

Abstract
A mosaic is an organism composed of two or more genetically distinct cell populations derived from a genetically homogeneous zygote. Cutaneous mosaicisms are the clinical expressions of these disorders. The main event which allows the existence of mosaicism is a genetic mutation, either structural or functional. Cutaneous mosaicisms usually manifest by specific patterns on the skin and the archetypic pattern is the system of Blaschko lines, but others include checkerboard, phylloid, large patches without midline separation and lateralization. Since 1901, when Blaschko lines were first described, the study of mosasicism has helped to elucidate the behavior of numerous genetic diseases, generating therapeutic perspectives for these pathologies, including the promising gene therapy.
Keywords: Focal dermal hypoplasia, Incontinentia pigmenti, Loss of heterozygosity, Mosaicism, Nevus
Abstract
Um mosaico é um organismo formado por duas ou mais populações de células geneticamente distintas originadas a partir de um mesmo zigoto geneticamente homogêneo. Os mosaicismos são as expressões clínicas dessa desordem, e a mutação gênica seu evento determinante, que pode ser tanto estrutural quanto funcional. Os mosaicismos cutâneos costumam se expressar em padrões específicos, dentre os quais podem ser mencionados as prevalentes linhas de Blaschko, o padrão "checkerboard", o padrão filóide, o padrão em placa sem separação na linha média e o padrão de lateralização, que serão abordados neste artigo. Desde 1901, momento da primeira descrição das linhas de Blaschko, o estudo dos mosaicismos tem contribuído para a elucidação do comportamento de numerosas desordens genéticas, de forma a criar perspectivas terapêuticas para essas doenças, incluindo a promissora terapia gênica.
INTRODUCTION
A mosaic is an organism composed of two or more genetically distinct cell populations derived from a genetically homogeneous zygote. Mosaicism is the clinical expression of these disorders, as first described by Blaschko. Numerous genetic cutaneous diseases reflect mosaicism and many represent an opportunity to study systemic pathologies that would otherwise be occult or even fatal.
Contrary to common belief, cutaneous mosaicisms are common phenomena in dermatological practice. For instance, it is currently believed that all nevi represent a form of mosaicism.1
Traditionally, cutaneous diseases that reflect mosaicism are distributed along certain patterns and forms. The prototype for patterns of cutaneous mosaicism is the Blaschko lines system, originally described in 1901 by Alfred Blaschko. The other existing patterns will also be described in this article.
BRIEF HISTORY
In 1901, the German dermatologist Alfred Blaschko examined systematically more than 150 patients with segmental cutaneous lesions and established specific linear patterns for the distribution of lesions - "S"-shapes on the lateral and anterior aspects of the trunk, linear streaks on extremities and "V"-shapes on the central back -, which later came to be known as the Blaschko lines (Figure 1).2
FIGURE 1.

Blaschko lines
In the 1970s, Rudolph Happle hypothesized that the Blaschko lines represented a contrast between genetically normal and abnormal skin, characterizing mosaicism. However, genetic demonstration of mosaicism was carried out for the first time in 1994 for epidermal nevi of epidermolytic hyperkeratosis.3 In 2001, exactly one century after Blaschko's initial observation, Happle described the Blaschko lines pattern in the cephalic and cervical regions, appearing in hour-glass shape, converging at the nasal root, with perpendicular intersections on several areas of the face, spiral intersections on the scalp, and V-shaped in the cervical region (Figure 1).2,4
CLASSIC PATTERNS OF CUTANEOUS MOSAICISMS
In 1993, Happle described several forms of segmental manifestation of cutaneous diseases. These forms include the classic Blaschko lines, in addition to four others (Figure 2).2,5,6
FIGURE 2.

Patterns of cutaneous mosaicisms
Type 1: Blaschko lines
This is the most common pattern of cutaneous mosaicism. During embryogenesis, when the presence of the primitive line gives the embryonic disk bilateral symmetry, precursory cells start to proliferate on the midline and grow in transversal direction from this line. As the longitudinal growth of the embryo progresses, these cells set up in a V shape on the back and an S shape on the anterolateral portion of the trunk (Figure 1). Such lines represent ectodermal development patterns. Hence, the Blaschko lines disorders typically affect the keratinocytes and melanocytes.2This pattern features in a wide variety of congenital and acquired diseases, and can be divided into two further types: 1a and 1b (Figure 2).
Type 1a: Blaschko lines, narrow bands
This pattern involves lesions distributed along the Blaschko lines, in narrow bands. Typically, it is seen in X-linked Incontinentia pigmenti and pigmentary disorders previously known as "hypomelanosis of Ito" (Figure 3).
FIGURE 3.
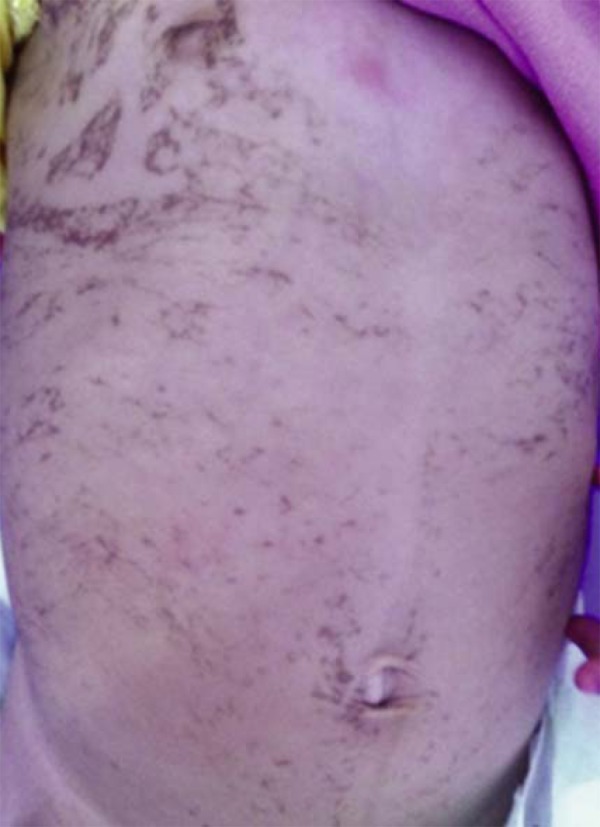
X-linked Incontinentia pigmenti. Pattern type 1a (Blaschko lines, narrow bands)
Type 1b: Blaschko lines, broad bands
With this variety, Blaschko lines appear as broad bands, as in the case of McCune-Albright syndrome, a disease that is characterized by polyostotic fibrous dysplasia, precocious puberty and hyperpigmentation in broad bands along the Blaschko lines.
