyme, 25-hydroxyvitamin D 1α-hydroxylase (1α-hydroxylase) [4]–[9]. 1α-hydroxylation is the rate-limiting step in the bioactivation of vitamin D. Human, rat and mouse 1α-hydroxylase (CYP27B1) genes have been cloned [10]–[15], and inactivating mutations in CYP27B1 have been identified in patients with autosomal recessive 1α-hydroxylase deficiency, also known as vitamin D dependent rickets type I A [10], [11], [16]–[18]. Vitamin D 24-hydroxylase (24-hydroxylase) catalyzes the 24-hydroxylation of 25OHD to 24,25(OH)2D and of 1,25(OH)2D to 1,24,25(OH)3D; both reactions initiate the metabolic inactivation of vitamin D via the C24-oxidation pathway [19], [20]. 24-hydroxylase activity is found in kidney, intestine, skin, macrophages and other tissues [21]. Circulating concentrations of 1,25(OH)2D primarily reflect its synthesis in the kidney; however, 1α-hydroxylase activity is also found in keratinocytes, macrophages, osteoblasts and other tissues [22]–[24]. Bilateral nephrectomy and chronic kidney disease result in low circulating 1,25(OH)2D concentrations, suggesting that extra-renal synthesis of 1,25(OH)2D contributes little to the maintenance of normal serum 1,25(OH)2D concentrations [25]. However, 1α-hydroxylase activity in extra-renal tissues is an important source of 1,25(OH)2D for its autocrine and paracrine functions in bone, skin, macrophages, prostate, parathyroid and several other tissues [26]–[30].
In the renal proximal tubule, 1α-hydroxylase activity is tightly regulated by parathyroid hormone (PTH), calcium (Ca), phosphorus (Pi) and 1,25(OH)2D [31], [32]. Recently, we and others demonstrated that fibroblast growth factor-23 (FGF-23), a bone-derived circulating peptide, is a critical determinant of the renal metabolism of vitamin D [33]–[37]. FGF-23 suppresses renal 1,25(OH)2D production by suppressing CYP27B1 and stimulating 24-hydroxylase (CYP24A1) mRNA expression in the kidney in normal mice [35], [37]. We showed in cultured human and mouse renal proximal tubule epithelia that FGF-23 can directly suppress CYP27B1 expression and that this effect is mediated by activation of the mitogen activated protein kinase signaling pathway (MAPK) [35]. However, whether FGF-23 regulates CYP27B1 expression by transcriptional or post-transcriptional mechanisms is unknown.
The human CYP27B1 gene is located on chromosome 12q13.1-q13.3 and consists of 9 exons spanning approximately 6.5 kb in length [11]. Regulatory elements within 1.5 kb of the 5′ flanking region of CYP27B1 are important for regulation by PTH, calcitonin, and 1,25(OH)2D [38]–[40] in renal tissue. Whether FGF-23 regulates CYP27B1 in extra-renal sites is unknown. To understand further the molecular mechanisms by which FGF-23 regulates CYP27B1 and the sites involved, we examined transcriptional regulation of CYP27B1 by FGF-23 both in vitro and in vivo. In the present study, we show that CYP27B1 transcription is regulated by FGF-23 in the kidney and in extra-renal sites and thus identify novel tissue targets for FGF-23 action.
Materials and Methods
Cell Culture and Transfection
Human embryonic kidney (HEK-293) cells stably transfected with the transmembrane (Tm) form of mouse klotho-pEF1 expression vector [41] (a kind gift from Makoto Kuro-o, University of Texas Southwestern, Dallas, TX), were grown and maintained as previously described [41]. Klotho is an obligatory co-factor for FGF-23 and confers tissue specificity for it actions in target tissues [42]. It is well established that FGF-23-dependent signal activation in HEK-293 cells is dependent on Tm klotho [41], [42]. Based on this prior work, we chose to perform all our experiments in HEK293 cells stably transfected with Tm klotho. HEK-293 cells were plated at 130,000/well in 24-well plates, in DMEM H-21 with 10% FBS (Hyclone, Waltham, MA). At 80% confluence, cells were transiently transfected with 1 µg DNA/well of pGL-3 basic vector or CYP27B1 promoter-driven firefly luciferase reporter plasmid using 2 µl/well of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) as per manufacturer’s protocol. Cells were co-transfected with 50 ng/well of pRL-CMV Vector (Promega, Madison, WI) containing the Renilla luciferase gene driven by the CMV promoter to normalize for transfection efficiency. HEK-293 cells were then treated with varying doses of FGF-23 for 21 hours in serum-free media. For the inhibitor experiments, CI-1040 (Pfizer, New York, NY), a selective MAPK kinase (MEK) inhibitor, that blocks phosphorylation of extracellular signal regulated kinase 1/2 (ERK1/2) was added 1 hour before treatment with FGF-23. Mouse aortic vascular smooth muscle cells (VSMC) were purchased from ATCC, VA, cultured in 6-well plates in DMEM H-21 with 10% FBS. At 80% confluence, cells were treated with FGF-23 for 5–30 minutes to determine activation of MAPK signaling pathway, and for 21 hours, to determine CYP27B1 mRNA expression.
Plasmid Constructs
The cloning and sequencing of the human CYP27B1 gene, and the localization of its transcriptional start site 62 bp upstream from ATG translational start site have been described [11]. A 5′ fragment extending from the EcoRI site at –345 bp to the translational start site was prepared by PCR and cloned into pGL-3-basic vector (Promega) containing the luciferase reporter gene, in-frame with the ATG site to obtain the 409 bp CYP27B1 promoter plasmid. To obtain longer fragments of 5′ flanking DNA, human genomic DNA was digested with BamHI, cloned into pcDNA 3.1 (Invitrogen Carlsbad, CA) and screened with the 5′ EcoRI/NheI to identify a clone with additional 5′ flanking DNA. The BamHI/EcoRI fragment of this clone extending from –1151 bp to –345 bp was inserted upstream from the –345 bp in the pGL-3-basic vector to obtain the “full-length” 1576 bp CYP27B1 promoter plasmid. Subsequent digestions with NheI, SmaI, HindIII, TthIII1 and Xho1 yielded additional deletion constructs containing −1171, −926, –789 and −200 bp, respectively. Egr-1 promoter driven luciferase plasmid was a kind gift from Kirin Pharma, Japan. pRL-CMV plasmid was purchased from Promega.
FGF-23
Recombinant human FGF-23(R176Q) (Genzyme Corporation, Framingham, MA) contains a mutation in its proprotein convertase (furin) proteolytic cleavage site in which arginine at position 176 is replaced by glutamine. This mutation is identical to that found in patients with autosomal dominant hypophosphatemic rickets and renders the FGF-23(R176Q) protein resistant to proteolytic processing [43]. FGF-23(R176Q) has enhanced biological potency in vivo and in vitro when compared with that of native FGF-23 [44]. Hereforth, recombinant human FGF-23(R176Q) will be referred to as FGF-23.
Animals
pCYP27B1(–1501 bp)-Luc transgenic mice were generated and bred as described previously [45]. The pGL3-pCYP27B1-luciferase (pGL3-pCYP27B1-luc) plasmid construct consists of the luciferase reporter gene flanked by the 1501 bp promoter region of the human 1α-hydroxylase gene. Briefly, the transgenic mice were generated by pro-nuclear injection of the purified pGL3-pCYP27B1-luc construct into CBA/C57 embryos to generate founder mice and further backcrossed more than 10 generations into a C57/Bl6 background. Hereforth, the pCYP27B1 (–1501 bp)-Luc transgenic mice will be referred to as 1α-Luc transgenic mice. The 1α-Luc transgenic mouse model is ideal to study the effect of various physiological stimuli; these mice demonstrate an appropriate increase in CYP27B1 promoter-driven luciferase activity in response to dietary restriction of calcium and vitamin D. In addition, promoter activity in these mice closely correlates with CYP27B1 mRNA and protein expression in renal and extra-renal tissues [46], [47].
Fgf-23 null mice (fgf-23−/−) were generated and bred as previously described [48], [49]. At an early age, fgf-23−/− mice develop hyperphosphatemia and increased serum 1,25 (OH)2D concentrations, the latter due to greatly increased renal expression of CYP27B1 mRNA and thereby increased renal 1,25(OH)2D synthesis. To generate the double mutant mouse, we crossbred mice heterozygous for fgf-23 gene (fgf-23+/−) and 1α-Luc gene (1α-Luc+/−) to obtain fgf-23+/−/1α-Luc+/− transgenic mice. Fgf-23+/−/1α-Luc+/− mice were mated with fgf-23+/−/1α-Luc−/− to obtain fgf-23−/−/1α-Luc+/− transgenic mice. Fgf-23+/+/1α-Luc+/−, fgf-23+/−/1α-Luc+/− and fgf-23−/−/1α-Luc+/− transgenic were studied at 28-35 days of age. To determine the effect of FGF-23 and MAPK inhibitor treatment on CYP27B1 expression in vivo, fgf-23−/−/1α-Luc+/− mice were fed a constant diet containing 0.6% phosphorus and 0.6% calcium (Teklad diet 98243, Harlan Laboratories, Madison, WI) and received the MEK inhibitor, PD0325901, 12.5 mg/kg/dose, or vehicle orally for 4 days. On day 5, a single intraperitoneal injection of FGF-23 (150 ng/g) was administered and mice were sacrificed 5 hours later and tissues were removed and frozen for subsequent preparation of total RNA and protein. To determine whether regulation of CYP27B1 expression was specific to FGF-23 action, fgf-23+/+/1α-Luc+/− mice were administered a single intraperitoneal injection of basic FGF (250 ng/g) and mice were sacrificed 5 hours later. All procedures were approved by the Committee on Animal Research, University of California San Francisco.
