

Abstract
Phytophthora capsici is a soilborne plant pathogen capable of infecting a wide range of plants, including many solanaceous crops. However, genetic resistance and fungicides often fail to manage P. capsici due to limited knowledge on the molecular biology and basis of P. capsici pathogenicity. To begin to rectify this situation, Illumina RNA-Seq was used to perform massively parallel sequencing of three cDNA samples derived from P. capsici mycelia (MY), zoospores (ZO) and germinating cysts with germ tubes (GC). Over 11 million reads were generated for each cDNA library analyzed. After read mapping to the gene models of P. capsici reference genome, 13,901, 14,633 and 14,695 putative genes were identified from the reads of the MY, ZO and GC libraries, respectively. Comparative analysis between two of samples showed major differences between the expressed gene content of MY, ZO and GC stages. A large number of genes associated with specific stages and pathogenicity were identified, including 98 predicted effector genes. The transcriptional levels of 19 effector genes during the developmental and host infection stages of P. capsici were validated by RT-PCR. Ectopic expression in Nicotiana benthamiana showed that P. capsici RXLR and Crinkler effectors can suppress host cell death triggered by diverse elicitors including P. capsici elicitin and NLP effectors. This study provides a first look at the transcriptome and effector arsenal of P. capsici during the important pre-infection stages.
Introduction
It has been found that more than 80 species of the genus Phytophthora can cause tens of billions of dollars of damage each year to a wide variety of agriculturally and ornamentally important crops worldwide [1], [2]. They also cause devastating plant diseases in forests, leading to severe losses [3]. These filamentous pathogens belong to the oomycetes, a class of eukaryotes that is phylogenetically related to heterokont brown algae instead of true fungi [4].
Phytophthora species are regarded as hemibiotrophs since for part of their life cycle they maintain a biotrophic relationship with their host. A series of cell types are formed prior to host cell penetration, including sporangia, zoospores, cysts, and germinating cysts with germ tubes, all of which are important for plant infection and disease development [5]. The infection by root pathogenic oomycetes is initiated by the release from sporangia of motile, biflagellate and wall-less zoospores, which encyst and adhere to the host surface following a chemotactically and electrotactically swimming stage. The cysts germinate by forming the germ tubes and start to penetrate the plant cuticle directly with the aid of secreted enzymes and colonize host tissues. Previous studies have shown that these pre-infection structures are rich in molecules involved in establishment of infection and elicitation of plant defenses [5]–[9]. The unraveling of the molecular processes regulating the life cycle of Phytophthora is therefore important to identify determinants of pathogenesis and develop appropriate control strategies.
The heterothallic oomycete Phytophthora capsici Leonian can cause seed, root, crown, foliar and fruit rot on a number of important crops, such as solanaceous crops (pepper, eggplant, tomato), cucurbits (cantaloupe, cucumber, squash, pumpkin, watermelon) and bean crops [1], [10], [11]. P. capsici is also recorded as a pathogen of Allium cepa, Nicotiana benthamiana and Arabidopsis thaliana [12]–[15]. To date, the diseases caused by P. capsici have become devastating ones of global economic importance. The cost that P. capsici inflicts upon worldwide vegetable production is estimated to be $1 billion every year [14]. Unfortunately, the control of P. capsici is a difficult task as there has been no available effective chemical or cultural strategy [14]. In order to improve methods for controlling Phytophthora diseases, it is essential to understand the molecular mechanisms by which the pathogen suppresses or escapes the host plant defenses.
Recent studies on plant pathogenic oomycetes have demonstrated that these pathogens accomplish their penetration and colonization of host plants by manipulating their hosts through a diverse arsenal of secreted proteins (effectors) [16]–[21]. According to their potential targeting sites in the host plant, these secreted effector proteins are classified into two classes, apoplastic and cytoplasmic effectors [16]. Apoplastic effectors, including elicitins, PcF/SCR-like proteins and NLPs (NPP1-like proteins), are located at the interface between pathogen and its host and fulfill a function on the outside of the host cell. Elicitins, one type of pathogen-associated molecular patterns (PAMPs), can trigger plant cell death response, normally known as hypersensitive reaction (HR) that is characteristic of plant defense response. These proteins share a conserved 98-amino-acid domain with a characteristic signature of six cysteine residues that form three distinct disulfide bonds [16]. Members of the PcF/SCR toxin family, small cysteine-rich proteins, are thought to be involved in the induction of plant cell death [16], [22]. Other toxins secreted by oomycetes belong to NLPs that elicit plant cell death in dicotyledonous plants [23], [24]. In contrast, cytoplasmic effectors are able to translocate inside host cells where they interfere with the host defense responses. Two important groups of translocated effectors are RXLR and CRN (Crinkler) protein families [16]. RXLR effectors are named after an N-terminal RXLR amino acid motif contained by the first characterized effectors of this class, where ‘X’ denotes a nonconserved amino acid residue [25], [26]. This motif assists in the translocation of the proteins into the host’s cytoplasm where the effectors function as virulence or avirulence factors depending on the host genotype. Host translocation may also occur with variations of the RXLR motif (such as QXLR, GXLR) or even in the absence of any such motif may [27]–[31]. A structural dissection of these proteins has revealed a signal peptide followed by an RXLR motif at the N-terminus, which are required for secretion and targeting of the proteins whereas the C-terminus executes the actual effector activity [27], [32], [33]. A second, well-studied class of host-translocated effectors is the Crinklers (CRNs), named for their ability to trigger host leaf-crinkling and necrosis accompanied by an induction of host defenses [34]. Similar to the RXLR effectors, the Crinklers are modular proteins with a signal peptide, conserved N-termini and highly diverse C-terminal domains [18]. The conserved Crinkler N-termini, harboring a distinct LXLFLAK motif and the conserved DWL domain, are functionally equivalent to the N termini of RXLR effectors and required for effector targeting and host translocation [18], [35], [36]. Deletion and expression analyses in N. benthamiana have defined important C-terminal domains that are required for induction of cell death [18]. Stable transformation-based gene silencing experiments demonstrated that two Phytophthora sojae CRNs are required for successful infection of soybean [37]. van Damme et al. [38] reported that CRN8, a host-translocated CRN effector of Phytophthora infestans is a functional RD kinase inside host cell and that heterologous expression of CRN8 in planta resulted in enhanced virulence by P. infestans.
Although the impact of crop loss caused by P. capsici has increased in recent years, little is known about the molecular basis of the pathogenicity of P. capsici. To explore recently-released P. capsici reference genome sequence and to accelerate research aiming to address the molecular mechanisms underlying host plant infection, defining the P. capsici transcriptome therefore is an important strategy to interpret the functional elements of the genome and to dissect the molecular events that accompany pathogenesis. Gene transcripts can be profiled by a number of techniques aimed at isolating differentially expressed genes (DEGs), such as serial analysis of gene expression [39], microarray [40], [41], and cDNA libraries [42]. During the past few years, however, next-generation sequencing (NGS) technology has been an efficient route for generating enormous sequence collections that represent expressed genes and quantitate expression levels in particular organs, tissues, or cells, under different treatments or conditions [43], [44]. In this study, we employed the P. capsici draft genome as a reference for a transcriptional study using Illumina RNA-Seq [43]. RNA samples from three important life cycle stages were deeply sequenced. P. capsici mycelia growing in culture medium (hereafter referred to as MY), zoospores (hereafter referred to as ZO) and germinating cysts with germ tubes (hereafter referred to as GC) were analyzed, and transcript abundances compared to characterize gene expression patterns during these pre-infection stages. We specifically focus on the identification of RXLRs, CRNs, NLPs and elicitins, predicting that they would be more highly expressed in pre-infection or infection stages versus vegetative growth stage. Our results supported our prediction, revealing more transcriptional changes in P. capsici during the host infection stages. Ectopic expression assays in N. benthamiana showed that P. capsici NLP and elicitin effectors can induce cell death whereas RXLR and CRN effectors can suppress host cell death triggered by them but also other different elicitors. This study provides a critical step to characterize the mechanisms of pathogenicity and virulence of P. capsici.
Materials and Methods
Oomycete materials
Strain Pc537 of P. capsici was isolated from diseased pepper in China, preserved in liquid nitrogen and routinely maintained on 10% V8 agar media at 25 °C in the dark [1]. Samples of mycelia, sporangia, zoospores and germinating cysts were prepared as the following steps. Hyphal tip plugs were used to inoculate 30 mL of sterile clarified 10% V8 broth in 90-mm Petri dishes. Stationary mycelial cultures were incubated at 25 °C in the darkness for 4 days. To induce the sporangium formation, the agar culture from each 1 to 2-week-old Petri dish was cut into six pieces, covered with sterile distilled water and kept in Petri dishes at 25 °C in darkness. Then, the water in dishes was replaced by new sterile distilled water every 12 h. After 3 to 4 times changes of distilled water, plenty of sporangia were produced. To encourage the release of zoospores, the culture with sporangia was placed at 4 °C for 30 min, and then incubated at room temperature for another 30 min. The sporangia and zoospores were checked under an Olympus light microscope. Harvesting of sporangia and zoospores were performed according to Randall et al. [45]. Cysts were obtained by rigorously vortexing the zoospore suspension for 30 s. After this step, a solution containing 20,000 encysted zoospores was dropped onto a cellophane membrane that was pre-treated and placed on a detached leaf of N. benthamiana following the previously described procedure [46]. This was established to mimic the cyst germinating on host surface but to avoid the inclusion of plant biomass into further analysis. A moist Whatman filter paper was already placed underneath the leaf to keep humidity in a sterile Petri dish. Incubation was conducted at 25 °C in the darkness. The cyst germination was regularly observed every 10 min under the microscope after 40 min post-inoculation. Germinating cysts were collected when the average length of germ tubes of 80% germinating cysts was twice the length of the average diameter of cysts (Figure 1A). Sporangia, zoospores or germinating cysts were collected by centrifugation at 1,500 g for 10 min and immediately preserved in liquid nitrogen for RNA isolation.
Figure 1. The Phytophthora capsici cysts germinating on different surfaces.