Type 2: the "Checkerboard" pattern
This type typically concerns alternate areas of pigmentary disturbance in each hemibody, with an abrupt interruption at the midline, resembling a checkerboard (Figure 2). Classic examples include systematized nevus spilus and X-linked congenital generalized hypertrichosis. Other lesions that present this pattern include the Becker nevus, café au lait spots, port-wine stains and cutis marmorata telangiectatica congenita, among others. Individuals known as human chimeras, with two original, genetically different cell ancestries, can also present pigmentary disorders in this pattern.
Type 3: the Phylloid pattern
This pattern was recently described and is characterized by a "leaf-like" appearance in the pigmentary disturbance. It is composed of oval, leaf/pearshaped, asymmetrical or elongated stains (Figure 2). All patients with this pattern of hypopigmentation also had other abnormalities, like mental handicaps, agenesis of the corpus callosum, conductive deafness, coloboma of the retina, craniofacial anomalies, as well as various musculoskeletal anomalies (brachydactyly, clinodactyly and campylodactyly). Phylloid hypomelanosis is the classic example of this pattern; it is a syndrome characterized by the association of cutaneous lesions with other aforementioned anomalies, due to chromosome 13 trisomy or tetrasomy mosaicism. The phylloid pattern can also manifest with hyperpigmentation.
Type 4: Patchy pattern without midline separation
The distribution of lesions is due to large plaques that do not respect the dorsal or ventral midline (Figure 2). Typically, it is found in giant congenital melanocytic nevi (Figure 4). It is held that this is because of a genetic mutation that would have been fatal but for the mosaicism, as complete cutaneous involvement has never been observed. Nonetheless, at present, there is no molecular or cytogenetic proof for this hypothesis.7
FIGURE 4.
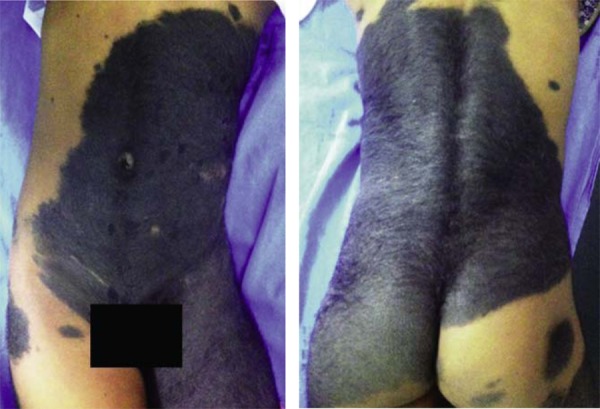
Giant congenital melanocytic nevus. Plaque pattern, crossing the dorsal and ventral midlines
Type 5: Lateralization pattern
The pattern is characterized by involvement of only one hemibody, with a sharp midline demarcation, due to abrupt interruption of lesions in this area (Figure 2). It is unique to CHILD syndrome, a rare Xlinked dominant genodermatosis that is fatal to males, characterized by congenital hemidysplasia, icthyosiform erythroderma and limb defetcs. With this disease, visceral abnormalities are always ipsilateral to cutaneous lesions. However, both contralateral and ipsilateral lesions can occur jointly, following the Blaschko lines.
CLASSIC MOSAICISM PATTERNS AND EMBRYOLOGY
Cutaneous mosaicism patterns correlate with mutated cell components.1Thus, mosaic lesions derived from epidermal components normally follow Blaschko line patterns and their subtypes, and virtually never appear in checkerboard form. On the other hand, mosaic lesions of mesodermal origin generally manifest in checkerboard patterns or diffuse plaques, as in vascular and collagenous nevi. Nevertheless, they may follow the Blaschko lines, as in focal dermal hypoplasia and atrophoderma of Moulin.1The socalled classic patterns of mosaicism usually exhibit greater predisposition to the simultaneous existence of extracutaneous abnormalities than the non-classic ones. Thus, precocious ectodermal mutations can lead to neurocutaneous syndromes, affecting the skin, central nervous system and eyes, as happens with epidermal nevus syndrome and the previously termed Hypomelanosis of Ito.1
ETIOPATHOGENESIS OF CUTANEOUS MOSAICISMS
Mosaicisms can originate from different mechanisms but genetic mutation is an essential condition. Genetic (or somatic) mosaicisms stem from gene mutations that occur during embryogenesis. Yet epigenetic mosaicism is due to posterior modifications in gene expression (inactivation of the X chromosome or autosomal genes). The former cannot be inherited, except in cases of gonadal genetic mosaicism; though epigenetic mosaicisms are passed on to the next generation of cells and can thus be inherited.2,7
CLASSIFICATION OF CUTANEOUS MOSAICISMS
Genetic mosaicism (somatic)
This type of mosaicism emerges when a cell undergoes a de novo postzygotic mutation during embryonic development and therefore, cells that are derived from this will carry the mutation. The resulting embryo will thus carry the two genetically distinct cell populations, one with the mutation, the other without it. Clinically, the mutated cells will express a different phenotype from the others, manifesting the characteristics of the disease in segmental fashion.1,2,7
It is subdivided into:
a) mosaicism in non-fatal autosomal dominant diseases;
b) mosaicism in fatal autosomal diseases; and
c) mosaicism in inflammatory polygenic diseases.1,5,7
A) Mosaicism in non-fatal autosomal dominant diseases
Type 1 segmental mosaicism:
It starts during embryonic development, due to a de novo postzygotic mutation in one of the alleles of a given gene, resulting in an altered allele. From this moment, the individual will have two cell populations, one normal, the other sick (Figure 5).1,2,7Thus, the characteristics of this disease will be distributed along the Balschko lines or other mosaic patterns, corresponding to cells containing the mutation.2,5,8The rest of the skin will be normal genotypically and phenotypically. In general, this type of mosaicism is not inherited, except when the mutation affects the gonads. Examples of type 1 segmental mosaicisms include epidermolytic hyperkeratosis, type 1 neurofibromatosis, Darier's disease, tuberous sclerosis, basal cell nevus syndrome, multiple syringomas and pachyonychia congenita type 1.1,5
FIGURE 5.

