

Abstract
Study Objectives
To determine the influence of the CYP2C8*2 polymorphism on pioglitazone pharmacokinetics in healthy African American volunteers.
Design
Prospective, open-label, single-dose pharmacokinetic study.
Setting
University of Colorado Hospital Clinical and Translational Research Center.
Patients
Healthy African-American volunteers between 21 to 60 years of age were enrolled in the study based on CYP2C8 genotype: CYP2C8*1/*1 (n=9), CYP2C8*1/*2 (n=7), and CYP2C8*2/*2 (n=1).
Intervention
Participants received a single 15 mg dose of pioglitazone in the fasted state, followed by a 48-hour pharmacokinetic study.
Measurements and Main Results
Plasma concentrations of pioglitazone and its M-III (keto) and M-IV (hydroxy) metabolites were compared between participants with the CYP2C8*1/*1 genotype and CYP2C8*2 carriers. Pioglitazone AUC0-∞ and t1/2 did not differ significantly between CYP2C8*1/*1 and CYP2C8*2 carriers (AUC0-∞,7331 ± 2846 versus 10431 ± 5090 ng*h/ml, p=0.15; t1/2, 7.4 ± 2.7 versus 10.5 ± 4.0 h, p=0.07). M-III and M-IV AUC0-48 also did not differ significantly between genotype groups. However, the M-III/pioglitazone AUC0-48 ratio was significantly lower in CYP2C8*2 carriers than CYP2C8*1 homozygotes (0.70 ± 0.15 versus 1.2 ± 0.37, p=0.006). Similarly, CYP2C8*2 carriers had a significantly lower M-III/M-IV AUC0-48 ratio than participants with the CYP2C8*1/*1 genotype (0.82 ± 0.26 versus 1.22 ± 0.26, p=0.006).
Conclusion
These data suggest that CYP2C8*2 influences pioglitazone pharmacokinetics in vivo, particularly the AUC0-48 ratio of M-III to parent drug, and the AUC0-48 ratio of M-III to M-IV. Additional, larger studies are needed to further investigate the impact of CYP2C8*2 on the pharmacokinetics of CYP2C8 substrates in individuals of African descent.
Keywords: pioglitazone, CYP, CYP2C8, pharmacokinetics, pharmacogenetics, African-American
Introduction
The cytochrome P450 (CYP) 2C8 isoenzyme plays a major role in the hepatic metabolism of a diverse group of pharmacologic agents such as pioglitazone (antidiabetic), repaglinide (antidiabetic), paclitaxel (chemotherapeutic), and amodiaquine (antimalarial).1 Interindividual variability exists in CYP2C8-mediated metabolism, and some of this variability is governed by polymorphisms in the CYP2C8 gene. 1, 2 To date, most CYP2C8 pharmacogenetic studies have focused on the CYP2C8*3 allele (Arg139Lys, Lys399Arg), which is common in Caucasians (allele frequency of 10-23%) but is rare in other race and ethnic groups.2 In contrast to CYP2C8*3, less is known about the impact of other CYP2C8 alleles, e.g., CYP2C8*2, on CYP2C8 substrate disposition, particularly in nonCaucasian populations.
CYP2C8*2 refers to an Ile to Phe change at codon 269 in exon 5 of the CYP2C8 gene. The CYP2C8*2 allele is present at a frequency of 18% in African American populations, but is rare or absent in Caucasians and Asians.3 In vitro, CYP2C8*2 has been associated with decreased intrinsic clearance of the CYP2C8 substrates paclitaxel and amodiaquine. 3-5 However, few studies have prospectively evaluated the influence of CYP2C8*2 on the pharmacokinetic disposition of CYP2C8 substrates in humans.