The cysts were germinated on a cellophane membrane that was placed on the top of a Nicotiana benthamiana leaf (A), or directly on N. benthamiana leaves (B-C), or directly in water (D). Photos were taken at 70 (A), 60 (B-C) and 90 (D) min post-inoculation. To make samples for observation in (B), free water around the inoculation sites was absorbed away by filter paper and germinating cysts with some leaf tissue were peeled off using the sticky tape method [79]. The cysts germinating directly on N. benthamiana leaves were also observed using Cryoscanning electron microscopy (Hitachi S-4800 SEM) according to the instruction manual (C). The germinating cysts (A, B and D) were observed under an Olympus microscope. Scale bar = 10 µm.
Plant growth and inoculation
N. benthamiana plants were grown in Styrofoam cups containing sterile soil and placed in greenhouse at 25 °C with a cycle of 16 h day and 8 h night, until 5 to 6 weeks old.
The plants were watered two to three hours prior to inoculation to insure that the soil was wet. Each tobacco plant was inoculated by directly decanting 9 mL of freshly prepared zoospore suspension (1×105 zoospores·mL−1) around the rhizosphere. The plants inoculated with sterile water served as controls. All the plants were moved into a growth chamber with the setting to a temperature of 25±1°C, a relative humidity of 98% ± 1% and a cycle of 16 h day and 8 h night. No evident macroscopic symptoms were observed in the roots until 36 h post-inoculation (hpi) (Figure S1); therefore, we cut the portion of the roots that had been in direct contact with the inocula for the earlier time points. The infected portions of the inoculated roots were harvested at 1.5, 3, 6, 12, 24, 36, 72 hpi (Figure S1). Additional plants, inoculated and mock-inoculated, kept for 5 days after inoculation were totally dead or not infected at all, respectively. This was used to ensure the success of the inoculations. All collected samples were flash frozen in liquid nitrogen and stored at –75°C until RNA extraction.
Isolation of nucleic acids
Genomic DNA of P. capsici isolate Pc537 was extracted from mycelia using CTAB (cetyltrimethylammonium bromide) method [47]. RNA was extracted from samples using RNAiso Plus reagent (Takara Biotechnology (Dalian), China) according to the manufacturer’s instructions. The quality and quantity of both DNA and RNA were checked by gel electrophoresis and spectrophotometrically. Prior to cDNA synthesis, RNA samples were treated with DNase I (TaKaRa). RT-PCR (without reverse transcriptase) running 45 cycles was used to check that RNA samples were not contaminated with genomic DNA. After that, all RNA samples were stored at –75°C until further use.
Library preparation and sequencing
Total RNA was isolated from each of three samples (MY, ZO and GC). Total RNA of each sample was processed for Illumina RNA-Seq. The RNA-Seq libraries were constructed according to the manufacturer’s instructions of Illumina Gene Expression Preparation Kit (Illumina, San Diego, CA, USA). Briefly, poly (A+) mRNA was isolated from total RNA using Magnetic Oligo(dT) Beads (Illumina). To avoid priming bias, mRNA was fragmented by the RNA fragmentation kit (Ambion, Austin, TX, USA) before cDNA synthesis. First-strand cDNA was synthesized from the cleaved RNA using SuperScript III reverse transcriptase and random hexamer primers (Invitrogen, Carlsbad, CA, USA), followed by second-strand cDNA synthesis using DNA polymerase I (New England BioLabs, Ipswich, MA, USA) (NEB). Residual RNA in the reaction was removed by using RNase H (Invitrogen). After purification by QIAquick PCR Purification Kit (QIAGEN, Dusseldorf, Germany), the double-strand cDNA was treated to repair ends by T4 DNA polymerase, Klenow Enzyme, and T4 polynucleotide kinase (NEB). A single ‘A’ base was then added to the 3′ end of cDNA by Klenow 3′ to 5′ exo-polymerase (NEB) for DNA ligation to PE Adapter Oligo Mix (Illumina) which had a single ‘T’ base overhang. After ligation, the DNA was purified by MinElute PCR Purification Kit (QIAGEN) and separated by electrophoresis on a 2% agarose gel. The range of cDNA fragments (200±25 bp) were excised from the gel and purified for downstream analysis. PCR amplification with 10 to 12 cycles using Phusion polymerase (Finnzymes, Espoo, Finland) was performed with primers complementary to the adapter sequences to enrich the samples for the desired fragments. The PCR products were purified using QIAquick PCR Purification Kit and dissolved in 30 µL of QIAGEN EB buffer. These DNA fragments represented the designated mRNA library. Subsequently, the purified DNA fragments were single-end sequenced on the Illumina Cluster Station and Illumina Genome Analyzer IIx sequencing platform. Image recognition and base calling were performed using the Illumina Pipeline.
Sequence bioinformatics and differential expression analysis
The raw reads were first cleaned by eliminating adapters and low-quality sequences (reads with ambiguous bases ‘N’), and reads with more than 10% Q <20 bases (those with a base quality less than 20). The minimum acceptance threshold in length was 60 bp because small reads might represent sequencing artifacts.
Several complementary approaches were utilized to annotate the sequences. The read sequences were initially annotated based on the P. capsici reference genome (LT1534 v11.0) (http://genome.jgi-psf.org/Phyca11/Phyca11.home.html). Searches were conducted using the BLAST-Like Alignment Tool (BLAT) [48] with all default parameters but -tileSize = 9 and -stepSize = 6. Gene names were assigned to the sequences based on the best hit (highest score). Each matched gene model sequence was then downloaded from the P. capsici genome database and used for the following comparisons. Due to the availability of the well-annotated P. infestans genome, comparison of expressed P. capsici genes was carried out against this genome database (http://www.broadinstitute.org/annotation/genome/phytophthora_infestans/MultiHome.html) using BLASTn (E-value cutoff of 1e-10) [49]. The P. capsici genes were also compared against the NCBI Nt database (BLASTn), Swiss-Prot, NCBI Nr, TrEMBL databases (BLASTx) with an E-value of 1e-5 [49]. Functional annotation by gene ontology terms (GO, http://www.geneontology.org) was analyzed by Blast2GO software (BLASTx, E-value cutoff of 1e-5). The sequences were also aligned to the Clusters of Orthologous Groups (COG) database (http://www.ncbi.nlm.nih.gov/COG/) to predict and classify functions (BLASTx, E-value cutoff of 1e-5). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were assigned to the sequences using the online KEGG Automatic Annotation Server (KAAS) (http://www.genome.jp/kegg/kaas/) (BLASTx, E-value cutoff of 1e-5). The bi-directional best hit (BBH) method was used to obtain KEGG Orthology (KO) assignment [50]. The output of KEGG analysis includes KO assignments and KEGG pathways that are populated with the KO assignments.
To compare the gene expression level in different libraries, the transcript level of each expressed gene was calculated and normalized to the reads per kilobase of exon model per million mapped reads (RPKM) [51]. The RPKM measure of read density reflects the molar concentration of a transcript in the starting sample by normalizing for RNA length and for the total read number in the measurement. Significance of differential gene expression was determined by using General Chi-squared test integrated in IDEG6 software (http://telethon.bio.unipd.it/bioinfo/IDEG6/) [52]. IDEG6 is a web tool designed to identify differentially expressed genes in multiple tag experiments and assigns a P-value by statistical analysis of matrices of expression data. An expression profile matrix was built representing the frequency of unique genes in libraries using RPKM integer values, and imported into the IDEG6 software to conduct General Chi-squared test. P values from this method were adjusted to account for multiple testing by use of the false discovery rate (FDR). Assume that we have picked out R DEGs in which Y genes really show differential expression and the other V genes are false positive. If we decide that the error ratio ‘‘Q = V/R’’ must stay below a cutoff (e.g. 5%), we should preset the FDR to a number no larger than 0.05 [53]. FDR was applied to determine the threshold of P value in analyses. Here, an FDR-adjusted P value < 0.01 was considered a statistically significant result. Genes with an adjusted P value < 0.01 and the absolute value of log2 (expression fold change) ≥ 1 were deemed to be differentially expressed.
RT-PCR analysis
RNA was reverse transcribed into cDNA using M-MLV Reverse Transcriptase (RNase H Minus) and random hexamer primers (TaKaRa) following the manufacturer’s protocol.
The primers used in the reactions are listed in Table S1. PCR reactions took place on a thermo cycler LS-96G (Thermofisher, Dubuque, IA, USA) with the following programs: 94 °C for 5 min, followed by 29 cycles of 95 °C for 15 s, 46 to 60 °C for 30 s, and 72 °C for 1 min, and a final extension of 72 °C for 2 min. The housekeeping gene encoding ubiquitin-conjugating enzyme (Ubc) (the DOE Joint Genome Institute [JGI] protein ID 510705) was used as the internal control [54]. Genomic DNA of P. capsici was used as a control template to ensure that PCR reactions worked properly while water control was included to minimize the misinterpretation of the results. When gene expression was monitored in infected N. benthamiana, cDNA from mock-inoculated plants was used as an additional negative control for each primer pair. This was to exclude the possibility that the amplification was due to homologous plant genes. All RT-PCRs were performed at least three times.
Gene cloning and construct development
To amplify the full-length genes, the gene models of P. capsici reference genome were used to design primers (Table S1). The genes were initially PCR amplified from P. capsici Pc537 genomic DNA and cloned into a TA vector pMD-18T (TaKaRa). Signal peptide scores for each gene model were predicted by the SignalP v3.0 server [55].
To construct the transient assay vectors, gene fragments were PCR amplified using high-fidelity DNA polymerase (PrimeSTAR® HS DNA Polymerase; TaKaRa) and individually sub-cloned directly into a binary potato virus X (PVX) vector pGR107 [56] predigested by restrict enzyme SmaI (NEB). Primers (Table S1) towards elicitin and NLP genes were designed to amplify the full-length gene sequences including signal peptide-encoding region from cDNA samples in which the genes were expressed. In contrast, the PVX::RXLR or PVX::CRN constructs were designed to express RXLR or CRN without the predicted N-terminal signal peptide. Their gene fragments were amplified directly from the genomic DNA since both categories don’t contain any introns. The sequencing was done using PVX-F/PVX-R primers [34] complementary to the vector sequence and flanking the insert to identify the insert orientation and PCR-derived errors.