Real-Time PCR
Total RNA was isolated from tissues using TRIzol reagent (Invitrogen). cDNA was synthesized using 1×PCR buffer, 7.5 mM MgCl2, 1 mM dNTP, 5 µM random primers, and 2.5 U/L Moloney murine leukemia virus reverse transcriptase (Invitrogen) at the following temperatures: 25°C for 10 min, 48°C for 40 min, and 95°C for 5 min. The probes and primers for mouse CYP27B1, CYP24A1 and β-glucuronidase (Gus) were custom designed as previously described [50]. The mRNA abundance of the gene of interest, expressed relative to that of Gus mRNA, was quantitated by real-time PCR using the ABI 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA) as previously described [35]. The threshold cycle (Ct) at which a statistically significant increase in signal above background fluorescence was determined, and the Ct values for the gene of interest were normalized to Ct values for Gus.
Western Blot Analysis
Mouse kidney total protein (25 µg), and renal mitochondrial protein (35 µg) were isolated, fractionated on 10% SDS-polyacrylamide gel and transferred to polyvinilidine difluoride membranes (PVDF) membranes (BioRad, Hercules, CA) as previously described [50]. Activation of MAP kinase signaling in the kidney was determined by detection of pERK1/2 protein using anti-phospho-ERK1/2 monoclonal antibody (Cell Signaling Technology, Danvers, MA) and visualized by chemiluminescence (Supersignal West Dura, Pierce Biotechnology, Rockford, IL). Equal protein loading was determined using a mouse anti-ERK 2 monoclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). For detection of 1α-hydroxylase protein, membranes were probed with rabbit anti-1α-hydroxylase polyclonal antibody (1∶1500 dilution) [51]. Equal protein loading was determined using a rabbit anti-β-actin polyclonal antibody (1∶6500) (Cell Signaling Technology, Danvers, MA). The membranes were subsequently blotted using an infrared (IR) labeled secondary antibody, IR Dye 800 CW and IR Dye 680 (1∶25,000) (Li-Cor Biosciences, Lincoln, NE). The bound complex was detected using Odyssey Infrared Imaging System (LiCor Biotechnology). The images were analyzed using the Odyssey Application Software to obtain the integrated fluorescence intensities from IR detection. The ratio of fluorescence intensities of the protein of interest was normalized to that of the loading control protein and the data were plotted on a bar graph.
Luciferase Activity
HEK 293 cells were lysed using passive lysis buffer from the dual luciferase assay kit (Promega, Madison, WI). 10 µl of lysate was used to quantify firefly luciferase activity using a TD 20/20 luminometer (Turner Design Instruments, Sunnyvale, CA). Luciferase activity was normalized by calculating the ratio of firefly luciferase to renilla luciferase activity per sample and results expressed as fold change of FGF-23-treated to vehicle-treated samples. Experiments were performed in triplicate and repeated 3 times. For mouse samples, tissues were homogenized in passive lysis buffer and centrifuged at 15,000 g for 5 min at 4°C as previously described [47]. Firefly luciferase activity was measured in the supernatant in duplicate using the Luciferase Assay System (Promega). A volume of 100 µl of supernatant was combined with 50 µl of Luciferase Assay Reagent and the luminescence signal was measured using the Victor luminometer (PERKinElmer, Waltham, MA). All luciferase measurements were corrected for auto-luminescence by subtracting the luminescence signal measured in a mixture of 100 µl of passive lysis buffer and 50 µl of Luciferase Assay Reagent. The total protein content of the tissue supernatants was measured using the BCA Protein Assay Kit (Bio-Rad). The activity of the 1α-hydroxylase promoter was expressed as the enzymatic luciferase activity per µg of total cellular protein.
Serum Biochemistry
Serum Ca and Pi concentrations were determined using kits from Stanbio Laboratories (San Antonio, TX). Serum intact PTH concentrations were determined using EIA kits from Immutopics International (San Clemente, CA).
Statistical Analysis
Data are expressed as mean ± SEM. The significance of differences between two groups was analyzed by student t test or between multiple groups by analysis of variance (ANOVA). Post-hoc analyses were performed using the Bonferroni method. P value <0.05 was considered as statistically significant.
Results
Basal Activity of the Human CYP27B1 Promoter
We generated a 1576 bp CYP27B1 promoter fragment from PCR reactions using appropriate primers and human genomic DNA as template and we tested the functional activity of the CYP27B1 promoter in transiently transfected HEK-293 cells. Luciferase activity of the full-length (−1576 bp) promoter was substantially (170-fold) higher than that of the promoterless vector, and increased luciferase activity was sustained in the −1.1 kb and −926 bp deletion constructs (P<0.05) (Fig. 1A). Promoter activity decreased by 70% in cells transfected with the 789bp deletion construct as compared to the full-length promoter, suggesting the presence of an enhancer between −926 bp and −789 bp. Promoter activity increased with further shortening of the promoter to −409 bp suggesting the presence of a silencer between −789 and −409 bp. With the −200 bp construct, promoter activity was 50-fold higher than the vector control but considerably reduced compared to the full-length promoter activity (P<0.05). We then determined the response of the CYP27B1 promoter to stimulation by forskolin which increases intracellular cAMP and stimulates 1α-hydroxylase promoter activity in opossum kidney cell cultures [52]. With forskolin treatment, activity of the full-length CYP27B1 promoter increased by 76% compared to the vehicle-treated cells. The stimulatory effect of forskolin was sustained in all the deletion constructs (Fig. 1B).
Figure 1. A. Basal activity of the human CYP27B1 promoter constructs (−1576 bp, −1170 bp, −926 bp, −789 bp, −409 bp, −200 bp) in HEK293 cells.
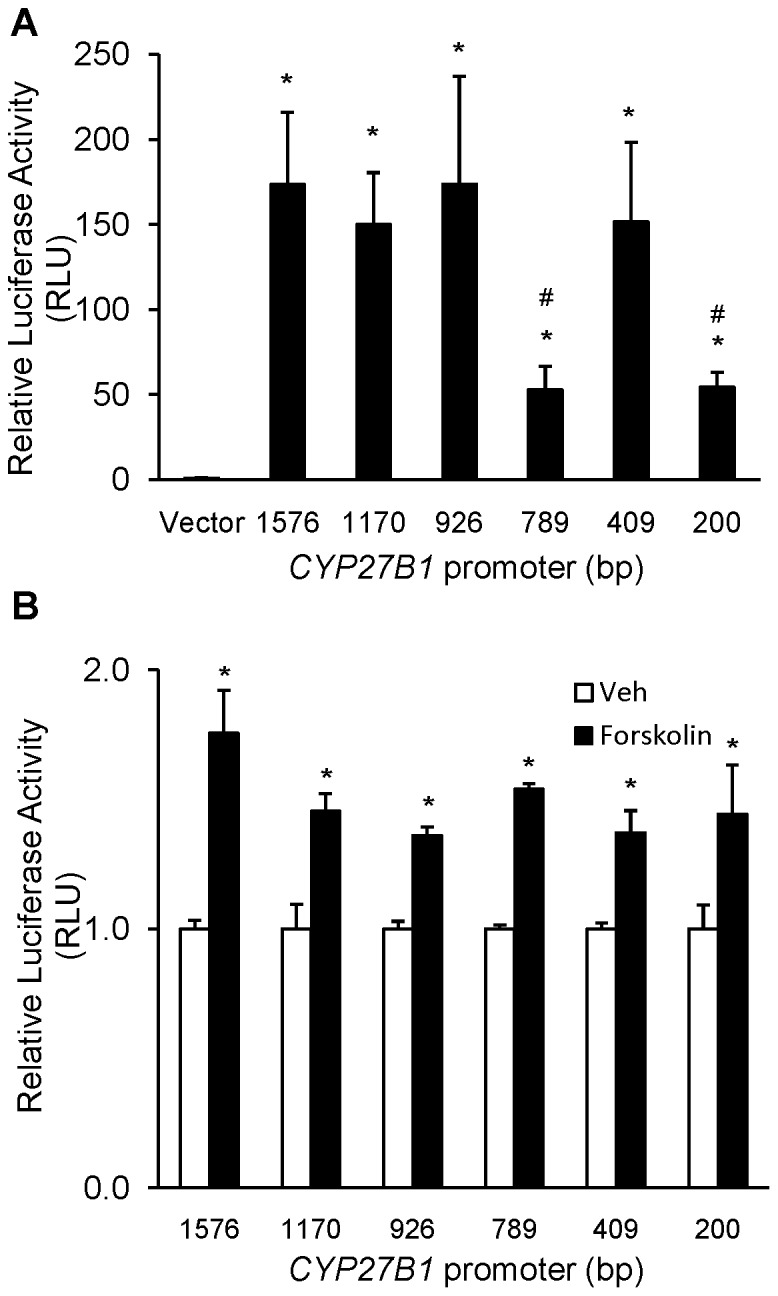
1α-hydroxylase promoter-driven firefly luciferase activity is normalized to renilla luciferase activity in each individual sample and expressed as relative luciferase activity units (RLU). *P<0.05 when compared to HEK293 cells transfected with empty vector. # P<0.05 when compared to HEK293 cells transfected with 1.6 kb CYP27B1 promoter construct. B. Effect of forskolin (10−5 M) on CYP27B1 promoter activity in HEK293 cells. Bars depict fold induction of relative luciferase activity expressed as mean±SEM. *P<0.05 when compared to HEK293 cells treated with vehicle for each promoter deletion construct. (n = 3 experiments, each performed in triplicate).