Pioglitazone is a peroxisome proliferator-activated receptor-γ agonist used in the treatment of type 2 diabetes. Pioglitazone is also used as a probe drug in clinical pharmacology studies because of its dependence on CYP2C8 for metabolism. 6, 7 CYP2C8 plays a major role in the metabolism of pioglitazone to its two major circulating active metabolites, M-IV (hydroxy) and M-III (keto) (Figure 1). 8, 9 M-III (keto) is a secondary metabolite that is formed from the M-IV (hydroxy) metabolite. 8, 10 To our knowledge, no data exist regarding the impact of CYP2C8*2 on pioglitazone pharmacokinetics in populations of African descent. As such, we prospectively set out to determine if CYP2C8*2 influences the pharmacokinetics of pioglitazone in healthy African-American volunteers. Based on in vitro data, we hypothesized that CYP2C8*2 would impair metabolism and result in increased plasma exposure of parent pioglitazone and decreased plasma exposure of the M-IV and M-III metabolites.
Figure 1.
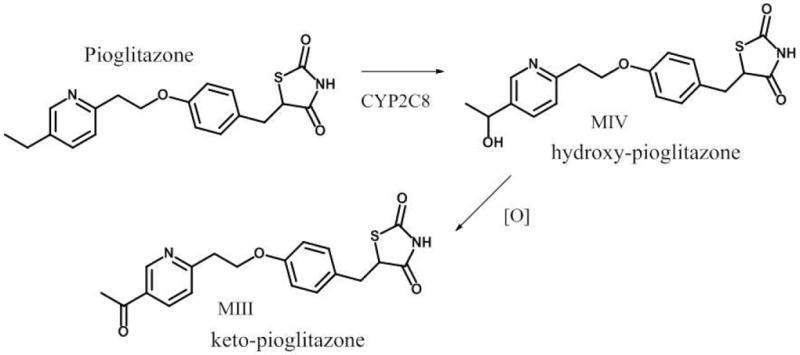
Metabolism of pioglitazone to its major active metabolites, M-IV and M-III
Methods
Study Design and Participants
The study was approved by the Colorado Multiple Institutional Review Board, and all participants provided written, informed consent. The investigation was conducted as an open-label, single-dose, pharmacokinetic study in healthy African-American volunteers between 21 to 60 years of age. Participants were prospectively screened and enrolled based on CYP2C8 genotype, i.e., CYP2C8*1/*1 or CYP2C8*2 carriers. Participants were excluded for any of the following: presence of the CYP2C8*3 and/or CYP2C8*4 alleles; body mass index <18 kg/m2 or ≥35 kg/m2; current or past history of cardiovascular, hepatic, renal, endocrine, gastrointestinal, hematologic, immunologic, or neurologic diseases; active malignancy; self-reported HIV positivity; active drug or alcohol abuse; or pregnancy. Laboratory exclusion criteria included fasting plasma glucose ≥ 126 mg/dL, serum potassium >5 mEq/L or <3.3 mEq/L, serum creatinine >1.2 mg/dL, liver function tests ≥ 2 times the upper limit of normal, hematocrit <36% in men or <34% in women, platelets <150 × 109/L, white blood cell count <4.0 × 109/L or >11.1 × 109/L, or any other laboratory abnormalities classified as grade 2 or higher per published grading criteria.11 Medication exclusions were: antidiabetic agents, systemic glucocorticoids, or any agent known to inhibit or induce the CYP2C8 and/or CYP3A4 metabolizing enzymes (e.g., gemfibrozil, trimethoprim, rifampin, grapefruit juice). A description of participant-reported concomitant medications is provided in the Supporting Information.
Study Protocol
The pharmacokinetic study took place at the University of Colorado Hospital Clinical and Translational Research Center (CTRC). Participants were admitted to the CTRC after an overnight fast. A single dose of 15 mg of pioglitazone was administered by mouth at 8:00 A.M. with 150 ml of water. Participants received a calorie-controlled breakfast (600 calories; 55% carbohydrate, 15% protein, 30% fat) 2 hours after pioglitazone ingestion. Meals were also given 6, 10, and 24 hours after pioglitazone intake. Participants were asked to abstain from smoking and caffeine during the study time period. Blood samples for the measurement of pioglitazone, M-III, and M-IV plasma concentrations were collected predose, and 0.5, 1, 2, 3, 4, 5, 7, 9, 12, 18, 24, and 48 hours post-pioglitazone dose. Plasma was harvested within 30 minutes of each blood draw and stored at -80°C for later bioanalytical processing.