Multiple sequence alignment of P. capsici
Sequence alignment was performed to investigate the relationship of the predicted effectors with other Phytophthora spp. using the publicly available program BioEdit (v7.2.0) [57]. Initially, five TA clones for each gene were sequenced by DNA Sanger sequencing using PVX primers. For genes larger than 1 kbp in length, additional gene-specific primers were designed at an interval of 700 bp. The full-length sequence, translated using BioEdit, was used to perform a protein–protein BLAST (BLASTP) search in FungiDB database (http://fungidb.org; v2.3) using default parameters, and the protein sequences of other Phytophthora species were downloaded from the database. These sequences were then employed for multiple sequence alignments using BioEdit.
Agrobacterium tumefaciens infiltration assay
The binary construct was introduced independently into A. tumefaciens strain GV3101 by electroporation and transformants were selected with rifampicin (25 µg·mL−1) and kanamycin (50 µg·mL−1). After growing at 28°C in Luria-Bertani (LB) agar plates supplemented with antibiotics for 2 days, individual colonies were extracted with NaOH (2 mM) lysis solution and PCR amplified to verify that the correct clones were selected for plant transient expression assay.
For infiltration, the recombinant strains were grown in LB broth plus appropriate antibiotics at 28 °C and 200 rpm for 2 days. The cells were collected by centrifugation at 3,000 g for 5 min, washed three times in 10 mM MgCl2, and resuspended in 10 mM MgCl2 to an optical density at 600 nm of 0.4 to 0.6, and then incubated at room temperature for 1 to 3 h prior to infiltration. Infiltration experiments were conducted on 5- to 6-week-old N. benthamiana plants. For cell death induction experiments, small nicks were made in each leaf with a needle and then 20 to 40 µL of cell suspension carrying the respective constructs was infiltrated through each nick using a blunt syringe. For cell death suppression experiments, A. tumefaciens cells carrying either of RXLR and CRN genes were infiltrated initially following the above method. Then, 24 h later the same infiltration site was challenged with A. tumefaciens cells carrying the cell death-inducing genes: Bax (triggering HR-mimicking cell death in plants) [58], a P. sojae CRN effector gene PsCRN63 [37], a P. sojae RXLR effector gene Avh241 [59], [60], PsojNIP [61], P. infestans INF1 [32], a combination of P. infestans Avr3a and potato R3a as an HR inducer [62], or the effector genes characterized in this study. A. tumefaciens strains carrying GFP gene alone were infiltrated in parallel as negative controls. Each assay consisted of at least four plants inoculated on three leaves (total of twelve leaves). Symptom development was monitored visually 3 to 10 d after infiltration, and photographs were taken at 5 d. All of the experiments were repeated at least three times.
Results
Gene expression profiling using Illumina sequencing
To understand the molecular bases of pathogenicity of P. capsici, three cDNA libraries representing mycelia grown in V8 liquid medium (MY), swimming zoospores (ZO) and germinating cysts with germ tubes (GC) were constructed. We used a cellophane membrane placed on the host leaf as a surface for the induction of cyst germination in order to carry out molecular investigation of the early infection events by P. capsici but without the involvement of plant biomass into later processing. The cysts germinating on both cellophane membrane (Figure 1A) and tobacco leaf (Figure 1B, 1C) were highly similar, demonstrating the germination processes on two different surfaces were nearly synchronous and therefore the validity of this system for inducing the cyst germination. We did not harvest the germinating cysts in water because without the induction of surface martrix or host-derived compounds, the cyst germination was slower and not uniform, and only about 30% cysts were germinated in water after 90 min incubation (Figure 1D). Hence, RNA from the cysts germinating on cellophane membrane was extracted at 70 min post inoculation and used for Illumina RNA-Seq together with the MY and ZO RNA samples.
Each sequenced sample yielded 100-bp reads from single-end sequencing of cDNA fragments. After stringent quality assessment and data clearance, 11 to 12 million (M) reads with ∼94% Q20 bases (those with a base quality greater than 20) were selected as high quality reads for each library and used in the later analysis (Table 1). An average ‘G+C’ content of above 50% (51.01%, 55.18%, 54.55% for MY, ZO, GC libraries, respectively) was observed for the P. capsici ESTs. The high quality reads produced in this study have been deposited in the NCBI Sequence Read Archive database, accessible through the accession number SRP024305.
Table 1. Description of three Phytophthora capsici RNA-Seq libraries.
| Library name | Cycle number | Total reads | Total bases | G+C content (%) | Cycle Q20 (% ) |
| MY | 100 | 11,047,203 | 1,104,720,300 | 51.01 | 94.67 |
| ZO | 100 | 11,696,377 | 1,169,637,700 | 55.18 | 95.87 |
| GC | 100 | 12,066,318 | 1,206,631,800 | 54.55 | 94.41 |
MY, vegetative growth mycelia; ZO, zoospores; GC, germinating cysts with germ tubes.
Sequence mapping to the P. capsici reference genome
The distinct reads were compared against the P. capsici LT1534 genome that contains 19,805 gene models. The results showed that a substantial proportion of reads of each library (88.39% in MY library, 83.78% in ZO library and 80.60% in GC library) were matched to P. capsici genes (Table 2). Of them, the majority of mapped reads were ‘Perfect mapped reads’ without one base pair mismatch or Indel (48.47% in MY library, 45.44% in ZO library and 44.86% in GC library). After mapping reads to the gene models of P. capsici reference genome, 13,901, 14,633 and 14,695 putative genes were identified from the reads of the MY, ZO and GC libraries, respectively (Material S1). These data indicated that approximately 70% of P. capsici genes predicted in reference genome were expressed during the pre-infection stages, with more transcripts present in the GC stage than in the other two stages.
Table 2. Summary of Illumina reads mapping to Phytophthora capsici reference genome by BLAT analysis.
| Library name | Total reads | Mapped readsa | Perfect Mapped readsb | Mismatched Readsb | Indel readsb | Indel and mismatched readsb | Identitiesc |
| MY | 11,047,203 | 9,764,549 | 4,732,924 | 2,414,413 | 1,991,724 | 625,488 | 99.41% |
| 100% | 88.39% | 48.47% | 24.73% | 20.40% | 6.41% | ||
| ZO | 11,696,377 | 9,799,624 | 4,453,292 | 4,565,083 | 410,353 | 370,896 | 98.94% |
| 100% | 83.78% | 45.44% | 46.58% | 4.19% | 3.78% | ||
| GC | 12,066,318 | 9,725,254 | 4,362,412 | 4,125,646 | 810,886 | 426,310 | 99.01% |
| 100% | 80.60% | 44.86% | 42.42% | 8.34% | 4.38% |
MY, vegetative growth mycelia; ZO, zoospores; GC, germinating cysts with germ tubes.
The percentages below numbers for each library indicate the ratios of ‘Mapped reads’ to ‘Total reads’.
The percentages below numbers in separate columns indicate the ratios of mapped reads in each case stated to ‘Mapped reads’.
The percentages under this term for each library indicate the ratio of the number of mapped nucleotides without mismatching to that of total nucleotides.
Sequencing depth of three libraries was determined by saturation analysis. The saturation analysis was performed to check whether the number of detected genes keep increasing when the total amount of sequencing reads increases. Figure S2 showed a similar trend of saturation for three libraries where the number of detected genes almost ceases to increase when the number of reads reaches 10 M or higher. This indicates that three libraries were sequenced to saturation, generating a full representation of the transcripts in the conditions tested.
Heterogeneity and redundancy are two significant characteristics of mRNA expression. Therefore, the distribution of gene expression levels was used to evaluate the normality of the library data. The level of gene expression was determined by calculating the number of reads for each gene and then normalizing this to the RPKM. As shown in Figure 2, the majority of mRNA is expressed at low levels whereas a small proportion of mRNA is highly expressed. As for the GC, the highest number of genes (2,753) distributes around 4 (≥4 and <5, and 4 refers to 16 RPKM) and 0.78% of the genes (115 of 14,695) are highly expressed at more than 10. The distribution showed the similar patterns for all three libraries.
Figure 2. The distribution of gene expression levels.
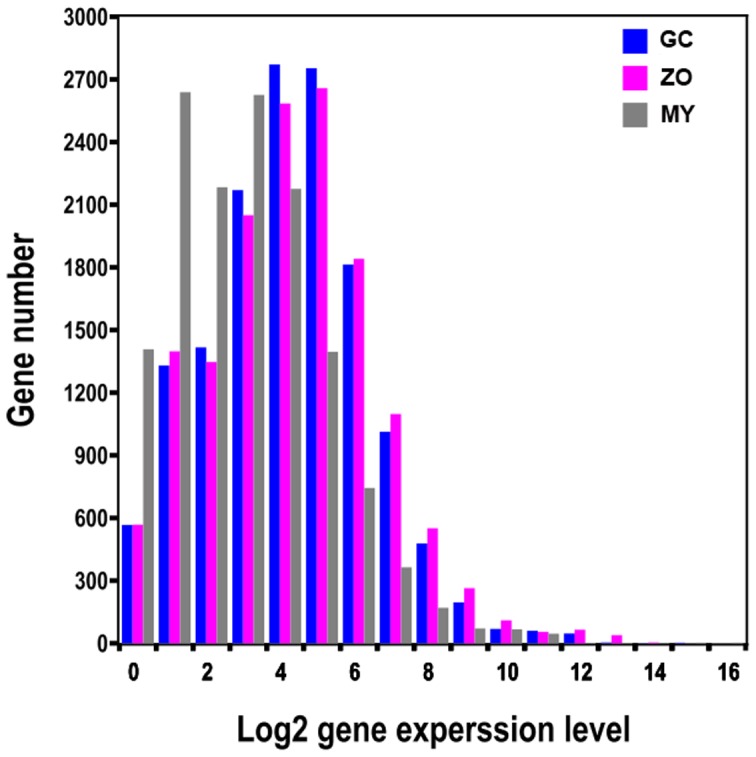
Total reads from each of three libraries that match to the 19,805 P. capsici gene models were plotted as integrated log2 values. The distribution is based on the number of genes falling in each log2 gene expression category.