Effect of FGF-23 on CYP27B1 Promoter Activity in vitro
To determine whether FGF-23 transcriptionally regulates the CYP27B1 gene, we used HEK-293 cells stably transfected with klotho and expressing a CYP27B1 promoter/luciferase-reporter construct. Treatment of cells with recombinant human FGF-23 elicited a dose-dependent suppression of luciferase activity that was maximal (70%) at a dose of 100 ng/ml (Fig. 2A). Higher concentrations of FGF-23 did not suppress promoter activity further. To identify the responsible regulatory region of the CYP27B1 promoter, we treated HEK 293 cells transfected with CYP27B1 promoter-deletion constructs with FGF-23 (100 ng/ml). FGF-23 suppressed CYP27B1 promoter activity in all the deletion constructs, although the fold-change of suppression varied with each construct (Fig. 2B). These findings provide evidence that FGF23 regulates CYP27B1 at the transcriptional level and suggests that the FGF-23-responsive regulatory region lies within 200 bp of the CYP27B1 transcription start site. To determine whether klotho is required for the regulation of CYP27B1 expression by FGF-23 in HEK-293 cells, we determined CYP27B1 mRNA expression and CYP27B1 promoter activity in HEK-293 cells that were not stably transfected with klotho. In the absence of klotho, treatment of HEK-293 cells with FGF-23 did not elicit a significant suppression of CYP27B1 mRNA expression (100.4±2.6 (vehicle) vs 108.0±7.0 (FGF-23 (10 ng/ml), P = 0.18) or promoter activity (104.2±23.4 (vehicle) vs 74.8±10.6 (FGF-23 (10 ng/ml), P = 0.17).
Figure 2. Effect of FGF-23 on CYP27B1 promoter activity in HEK293 cells.
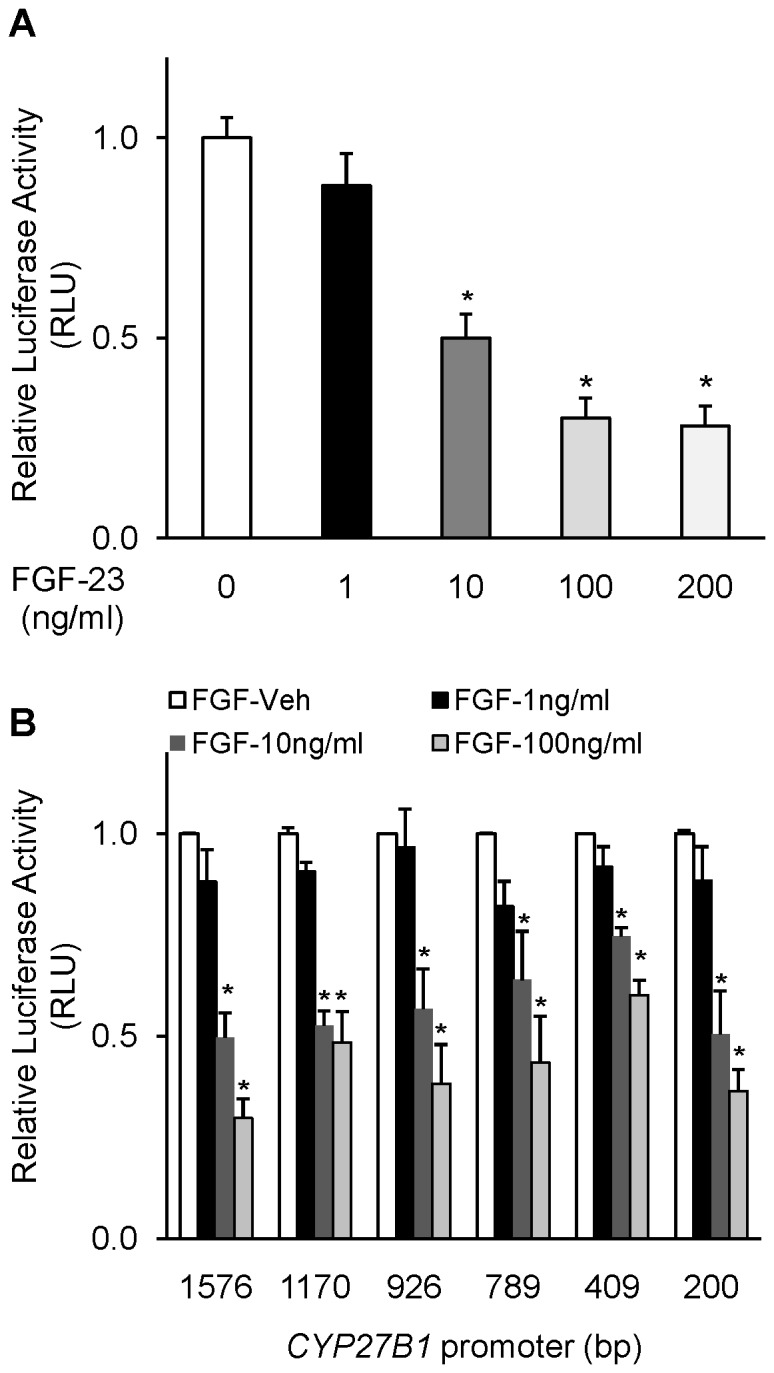
HEK293 cells were transfected with CYP27B1 promoter constructs and treated with FGF-23 (1–200 ng/ml) or vehicle for 21 hours. A. 1576 bp full-length CYP27B1 promoter-driven luciferase activity normalized to renilla luciferase activity and expressed as fold change to vehicle-treated samples. B. CYP27B1 promoter activity of the deletion constructs (−1576 bp, −1170 bp, −926 bp, −789 bp, −409 bp, −200 bp), normalized to renilla luciferase activity. Bars depict fold change of luciferase activity relative to vehicle-treated samples, expressed as mean±SEM (n = 3 experiments, each performed in triplicate). *P<0.05 when compared to the vehicle group.
Role of MAP Kinase Signaling Pathway in the Transcriptional Regulation of CYP27B1 by FGF-23 in vitro
We previously demonstrated that the suppression of CYP27B1 mRNA expression by FGF-23 depends upon MAPK signaling via ERK1/2 [35], [50], [53]. Acute activation of this pathway is evidenced by phosphorylation of ERK1/2 and increased expression of the transcription factor, early growth response-1 (egr-1). When we treated HEK-293 cells transfected with egr-1 promoter-driven luciferase plasmid with FGF-23 (100 ng/ml), egr-1 promoter activity was stimulated by 7-fold compared to vehicle-treated cells. Pre-treatment with a specific MEK inhibitor, CI-1040, blocked the stimulation of egr-1 promoter activity by FGF-23 at higher doses (5 and 10 µM) but showed no effect at lowest dose (1 µM) (Fig. 3A). These findings confirm the activation of ERK1/2 signaling in HEK-293 cells by FGF-23. To determine whether transcriptional regulation of CYP27B1 by FGF-23 depends upon activation of MAPK signaling, we treated HEK-293 cells transfected with the −200 bp CYP27B1 promoter construct with FGF-23, with and without prior treatment with CI-1040. FGF-23 suppressed CYP27B1 promoter activity by 43%, but the suppressive effect was completely blocked by pre-treatment with CI-1040 (Fig. 3B).
Figure 3. Role of MEK/ERK1/2 pathway in FGF-23-mediated signaling in HEK293 cells.