Genetic analyses
For the genetic screening process, a buccal cell sample was collected from each participant using a published mouthwash method.12 Genomic DNA was isolated from buccal cells using a QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA). Subjects were genotyped for CYP2C8*2 (Ile269Phe, rs11572103), CYP2C8*3 (Arg139Lys, rs11572080; Lys399Arg, rs10509681), and CYP2C8*4 (Ile264Met, rs1058930) using PCR-pyrosequencing assays (PSQ 96 MA, Qiagen, Valencia, CA, USA), according to standard manufacturer protocol. Genotype and allele frequencies for the screening population are provided in the Supporting Information.
Drug Concentration Analyses
Plasma concentrations of pioglitazone, M-III, and M-IV were measured using a validated LC/MS-MS assay. A detailed description of plasma sample preparation, chromatography, mass spectrometry (MS) conditions, and quality control data are provided in the Supporting Information. Briefly, pioglitazone hydrochloride, hydroxy-pioglitazone, keto-pioglitazone, and pioglitazone-d4 (internal standard) were purchased from Toronto Research Chemicals, Inc. (North York, Ontario, Canada). Plasma samples were prepared using a protein precipitation method with acetonitrile, methanol, and water.13 Chromatographic separation was conducted on a 50 × 4.6 mm, 5 micron, Zorbax extended C18 column (Agilent Technologies, Santa Clara, CA, USA) equipped with a guard column. The two mobile phases consisted of (A) 10 mM ammonium acetate, 0.1% formic acid in water, and (B) 50:50 acetonitrile:methanol (1:1), delivered at a flow rate of 0.4 ml/min. The retention times for M-IV, M-III, and pioglitazone were 4.7 min, 5.1 min, and 5.5 min, respectively. For MS analysis, an Applied Biosystems Sciex 4000® (Applied Biosystems, Foster City, CA, USA) was used in ESI positive ion mode. The lower limit of quantification was 1.2 ng/ml for pioglitazone, M-III, and M-IV. The assays were linear over the range of 0.04 ng/ml to 1850-2000 ng/ml, but the lower limit of quantification from extracted plasma was 1.2 ng/ml for pioglitazone, M-III, and M-IV. The method accuracies were within 5.3%, 4.0%, and 5% for pioglitazone, M-IV, and M-III, respectively. Interday precision was ±9.1%, ±12%, and ±11.1% for pioglitazone, M-III, and M-IV, respectively.
Sample Size Calculation
Sample size was calculated by statistical power analysis using NCSS PASS software, assuming a two-tailed α of 0.05. The study was powered on the expected difference in pioglitazone AUC0-∞ between individuals with the CYP2C8*1/*1 genotype and CYP2C8*2 allele carriers. A two-sample, two-tailed t test with a sample size of 18 subjects (n=9 CYP2C8*1/*1 genotype and n=9 CYP2C8*2 carriers) would provide 88% power to detect a 50% difference in mean pioglitazone AUC0-∞ between the two genotype groups, assuming a coefficient of variation of 25%.14, 15
Pharmacokinetic and Statistical Analyses
Plasma concentration-time curves of pioglitazone, M-III, and M-IV were generated. Maximum plasma concentrations (Cmax) and time to maximum plasma concentration (Tmax) were taken from these curves. Noncompartmental analysis (WinNonlin version 5.2.1, Pharsight Corporation, Mountain View, CA, USA) was used to determine other pharmacokinetic parameters. The terminal elimination rate constant (λZ) was obtained by regression of the log-linear portion of the pioglitazone plasma concentration-time curves. Half-life (t1/2) was calculated as 0.693 divided by λZ. Pioglitazone area under the plasma concentration-time curve from 0 to infinity (AUC0-∞) and 0-48 hours (AUC0-48), M-III AUC0-48, and M-IV AUC0-48 were calculated using the linear-log trapezoidal rule. Weight-adjusted AUC0-∞ was calculated as AUC0-α divided by each subject’s weight (in kg).