Type 1 and type 2 segmental mosaicism in autosomal dominant diseases
Type 2 segmental mosaicism:
Type 2 segmental mosaicism occurs in individuals carrying the autosomal dominant disease caused by a mutation in one of the alleles in one gene. In this case, a new postzygotic mutation takes place during embryonic development, inactivating the other allele that was normal, causing what is called a loss of heterozygosity (Figure 5).1,2,5
As a result of this, an individual who is diffusely and mildly affected by the disease will also present an earlier onset and a worst presentation of the same disease in a mosaic form.1,5Proven examples of type 2 segmental mosaicisms include once again epidermolytic hyperkeratosis, type 1 neurofibromatosis, tuberous sclerosis, cutaneous leiomyomatosis, multiple syringomas, as well as Buschke-Ollendorf syndrome, Darier's disease, Hailey-Hailey disease and disseminated superficial actinic porokeratosis, among others.1,2
B) Mosaicism in fatal autosomal diseases
This type of mosaicism involves dominant mutations which, if present in the zygote, would be fatal to the organism.1,5However, since the mutation occurs after the formation of the zygote, cells carrying the fatal mutation survive as a mosaic, presumably on account of the proximity to normal cells.1,5,8,9
Fatal autosomal recessive diseases can also manifest as mosaicisms. This happens when higid, heterozygotic individuals suffer a postzygotic mutation or another genetic event that inactivates the normal allele during uterine development, resulting in distribution of mosaics in affected tissue. This mechanism can be explained using the concept of paradominance, which is also responsible for family aggregation of primarily sporadic disorders. Heterozygotic carriers of paradominant mutations are phenotypically normal and transmit the mutation to their offspring without clinical expression. This explains the inheritance pattern of cutis marmorata telangiectatica congenita, Sturge Weber syndrome, and certain syndromes involving melanocytes (like Becker nevi and speckled lentiginous nevus syndrome).
This section will focus on hypomelanosis of Ito and verrucous epidermal nevi as examples of fatal autosomal disorders. Other examples of fatal autosomal diseases that survive through mosaicism are outlined in chart 1.1,5
CHART 1.
Examples of surviving fatal autosomal mutations from the mosaicism
| • Pigmentary mosaicism (including phylloid hypomela-nosis and the previosuly termed hypomelanosis of Ito) |
| • Verrucous epidermal nevus syndrome |
| • Nevus comedonicus syndrome |
| • McCune-Albright syndrome |
| • Multiple syringomas |
| • Buschke-Olendorff syndrome |
| • Schimmelpenning syndrome |
| • Cutis marmorata telangiectatica congenita |
| • Giant congenital melanocytic nevus |
Hypomelanosis of Ito
Hypomelanosis of Ito is a generic term for hypopigmentation along the lines of Blaschko, which is sometimes used wrongly to define a specific entity. The difficulty in characterizing precisely hypomelanosis of Ito has led certain authors to reserve this term for patients with associated extracutaneous anomalies.2
Hypopigmentation along the Blaschko lines can be caused by several mutations, such as translocations, trisomy, triploidy or chromosomal aberrations, which would otherwise be incompatible with life.7,10
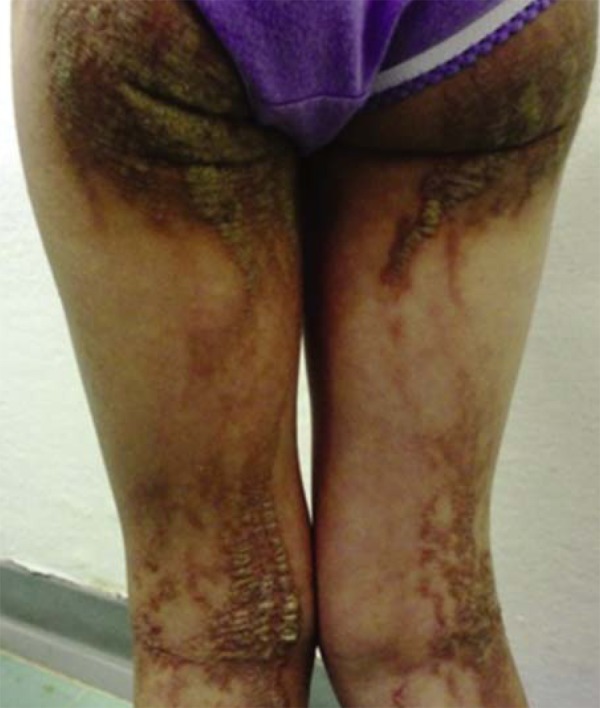
Hypochromic macules can appear linearly or in swirls, along the Blaschko lines, unilaterally or bilaterally, and can be present from birth or appear during infancy (Figure 6). Exposure to sun can precipitate the development or accentuation of lesions, by increasing the contrast with normal skin. Together with the cutaneous condition, there can be abnormalities in the central nervous system, convulsions, psychomotor delay and ocular, skeletal and dental anomalies.2,10,11
FIGURE 6.