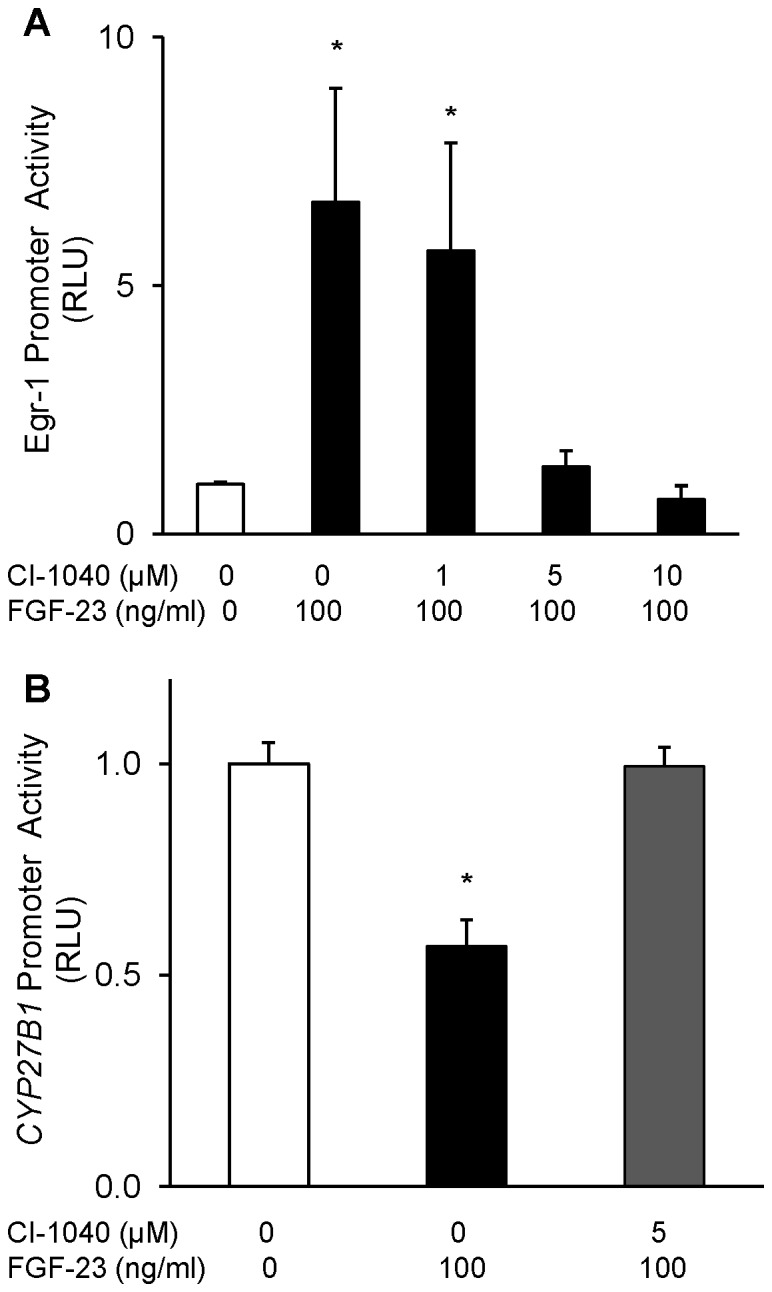
HEK293 cells were transfected with Egr-1 promoter or −200 bp CYP27B1 promoter plasmid and treated with FGF-23 (100 ng/ml) or vehicle for 21 hours. For MEK inhibition, HEK-293 cells were pre-treated with CI-1040 (0–10 µM) 30 min prior to FGF-23 treatment. A. Egr-1 promoter-driven luciferase activity normalized to renilla luciferase activity and expressed as fold change to vehicle-treated samples. B. CYP27B1 promoter activity of the −200 bp deletion construct normalized to renilla luciferase activity and expressed as fold change to vehicle-treated samples. Bars depict mean±SEM (n = 3 experiments, each performed in triplicate). *P<0.05 when compared to the vehicle group.
Role of FGF-23 in the Transcriptional Regulation of CYP27B1 Gene in Mice
Targeted ablation of the fgf-23 gene in mice disrupts calcium, phosphorus and 1,25 (OH)2D homeostasis [34], [48], giving rise to hypercalcemia, hyperphosphatemia and increased serum 1,25(OH)2D concentrations, the latter due to significantly increased renal expression of CYP27B1 mRNA and protein. To determine whether the increased expression of CYP27B1 mRNA and protein in fgf23 null mice is due to an increase in gene transcription, we measured CYP27B1 promoter-driven luciferase activity, mRNA and protein expression in the kidney in fgf-23+/+/1α-Luc+/−, fgf-23+/−/1α-Luc+/− and fgf-23−/−/1α-Luc+/− transgenic mice at 3 weeks of age. Luciferase activity in the kidney was 2.5-fold higher in fgf-23−/−/1α-Luc+/− mice than in control mice (fgf-23+/+/1α-Luc+/−) (Fig. 4A). We detected no significant change in promoter activity in the heterozygous (fgf-23+/−/1α-Luc+/−) mice compared to the control group. Renal expression of CYP27B1 mRNA and protein were upregulated by 300- and 50-fold, respectively, in fgf-23−/−/1α-Luc+/− transgenic mice (Fig. 4B and C). These findings provide evidence that up-regulation of CYP27B1 in the kidney in fgf-23 null mice is mediated at least in part by an increase in gene transcription.
Figure 4. Renal expression of CYP27B1.

Fgf-23+/+/1α-Luc+/− (wt), fgf-23+/−/1α-Luc+/− (het) and fgf-23−/−/1α-Luc+/− (ko) mice were bred as described in Methods. Mice were sacrificed at 4 weeks of age and the kidneys removed and divided into three parts for determination of A. CYP27B1 promoter activity, expressed as luciferase activity per mg of tissue. Graph depicts fold change with respect to luciferase activity in fgf-23+/+/1α-Luc+/− mice. B. CYP27B1 mRNA expression, quantitated by real-time PCR, normalized to that of gus mRNA, and expressed as a percent relative to fgf-23+/+/1α-Luc+/− mice. C. Renal mitochondrial 1α-hydroxylase protein abundance normalized to β-actin (for western blotting, n = 1 mouse/lane). Lane 1–2 (fgf-23+/+/1α-Luc+/− mice), 3–4 (fgf-23+/−/1α-Luc+/− mice), 5–6 (fgf-23−/−/1α-Luc+/− mice). Bars depict mean±SEM (n = 5 mice/group), *P<0.05, compared to fgf-23+/+/1α-Luc+/− mice.
Role of MAP Kinase Signaling in the Transcriptional Regulation of CYP27B1 by FGF-23 in Mice
In a mouse model of FGF-23 excess, we showed that FGF-23-induced suppression of renal CYP27B1 mRNA and protein was critically dependent upon MAPK signaling [50], [53]. To determine in vivo whether suppression of CYP27B1 gene transcription by FGF-23 depends upon MAPK signaling, we administered FGF-23 to fgf-23−/−/1α-Luc+/− transgenic mice that were pre-treated with the MEK inhibitor, PD0325901. FGF-23 treatment suppressed CYP27B1 promoter-driven luciferase activity in fgf-23−/−/1α-Luc+/− transgenic mice by 26%, and this suppressive effect was blocked by pre-treatment with PD0325901 (Fig. 5A). Simultaneously, FGF-23 treatment suppressed renal mitochondrial 1α-hydroxylase protein by 60%, and this effect was also blocked by PD0325901 (Fig. 5B). These studies provide evidence that the suppression of CYP27B1 transcription by FGF-23 in vivo is dependent on activation of MAPK signaling via ERK1/2.
Figure 5. Effects of MEK/ERK1/2 signaling blockade on CYP27B1 in the kidney.

Fgf-23+/+/1α-Luc+/− (wt) and fgf-23−/−/1α-Luc+/− (ko) transgenic mice were treated with vehicle or PD0325901 and administered a single injection of FGF-23 as described in Methods. A. Renal CYP27B1 activity, expressed as luciferase activity per mg of tissue. Graph depicts fold change with respect to luciferase activity in vehicle-treated fgf-23+/+/1α-Luc mice. B. Renal mitochondrial 1α-hydroxylase protein abundance normalized to β-actin (for western blotting, n = 1 mouse/lane). Lane 1–2 (vehicle-treated fgf-23+/+/1α-Luc+/− mice), 3–4 (vehicle-treated fgf-23−/−/1α-Luc+/− mice), 5–6 (FGF-23-treated fgf-23−/−/1α-Luc+/− mice), 7–8 (FGF-23+PD0325901-treated fgf-23−/−/1α-Luc+/− mice). Bars depict mean±SEM (n = 5 mice/group. *P<0.05, compared to vehicle-treated fgf-23+/+/1α-Luc+/− mice; # P<0.05, compared to vehicle-treated fgf-23−/−/1α-Luc+/− mice.
Role of FGF-23 in the Regulation of CYP27B1 in Extra-renal Sites
Cardiovascular
Autocrine and paracrine effects of 1,25(OH)2D have been well described in several tissues [26]–[28]. CYP27B1 mRNA expression is detected in the heart [54], although its role in normal cardiac function or disease states is unknown. FGF-23 directly induces left ventricular hypertrophy in normal mice and stimulates hypertrophy of isolated cardiac myocytes, suggesting that the heart is a target tissue for FGF-23 action [55]. To determine whether FGF-23 regulates CYP27B1 in extra-renal tissues, we utilized the fgf-23−/−/1α-Luc+/− transgenic mouse model. We determined 1α-hydroxylase promoter activity and mRNA expression in the heart of fgf-23+/+/1α-Luc+/−, fgf-23+/−/1α-Luc+/− and fgf-23−/−/1α-Luc+/− transgenic mice at 4 weeks of age. In fgf-23−/−/1α-Luc+/− transgenic mice, CYP27B1 promoter-driven luciferase activity in the heart was 5-fold higher (Fig. 6A), and CYP27B1 mRNA abundance was 2-fold higher (Fig. 6C) than in fgf-23+/+/1α-Luc+/− mice. To demonstrate that FGF-23 can suppress cardiac CYP27B1 mRNA expression, fgf-23−/−/1α-Luc+/− mice received a single injection of FGF-23 (150ng/g) or vehicle. Cardiac CYP27B1 mRNA expression decreased by 30% in the heart in FGF-23-treated mice compared to vehicle treated mice (194.30±84.35 vs 136.0±38.84, P<0.05). We also determined CYP27B1 promoter activity and gene expression in the aorta. We demonstrate a 3- and 7-fold increase in CYP27B1 promoter activity and mRNA expression, respectively, in the aorta of fgf-23−/−/1α-Luc+/− transgenic mice compared to fgf-23+/+/1α-Luc+/− mice (Fig. 6B and D). These studies provide evidence in mice lacking circulating FGF-23 that CYP27B1 gene expression is transcriptionally upregulated in both the heart and aorta. Of note, we detected no significant changes in either CYP27B1 promoter activity, or mRNA abundance in the heart and aorta in heterozygous (fgf-23+/−/1α-Luc+/−) mice compared to the control group. To determine whether regulation of CYP27B1 expression was specific to FGF-23 action in target tissues, we administered basic FGF to fgf-23+/+/1α-Luc+/− mice and determined CYP27B1 mRNA abundance in the kidney and heart. Basic FGF is another member of the FGF family of proteins that can activate MAPK signaling in several target tissues. We observed no significant change in CYP27B1 mRNA abundance in the kidney (89±20 in basic FGF-treated group vs 49±12 in vehicle-treated group, n = 4 mice/group, P = 0.22) or in the heart (98±20 in FGF2-treated group vs 108±28 in vehicle-treated group, n = 4 mice/group, P = 0.77). There was an upward trend (but not statistically significant) for CYP27B1 mRNA abundance in the kidney of mice treated with basic FGF which was opposite in direction to that observed in mice treated with FGF-23.