The primary endpoint of the study was the difference in pioglitazone AUC0-∞ between CYP2C8 genotype groups (CYP2C8*1/*1 versus CYP2C8*2 carriers). Secondary endpoints compared between CYP2C8 genotype groups included other pioglitazone, M-III, and M-IV pharmacokinetic parameters (e.g., pioglitazone AUC0-48, M-III/pioglitazone AUC0-48, M-IV/pioglitazone AUC0-48, and M-III/M-IV AUC0-48). Baseline demographics were compared between CYP2C8*1/*1 homozygotes and CYP2C8*2 carriers by Fisher exact test for categorical data or by independent t tests for continuous data. Data that did not follow a normal distribution were log-transformed prior to analysis, and then back-transformed for data presentation. Pharmacokinetic data were compared between CYP2C8 genotype groups (*1/*1 versus *2 carriers) using independent t tests, Mann Whitney U tests (for time data), or generalized linear model analysis (for assessment of covariates). Statistical analyses were conducted using SPSS version 18.0 software. A p value of <0.05 was used as the level of significance.
Results
In this study, 68 healthy volunteers were prospectively genotyped for CYP2C8*2, and 19 subjects were enrolled in the study. Two subjects were subsequently withdrawn because one possessed the CYP2C8*3 alleleand one was related to another person in the cohort. Results are presented for the remaining 17 subjects who completed the study.
The study cohort consisted of 10 women and 7 men, mean age of 42 ± 10 years, mean weight of 77.7 ± 11.9 kg. The CYP2C8 genotype distributions were: *1/*1(n=9); *1/*2(n=7); and *2/*2, (n=1). Baseline demographics did not differ significantly between CYP2C8*1/*1 and CYP2C8*2 carrier groups (Table 1). Pioglitazone AUC0-∞ and Cmax varied 5.9- and 3.2-fold, respectively, between individuals. Pioglitazone plasma concentration-time curves by CYP2C8 genotype are shown in Figure 2. A comparison of pioglitazone pharmacokinetic parameters between CYP2C8 genotype groups is shown in Table 2. Among CYP2C8*2 carriers, mean pioglitazone AUC0-∞ was 42% higher and mean t1/2 was 1.4 times longer, than in CYP2C8*1 homozygotes. However, these results did not reach statistical significance (p= 0.15 and 0.07, respectively). When body weight was taken into account, mean weight-adjusted AUC0-∞ was 59% higher in CYP2C8*2 carriers versus CYP2C8*1/*1 (p=0.10). Mean pioglitazone Cmax and median pioglitazone Tmax did not differ significantly between CYP2C8 genotype groups. Given the high percentage of smokers in the study, a generalized linear model analysis was conducted to account for smoking status (yes/no). Pioglitazone AUC0-∞, AUC0-48, Cmax, and t1/2 did not differ significantly between CYP2C8 genotype groups when controlling for smoking status (p=0.20, p=0.26, p=0.94, and p=0.12, respectively).
Table 1.
Baseline Demographics by CYP2C8 Genotype Group (n=17)
| Variable |
CYP2C8*1/*1 (n=9) |
CYP2C8*2 carriers (n=8) |
P value |
|---|---|---|---|
| Age (years) | 43 ± 10 | 41 ± 11 | 0.77 |
| Weight (kg) | 81.1 ± 11.3 | 73.9 ± 12.1 | 0.23 |
| Male, n (%) | 4 (44.4%) | 3 (37.5%) | 1.0 |
| Current smoker, n (%) | 5 (55.6%) | 3 (37.5%) | 0.64 |
| Hormonal contraceptives, n (%) | 1 (11.1%) | 1 (12.5%) | 1.0 |
Data are expressed as mean ± SD, or number (%)
Figure 2.