Gene expression during the pre-infection stages
We analyzed the gene expression variations during the pre-infection stages of P. capsici. To study the differentially expressed genes between each library pair (GC-MY, ZO-MY and GC-ZO), we performed filtering using IDEG6 program to identify twofold up-regulated and twofold down-regulated genes with P value < 0.01, employing General Chi-squared test and FDR correction. The results, shown in Figure 3, indicate that the greatest changes in gene expression occurred during cyst germination (GC compared with MY and ZO). Between GC and MY libraries, a total of 2,578 DEGs were detected with 2,302 up-regulated and 276 down-regulated genes (Material S2). Between ZO and MY libraries, there were 1,898 DEGs detected with 1,742 up-regulated and 155 down-regulated genes (Material S2). By contrast to ZO, at GC there were more differentially expressed genes when compared with MY. This also confirmed that the GC was a distinct group. For GC–ZO pair, the gene-expression pattern changed steadily but not dramatically (1,552 up-regulated and 1,265 down-regulated genes) (Material S2; Figure 3). The DEGs with five-fold or greater differences in accumulation were shown in Figure 4. The distribution of genes corresponding to different fold change categories showed that the expression of 7.01–14.35% of genes changed by at least five fold while the expression level of the majority of genes was within fivefold difference.
Figure 3. Changes in gene expression profile among the different developmental stages.
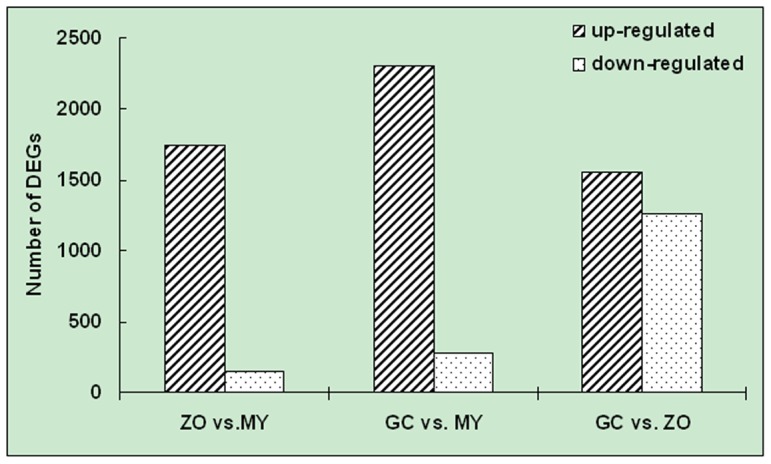
The number of up-regulated and down-regulated genes between library pairs is summarized.
Figure 4. Differentially expressed genes in different stages.
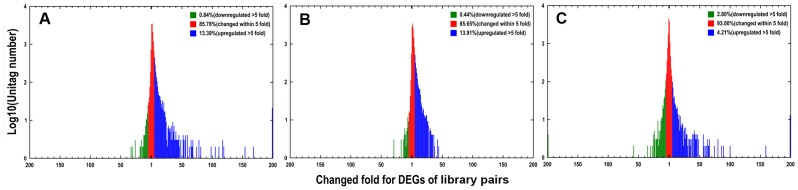
The ‘x’ axis represents fold-change of differentially expressed unique reads in different library pairs. The ‘y’ axis represents the number of unique reads (log10). Differentially accumulating unique reads within 5-fold difference between pairs are shown in the red region. The blue and green regions represent unique reads that are up- and down-regulated more than 5 fold in the stages, respectively. Library pairs: (A), GC vs. MY; (B) ZO vs. MY; (C) GC vs. ZO.
The P. capsici genome sequence is publicly available. However, the annotation of Phyca11 gene models has not been completed. To get annotations for DEGs of three library pairs, we mapped them to additional 8 databases (NCBI Nr and Nt databases, Swiss-Prot, TrEMBL, COG, GO, KEGG and P. infestans genome databases). Altogether, the comparisons against these databases achieved annotations for most of the DEGs (1,893 of 1,898 for ZO-MY, 2,567 of 2,578 for GC-MY and 2,803 of 2,817 for GC-ZO) (Material S3).
A total of 98 P. capsici effector genes with known or putative roles in virulence [reviewed by 16, 17, 63] for library pairs of samples were identified (Table 3). These genes were grouped into classes defined as RXLRs, CRNs, elicitins, transglutaminase elicitors, NLPs, CBELs (cellulose binding elicitor lectins) and enzyme inhibitors (Table 3; Material S4). Most of the genes in these families were differentially expressed with a significant P value (P < 0.01) in GC versus MY and ZO. Of the RXLRs, 20 showed increased expression at GC, whereas four decreased, compared with MY. Of the CRNs, three increased at GC, whereas no genes decreased, compared with MY. Surprisingly, all 13 CRNs showed increased expression at ZO, compared with MY (Table 3).
Table 3. The effector genes differentially expressed during the pre-infection stages of Phytophthora capsici.
| Differentially expressed genesb | ||||||||||
| Pair GC - MY | Pair ZO - MY | Pair GC - ZO | ||||||||
| Gene category | Totala | GC | MY | No change | ZO | MY | No change | GC | ZO | No change |
| RXLR | 32 | 20 | 4 | 8 | 7 | 0 | 25 | 22 | 5 | 5 |
| Crinker (CRN) | 13 | 3 | 0 | 10 | 13 | 0 | 0 | 0 | 2 | 11 |
| Elicitin | 24 | 13 | 7 | 4 | 9 | 2 | 13 | 9 | 10 | 5 |
| Transglutaminase elicitor | 7 | 4 | 0 | 3 | 1 | 3 | 3 | 5 | 0 | 2 |
| NLP | 8 | 3 | 0 | 5 | 0 | 0 | 8 | 7 | 0 | 1 |
| CBEL | 4 | 3 | 0 | 1 | 0 | 0 | 4 | 2 | 1 | 1 |
| Enzyme inhibitor | 10 | 7 | 1 | 2 | 3 | 0 | 7 | 2 | 3 | 5 |
| Total | 98 | 53 | 12 | 33 | 33 | 5 | 60 | 47 | 21 | 30 |
MY, vegetative growth mycelia; ZO, zoospores; GC, germinating cysts with germ tubes. Crinker (CRN), crinking and necrosis-inducing protein; NLP, NPP1-like protein; CBEL, cellulose binding elicitor lectin.
Total number of P. capsici effector genes, homologous to P. infestans genes.
Genes had a FDR < 0.05 and P < 0.01.
Pathway enrichment analysis of DEGs
KEGG pathway enrichment analysis was performed to categorize the biological functions of DEGs. We mapped all the genes to terms in KEGG database. Specific enrichment of genes was observed for 264 pathways in the comparison of the GC and the MY, including 229, 35 pathways affected by up- and down-regulated DEGs, respectively (Material S5). A total of 208, 33 pathways were affected by up- and down-regulated DEGs, respectively, between the pair ZO vs. MY (Material S5). In contrast, a total of 155, 162 pathways were affected by up- and down-regulated DEGs, respectively, between the pair GC vs. ZO (Material S5). The majority of the DEGs were significantly up-regulated in the GC and ZO. The first ten enriched pathways between library pairs were reported in Table 4. Notably, ribosomal-associated proteins constituted the top pathway affected by the up-regulated DEGs among the three library pairs. The other pathways for up-regulated DEGs are mainly involved in secondary metabolism (such as purine metabolism, pyrimidine metabolism and glycolysis), energy metabolism, splicing, and protein synthesis. There were enriched pathways affected by the down-regulated DEGs, which were also involved in the secondary metabolism and energy conversion. However, up-regulated DEGs involved in the pathways were more than down-regulated DEGs. These results agree with the findings from the DEG analysis and suggest that the metabolism and signal transduction pathways might be more active in the GC and ZO.
Table 4. List of first ten pathways for up- and down-regulated DEGs between library pairs.
| Pathway term | Pathway ID | DEGs tested | P value |
| Library GC vs. MY | |||
| Pathways for up-regulated DEGs | |||
| Ribosome | ko03011 | 59 | 3.63e-14 |
| Ribosome biogenesis in eukaryotes | ko03008 | 44 | 1.35e-10 |
| Purine metabolism | ko00230 | 40 | 2.15e-04 |
| Translation factors | ko03012 | 33 | 1.38e-08 |
| Chaperones and folding catalysts | ko03110 | 31 | 8.17e-03 |
| RNA transport | ko03013 | 28 | 0.3723 |
| Energy production and conversion | ko05016 | 26 | 0.0770 |
| Spliceosome | ko03041 | 25 | 0.9999 |
| Pyrimidine metabolism | ko00240 | 24 | 0.0074 |
| Chromosome | ko03036 | 21 | 0.9999 |
| Pathways for down-regulated DEGs | |||
| Peptidases | ko01002 | 4 | 0.9999 |
| Lipid biosynthesis proteins | ko01004 | 4 | 0.4758 |
| Glycolysis / Gluconeogenesis | ko00010 | 3 | 0.0888 |
| Biosynthesis of unsaturated fatty acids | ko01040 | 3 | 0.0466 |
| Nitrogen metabolism | ko00910 | 3 | 0.0263 |
| Lysosome | ko04142 | 3 | 0.9003 |
| Lipid transport and metabolism | ko03320 | 2 | 0.0269 |
| Arginine and proline metabolism | ko00330 | 2 | 0.0924 |
| Chaperones and folding catalysts | ko03110 | 2 | 0.0082 |
| Oxidative phosphorylation | ko00190 | 2 | 0.9915 |
| Library ZO vs. MY | |||
| Pathways for up-regulated DEGs | |||
| Ribosome | ko03011 | 82 | 4.43e-42 |
| Energy production and conversion | ko05016 | 38 | 2.75e-07 |
| Oxidative phosphorylation | ko00190 | 28 | 7.35e-07 |
| Mitochondrial electron transport | ko05010 | 26 | 1.91e-06 |
| Energy metabolism | ko05012 | 25 | 1.09e-07 |
| Ubiquitin system | ko04121 | 24 | 0.8363 |
| Spliceosome | ko03041 | 24 | 0.9994 |
| GTP-binding proteins | ko04031 | 23 | 1.12e-12 |
| Peptidases | ko01002 | 19 | 0.9892 |
| DNA repair and recombination proteins | ko03400 | 18 | 0.9823 |
| Pathways for down-regulated DEGs | |||
| Amino acid related enzymes | ko01007 | 5 | 0.9776 |
| Amino acid transport and metabolism | ko00710 | 3 | 0.4536 |
| Propanoate metabolism | ko00640 | 3 | 0.7245 |
| Alanine, aspartate and glutamate metabolism | ko00250 | 3 | 0.6568 |
| Valine, leucine and isoleucine biosynthesis | ko00290 | 2 | 0.7885 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | ko00400 | 2 | 0.6987 |
| Glycine, serine and threonine metabolism | ko00260 | 2 | 0.9539 |
| Cysteine and methionine metabolism | ko00270 | 2 | 0.7632 |
| Butanoate metabolism | ko00650 | 2 | 0.1886 |
| Oxidative phosphorylation | ko00190 | 2 | 7.35e-07 |
| Library GC vs. ZO | |||
| Pathways for up-regulated DEGs | |||
| Ribosome biogenesis in eukaryotes | ko03008 | 39 | 6.31e-09 |
| Purine metabolism | ko00230 | 20 | 0.3194 |
| Chaperones and folding catalysts | ko03110 | 19 | 0.0102 |
| Protein processing in endoplasmic reticulum | ko04141 | 15 | 0.1253 |
| Nitrogen metabolism | ko00910 | 15 | 0.0001 |
| Amino acid related enzymes | ko01007 | 14 | 0.2509 |
| Spliceosome | ko03041 | 12 | 0.9976 |
| Glycolysis / Gluconeogenesis | ko00010 | 11 | 0.0255 |
| Alanine, aspartate and glutamate metabolism | ko00250 | 11 | 0.0357 |
| Proteasome | ko03051 | 10 | 3.31e-05 |
| Pathways for down-regulated DEGs | |||
| Ribosome | ko03011 | 28 | 0.1461 |
| Peptidases | ko01002 | 27 | 0.3993 |
| Ubiquitin system | ko04121 | 20 | 0.9881 |
| Energy production and conversion | ko05016 | 18 | 0.4984 |
| Proteasome | ko03051 | 18 | 3.31e-05 |
| Oxidative phosphorylation | ko00190 | 18 | 0.0780 |
| GTP-binding proteins | ko04031 | 16 | 6.05e-05 |
| Spliceosome | ko03041 | 16 | 0.9976 |
| Mitochondrial electron transport | ko05010 | 16 | 0.1487 |
| Energy metabolism | ko05012 | 15 | 0.0818 |
MY, vegetative growth mycelia; ZO, zoospores; GC, germinating cysts with germ tubes.