Figure 6. Cardiac and aortic expression of CYP27B1.

Fgf-23+/+/1α-Luc+/− (wt), fgf-23+/−/1α-Luc+/− (het) and fgf-23−/−/1α-Luc+/− (ko) mice were bred as described in Methods. Mice were sacrificed at 4 weeks of age, the heart and aorta removed and divided into two parts for determination of CYP27B1 promoter activity in heart (A), and aorta (B), expressed as luciferase activity per mg of tissue. Graph depicts fold change with respect to luciferase activity in fgf-23+/+/1α-Luc+/− mice. CYP27B1 mRNA expression in heart (C), and aorta (D), quantitated by real-time PCR, normalized to that of gus mRNA, and expressed as a percent relative to fgf-23+/+/1α-Luc+/− mice. Bars depict mean±SEM (n = 5 mice/group) *P<0.05, compared to fgf-23+/+/1α-Luc+/− mice.
To demonstrate whether FGF-23 activates MAPK signaling via ERK1/2 and directly suppresses CYP27B1 expression in the vascular system, we treated cultured mouse aortic vascular smooth muscle cells (VSMC) with FGF-23. We detected phosphorylation of ERK1/2 protein at 5–30 minutes in FGF-23-treated VSMC when compared to vehicle-treated cells (Fig. 7A). Furthermore, FGF-23 significantly suppressed CYP27B1 mRNA abundance in cultured VSMC when compared to the vehicle-treated group (Fig. 7B). These studies provide evidence for a direct suppression of CYP27B1 gene expression by FGF-23 in the aorta.
Figure 7. Effect of FGF-23 on CYP27B1 mRNA expression in cultured mouse aortic vascular smooth muscle cells (VSMC).
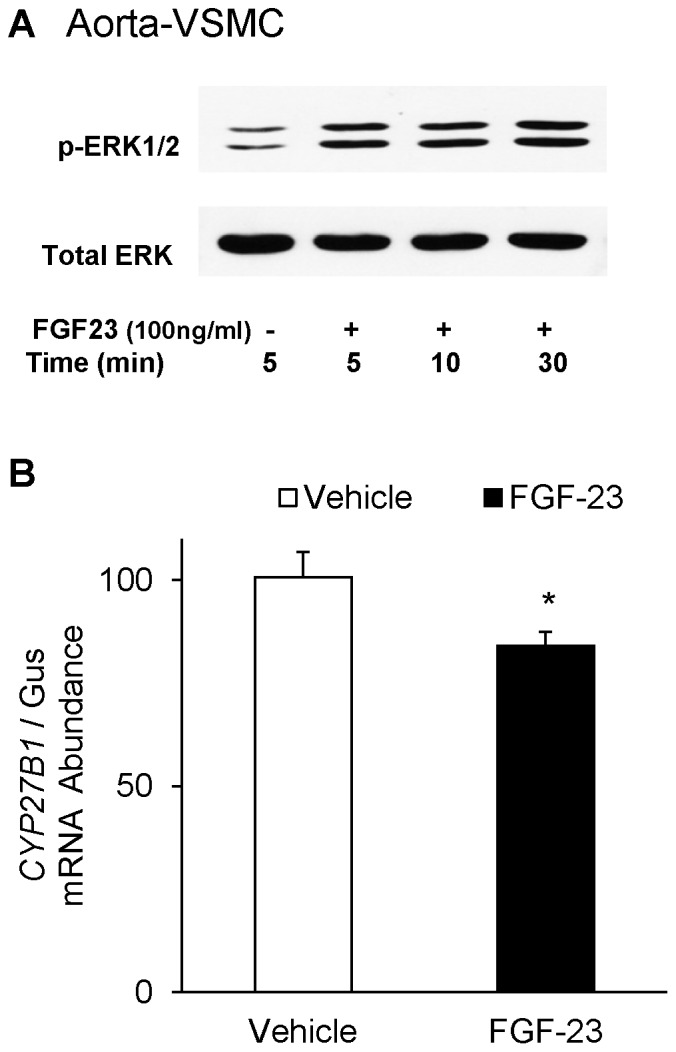
A. Activation of ERK1/2 signaling pathway was demonstrated in VSMC treated with FGF-23 (100 ng/ml) for 5–60 min. Phosphorylated ERK1/2 protein expression was detected by western blot analysis. Total Erk2 protein expression was used as loading control. B. CYP27B1 mRNA expression in VSMC treated with FGF-23 (100 ng/ml) for 21 hrs. mRNA expression was quantitated by real-time PCR, normalized to that of gus mRNA, and expressed as a percent relative to vehicle-treated group. Bars depict mean±SEM (n = 3 separate experiments in triplicate) *P<0.05, compared to vehicle-treated group.
Other extra-renal sites
To identify novel target tissues in which CYP27B1 expression might be up regulated in the absence of circulating FGF-23, we determined CYP27B1 mRNA expression in fgf-23−/−/1α-Luc+/− mice in several extra-renal tissues. In the lung, CYP27B1 promoter-driven luciferase activity was 35% higher (Fig. 8A) and CYP27B1 mRNA was 3-fold higher (Fig. 8B) in fgf-23−/−/1α-Luc mice compared to fgf-23+/+/1α-Luc+/− mice. In the spleen, CYP27B1 mRNA was 3-fold higher and in the testis was 10-fold higher (Fig. 9) in fgf-23−/−/1α-Luc+/− mice than in control mice; there were no significant difference in CYP27B1 mRNA in these tissues in the heterozygous mice (fgf-23+/−/1α-Luc+/−) when compared to the control group (data not shown). In contrast to the increased CYP27B1 expression in lung, spleen and testis of fgf-23−/−/1α-Luc+/− mice, CYP27B1 mRNA in skin and brain in these animals were lower by 80% and 28%, respectively, compared to control mice (Fig. 9). There was no significant change in CYP27B1 mRNA expression in the small and large intestine and femur (Fig. 9), extra-renal tissues reported to synthesize 1,25(OH)2D [47]. In the kidney, expression of CYP24A1 mRNA was 63% lower in fgf-23−/−/1α-Luc+/− mice compared to control mice (110.46±23.84 vs 40.43±6.44, P<0.05). CYP24A1 mRNA expression increased by 13-fold (1574.42±539.59 vs118.98±45.93, P<0.05) in the small intestine, by 21-fold (2441.99±603.56 vs 113.30±33.01, P<0.05) in the large intestine, and by 3-fold (655.80±141.46 vs 222.99±36.13, P<0.05) in the testis in fgf-23−/−/1α-Luc+/− mice compared to the control group. No significant differences in CYP24A1 mRNA were observed in the other tissues examined.
Figure 8. Pulmonary expression of CYP27B1.

Fgf-23+/+/1α-Luc+/− (wt), fgf-23+/−/1α-Luc+/− (het) and fgf-23−/−/1α-Luc+/− (ko) mice were bred as described in Methods. Mice were sacrificed at 4 weeks of age and the lung removed and divided into two parts for determination of A. CYP27B1 promoter activity, expressed as luciferase activity per mg of tissue. Graph depicts fold change with respect to luciferase activity in fgf-23+/+/1α-Luc+/− mice. B. CYP27B1 mRNA expression, quantitated by real-time PCR, normalized to that of gus mRNA, and expressed as a percent relative to fgf-23+/+/1α-Luc+/− mice. Bars depict mean±SEM (n = 5 mice/group), *P<0.05, compared to fgf-23+/+/1α-Luc+/− mice.
Figure 9. CYP27B1 expression in extra-renal tissues of fgf-23+/+/1α-Luc+/− (wt) and fgf-23−/−/1α-Luc+/− (ko) mice.

Mice were sacrificed at 4 weeks of age and the organs removed for determination of CYP27B1 mRNA expression, quantitated by real-time PCR, normalized to that of gus mRNA, and expressed as a percent relative to fgf-23+/+/1α-Luc+/− mice. Bars depict mean±SEM (n = 5 mice/group), *P<0.05, compared to fgf-23+/+/1α-Luc+/− mice. Sm.Int- small intestine, Lg.Int-large intestine.