Hypomelanosis of Ito. Linear hypopigmentation along the Blaschko lines. (Image courtesy of Dr. Roberto Dóglia Azambuja, University Hospital of Brasilia, Brasilia, Federal District)
Verrucous epidermal nevus
Hamartomas are abnormal accumulations of tissue components. Thus, all epidermal nevi are epidermal hamartomas, which can be derived from keratinocytes, hair follicles, sebaceous or sweat glands.1
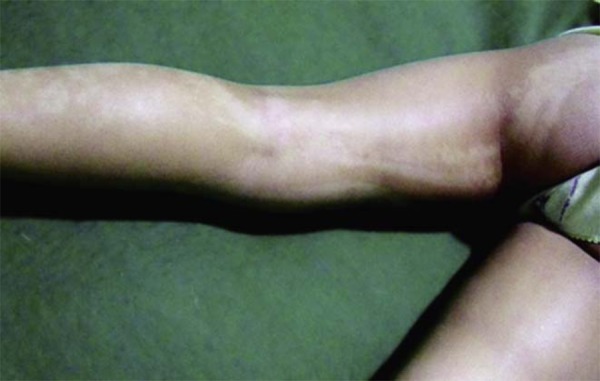
Verrucous epidermal nevus originate from keratinocyte hyperplasia, and are characterized by brown or skin-colored papules and/or plaques, with a verrucous or velvety surface, appearing linearly, following the Blaschko lines (Figures 7A and 7B). On flexor surfaces and osseous prominences, these nevi can become more hyperkeratotic (Figure 8). In rare cases, it is possible for basal cell carcinomas, keratocanthomas, spinocellular carcinomas, and malignant eccrine poromas to develop, though these are rarer than with the other epidermal nevi (sebaceous and apocrine). Today, it is known that up to 33% of verrucous epidermal nevi are due to mutations in the FGFR3 gene, which is also responsible for the development of seborrheic keratoses.1
FIGURE 7.

Verrucous epidermal nevus: A) Brown verrucous plaques following the Blaschko lines (typo 1b); B) Brown papules and plaques distributed linearly along the Blaschko lines
FIGURE 8.
Verrucous epidermal nevus. Accentuation of hyperkeratosis in flexor areas
When lesions are diffuse, the condition is named ichthyosis hystrix and, in this case, it can be accompanied by neurological, ocular and skeletal abnormalities, constituting the verrucous epidermal nevus syndrome.
C) Mosaicism in inflammatory polygenic diseases
Many polygenic diseases can also manifest in segmental form.1,12,13The distribution of these diseases tends to be symmetrical and diffuse. However, it is possible to have linear or unilateral presentation, as well as other superimposed segmental arrangements in relation to the classic manifestation of the disease.
Such cases should not be categorized as type 2 segmental mosaicism because this term applies exclusively to monogenic traits. For polygenic diseases, the term "superimposed segmental manifestation" seems more appropriate.12,13
This pronounced segmental involvement has been explained by the loss of heterozygosity concerning one of the genes that predisposes people to the disease, during a precocious stage of development.5The loss of heterozygosity can stem from several mechanisms like mitotic recombination, gene conversion, punctual mutations, deletions and mitotic nondisjunctions.12,13
Examples of polygenic diseases that can entail segmental presentation include: psoriasis, lichen planus, dermatomyositis, atopic dermatitis, systemic lupus erythematosus, granuloma annulare, graft versus host disease, erythema multiforme, drug eruptions, pemphigus vulgaris, and vitiligo, among others (Figure 9).1,5,12,13
FIGURE 9.
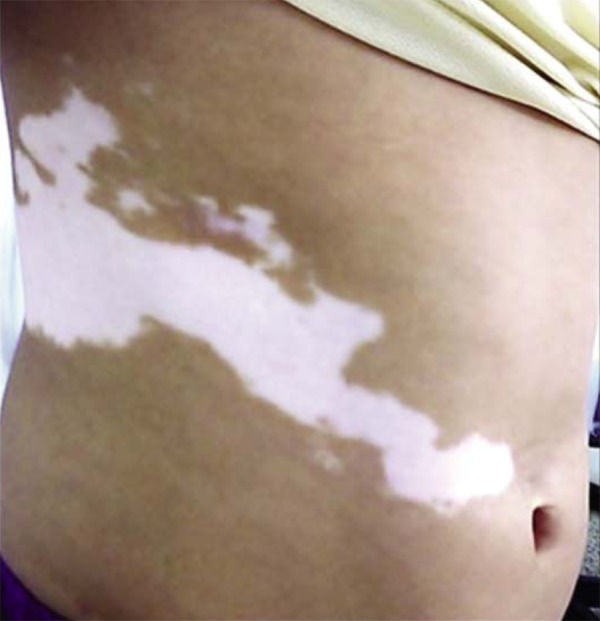
Segmental vitiligo
This distribution pattern has already been described as zosteriform. However, this term is inaccurate, given that lesions do not follow the dermatomes, but rather, the Blaschko lines.5
Epigenetic (functional) mosaicism
Functional mosaicism does not entail gene mutations per se, with structural or sequential DNA modifications, but rather, changes in gene expression (gene activation or silencing). An example of functional mosaicism is the deactivation of one of the X chromosomes in females during embryonic development, a phenomenon known as lyonization. It occurs particularly in X-linked disorders.
Retrotransposons are genetic sequences of viral origin that interpose themselves to the human genome, provoking changes in gene expression, and which are perhaps involved in this type of mosaicism.1,2
Gene changes related to functional mosaicism can be autosomal or X-linked, and dominant or recessive.1 X-linked disorders can occur in three patterns: X-linked recessive diseases, predominant in males; non-fatal X-linked dominant diseases, which affect both sexes; and fatal X-linked dominant diseases affecting males.2
In the case of X-related recessive diseases, male patients present the generalized form of the disease, while female patients present variable mild phenotypes, since only cells where the normal X has been inactivated will exhibit abnormal phenotypes.1
On the other hand, in fatal X-linked dominant diseases, female patients will have mosaic phenotypes, and survive due to the concomitant presence of normal cells, since only cells in which the normal X is inactivated will be sick. These diseases rarely affect men, as the embryo would probably be unviable. When they are found in men, it is due to the karyotype XXY, and they survive on account of the same mechanism as women. Another possible survival mechanism for men happens via somatic, postzygotic mutation, as some cells are saved from the mutation.1,14
A) Functional mosaicisms in X-linked diseases
Cutaneous lesions tend to be distributed along the Blaschko lines pattern, in narrow bands. Exceptions include CHILD syndrome, which has pattern type 5.2
Below, detailed descriptions are provided of Goltz-Gorlin syndrome and Bloch-Sulzberger syndrome, examples of X-linked genodermatoses that manifest as mosaics.