Expression analysis by RT-PCR
Nineteen genes representing the families described above (8 RXLRs, 2 CRNs, 6 elicitins and 3 NLPs) were selected for confirmation as well as to monitor their expression during the host infection with RT-PCR (Table S1). Hereafter, we refer to them by their JGI protein designations, as that is what they are related with, with ‘Pc’ (for P. capsici) at the beginning of each name.
All the genes were examined for expression in four developmental stages (mycelia, sporangia, zoospores and germinating cysts) and in N. benthamiana root samples at seven time-points after inoculation (1.5, 3, 6, 12, 24, 36 and 72 hpi) as well (Figure 5). The setup of these inoculation time-points assures that the whole infection period of P. capsici in N. benthamiana could be investigated. The disease progressed quickly in the plants, and by 5 dpi all inoculated plants were collapsed. As shown in Figure S1, N. benthamiana root rot was evident at 36 hpi. Brown lesions appeared in the middle of the roots and stems (Figure S1), but no evident macroscopic symptoms were observed on the leaves until 72 hpi. No symptoms developed in the control plants.
Figure 5. Expression patterns of 19 Phytophthora capsici genes during pre-infection and infection stages.

Lanes: MY, plate-grown mycelia; SP, sporangia; ZO, zoospores; GC, germinating cysts; Mock, mock-inoculated plants; 1.5 – 72 hpi, Nicotiana benthamiana roots at 1.5, 3, 6, 12, 24, 36, 72 h post-inoculation (hpi); W, sterile distilled water. Gene names under different categories are listed on the left and sizes are listed on the right. All sizes were as expected, and the experiment was performed with at least three biological replicates.
Consistent with the RNA-Seq results, the majority of the genes (15/19) were expressed during at least one of the pre-infection stages. The remaining four genes (Pc109174, Pc508761, Pc20942 and Pc97196) showed low expression during these pre-infection stages. However, all tested genes showed increased expression in infection stages compared with developmental stages. Noticeably, most RXLR genes were expressed most highly at early infection time points, except Pc536383 and Pc107349, which showed highly expression at later infection stages (Figure 5).
P. capsici elicitin and NLP effectors cause cell death in N. benthamiana
Expression studies indicated that we had identified potentially important infection-related genes in P. capsici, including RXLRs, CRNs, elicitins and NLPs. To determine functionality, initially we cloned and performed multiple sequence alignment of four effector genes: Pc22053 (RXLR), Pc506611 (CRN), Pc508761 (elicitin) and Pc118548 (NLP). Full-length copies of selected genes were obtained by designing primers based on the P. capsici LT1534 gene models, and cloned into TA vector pMD-18T. The SignalP hidden Markov model (HMM) probability values of the putative proteins deduced from the selected genes were higher than 0.98. For SignalP NN mean S score, all the effectors had a value higher than 0.700, except Pc506611 (0.584) (Table 5). The resulting translated amino acid sequences were used for alignments (Figure 6). A single species was chosen to represent each of five Phytophthora clades [64]. BLAST results with the predicted proteins showed that P. capsici Pc22053, Pc506611, Pc508761 and Pc118548 have 18%, 91%, 50% and 78% identity to P. infestans homologues, respectively (Table 5). The open reading frame (ORF) of RXLR gene Pc22053 is 519-bp long without any introns, which was mis-annotated as containing a 120-bp intron in the reference genome. The predicted protein showed 15 amino acids including RXLR-dEER motif conserved across three different Phytophthora species whereas 32 amino acids were shared with P. infestans. The CRN effector Pc506611 shared 100 amino acids including the conserved LXLFLAK and HVLVVVP motifs with three different Phytophthora species, whereas 368 of 402 amino acids (91%) were conserved between both P. capsici and P. infestans. The elicitin Pc508761 revealed that, of 182 amino acids in P. capsici, 54 were conserved across six different Phytophthora species. Conversely, 92 amino acids were conserved when aligned with only P. infestans. One NLP effector of interest, Pc118548, showed 86 of 246 amino acids conserved across three different Phytophthora species and an additional oomycete, Pythium ultimum, whereas 193 were shared with P. infestans, corresponding to 78% identity. The data showed that all aligned NLPs have two conserved cysteine residues, which define these proteins as type I NLP [23], [65]. We also noted that the central conserved GHRHDWE heptapeptide motif is present in all NLPs, but has changed in sequence in NLPs from the selected Phytophthora species but not from Py. ultimum (Figure 6).
Table 5. Summary and comparison of putative RXLR, CRN, elicitin and NLP protein sequences from Phytophthora capsici with Phytophthora infestans.
| Gene | SignalP | SignalP | SignalP | % IDc | Number of amino acids (total) | Number of amino acids (conserved)d | ||||
| Gene | GenBank accession no. | Genome positiona | length (bp) | HMMb | NNb | lengthb | P. capsici | P. infestans | ||
| Pc22053 | KF220580 | scaffold_535:1187–1705 | 519 | 1.000 | 0.906 | 22 | 18 | 172 | 102 | 32 |
| Pc506611 | KF220579 | scaffold_20:748014–749251 | 1209 | 0.988 | 0.584 | 17 | 91 | 402 | 404 | 368 |
| Pc508761 | KF220578 | scaffold_38:51106–51767 | 549 | 1.000 | 0.771 | 20 | 50 | 182 | 188 | 92 |
a Genome position refers to the above genes’ position in the P. capsici genome sequence assembly database v11.0.
Hidden Markov model (HMM) probability, NN mean S score, and signal peptide length were predicted using SignalP v3.0.
Blasted against the P. infestans genome sequence.
Conserved between both P. capsici and P. infestans.
Figure 6. Alignment of four protein sequences from Phytophthora capsici against different Phytophthora species and Pythium ultimum.

Full-length P. capsici homologous sequences were translated using BioEdit. (A) Pc22053. Red and blue boxes denote putative RXLR and dEER motifs, respectively. (B) Pc506611. Red and blue boxes denote putative LXLFLAK and HVLVVVP motifs, respectively. (C) Pc508761; (D) Pc118548. Red arrows indicate conserved cysteines and key functional residues previously described [24]. The putative GHRHDWE motif was denoted by red box. All motifs mentioned above were also marked by black asterisks for visualization.
The selected genes then were sub-cloned into the PVX vector pGR107. Transient expression assays with these four genes were performed on N. benthamiana plants. We tested whether they could individually induce necrosis in N. benthamiana using infiltration of A. tumefaciens cells with pGR107 carrying each of the genes. These methods have been widely used for transient expression of genes in N. benthamiana and for determining whether particular genes can induce or suppress cell death [18], [32], [34], [37], [59], [66]–[68]. The known elicitor of cell death, P. infestans INF1, was used as a positive control. Agro-infiltration of INF1, Pc508761 and Pc118548 induced hypersensitive response (HR)-like host cell death, as indicated by the macroscopic necrosis, whereas only mosaic symptoms due to PVX were observed following agro-infiltration of Pc22053, Pc506611 or GFP under the same conditions (Figure 7A). Pc508761 induced an HR within 3 days like INF1 whereas Pc118548 triggered an HR at 4 days post-infiltration.
Figure 7. Transient assays of Phytophthora capsici RXLR, CRN, NLP and elicitin effectors in Nicotiana benthamiana.

(A) Pc118548 (NLP) and Pc508761 (elicitin) trigger cell death in N. benthamiana. The plant leaves were infiltrated with A. tumefaciens (strain GV3101) cells to express Pc22053 (RXLR), Pc506611 (CRN), Ps118548, Pc508761, P. infestans INF1 (positive control) or GFP (negative control). Photographs were taken 5 d after infiltration. (B) – (I), Pc22053 and Pc506611 suppress cell death caused by different cell death inducers in N. benthamiana. Agro-infiltration sites in each N. benthamiana leaf expressing Pc22053 (top left), Pc506611 (top right), or GFP (middle right) were challenged with A. tumefaciens expressing Bax (B), INF1 (C), Pc508761 (D), PsojNIP (E), Pc118548 (F), PsAvh241 (G), PsCRN63 (H) and Avr3a/R3a (I). The other three sites in each leaf expressing GFP only (middle left), inducer itself (bottom right) or infiltrated by 10 mM MgCl2 buffer only (bottom left) served as controls. Photographs were taken 5 d after cell death inducer infiltration.