Serum Biochemical Values in fgf-23−/−/1α-Luc+/− Mice
Hypocalcemia, hypophosphatemia and increased PTH concentrations are known inducers of renal CYP27B1 gene expression in mice. To determine whether changes in serum concentrations of Ca, Pi, or PTH might explain the difference in CYP27B1 gene expression between fgf-23−/−/1α-Luc+/− and fgf-23+/+/1α-Luc+/− mice, we determined their serum concentrations in these animals. In fgf-23−/−/1α-Luc+/− mice, the mean concentrations of serum Ca and Pi were significantly higher when compared to values in control mice (Table 1). PTH concentrations did not differ between the groups of mice. Moreover, administration of FGF-23 to fgf-23−/−/1α-Luc+/− mice did not significantly alter serum Ca, Pi or PTH concentrations suggesting that the changes in CYP27B1 gene expression in these mice are due to the direct action of FGF-23 in target tissues and not due to changes in serum Ca, Pi or PTH.
Table 1. Serum biochemical values in fgf-23+/+/1α-Luc+/− (wt) and fgf-23−/−/1α-Luc+/− (ko) transgenic mice.
| Mouse | Serum Ca (mg/dl) | Serum Pi (mg/dl) | Serum PTH (pg/ml) |
| Fgf-23+/+/1α-Luc+/− (wt) | 9.0±0.3 | 9.5±0.5 | 40.1±7.8 |
| Fgf-23−/−/1α-Luc+/− (ko) (vehicle-treated) | 10.9±0.3* | 16.0±0.4* | 29.9±3.8 |
| Fgf-23−/−/1α-Luc+/− (ko) (FGF-23-treated) | 11.1±0.7* | 17.1±0.9* | 31.2±5.2 |
P<0.05 when compared to fgf-23+/+/1α-Luc+/− (wt) mice. Values expressed as Mean±SEM, n = 7 mice per group.
Discussion
We provide evidence that CYP27B1 is transcriptionally regulated by FGF-23 in the kidney, using both an in vitro and in vivo promoter-driven luciferase reporter system. We demonstrate that MAPK signaling via MEK/ERK1/2 plays a critical role in the transcriptional regulation of CYP27B1 in the kidney. In addition, we identify novel tissue targets for FGF-23-dependent regulation of CYP27B1 expression, specifically, the heart, aorta, spleen, lung, skin, brain and testis.
After the human CYP27B1 gene was cloned [10], [11], promoter activity of the 5′ flanking region (∼1.5 kb length) was examined in vitro in modified pig kidney (AOK-B50) [38],[40] mouse renal proximal tubule (MCT, MPCT) [32], [39], [56] and HEK-293 [57], [58] cells. Here, we studied transcriptional activity of the human CYP27B1 gene in HEK-293 cells transfected with 1.6 kb of 5′ flanking DNA. We chose HEK-293 cells because they are more easily transfected than other cell lines, and signaling by FGF-23 has been well described in this system [41], [59]. In HEK-293 cells, basal activity of the 1.6 kb human CYP27B1 promoter was 170-fold higher than that of the promoterless vector, and the activity remained high with −1.1 kb, −926 bp and −409 bp deletion constructs. We observed a 3-fold reduction in basal activity of the −789 bp and −200 bp deletion constructs when compared to the full-length 1.6 kb promoter, suggesting the presence of enhancers in the regions from −926 bp to −789 bp and −409 bp to −200 bp. Our findings are similar to those described previously in HEK-293 cells [57], although the CYP27B1 promoter deletion constructs differed by a few base pairs in the two studies. We demonstrate that FGF-23 induced a dose-dependent suppression of CYP27B1 promoter activity with a maximum suppression of 70%. The suppression of CYP27B1 promoter activity by FGF-23 was seen in all the deletion constructs examined, suggesting that the regulatory region for FGF-23 lies within the first 200 bp of 5′ flanking DNA. Thus, regulation of renal CYP27B1 expression by FGF-23 occurs at least in part by transcriptional mechanisms, similar to the regulation induced by PTH, calcitonin and 1,25(OH)2D in the kidney [38]–[40], [56], [58].
Having demonstrated transcriptional regulation of CYP27B1 by FGF-23 in vitro, we sought to demonstrate regulation in vivo by utilizing 1α-Luc transgenic mice, in which dietary restriction of calcium and vitamin D [45]–[47] induces appropriate increases in renal CYP27B1 transgene expression. We found that in the fgf-23−/−/1α-Luc+/− mice, renal CYP27B1 promoter activity in the kidney was increased by 250% compared with that in mice bearing an intact fgf-23 gene. The induction of CYP27B1 promoter activity was accompanied by concomitant increases in renal expression of CYP27B1 mRNA and protein. Of note, the increased CYP27B1 expression in fgf-23 null mice cannot be explained by their increased serum concentrations of phosphorus, calcium or 1,25(OH)2D (which are characteristic features of fgf-23 null mice), as these factors suppress CYP27B1 expression. With administration of FGF-23, renal CYP27B1 promoter activity and protein abundance decreased significantly, and the suppressive effect was blocked by pre-treatment with a MEK inhibitor, PD0325901. We also observed that FGF-23-induced suppression of CYP27B1 promoter activity was blocked by MEK inhibition in HEK 293 cells transfected with the 200 bp deletion construct. Thus, the present findings provide evidence that regulation of renal CYP27B1 expression by FGF-23 is mediated at least in part by transcriptional mechanisms via activation of the ERK1/2 signaling pathway, both in vivo and in vitro.
Circulating 1,25(OH)2D has effects on cardiovascular tissue: 1,25(OH)2D can decrease myocardial proliferation and inhibit ventricular hypertrophy in cultured cells and in experimental animals [60]–[63]. In patients with chronic kidney disease, treatment with 1,25(OH)2D is associated with reduced risk of cardiovascular death [64], [65]. CYP27B1 expression is detected in the heart [54], [66] but whether local 1,25(OH)2D synthesis contributes to normal functioning of the myocardium is not known. 1α-hydroxylase enzyme activity is present in vascular smooth muscle cells and is regulated by PTH [67]; however, whether local 1,25(OH)2D synthesis contributes to the normal function of vascular smooth muscle cells is unknown. Recently, the myocardium [55] and vascular smooth muscle cells [68] were identified as targets for FGF-23 action. In the heart, FGF-23 induces left ventricular hypertrophy independent of its co-factor, klotho, and in vascular smooth muscle cells, FGF23 inhibits vascular calcification, an effect that is klotho-dependent. In the present study, we provide evidence that FGF-23 down-regulates CYP27B1 in the heart and aorta in fgf-23 null mice. In fgf-23−/−/1α-Luc+/− transgenic mice, we demonstrate in the heart a 500% increase in CYP27B1 promoter activity and 200% increase in mRNA expression, and in the aorta, we demonstrate a 300% increase in CYP27B1 promoter activity and a 700% increase in mRNA expression when compared to those values in tissues from control mice. Furthermore, we demonstrate in cultured mouse aortic vascular smooth muscle cells that FGF-23 activates ERK1/2 signaling and directly suppresses CYP27B1 mRNA expression. We thus demonstrate for the first time, that FGF-23 suppresses CYP27B1 gene transcription in cardiovascular tissue. Whether regulation of CYP27B1 gene expression by FGF-23 in cardiovascular tissue plays a role in the myocardial or vascular structure or function requires further investigation.
The parathyroid gland expresses klotho and is a target for FGF-23 action which is to inhibit PTH secretion and stimulate CYP27B1 expression [29], [69], [70]. The stimulation of CYP27B1 expression by FGF-23 in the parathyroid gland is in sharp contrast with its suppressive effect on CYP27B1 expression in the kidney. Similar to its effect in the parathyroid gland, we found that in the brain and skin of fgf-23−/− mice, absence of circulating FGF-23 down-regulated CYP27B1 mRNA expression which is opposite to the direction of change in CYP27B1 mRNA expression in the kidney, heart, aorta, lung, spleen and testis in these mice. Since 1α-hydroxylase enzyme activity in extra-renal tissues is an important source of 1,25(OH)2D for its autocrine and paracrine functions [26]–[30], further investigation is necessary to determine whether FGF-23-induced regulation of CYP27B1 in extra-renal sites plays a role in the normal function of these tissues. In the lung, increased CYP27B1 mRNA expression in alveolar macrophages of patients with sarcoidosis [22], [71], [72] contributes to increased pulmonary production of 1,25(OH)2D. However, whether pulmonary 1,25(OH)2D production plays a role in regulating normal biological functions in the lung is unknown. Similarly, the CYP27B1 gene is expressed in the testis [73] but whether testicular 1,25(OH)2D production plays a role in regulating biological functions in the testis is unknown. Here, for the first time we show a significant up-regulation of CYP27B1 expression in the lung and testis in mice lacking circulating fgf-23. It is of interest that fgf-23 null mice demonstrate pathological changes in the lung and testes that are rescued by global deletion of the CYP27B1 in the double mutant fgf-23−/−/1α- hydroxylase−/− mouse [49]. Specifically, fgf-23−/− mice develop pulmonary emphysema and significant testicular atrophy and infertility that are notably ameliorated in the double mutant fgf-23−/−/1α- hydroxylase−/− mouse [49]. Whether the improvement in pathological changes in fgf-23−/−/1α- hydroxylase−/− mice are induced by significant reductions in circulating serum 1,25(OH)2D concentrations or decreased local synthesis of 1,25(OH)2D in the lung and testis could not be determined in the above mentioned study. Thus, the contribution of local 1,25(OH)2D synthesis to the biological functions of the lung and testis remain to be determined.