Focal dermal hypoplasia (Goltz-Gorlin or Goltz syndrome):
This is a rare kind of X-linked, dominant mesoectodermal genodermatosis, fatal in men, while 90% of affected patients are female. It affects multiple organs, in addition to the skin.15
The main cutaneous alterations include atrophic lesions, with erythema, hyperpigmentation or hypopigmentation, or even vitiligoid spots, in a reticular pattern, which are present from birth and usually follow the Blaschko lines (Figure 10A).15,16,17Yellow-brown nodules are also characteristic, stemming from the herniation of subcutaneous tissue (Figure 10B). There can also be vegetative fibrovascular periorificial lesions (oral, perineal, vulvar), which can easily be mistaken for lesions stemming from the human papillomavirus (Figure 10B and 10C).15
FIGURE 10.

Goltz syndrome. A) Dyschromic areas of reticular nature following the Blaschko lines; B) Yellow nodules corresponding to herniation of subcutaneous tissue and periorificial papillomatosis lesions; C) Genital papillomatosis lesions; D) Syndactyly, representing "lobster foot"
Other manifestations include adnexal alterations, like rarefaction and capillary fragility, nail deformities, asymmetrical skeletal, ocular, neurological, pulmonary, cardiovascular and dental anomalies15,16,18Classic radiological characteristics are striated osteopathy, shortening of limbs and syndactyly, including "lobster hand/foot" deformities (Figure 10D). The disease has been associated with PORCN gene mutations, located in the Xp11.23 locus, which codifies proteins of the endoplasmic reticulum associated with the secretion of Wnt proteins.16,17
Incontinentia Pigmenti (Bloch-Sulzberger syndrome):
Incontinentia pigmenti is a rare, X-linked dominant genodermatosis, caused by a NEMO gene mutation (nuclear factor kappa b essential modulator), located in the Xq28 locus. This gene acts in the transcription of nuclear factor kappa b (NFκB), which protects against apoptosis induced by TNF α.6,19
The mutation is fatal in males, who only survive in the context of Klinefelter syndrome or postzygotic mutations. It is a multisystem disorder, affecting tissues derived from the ectoderm (neurological, ocular, skeletal and skin tissues).19
The cutaneous findings are specific to the syndrome and occur in 96% of cases. They are usually divided into four stages, which can be concomitant or sequential: stage 1- during birth or the first months of life, characterized by linear inflammatory vesicles and bullae that can last weeks to months; stage 2- linear verrucous hyperkeratotic plaques appear (they can last several months); stage 3- brown or grey-blue, superimposed pigmentation can emerge, distributed along the Blaschko lines or appearing as "Chinese characters", which tends to fade slowly until it disappears in adulthood; and lastly, stage 4- linear hypopigmented macules, with loss of cutaneous appendages in the midsection and limbs, in adulthood (Figure 11).19,20
FIGURE 11.

Incontinentia pigmenti. A) Inflammatory vesicle in genital region (stage 1); B and C) Brown pigmentation on the trunk and lower limbs, distributed linearly along the Blaschko lines appearing as "Chinese characters" (stage 3)
Extracutaneous manifestations occur in 70-80% of cases, affecting the central nervous system (convulsions, mental retardation, hydrocephalus), eyes (squint eyes, cataract, anophthalmia, microphthalmia), teeth (hypodontia, partial anodontia), and the musculoskeletal system (syndactyly, cranial deformities, hemiatrophy of limbs).19
Other X-linked disorders that are fatal to males include CHILD syndrome, type 1 oral-facial-digital syndrome and Conradi-Hunermann-Happle syndrome.19,21Nonfatal disorders include X-linked recessive hypohidrotic ectodermal dysplasia, Menkes disease, Xlinked congenital dyskeratosis, ichthyosis follicularis, alopecia and photophobia (IFAP), Partington syndrome and X-linked hypertrichosis.21
Reverse mosaicism
Reverse mosaicism occurs when a previously faulty gene undergoes spontaneous repair. Clinically, healthy areas are found in segmental distribution among affected skin areas.1
The correction mechanisms involved include reverse mutation, gene conversion, gene deletion, intragenic recombination and second-site mutation.1Reverse mutation occurs when the pathogenic mutation changes the wild-type sequence, restoring the transcription of the original protein. Gene conversion and intragenic recombination both involve homologous recombination and cannot be confused with a potential reversion mechanism in heterozygotes. Gene conversion involves a unidirectional, not reciprocal, transfer of genetic material from a donor sequence to a receptor sequence. Second-site mutations refer to the presence of a compensatory mutation above or below the faulty sequence, resulting in restoration of the sequence reading. Other, less characteristic reversions are retrotransposons and DNA slippage.22
Reverse mosaicism has been described in several genetic disorders, for instance Kindler syndrome, epidermolysis bullosa, fanconi anemia and Wiskott-Aldrich syndrome.22-24
Twin spotting (didymosis)
Twin spots are plaques from mutated tissue that differ among themselves and from the rest of the skin. Mutant areas can be paired or interspersed in the same hemibody, or they can be located on opposite sides, following (or not) the Blaschko lines.25,26
This form of cutaneous mosaicism occurs when an embryo that presents two distinct recessive mutations in each homologous chromosome undergoes "crossing-over" during the process of cell division. Thus, it originates two homozygous cells for different phenotypes. Hence, two stem-cells are formed, with distinct characteristics, which will originate the two adjacent clonal lineages, precursors of twin spots. The other cells in the embryo will remain heterozygous, with a normal phenotype.26
Two types of twin spots have been described: allelic and non-allelic twin spotting. With allelic twin spotting, areas with an excess or lack of skin characteristics are paired. For instance, this applies to cutis tricolor, as hypopigmented and hyperpigmented macules are paired; vascular twin nevi (telangiectatic nevus associated with anemic nevus); and Proteus syndrome, where segmental areas of hypertrophy and hypotrophy are present.1,24,25
In non-allelic twin spotting, the loss of heterozygosity involves more than one gene locus. There are areas of mutated tissue with different cell components. Examples of this type of twin spotting include phakomatosis pigmentokeratotica and phakomatosis pigmentovascularis.1,24,25
CONCLUSION
A century on from the description of the Blaschko lines, more detailed studies are still needed on the dermatosis that make up cutaneous mosaicisms and their presentation patterns. The discovery of many of the mechanisms involved in the mosaicisms has been crucial in elucidating fundamental aspects of human genetics and the behavior of the diseases and their types of inheritance. Therefore, there is potential for more complete understanding of various pathologies, as well as for hope as regards the use of gene therapy in managing these diseases.