P. capsici RXLR and CRN effectors suppress host cell death
To further explore the functions of Pc22053 (RXLR) and Pc506611 (CRN), we tested whether they could suppress cell death like other bacterial and oomycete effectors [32], [37], [59], [66], [69]. Pc508761, Pc118548 plus six additional cell death inducers were used in this assay: BAX, INF1, PsojNIP, a combination of Avr3a and R3a, Avh241 and PsCRN63. Cell death symptoms were observed when A. tumefaciens cells carrying the above genes were infiltrated into N. benthamiana leaves (Figure 7B-7I). However, when Pc22053 was infiltrated in N. benthamiana leaves 24 h prior to infiltration of each of the inducers, this pretreatment blocked cell death triggered by all 8 cell death inducers. By comparison, Pc506611 could protect N. benthamiana tissue from cell death triggered by all tested cell death inducers but R3a/Avr3a (Figure 7I). Furthermore, prior infiltration with A. tumefaciens cells containing the GFP gene, a negative control, did not protect against cell death (Figure 7B-7I).
Discussion
We present a deep transcriptional study of the oomycete P. capsici, a pathogen of interest because of its damage to diverse crops and its genetic relatedness to other important oomycete pathogens [14]. We anchored the P. capsici transcriptome using its draft genome as a reference [70], investigated the global gene expression in three important life cycle stages (MY, ZO and GC) and identified a large number of genes associated with specific stages and pathogenicity. We identified 13,901, 14,633 and 14,695 genes from the reads of the MY, ZO and GC libraries, respectively (Material S1). We showed that about 70% of the P. capsici genes in LT1534 genome are represented in our study. The remaining 30% of conserved eukaryotic genes may not have been sampled because not all developmental and infection stages of P. capsici were represented in our material (e.g. sporangium, but also sexual reproduction).
Comparative analysis between two of samples showed major differences between the expressed gene content of MY, ZO and GC stages. A suite of genes associated with specific stages and pathogenicity were identified between two of samples (Material S2 and S3). These transcriptional shifts generally agreed with the biological process of the pathogen. This was confirmed by the analysis of differentially expressed genes, which showed major transitional shifts between the libraries from the different groups. The distinction of ZO and GC from MY is probably due to its divergence from non-mycelium status (e.g., they are either swimming in water or attached to the host surface to germinate). The GC was an especially distinct group with more differentially expressed genes detected, probably because the initial host-pathogen interaction occurred during this stage can reasonably be expected to modify the pathogen’s expression profile. Additionally the GC stage is transitory between the ZO and MY stages. The results of KEGG pathway enrichment analysis (Table 4; Material S5) lend support to the biological significance of gene expression profiles derived from the deep sequencing, which will assist in the discovery and annotation of P. capsici genes playing key roles in development and particularly in pre-infection stages. It is not surprising that the ‘ribosome-related’ pathways were the most affected for the DEGs more common in ZO and GC libraries. This finding implies that the oomycete utilizes new ribosomes or changes in ribosome components to help synthesize additional proteins to facilitate its swimming towards potential host plants and subsequent germination on host surface.
Sequencing of plant pathogenic oomycetes has revealed a variable number of effectors in different species. In the present study, 99 P. capsici effector genes were identified with known or putative roles in virulence (Table 3; Material S4). We predicted that these RXLRs, CRNs, NLPs and elicitins would be highly expressed during pre-infection and infection stages. Our results partially supported our hypothesis, in that these effectors were mostly induced in planta (Figure 5). A study by Haas et al. [18], in which the authors examined genome-wide expression in P. infestans over four time points from 2 to 5 dpi of potato, revealed similar results that in general RXLRs showed early expression. Interestingly, the elicitin class showed the induction at GC and infection stages for P. capsici, whereas there were no elicitins induced at all during infection by P. infestans. DEG analysis showed that in contrast to ZO (13), only 3 CRNs showed increased expression at GC compared with MY. This implied that some CRNs were probably repressed at GC stage (an important pre-infection stage). In a recent study on P. capsici CRNs, based on contrasting gene expression profiles, Stam et al. [36] defined two classes of CRN effectors (Class 1 and 2). We noted from the RT-PCR result that two tested CRN genes fell into Class 1 featuring high levels of expression at the early time points, a decrease during subsequent biotrophic stages and expression in the later stages (Figure 5). However, another similar study by Kunjeti et al. [68] showed Phytophthora phaseoli CRNs were mostly repressed in planta. A transcriptional study on P. sojae showed induction of RXLRs, NLPs, CRNs and elicitins was high at the GC stage and another peak later in infection stage on soybean [71]. These data suggest that P. capsici expresses some genes in common with other Phytophthora pathogens, but the timing may differ.
Our RXLR dataset includes several predicted orthologs to known avirulence genes such as P. sojae Avr1b and P. infestans Avr3a [62], [72]. For instance, the predicted protein Pc22053 showed 33% identity with P. sojae Avr1b (BLASTx, E-value = 2e-05). However, Pc22053 can not cause cell death in N. benthamiana during the transient expression assays (Figure 7A). This is not surprising as data from P. sojae indicate that Avr1b induced cell death occurs only on soybean plants carrying Rps1b [73]. To explore this further, we tested for transcripts of Pc22053 by RT-PCR and observed its highly expression during the host infection (Figure 5). Furthermore, we observed that Pc22053 can suppress the HR induced by all 8 different effectors (Figure 7B-7I). Similarly, selected RXLR effectors and the RXLR-like SNE1 protein from other Phytophthora species have demonstrated their ability to suppress cell death and defense [27], [29], [32], [59]. We demonstrated that one P. capsici CRN gene Pc506611 does not cause plant cell death when transiently expressed in N. benthamiana (Figure 7A). This is not a rare case because recent studies show that cell death induction is not a universal feature of CRN proteins. Over-expression of Phytophthora CRN domains only induce necrosis in a few cases [18], [36], [37]. Intriguingly, when the putative Pc506611 CRN protein sequence was compared against NCBI Nr database, a ubiquitin-like (UBL) domain was detected in this protein (Position 13–58; BLASTP, E-value = 3.99e-03). The family of proteins containing UBL and ubiquitin-associated domains has been implicated in proteasomal degradation [74]. Whether the protein contributed to the pathogenicity through ubiquitin pathway during the host-oomycete interaction is yet to be determined. Furthermore, the high expression of Pc506611 during the GC and early infection stages (Figure 5) leads us to suspect that it contributes positively to the pathogen’s virulence. Ectopic expression showed that this gene can suppress plant cell death caused by all tested cell death inducers but R3a/Avr3a (Figure 7B-7I). Data from a recent study on P. sojae CRNs indicate that one CRN, PsCRN115, also suppresses cell death elicited by e.g. PsojNIP and PsCRN63 [37]. The similar results indicated that this family of effectors also has similar abilities to RXLR effectors in suppressing plant defense. Host cell death or the HR is an effective and ultimate defense mechanism against obligate biotrophic and hemibiotrophic pathogens [75]. However, delayed HR could be counterproductive and benefit hemibiotrophic pathogens in necrotrophic growth stages. P. capsici is a hemibiotroph and switches from biotrophic to necrotrophic growth 18 to 42 h following the invasion of N. benthamiana leaves [14]. Therefore, the ability of P. capsici to suppress or delay the HR of host tissue is likely a major component of its pathogenic strategy, as is for P. sojae [59], [76].
We observed a small number of NLPs and elicitins with differential expression (Table 3; Material S4; Figure 5). DEG analysis showed that three NLPs showed increased expression at GC compared with MY. RT-PCR analysis demonstrated that they are highly expressed during the infection stages. Similarly, a study on P. sojae NLPs found that most NLP are highly expressed during cyst germination and infection stages [67]. Ectopic expression showed that one of our P. capsici NLP genes (Pc118548) triggered cell death in N. benthamiana (Figure 7A). Although it is accepted that NLPs can contribute to virulence as toxins, it has been reported that many members of this family do not possess obvious toxic activity [24], [67]. The expansion of NLP gene families in Phytophthora spp. has been noted in whole-genome analyses and the size of the NLP family in P. infestans (n = 27), P. sojae (n = 39), and Phytophthora ramorum (n = 59) has been estimated through annotation [17], [18]. Recently, 18 putative NLP genes have been cloned from P. capsici strain SD33 [77]. However, the exact number of functional or ‘real’ genes encoding NLPs in P. capsici is yet to be determined and their exact functional roles in P. capsici virulence remain to be elucidated.
In total 24 elicitin genes were detected in the present study by Illumina sequencing. DEG analysis showed that 13 and 9 elicitin genes were up-regulated at GC and ZO stages compared with MY, respectively (Table 3; Material S4). Gene expression analyses showed that P. capsici elicitin genes were mostly induced during the host infection (Figure 5). A study on infection-related gene changes in P. phaseoli by Kunjeti et al. [68] revealed similar results. They observed that the expression of four elicitin genes was increased while that of 2 decreased. Ye et al. [71] also reported the induction of elicitins was high at the germinated-cyst stage and infection stage on soybean. Transient assay showed that one of P. capsici elicitin genes (Pc508761) induced plant cell death (Figure 7A). Future studies will involve an exploration of whether these elicitins are required for host infection or sterol acquisition [78].
Conclusions
P. capsici is a widespread and destructive plant pathogen and in particular presents an attractive model for understanding oomycetes with broad-host-ranges. Here, we have demonstrated that deep sequencing of the transcriptome combined with computational tools such as DEG profiling has provided a wealth of information about the genes differentially expressed during the pre-infection stages of P. capsici. Our findings point to potentially important P. capsici RXLR and CRN effectors that can modulate host defense circuitry and benefit parasite colonization. An important challenge in the next step is to determine how these effector proteins manipulate host processes. This dataset will ultimately help to elucidate the mechanisms underlying the pathogenicity and to design novel control strategies for the diseases caused by P. capsici.
Supporting Information
The disease progressions in Nicotiana benthamiana roots caused by Phytophthora capsici isolate Pc537. Each plant root was cut open before macroscopic observations. Arrows indicate the inoculation part or browning, necrosis and rot symptoms. (A) controls (mock-inoculated); (B-D) the plants at 24, 36, 72 h post-inoculation, respectively.