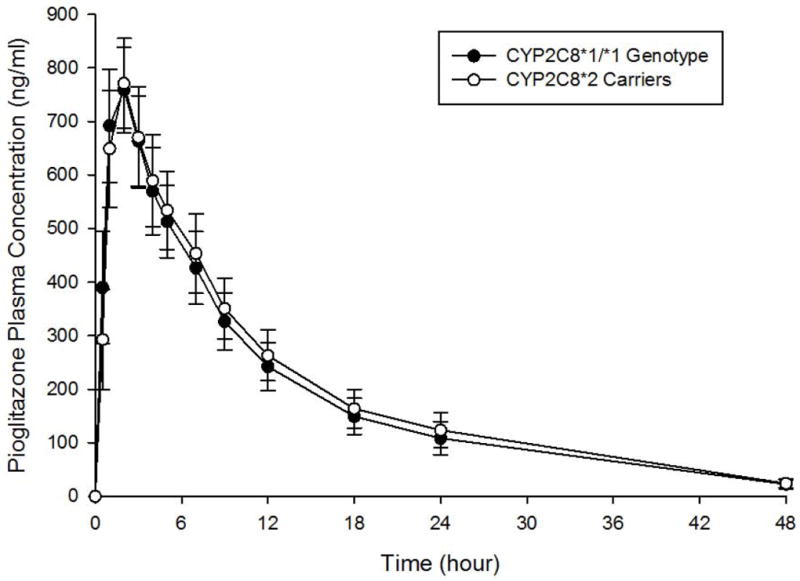
Pioglitazone plasma concentration-time curves by CYP2C8 genotype. Closed circles represent the CYP2C8*1/*1 genotype. Open circles represent CYP2C8*2 carriers. Data are shown as mean ± SEM.
Table 2.
Pioglitazone Pharmacokinetic Parameters by CYP2C8 Genotype Group
| Parameter |
CYP2C8*1/*1 (n = 9) |
CYP2C8*2 carriers (n = 8) |
P value |
|---|---|---|---|
| AUC0-∞ (ng*h/ml) | 7331 ± 2846 | 10431 ± 5090 | 0.15 |
| AUC0-∞/kg (ng*h/ml/kg) | 93 ± 43 | 148 ± 88 | 0.10 |
| AUC0-48 (ng*h/ml) | 7125 ± 2708 | 9923 ± 4797 | 0.18 |
| Cmax (ng/ml) | 808 ± 223 | 831 ± 290 | 0.90 |
| t1/2 (h) | 7.4 ± 2.7 | 10.5 ± 4.0 | 0.07 |
| Tmax (h) | 1.0 (0.5-2.0) | 1.5 (1.0-3.0) | 0.37 |
Data are expressed as mean ± SD or median (range).