QUESTIONS
1. It is known that distribution patterns along the Blaschko lines take the following forms:
a) "V"-shaped on the chest, arch-shaped on the abdomen and "S"shaped on the central back;
b) "S"-shaped on anterolateral aspects of the trunk and "V"-shaped on the central back;
c) Linear streaks on the chest and abdomen and "V"-shaped on the central back;
d) "V"-shaped on the chest and "S"-shaped on the abdomen and central back.
2. Regarding classic patterns of cutaneous mosaicism, it can be stated that:
a) the pattern along the Blaschko lines is the most common;
b) the X-linked pattern of Incontinentia pigmenti is pattern type 1b;
c) the phylloid pattern is never associated with neurological abnormalities;
d) CHILD syndrome exhibits a "checkerboard" pattern - type 3.
3. The following can be stated about the etiopathogenesis of cutaneous mosaicisms:
a) the mechanisms related to epigenetic mosaicism include mutations, deletions and translocations;
b) with genetic mosaicism, there is a postzygotic mutation in one of the alleles, giving rise to two genetically different cell populations, one with the mutation, the other without;
c) genetic mosaicism involves mutations only in genetic expression, like the activation or silencing of genes;
d) retrotransposons are viral genetic sequences that interpose themselves to the human genome and are perhaps involved in genetic mosaicism.
4. Type 1 segmental mosaicism:
a) is a type of genetic mosaicism that emerges in individuals already carrying an autosomal dominant disease when there is a new postzygotic mutation, resulting in more serious manifestations of the disease along the Blaschko lines;
b) in most cases, is passed on through the generations;
c) can never be inherited;
d) is a type of genetic mosaicism involving a mutation in one of the alleles during embryonic development, provoking the emergence of abnormal cell populations that are usually distributed along the Blaschko lines, while the rest of the skin remains normal.
5. The following can be said about hypomelanosis of Ito, except:
a) it is caused by a great variety of mutations such as translocations, trisomy, triploidy or chromosomal aberrations;
b) it is a generic term referring to hypopigmentation along the Blaschko lines, which generally manifests at birth or during infancy;
c) it is a well-defined entity, characterized by hypochromic stains that follow the Blaschko lines;
d) it can be associated with abnormalities of the central nervous system, and ocular, osseous and dental anomalies.
6. As regards epigenetic mosaicisms:
a) X-linked recessive diseases are more common in females;
b) X-linked recessive diseases usually manifest in men in generalized form, while in women, the phenotype is variable, as only cells in which the normal X is inactivated will be sick; c) fatal X-linked dominant diseases are almost always fatal in females;
d) focal dermal hypoplasia is fatal to women.
7. Indicate the correct statement regarding types of mosaicism:
a) somatic mosaicism stems from a postzygotic mutation, giving rise to two distinct cell populations. This mutation can be transmitted to offspring via mechanisms that are autosomal dominant, recessive or X-linked, depending on the inheritance pattern in question.
b) somatic mosaicism stems from a postzygotic mutation, giving rise to two genetically distinct populations. This mutation can be transmitted to offspring if the progenitor's genitals are affected;
c) somatic mosaicism stems from a postzygotic mutation, giving rise to two genetically distinct populations. Since this mutation is postzygotic, it cannot be transmitted to offspring;
d) somatic mosaicism stems from a postzygotic mutation, giving rise to two genetically distinct populations, and is equivalent to gonosomal mosaicism.
8. Indicate the correct statement regarding epigenetic mosiacism:
a) epigenetic mosaicism, just like somatic mosaicism, depends on structural genetic mutations;
b) epigenetic mosaicism can occur through translocations, punctual mutations and second-hit mutations;
c) epigenetic mosaicism is dependent on changes in the mechanisms that control genetic expression;
d) epigenetic mosaicism occurs commonly in X-linked diseases and thus only affects males.
9. Indicate the correct answer regarding mosaicism in fatal diseases:
a) the survival of the embryo carrying the fatal mutation depends on the coexistence of normal cells and mutated cells in one same tissue;
b) Along the letal diseases that manifest with mosaicism there are Incontinentia pigmenti, hypomelanosis of Ito and Sturge Weber syndrome;
c) in autosomal dominant diseases, the survival mechanism of the fetus is due to a postzygotic mutation of a part of the affected cells, which lose the mutation and become normal;
d) with autosomal recessive diseases, mutation of the second allele is not necessary for an individual to develop with mosaicism, due to the phenomenon of paradominance.
10. Indicate the incorrect statement:
a) hypomelanosis of Ito can be identified by the presence of hypochromia following the Blaschko lines but phylloid and checkerboard patterns are also typical indicators;
b) the term hypomelanosis of Ito has been used generically to refer to hypomelanosis following the Blaschko lines;
c) there is no consensus in the literature on whether or not hypomelanoses following the Blaschko lines can be called hypomelanosis of Ito;
d) not all hypomelanoses that follow the Blaschko lines come with neurological disorders.