In our study, absence of FGF-23 did not affect expression of CYP27B1 mRNA in the intestine and bone, which are important extra-renal sites for local 1,25(OH)2D synthesis. However, absence of FGF-23 was associated with a dramatic increase in intestinal expression of CYP24A1 mRNA. Such up-regulation of CYP24A1 mRNA expression might be mediated by the high circulating 1,25(OH)2D concentrations and could mitigate 1,25(OH)2D-mediated intestinal absorption of calcium and phosphorus. In summary, we demonstrate that regulation of CYP27B1 by FGF-23 occurs at least in part via transcriptional mechanisms in kidney and extra-renal tissues and that ERK1/2 signaling plays an important role in mediating FGF-23 action. We have identified the heart, aorta, and several other sites as novel target tissues for FGF-23-induced regulation of CYP27B1. Further studies are necessary to understand the role of FGF-23-mediated regulation of CYP27B1 in these extra-renal sites.
Acknowledgments
We would like to thank Dr. Beate Lanske, Harvard School of Dental Medicine and Dr. Howard Morris, University of South Australia for their advice about manuscript preparation.
Funding Statement
This work was supported by National Institutes of Health Grant DK-073092 (to Farzana Perwad), Norman S. Coplon Grant, Satellite Health Care Inc (to Farzana Perwad) (http://www.satellitehealth.com/about_satellite/philanthropy/coplon_grants), a VA merit grant from the Department of Veterans Affairs (to Harvey J. Armbrecht), and the Pediatric Nephrology Innovative Research Fund (to Anthony A. Portale). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The funders provided financial support for research technician, mice, lab supplies and the salary of the investigators.
References
- 1.Portale AA, Perwad F, Miller WL (2011) Rickets due to hereditary abnormalities of vitamin D synthesis or action. In: Pediatric Bone - Biology and Diseases. New York: Elsevier Science. 679–698.
- 2. Cali JJ, Russell DW (1991) Characterization of human sterol 27-hydroxylase. J Biol Chem 266: 7774–7778. [PubMed] [Google Scholar]
- 3. Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW (2004) Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci U S A 101: 7711–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fraser DR, Kodicek E (1970) Unique biosynthesis by kidney of a biologically active vitamin D metabolite. Nature 228: 764–766. [DOI] [PubMed] [Google Scholar]
- 5. Gray R, Boyle I, DeLuca HF (1971) Vitamin D metabolism: The role of kidney tissue. Science 172: 1232–1234. [DOI] [PubMed] [Google Scholar]
- 6. Gray RW, Omdahl JL, Ghazarian JG, DeLuca HF (1972) 25-hydroxycholecalciferol-1a-hydroxylase. J Biol Chem 247: 7528–7532. [PubMed] [Google Scholar]
- 7. Brunette MG, Chan M, Ferriere C, Roberts KD (1978) Site of 1,25(OH)2 vitamin D3 synthesis in the kidney. Nature 276: 287–289. [DOI] [PubMed] [Google Scholar]
- 8. Kawashima H, Torikai S, Kurokawa K (1981) Localization of 25-hydroxyvitamin D 1a-hydroxylase and 24-hydroxylase along the rat nephron. Proc Natl Acad Sci USA 78: 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paulson SK, DeLuca HF (1985) Subcellular location and properties of rat renal 25-hydroxyvitamin D3–1a-hydroxylase. J Biol Chem 260: 11488–11492. [PubMed] [Google Scholar]
- 10. Fu GK, Lin D, Zhang MYH, Bikle DD, Miller WL, et al. (1997) Cloning of human 25-hydroxyvitamin D-1a-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol 11: 1961–1970. [DOI] [PubMed] [Google Scholar]
- 11. Fu GK, Portale AA, Miller WL (1997) Complete structure of the human gene for the vitamin D 1a-hydroxylase, P450c1a. DNA Cell Biol 16: 1499–1507. [DOI] [PubMed] [Google Scholar]
- 12. Monkawa T, Yoshida T, Wakino S, Shinki T, Anazawa H, et al. (1997) Molecular cloning of cDNA and genomic DNA for human 25-hydroxyvitamin D3 1a-hydroxylase. Biochem Biophys Res Commun 239: 527–533. [DOI] [PubMed] [Google Scholar]
- 13. Shinki T, Shimada H, Wakino S, Anazawa H, Hayashi M, et al. (1997) Cloning and expression of rat 25-hydroxyvitamin D3–1a-hydroxylase cDNA. Proc Natl Acad Sci U S A 94: 12920–12925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. St-Arnaud R, Messerlian S, Moir JM, Omdahl JL, Glorieux FH (1997) The 25-hydroxyvitamin D 1-alpha-hydroxylase gene maps to the pseudovitamin D-deficiency rickets (PDDR) disease locus. J Bone Miner Res 12: 1552–1559. [DOI] [PubMed] [Google Scholar]
- 15. Takeyama K, Kitanaka S, Sato T, Kobori M, Yanagisawa J, et al. (1997) 25-hydroxyvitamin D3 1a-hydroxylase and vitamin D synthesis. Science 277: 1827–1830. [DOI] [PubMed] [Google Scholar]
- 16. Wang X, Zhang MYH, Miller WL, Portale AA (2002) Novel gene mutations in patients with 1a-hydroxylase deficiency that confer partial enzyme activity in vitro . J Clin Endocrinol Metab 87: 2424–2430. [DOI] [PubMed] [Google Scholar]
- 17. Wang JT, Lin CJ, Burridge SM, Fu GK, Labuda M, et al. (1998) Genetics of vitamin D 1a-hydroxylase deficiency in 17 families. Am J Hum Genet 63: 1694–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim CJ, Kaplan LE, Perwad F, Huang N, Sharma A, et al. (2007) Vitamin D 1a-hydroxylase gene mutations in patients with 1a-hydroxylase deficiency. J Clin Endocrinol Metab 92: 3177–3182. [DOI] [PubMed] [Google Scholar]
- 19. Xie Z, Munson SJ, Huang N, Schuster I, Portale AA, et al. (2002) The mechanism of 1,25-dihydroxyvitamin D3 auto-regulation in keratinocytes. J Biol Chem 277: 36987–36990. [DOI] [PubMed] [Google Scholar]
- 20. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, et al. (2011) Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med 365: 410–421. [DOI] [PubMed] [Google Scholar]
- 21. Armbrecht HJ, Okuda K, Wongsurawat N, Nemani RK, Chen ML, et al. (1992) Characterization and regulation of the vitamin D hydroxylases. J Steroid Biochem Mol Biol 43: 1073–1081. [DOI] [PubMed] [Google Scholar]
- 22. Adams JS, Sharma OP, Gacad MA, Singer FR (1983) Metabolism of 25-hydroxyvitamin D3 by cultured pulmonary alveolar macrophages in sarcoidosis. J Clin Invest 72: 1856–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Howard GA, Turner RT, Sherrard DJ, Baylink DJ (1981) Human bone cells in culture metabolize 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 and 24,25-dihydroxyvitamin D3 . J Biol Chem 256: 7738–7740. [PubMed] [Google Scholar]
- 24. Bikle DD, Nemanic MK, Gee E, Elias P (1986) 1,25-dihydroxyvitamin D3 production by human keratinocytes. J Clin Invest 78: 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gray RW, Weber HP, Dominguez JH, Lemann J Jr (1974) The metabolism of vitamin D3 and 25-hydroxyvitamin D3 in normal and anephric humans. J Clin Endocrinol Metab 39: 1045–1056. [DOI] [PubMed] [Google Scholar]
- 26. Morris HA, Anderson PH (2010) Autocrine and paracrine actions of vitamin d. Clin Biochem Rev 31: 129–138. [PMC free article] [PubMed] [Google Scholar]
- 27. Bikle DD (2010) Vitamin D and the skin. J Bone Miner Metab 28: 117–130. [DOI] [PubMed] [Google Scholar]
- 28. Verstuyf A, Carmeliet G, Bouillon R, Mathieu C (2010) Vitamin D: a pleiotropic hormone. Kidney Int 78: 140–145. [DOI] [PubMed] [Google Scholar]
- 29. Ritter CS, Haughey BH, Armbrecht HJ, Brown AJ (2012) Distribution and regulation of the 25-hydroxyvitamin D(3) 1alpha-hydroxylase in human parathyroid glands. J Steroid Biochem Mol Biol 130: 73–80. [DOI] [PubMed] [Google Scholar]
- 30. Ritter CS, Armbrecht HJ, Slatopolsky E, Brown AJ (2006) 25-Hydroxyvitamin D(3) suppresses PTH synthesis and secretion by bovine parathyroid cells. Kidney Int 70: 654–659. [DOI] [PubMed] [Google Scholar]
- 31.Feldman D, Malloy PJ, Gross C (1996) Vitamin D: Metabolism and action. In: Marcus R, Feldman D, Kelsey J, editors. Osteoporosis. San Diego: Academic Press. 205–235.