(TIF)
Accumulation of Illumina total reads and unique genes in the three libraries. New unique genes (‘y’ axis) of MY, ZO, GC libraries (different color lines) decreased as the depth of sequencing (‘x’ axis) increased.
(TIF)
List of primers used for RT-PCR and cloning of the Phytophthora capsici genes.
(XLS)
Phytophthora capsici genes expressed during mycelium, zoospore and germinating cyst stages. (listed in separate sheets for each stage).
(XLS)
Differentially expressed genes between two samples. (3 sample-pairs listed in separate sheets).
(XLS)
Differentially expressed genes between two samples with annotation. (3 sample-pairs listed in separate sheets).
(XLS)
The effector genes differentially expressed during the pre-infection stages of Phytophthora capsici. (7 different categories listed in separate sheets with annotation).
(XLS)
Complete list of involved pathways for regulated DEGs between two samples. (3 sample-pairs listed in separate sheets).
(XLS)
Acknowledgments
We gratefully acknowledge Mark Gijzen (Agriculture and Agri-Food Canada) and two anonymous reviewers for their helpful comments on the manuscript. Thanks are also due to David Baulcombe (University of Cambridge, UK) for the PVX expression vector pGR107, Sophien Kamoun (The Sainsbury Laboratory, UK) for the constructs of R3a/Avr3a, Yuanchao Wang and Daolong Dou (Nanjing Agricultural University, China) for the constructs of BAX, INF1, PsAvh241, PsCRN63 and PsojNIP, and Qinghe Chen (Fujian Academy of Agricultural Sciences, China) for P. capsici isolates.
Funding Statement
This work is supported by National Natural Science Foundation of China (Grant no. 31101395), the Jiangsu Province Basic Research Program (Natural Science Foundation) of China (Grant no. BK2011443) and the Special Fund for Agro-scientific Research in the Public Interest of China (Grant no. 201303018). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Erwin DC, Ribeiro OK (1996) Phytophthora Diseases Worldwide. St Paul MN: APS Press. 562 p. [Google Scholar]
- 2. Fry W (2008) Phytophthora infestans: the plant (and R gene) destroyer. Mol Plant Pathol 9: 385–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rizzo DM, Garbelotto M, Hansen EM (2005) Phytophthora ramorum: integrative research and management of an emerging pathogen in California and Oregon forests. Annu Rev Phytopathol 43: 309–335. [DOI] [PubMed] [Google Scholar]
- 4.Bouwmeester K, van Poppel PMJA, Govers F (2009) Genome biology cracks enigmas of oomycete plant pathogens. In: Parker JE, editor. Molecular aspects of plant disease resistance. Oxford: Wiley-Blackwell. pp. 102–133.
- 5. Hardham AR (2007) Cell biology of plant–oomycete interactions. Cell Microbiol 9: 31–39. [DOI] [PubMed] [Google Scholar]
- 6. Shan W, Marshall JS, Hardham AR (2004) Gene expression in germinated cysts of Phytophthora nicotianae . Mol Plant Pathol 5: 317–330. [DOI] [PubMed] [Google Scholar]
- 7. Judelson HS, Ah-Fong AM, Aux G, Avrova AO, Bruce C, et al. (2008) Gene expression profiling during asexual development of the late blight pathogen Phytophthora infestans reveals a highly dynamic transcriptome. Mol Plant-Microbe Interact 21: 433–447. [DOI] [PubMed] [Google Scholar]
- 8. Savidor A, Donahoo RS, Hurtado-Gonzales O, Land ML, Shah MB, et al. (2008) Cross-species global proteomics reveals conserved and unique processes in Phytophthora sojae and Phytophthora ramorum . Mol Cell Proteomics 7: 1501–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen X, Klemsdal SS, Brurberg MB (2011) Identification and analysis of Phytophthora cactorum genes up-regulated during cyst germination and strawberry infection. Curr Genet 57: 297–315. [DOI] [PubMed] [Google Scholar]
- 10.Gevens AJ, Roberts PD, McGover RJ, Kucharek TA (2004) Vegetable diseases caused by Phytophthora capsici in Florida. University of Florida IFAS Extension. Available: http://edis.ifas.ufl.edu/pdffiles/VH/VH04500.pdf. Accessed 30 July 2013.
- 11. Gevens AJ, Donahoo RS, Lamour KH, Hausbeck MK (2008) Characterization of Phytophthora capsici causing foliar and pod blight of snap bean in Michigan. Plant Dis 92: 201–209. [DOI] [PubMed] [Google Scholar]
- 12. Ho HH (1990) Taiwan Phytophthora . Bot Bull Acad Sinica 31: 89–106. [Google Scholar]
- 13.Babadoost M, Tian D, Islam SZ, Pavon C (2008) Challenges and options in managing Phytophthora blight (Phytophthora capsici) of cucurbits. In: Pitrat M, editor. Cucurbitaceae 2008, Proceedings of the IXth EUCARPIA meeting on genetics and breeding of Cucurbitaceae. Avignon (France), May 21-24th, 2008. pp. 399–406.
- 14. Lamour KH, Stam R, Jupe J, Huitema E (2012) The oomycete broad-host-range pathogen Phytophthora capsici . Mol Plant Pathol 13: 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y, Bouwmeester K, van de Mortel JE, Shan W, Govers F (2013) A novel Arabidopsis-oomycete pathosystem: differential interactions with Phytophthora capsici reveal a role for camalexin, indole glucosinolates and salicylic acid in defence. Plant Cell Environ 36: 1192–1203. [DOI] [PubMed] [Google Scholar]
- 16. Kamoun S (2006) A catalogue of the effector secretome of plant pathogenic oomycetes. Annu Rev Phytopathol 44: 41–60. [DOI] [PubMed] [Google Scholar]
- 17. Tyler BM, Tripathy S, Zhang X, Dehal P, Jiang RHY, et al. (2006) Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science 313: 1261–1266. [DOI] [PubMed] [Google Scholar]
- 18. Haas BJ, Kamoun S, Zody MC, Jiang RHY, Handsaker RE, et al. (2009) Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans . Nature 461: 393–398. [DOI] [PubMed] [Google Scholar]
- 19. Baxter L, Tripathy S, Ishaque N, Boot N, Cabral A, et al. (2010) Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science 330: 1549–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lévesque CA, Brouwer H, Cano L, Hamilton JP, Holt C, et al. (2010) Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol 11: R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bozkurt TO, Schornack S, Banfield MJ, Kamoun S (2012) Oomycetes, effectors, and all that jazz. Curr Opin Plant Biol 15: 483–492. [DOI] [PubMed] [Google Scholar]
- 22. Orsomando G, Lorenzi M, Raffaelli N, Dalla Rizza M, Mezzetti B, et al. (2001) Phytotoxic protein PcF, purification, characterization, and cDNA sequencing of a novel hydroxyproline-containing factor secreted by the strawberry pathogen Phytophthora cactorum . J Biol Chem 276: 21578–21584. [DOI] [PubMed] [Google Scholar]
- 23. Gijzen M, Nurnberger T (2006) Nep1-like proteins from plant pathogens: recruitment and diversification of the NPP1 domain across taxa. Phytochem 67: 1800–1807. [DOI] [PubMed] [Google Scholar]
- 24. Ottmann C, Luberacki B, Kufner I, Koch W, Brunner F, et al. (2009) A common toxin fold mediates microbial attack and plant defense. Proc Natl Acad Sci USA 106: 10359–10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rehmany AP, Gordon A, Rose LE, Allen RL, Armstrong MR, et al. (2005) Differential recognition of highly divergent downy mildew avirulence gene alleles by RPP1 resistance genes from two Arabidopsis lines. Plant Cell 17: 1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Birch PR, Rehmany AP, Pritchard L, Kamoun S, Beynon JL (2006) Trafficking arms: oomycete effectors enter host plant cells. Trends Microbiol 14: 8–11. [DOI] [PubMed] [Google Scholar]
- 27. Dou D, Kale SD, Wang X, Jiang RHY, Bruce NA, et al. (2008) RXLR-mediated entry of Phytophthora sojae effector Avr1b into soybean cells does not require pathogen-encoded machinery. Plant Cell 20: 1930–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kale SD, Gu B, Capelluto DG, Dou D, Feldman E, et al. (2010) External lipid PI3P mediates entry of eukaryotic pathogen effectors into plant and animal host cells. Cell 142: 284–295. [DOI] [PubMed] [Google Scholar]
- 29. Kelley BS, Lee S, Damasceno CMB, Chakravarthy S, Kim B, et al. (2010) A secreted effector protein (SNE1) from Phytophthora infestans is a broadly acting suppressor of programmed cell death. Plant J 62: 357–366. [DOI] [PubMed] [Google Scholar]
- 30. Bailey K, Cevik V, Holton NJ, Byrne-Richardson J, Sohn KH, et al. (2011) Molecular cloning of ATR5(Emoy2) from Hyaloperonospora arabidopsidis, an avirulence determinant that triggers RPP5-mediated defense in Arabidopsis . Mol Plant–Microbe Interact 24: 827–838. [DOI] [PubMed] [Google Scholar]
- 31. Tian M, Win J, Savory E, Burkhardt A, Held M, et al. (2011) 454 genome sequencing of Pseudoperonospora cubensis reveals effector proteins with a putative QXLR translocation motif. Mol Plant–Microbe Interact 24: 543–553. [DOI] [PubMed] [Google Scholar]
- 32. Bos JIB, Kanneganti TD, Young C, Cakir C, Huitema E, et al. (2006) The C-terminal half of Phytophthora infestans RXLR effector AVR3a is sufficient to trigger R3a-mediated hypersensitivity and suppress INF1-induced cell death in Nicotiana benthamiana . Plant J 48: 165–176. [DOI] [PubMed] [Google Scholar]