11. Many inflammatory polygenic diseases that present in mosaic form have been described. Choose the correct statement on these diseases:
a) the manifestation of these diseases in mosaic form can be explained by type 2 segmental mosaicism, with more severe manifestation following the Blaschko lines superimposed over the lighter, generalized disease;
b) segmental manifestation of these diseases can be explained by the loss of heterozygocity of the affected genes;
c) loss of heterozygocity occurs through epigenetic mutations;
d) the most common manifestation of mosaicism with these diseases is the zosteriform pattern.
12. Indicate the incorrect statement regarding Goltz syndrome:
a) it is an X-linked autosomal dominant disease that is fatal to females;
b) the disease affects multiple organs as well as the skin;
c) yellow-brown nodules are characteristic, stemming from the herniation of subcutaneous tissue;
d) classic radiological characteristics are striated osteopathy, shortening of limbs and syndactyly, including "lobster hand" deformities.
13. Reverse mosaicism has been described for several genetic disorders and has signified hope in the treatment of these diseases. Indicate the incorrect statement:
a) reverse mosaicism occurs when a previously faulty gene undergoes spontaneous reparation;
b) diagnosis can be considered if healthy areas are found in segmental distribution among affected skin areas;
c) the correction mechanisms involved include reverse mutation, gene conversion, gene deletion, intragenic recombination and second-site mutation.
d) reparation can only happen through primary correction of the faulty gene.
14. Indicate the correct statement in relation to twin spotting:
a) this mechanism that presents segmentally was based on an experiment with plants;
b) the concept of twin spotting is based exclusively on the concept of paradominance and does not need structural chromosomal mutations to occur;
c) this model has been used to explain the occurrence of many nevi, namely: anemic, verrucous and sebaceous nevi;
d) three types of twin spotting have been described: allelic, nonallelic and episomal;
15. Following the description of the Blaschko lines, Happle described several other types of mosaicism. Indicate the incorrect statement:
a) phylloid hypomelanosis is an example of type 3 manifestation (phylloid type) and it occurs through chromosome 13 trisomy or tetrasomy;
b) giant congenital nevi also present segmentally and are examples of type 4 manifestation;
c) Blaschko lines disorders affect keratinocytes, melanocytes and mesodermal elements
d) the type 1a Blaschko lines pattern is typically seen in X-linked Incontinentia pigmenti and hypomelanosis of Ito.
16. Indicate the correct statement regarding verrucous epidermal nevi:
a) in rare cases, it is possible for basal cell carcinomas, keratoacanthomas, spinocellular carcinomas, and malignant eccrine poromas to develop, which is more common than with the other epidermal nevi (sebaceous and apocrine);
b) verrucous epidermal nevi stem from keratinocyte hyperplasia;
c) up to 33% of verrucous epidermal nevi are due to mutations in the FGFR3 gene, which is also responsible for the development of basal cell carcinoma;
d) with ichthyosis hystrix, malformations occur in the genitourinary tract.
17. Indicate the correct statement:
a) a chimera organism emerges from two genetically distinct cell lineages;
b) a chimera organism emerges from a zygote that has undergone a postzygotic mutation, giving rise to two different cell lines;
c) a chimera organism emerges from a progenitor who has undergone mutations in their gonadal cells and transmitted the mutation to their offspring;
d) a chimera organism emerges from serial mutations from the original embryonic cell.
18. Indicate the correct statement regarding the Blaschko lines:
a) they are lines that follow the dermatomes, and can be a presentation pattern for inflammatory cutaneous diseases;
b) they can also be described as a zosteriform pattern;
c) the term "Blaschkitis" has been used to describe any inflammatory disease following the Blaschko lines;
d) they can be classed into typo 1a and type 1b.
19. Indicate the incorrect statement regarding Incontinentia pigmenti:
a) it is an X-linked dominant genodermatosis;
b) it is an X-linked recessive genodermatosis;
c) it is a multisystem disorder, affecting tissues derived from the ectoderm (nerve, ocular, skeletal and skin tissues);
d) cutaneous findings are specific to the syndrome and are usually divided into four stages, which can be concomitant or sequential.
20. Cutaneous mosaicism patterns usually vary according to the mutated cell type. Regarding this topic, indicate the incorrect statment:
a) mosaic lesions derived from epidermal components usually acquire the Blaschko lines pattern;
b) mosaic lesions of mesodermal origin generally manifest in checkerboard patterns or as diffuse plaques, as happens with vascular and collagenous nevi;
c) mosaic lesions of mesodermal origin never follow the Blaschko lines;
d) precocious ectodermal mutations can lead to neurocutaneous syndromes.
Answer key.
Neurofibromatosis: chronological history and current issues. An Bras Dermatol. 2013;88(3):329-43.
| 1) d | 6) b | 11) b | 16) d | |||
| 2) b | 7) d | 12) c | 17) c | |||
| 3) b | 8) a | 13) d | 18) a | |||
| 4) c | 9) c | 14) d | 19) c | |||
| 5) b | 10) c | 15) b | 20) c |
Papers
Information for all members: The EMC-D questionnaire is now available at the homepage of the Brazilian Annals of Dermatology: www.anaisdedermatologia.org.br. The deadline for completing the questionnaire is 30 days from the date of online publication.
Footnotes
Study carried out at the University Hospital of Brasilia - University of Brasilia (HUB-UnB) - Brasilia (DF), Brazil.
Financial Support: None.
Conflict of Interest: None.