- 32. Zhang MYH, Wang X, Wang JT, Compagnone NA, Mellon SH, et al. (2002) Dietary phosphorus transcriptionally regulates 25-hydroxyvitamin D-1a-hydroxylase gene expression in the proximal renal tubule. Endocrinology 143: 587–595. [DOI] [PubMed] [Google Scholar]
- 33. Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, et al. (2004) FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 314: 409–414. [DOI] [PubMed] [Google Scholar]
- 34. Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, et al. (2004) Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113: 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perwad F, Zhang MY, Tenenhouse HS, Portale AA (2007) Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1alpha-hydroxylase expression in vitro. Am J Physiol Renal Physiol 293: F1577–F1583. [DOI] [PubMed] [Google Scholar]
- 36. Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, et al. (2005) Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology 146: 5358–5364. [DOI] [PubMed] [Google Scholar]
- 37. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, et al. (2004) FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19: 429–435. [DOI] [PubMed] [Google Scholar]
- 38. Kong XF, Zhu XH, Pei YL, Jackson DM, Holick MF (1999) Molecular cloning, characterization, and promoter analysis of the human 25-hydroxyvitamin D3–1a-hydroxylase gene. Proc Natl Acad Sci U S A 96: 6988–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murayama A, Takeyama K, Kitanaka S, Kodera Y, Hosoya T, et al. (1998) The promoter of the human 25-hydroxyvitamin D3 1a- hydroxylase gene confers positive and negative responsiveness to PTH, calcitonin, and 1a,25(OH)2D3 . Biochem Biophys Res Commun 249: 11–16. [DOI] [PubMed] [Google Scholar]
- 40. Gao XH, Dwivedi PP, Choe S, Alba F, Morris HA, et al. (2002) Basal and parathyroid hormone induced expression of the human 25-hydroxyvitamin D 1a-hydroxylase gene promoter in kidney AOK-B50 cells: role of Sp1, Ets and CCAAT box protein binding sites. Int J Biochem Cell Biol 34: 921–930. [DOI] [PubMed] [Google Scholar]
- 41. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, et al. (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281: 6120–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, et al. (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444: 770–774. [DOI] [PubMed] [Google Scholar]
- 43. Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, et al. (2002) Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo . Endocrinology 143: 3179–3182. [DOI] [PubMed] [Google Scholar]
- 44. Bai XY, Miao D, Goltzman D, Karaplis AC (2003) The autosomal dominant hypophosphatemic rickets R176Q mutation in FGF23 resists proteolytic cleavage and enhances in vivo biological potency. J Biol Chem 278: 9843–9849. [DOI] [PubMed] [Google Scholar]
- 45. Hendrix I, Anderson P, May B, Morris H (2004) Regulation of gene expression by the CYP27B1 promoter-study of a transgenic mouse model. J Steroid Biochem Mol Biol 89–90: 139–142. [DOI] [PubMed] [Google Scholar]
- 46. Hendrix I, Anderson PH, Omdahl JL, May BK, Morris HA (2005) Response of the 5′-flanking region of the human 25-hydroxyvitamin D 1alpha-hydroxylase gene to physiological stimuli using a transgenic mouse model. J Mol Endocrinol 34: 237–245. [DOI] [PubMed] [Google Scholar]
- 47. Anderson PH, Hendrix I, Sawyer RK, Zarrinkalam R, Manavis J, et al. (2008) Co-expression of CYP27B1 enzyme with the 1.5kb CYP27B1 promoter-luciferase transgene in the mouse. Mol Cell Endocrinol 285: 1–9. [DOI] [PubMed] [Google Scholar]
- 48. Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, et al. (2004) Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol 23: 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B (2006) Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J 20: 720–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ranch D, Zhang MYH, Portale AA, Perwad F (2011) Fibroblast Growth Factor-23 Regulates Renal 1,25(OH)2D and Phosphate Metabolism via the MAP Kinase Signaling Pathway in Hyp Mice. J Bone Miner Res 26: 1883–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Armbrecht HJ, Boltz MA, Ritter CS, Brown AJ (2007) Parathyroid hormone stimulation of the renal 25-hydroxyvitamin D-1alpha-hydroxylase–effect of age and free radicals. J Steroid Biochem Mol Biol 103: 330–333. [DOI] [PubMed] [Google Scholar]
- 52. Armbrecht HJ, Hodam TL, Boltz MA (2003) Hormonal regulation of 25-hydroxyvitamin D3–1alpha-hydroxylase and 24-hydroxylase gene transcription in opossum kidney cells. Arch Biochem Biophys 409: 298–304. [DOI] [PubMed] [Google Scholar]
- 53. Zhang MY, Ranch D, Pereira RC, Armbrecht HJ, Portale AA, et al. (2012) Chronic Inhibition of ERK1/2 Signaling Improves Disordered Bone and Mineral Metabolism in Hypophosphatemic (Hyp) Mice. Endocrinology 153: 1806–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen S, Glenn DJ, Ni W, Grigsby CL, Olsen K, et al. (2008) Expression of the vitamin d receptor is increased in the hypertrophic heart. Hypertension 52: 1106–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, et al. (2011) FGF23 induces left ventricular hypertrophy. J Clin Invest 121: 4393–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bajwa A, Forster MN, Maiti A, Woolbright BL, Beckman MJ (2008) Specific regulation of CYP27B1 and VDR in proximal versus distal renal cells. Arch Biochem Biophys 477: 33–42. [DOI] [PubMed] [Google Scholar]
- 57. Ebert R, Jovanovic M, Ulmer M, Schneider D, Meissner-Weigl J, et al. (2004) Down-regulation by nuclear factor kappaB of human 25-hydroxyvitamin D3 1alpha-hydroxylase promoter. Mol Endocrinol 18: 2440–2450. [DOI] [PubMed] [Google Scholar]
- 58. Turunen MM, Dunlop TW, Carlberg C, Vaisanen S (2007) Selective use of multiple vitamin D response elements underlies the 1 alpha,25-dihydroxyvitamin D3-mediated negative regulation of the human CYP27B1 gene. Nucleic Acids Res 35: 2734–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Farrow EG, Davis SI, Summers LJ, White KE (2009) Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol 20: 955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. O’Connell TD, Berry JE, Jarvis AK, Somerman MJ, Simpson RU (1997) 1,25-Dihydroxyvitamin D3 regulation of cardiac myocyte proliferation and hypertrophy. Am J Physiol 272: H1751–H1758. [DOI] [PubMed] [Google Scholar]
- 61. Weishaar RE, Simpson RU (1987) Vitamin D3 and cardiovascular function in rats. J Clin Invest 79: 1706–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU (2007) 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 103: 533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Weishaar RE, Kim S-N, Saunders DE, Simpson RU (1990) Involvement of vitamin D3 with cardiovascular function. III. Effects on physical and morphological properties. Am J Physiol 258: E134–E142. [DOI] [PubMed] [Google Scholar]
- 64. Teng M, Wolf M, Lowrie E, Ofsthun N, Lazarus JM, et al. (2003) R (2003) Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med 349: 446–456. [DOI] [PubMed] [Google Scholar]
- 65. Teng M, Wolf M, Ofsthun MN, Lazarus JM, Hernan MA, et al. (2005) Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J Am Soc Nephrol 16: 1115–1125. [DOI] [PubMed] [Google Scholar]
- 66. Zhou C, Lu F, Cao K, Xu D, Goltzman D, et al. (2008) Calcium-independent and 1,25(OH)2D3-dependent regulation of the renin-angiotensin system in 1alpha-hydroxylase knockout mice. Kidney Int 74: 170–179. [DOI] [PubMed] [Google Scholar]
- 67. Somjen D, Weisman Y, Kohen F, Gayer B, Limor R, et al. (2005) 25-hydroxyvitamin D3–1alpha-hydroxylase is expressed in human vascular smooth muscle cells and is upregulated by parathyroid hormone and estrogenic compounds. Circulation 111: 1666–1671. [DOI] [PubMed] [Google Scholar]
- 68. Lim K, Lu TS, Molostvov G, Lee C, Lam FT, et al. (2012) Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 125: 2243–2255. [DOI] [PubMed] [Google Scholar]
- 69. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, et al. (2007) The parathyroid is a target organ for FGF23 in rats. J Clin Invest 117: 4003–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Krajisnik T, Bjorklund P, Marsell R, Ljunggren O, Akerstrom G, et al. (2007) Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol 195: 125–131. [DOI] [PubMed] [Google Scholar]
- 71. Reichel H, Koeffler HP, Barbers R, Norman AW (1987) Regulation of 1,25-dihydroxyvitamin D3 production by cultured alveolar macrophages from normal human donors and from patients with pulmonary sarcoidosis. J Clin Endocrinol Metab 65: 1201–1209. [DOI] [PubMed] [Google Scholar]
- 72. Mason RS, Frankel T, Chan YL, Lissner D, Posen S (1984) Vitamin D conversion by sarcoid lymph node homogenate. Ann Intern Med 100: 59–61. [DOI] [PubMed] [Google Scholar]
- 73. Blomberg JM (2012) Vitamin D metabolism, sex hormones, and male reproductive function. Reproduction 144: 135–152. [DOI] [PubMed] [Google Scholar]