- 33. Van Poppel PM, Jiang RHY, Sliwka J, Govers F (2009) Recognition of Phytophthora infestans Avr4 by potato R4 is triggered by C-terminal domains comprising W motifs. Mol Plant Pathol 10: 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Torto TA, Li S, Styer A, Huitema E, Testa A, et al. (2003) EST mining and functional expression assays identify extracellular effector proteins from the plant pathogen Phytophthora . Genome Res 13: 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schornack S, van Damme M, Bozkurt TO, Cano LM, Smoker M, et al. (2010) Ancient class of translocated oomycete effectors targets the host nucleus. Proc Natl Acad Sci USA 107: 17421–17426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stam R, Jupe J, Howden AJM, Morris JA, Boevink PC, et al. (2013) Identification and characterisation CRN Effectors in Phytophthora capsici shows modularity and functional diversity. PLoS ONE 8: e59517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu T, Ye W, Ru Y, Yang X, Gu B, et al. (2011) Two host cytoplasmic effectors are required for pathogenesis of Phytophthora sojae by suppression of host defenses. Plant Physiol 155: 490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van Damme M, Bozkurt TO, Cakir C, Schornack S, Sklenar J, et al. (2012) The Irish potato famine pathogen Phytophthora infestans translocates the CRN8 kinase into host plant cells. PLoS Pathog 8: e1002875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Velculescu VE, Zhang L, Vogelstein B, Kinzler KW (1995) Serial analysis of gene expression. Science 270: 484–487. [DOI] [PubMed] [Google Scholar]
- 40. Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative monitoring of gene-expression patterns with a complementary DNA microarray. Science 270: 467–470. [DOI] [PubMed] [Google Scholar]
- 41. Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, et al. (1996) Expression monitoring by hybridization to highdensity oligonucleotide arrays. Nat Biotechnol 14: 1675–1680. [DOI] [PubMed] [Google Scholar]
- 42. Boguski MS, Tolstoshev CM, Bassett DE (1994) Gene discovery in dbEST. Science 265: 1993–1994. [DOI] [PubMed] [Google Scholar]
- 43. Asmann YW, Wallace B, Thompson EA (2008) Transcriptome profiling using next-generation sequencing. Gastroenterology 135: 1466–1468. [DOI] [PubMed] [Google Scholar]
- 44. Mardis ER (2008) The impact of next-generation sequencing technology on genetics. Trends Genet 24: 133–141. [DOI] [PubMed] [Google Scholar]
- 45. Randall TA, Dwyer RA, Huitema E, Beyer K, Cvitanich C, et al. (2005) Large-scale gene discovery in the oomycete Phytophthora infestans reveals likely components of phytopathogenicity shared with true fungi. Mol Plant-Microbe Interact 18: 229–243. [DOI] [PubMed] [Google Scholar]
- 46. Araki F, Miyagi Y (1977) Effects of fungicides on penetration by Pyricularia oryzae as evaluated by an improved cellophane method. J Pestic Sci 2: 457–461. [Google Scholar]
- 47. Winnepenninckx B, Backeljau T, de Wachter R (1996) Investigation of molluscan phylogeny on the basis of 18S rRNA sequences. Mol Biol Evol 13: 1306–1313. [DOI] [PubMed] [Google Scholar]
- 48. Kent WJ (2002) BLAT––the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35: W182–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- 52. Romualdi C, Bortoluzzi S, D'Alessi F, Danieli GA (2003) IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiol Genomics 12: 159–162. [DOI] [PubMed] [Google Scholar]
- 53. Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29: 1165–1188. [Google Scholar]
- 54. Yan HZ, Liou RF (2006) Selection of internal control genes for real-time quantitative RT-PCR assays in the oomycete plant pathogen Phytophthora parasitica . Fungal Genet Biol 43: 430–438. [DOI] [PubMed] [Google Scholar]
- 55. Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340: 783–795. [DOI] [PubMed] [Google Scholar]
- 56. Jones L, Hamilton AJ, Voinnet O, Thomas CL, Maule AJ, et al. (1999) RNA-DNA interactions and DNA methylation in post-transcriptional gene silencing. Plant Cell 11: 2291–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95–98. [Google Scholar]
- 58. Lacomme C, Santa Cruz S (1999) Bax-induced cell death in tobacco is similar to the hypersensitive response. Proc Natl Acad Sci USA 96: 7956–7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang Q, Han C, Ferreira AO, Yu X, Ye W, et al. (2011) Transcriptional programming and functional interactions within the Phytophthora sojae RXLR effector repertoire. Plant Cell 23: 2064–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yu X, Tang J, Wang Q, Ye W, Tao K, et al. (2012) The RxLR effector Avh241 from Phytophthora sojae requires plasma membrane localization to induce plant cell death. New Phytol 196: 247–260. [DOI] [PubMed] [Google Scholar]
- 61. Qutob D, Kamoun S, Gijzen M (2002) Expression of a Phytophthora sojae necrosis-inducing protein occurs during transition from biotrophy to necrotrophy. Plant J 32: 361–373. [DOI] [PubMed] [Google Scholar]
- 62. Armstrong MR, Whisson SC, Pritchard L, Bos JI, Venter E, et al. (2005) An ancestral oomycete locus contains late blight avirulence gene Avr3a, encoding a protein that is recognized in the host cytoplasm. Proc Natl Acad Sci USA 102: 7766–7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hogenhout SA, Van der Hoorn RAL, Terauchi R, Kamoun S (2009) Emerging concepts in effector biology of plant-associated organisms. Mol Plant-Microbe Interact 22: 115–122. [DOI] [PubMed] [Google Scholar]
- 64. Blair JE, Coffey MD, Park S-Y, Geiser DM, Kang S (2008) A multi-locus phylogeny for Phytophthora utilizing markers derived from complete genome sequences. Fungal Genet Biol 45: 266–277. [DOI] [PubMed] [Google Scholar]
- 65. Fellbrich G, Romanski A, Varet A, Blume B, Brunner F, et al. (2002) NPP1, a Phytophthora-associated trigger of plant defense in parsley and Arabidopsis . Plant J 32: 375–390. [DOI] [PubMed] [Google Scholar]
- 66. Dou D, Kale SD, Wang X, Chen Y, Wang Q, et al. (2008) Conserved C-terminal motifs required for avirulence and suppression of cell death by Phytophthora sojae effector Avr1b. Plant Cell 20: 1118–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dong S, Kong G, Qutob D, Yu X, Tang J, et al. (2012) The NLP toxin family in Phytophthora sojae includes rapidly evolving groups that lack necrosis-inducing activity. Mol Plant-Microbe Interact 25: 896–909. [DOI] [PubMed] [Google Scholar]
- 68. Kunjeti SG, Evans TA, Marsh AG, Gregory NF, Kunjeti S, et al. (2012) RNA-Seq reveals infection-related global gene changes in Phytophthora phaseoli, the causal agent of lima bean downy mildew. Mol Plant Pathol 13: 454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Abramovitch RB, Kim YJ, Chen S, Dickman MB, Martin GB (2003) Pseudomonas type III effector AvrPtoB induces plant disease susceptibility by inhibition of host programmed cell death. EMBO J 22: 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lamour KH, Mudge J, Gobena D, Hurtado-Gonzales OP, Schmutz J, et al. (2012) Genome sequencing and mapping reveal loss of heterozygosity as a mechanism for rapid adaptation in the vegetable pathogen Phytophthora capsici . Mol Plant-Microbe Interact 25: 1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ye W, Wang X, Tao K, Lu Y, Dai T, et al. (2011) Digital gene expression profiling of the Phytophthora sojae transcriptome. Mol Plant-Microbe Interact 24: 1530–1539. [DOI] [PubMed] [Google Scholar]
- 72. Shan W, Cao M, Leung D, Tyler BM (2004) The Avr1b locus of Phytophthora sojae encodes an elicitor and a regulator required for avirulence on soybean plants carrying resistance gene Rps1b . Mol Plant-Microbe Interact 17: 394–403. [DOI] [PubMed] [Google Scholar]
- 73. Qutob D, Tedman-Jones J, Dong S, Kuflu K, Pham H, et al. (2009) Copy number variation and transcriptional polymorphisms of Phytophthora sojae RXLR effector genes Avr1a and Avr3a . PLoS ONE 4: e5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Grabbe C, Dikic I (2009) Functional roles of ubiquitin-like domain (ULD) and ubiquitin-binding domain (UBD) containing proteins. Chem Rev 109: 1481–1494. [DOI] [PubMed] [Google Scholar]
- 75. Clem RJ (2007) Baculoviruses and apoptosis: a diversity of genes and responses. Curr Drug Targets 8: 1069–1074. [DOI] [PubMed] [Google Scholar]
- 76. Tyler BM (2009) Entering and breaking: virulence effector proteins of oomycete plant pathogens. Cell Microbiol 11: 13–20. [DOI] [PubMed] [Google Scholar]
- 77. Feng BZ, Li PQ, Fu L, Sun BB, Zhang XG (2011) Methodology identification of 18 genes encoding necrosis-inducing proteins from the plant pathogen Phytophthora capsici (Pythiaceae: Oomycetes). Genet Mol Res 10: 910–922. [DOI] [PubMed] [Google Scholar]
- 78. Yousef LF, Yousef AF, Mymryk JS, Dick WA, Dick RP (2009) Stigmasterol and cholesterol regulate the expression of elicitin genes in Phytophthora sojae . J Chem Ecol 35: 824–832. [DOI] [PubMed] [Google Scholar]
- 79.Forbes BA, Sahm DF, Weissfeld AS (2002) Bailey & Scott’s diagnostic microbiology. St LouisMO: Mosby. 789 p. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The disease progressions in Nicotiana benthamiana roots caused by Phytophthora capsici isolate Pc537. Each plant root was cut open before macroscopic observations. Arrows indicate the inoculation part or browning, necrosis and rot symptoms. (A) controls (mock-inoculated); (B-D) the plants at 24, 36, 72 h post-inoculation, respectively.
(TIF)
Accumulation of Illumina total reads and unique genes in the three libraries. New unique genes (‘y’ axis) of MY, ZO, GC libraries (different color lines) decreased as the depth of sequencing (‘x’ axis) increased.
(TIF)
List of primers used for RT-PCR and cloning of the Phytophthora capsici genes.
(XLS)
Phytophthora capsici genes expressed during mycelium, zoospore and germinating cyst stages. (listed in separate sheets for each stage).
(XLS)
Differentially expressed genes between two samples. (3 sample-pairs listed in separate sheets).
(XLS)
Differentially expressed genes between two samples with annotation. (3 sample-pairs listed in separate sheets).
(XLS)
The effector genes differentially expressed during the pre-infection stages of Phytophthora capsici. (7 different categories listed in separate sheets with annotation).
(XLS)
Complete list of involved pathways for regulated DEGs between two samples. (3 sample-pairs listed in separate sheets).
(XLS)