REFERENCES
- 1.Torrelo A. Schachner LA, Hansen R. Pediatric Dermatology. London: Mosby; 2010. Cutaneous Mosaicism; pp. 529–556. [Google Scholar]
- 2.Molho-Pessach V, Schaffer JV. Blaschko lines and other patterns of cutaneous mosaicismo. Clin Dermatol. 2011;29:205–225. doi: 10.1016/j.clindermatol.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 3.Paller AS, Syder AJ, Chan YM, Yu QC, Hutton E, Tadini G, et al. Genetic and Clinical Mosaicism in a Type of Epidermal Nevus. N Engl J Med. 1994;331:1408–1415. doi: 10.1056/NEJM199411243312103. [DOI] [PubMed] [Google Scholar]
- 4.Happle R, Assim A. The lines of Blaschko on the head and neck. J Am Acad Dermatol. 2001;44:6125–6125. doi: 10.1067/mjd.2001.112386. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg I, Sprecher E. Patterned disorders in dermatology. Clin Dermatol. 2011;29:498–503. doi: 10.1016/j.clindermatol.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 6.Siegel DH. Cutaneous mosaicism: a molecular and clinical review. Adv Dermatol. 2008;24:223–244. doi: 10.1016/j.yadr.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Happle R. Harper J, Oranje A, Prose N. Textbook of Pediatric Dermatology. Oxford: Blackwell Publishing; 2006. Principles of Genetics, Mosaicism and Molecular Biology; pp. 1228–1246. [Google Scholar]
- 8.Lipsker D, Flory E, Wiesel ML, Hanau D, de la Salle H. Between light and dark, the chimera comes out. Arch Dermatol. 2008;144:327–330. doi: 10.1001/archderm.144.3.327. [DOI] [PubMed] [Google Scholar]
- 9.Hamm H. Cutaneous mosaicism of lethal mutations. Am J Med Genet. 1999;85:342–345. doi: 10.1002/(sici)1096-8628(19990806)85:4<342::aid-ajmg6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 10.Devillers C, Quatresooz P, HermannsLe T, Szepetiuk G, Lemaire R, Pierard Franchimont C, et al. Hipomelanosis of Ito: pigmentary mosaicism with immature melanosome in keratinocytes. Int J Dermatol. 2011;50:1234–1239. doi: 10.1111/j.1365-4632.2011.04974.x. [DOI] [PubMed] [Google Scholar]
- 11.Almeida AS, Cechin WE, Ferraz J, Rodriguez R, Moro A, Jorge R, et al. Hipomelanose de Ito - relato de um caso. J Pediatr. 2001;77:59–62. doi: 10.2223/jped.114. [DOI] [PubMed] [Google Scholar]
- 12.Happle R. Superimposed segmental manifestation of polygenic skin disorders. J Am Acad Dermatol. 2007;57:690–699. doi: 10.1016/j.jaad.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 13.Happle R. Superimposed Segmental Manifestation of Both Rare and Common Cutaneous Disorders: A New Paradigm. Actas Dermosifiliogr. 2009;100:S77–S85. doi: 10.1016/s0001-7310(09)73171-0. [DOI] [PubMed] [Google Scholar]
- 14.Siegel DH. Cutaneous mosaicism: a molecular and clinical review. Adv Dermatol. 2008;24:223–244. doi: 10.1016/j.yadr.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 15.Souza e Souza I, Cunha PCAS. Goltz syndrome: report of two cases. An Bras Dermatol. 2003;78:91–97. [Google Scholar]
- 16.Urbano LMF, Leal IIR, Costa IMC. Do you know this syndrome? Focal Dermal Hypoplasia (Goltz syndrome) An Bras Dermatol. 2011;86:391–440. doi: 10.1590/s0365-05962011000200036. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Reid Sutton V, Omar PerazaLlanes J, Yu Z, Rosetta R, Kou YC, et al. Mutations in Xlinked PORCN, a putative regulator of Wnt signaling, cause focal dermal hypoplasia. Nat Genet. 2007;39:836–838. doi: 10.1038/ng2057. [DOI] [PubMed] [Google Scholar]
- 18.Grzeschik KH, Bornholdt D, Oeffner F, König A, del Carmen Boente M, Enders H, et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet. 2007;39:833–835. doi: 10.1038/ng2052. [DOI] [PubMed] [Google Scholar]
- 19.Pereira MAC, Mesquita LAF, Budel AR, Cabral CSP, Feltrim AS. X-linked incontinentia pigmenti or Bloch-Sulzberger syndrome: a case report. An Bras Dermatol. 2010;85:372–375. doi: 10.1590/s0365-05962010000300013. [DOI] [PubMed] [Google Scholar]
- 20.LlanoRivasa I, SolerSánchezb T, MálagaDiéguezc I, FernándezToral J. Incontinencia igmenti. Cuatro pacientes con diferentes manifestaciones clínicas. An Pediatr. 2011;76:156–160. doi: 10.1016/j.anpedi.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Happle R. X-chromosome inactivation: role in skin disease expression. Acta Paediatr. 2006;95:16–23. doi: 10.1111/j.1651-2227.2006.tb02384.x. [DOI] [PubMed] [Google Scholar]
- 22.Lai-Cheong JE, McGrath JA, Uitto J. Revertant mosaicism in skin: natural gene therapy. Trends Mol Med. 2011;17:140–148. doi: 10.1016/j.molmed.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiritsi D, He Y, Pasmooij AM, Onder M, Happle R, Jonkman MF, et al. Revertant mosaicism in a human skin fragility disorder results from slipped mispairing and mitotic recombination. J Clin Invest. 2012;122:1742–1746. doi: 10.1172/JCI61976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasmooij AM, Pas HH, Bolling MC, Jonkman MF. Revertant mosaicism in junctional epidermolysis bullosa due to multiple correcting secondsite mutations in LAMB3. J Clin Invest. 2007;117:1240–1248. doi: 10.1172/JCI30465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Froes GC, Bittencourt FV, Resende ACB, Brandão FV. Case for diagnosis. Twin spotting. An Bras Dermatol. 2008;83:90–92. [Google Scholar]
- 26.Happle R. Mosaicism in human skin. Understanding the patterns and mechanisms. Arch Dermatol. 1993;129:1460–1470. [PubMed] [Google Scholar]