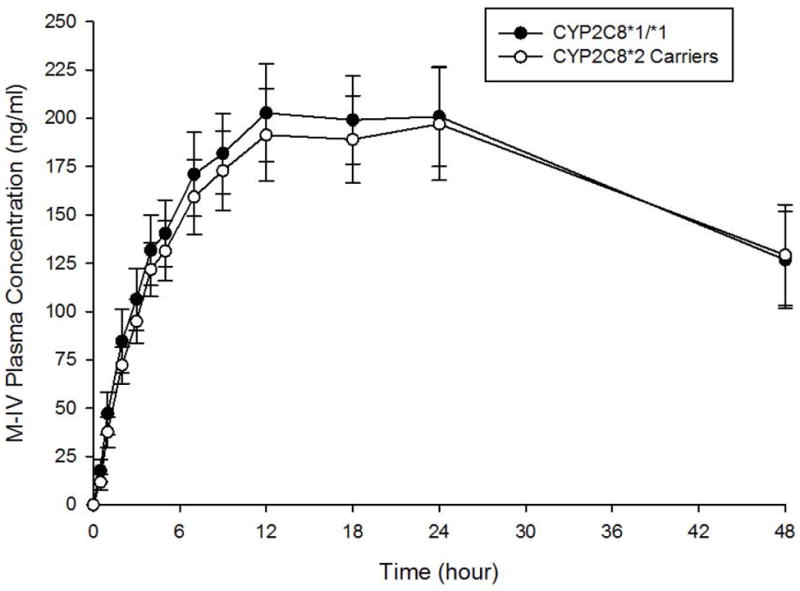
M-III and M-IV plasma concentration-time curves by CYP2C8 genotype are shown in Figures 3A and 3B, respectively. A comparison of M-III and M-IV pharmacokinetic parameters between CYP2C8 genotype groups is shown in Table 3. Mean M-III and M-IV AUC0-48 and Cmax did not differ significantly between genotype groups, although M-III AUC0-48 was 13% lower in CYP2C8*2 carriers than in CYP2C8*1 homozygotes (p=0.11). No significant differences in M-III AUC0-48, M-III Cmax, M-IV AUC0-48, and M-IV Cmax were evident between genotype groups after controlling for smoking status (p=0.22, p=0.12, p=0.33, and p=0.42, respectively). In terms of metabolite to parent ratios, the mean M-III/pioglitazone AUC0-48 ratio was 42% lower (P=0.006) in CYP2C8*2 carriers than in subjects with the CYP2C8*1/*1 genotype. However, the mean M-IV/pioglitazone AUC0-48 ratio did not differ based on genotype. When the metabolites were compared with each other, the mean M-III/M-IV AUC0-48 ratio was 33% lower in CYP2C8*2 carriers than in CYP2C8*1 homozygotes (p=0.006). After controlling for smoking status, similar results were obtained between genotype groups for M-III/pioglitazone AUC0-48 ratio (p=0.009), M-IV/pioglitazone AUC0-48 ratio (p=0.72), and M-III/M-IV AUC0-48 ratio (p=0.01).
Figure 3.
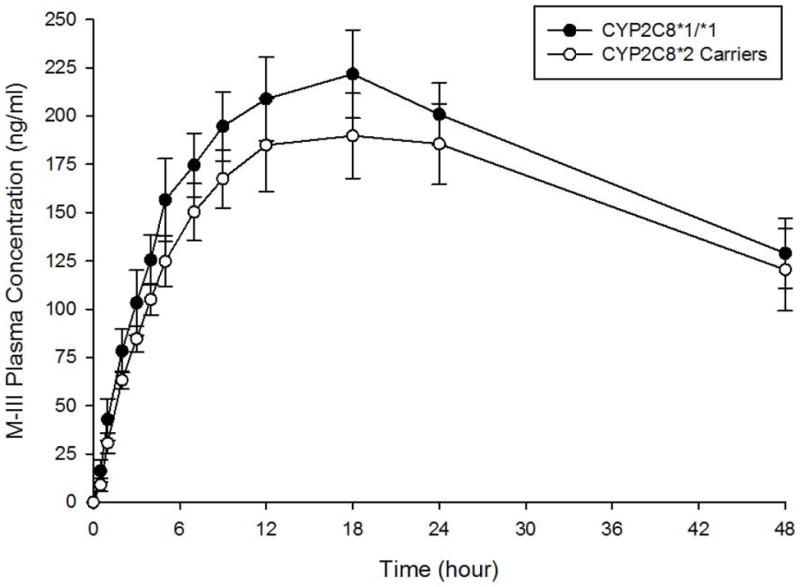
A. M-III metabolite plasma concentration-time curves by CYP2C8 genotype. Closed circles represent the CYP2C8*1/*1 genotype. Open circles represent CYP2C8*2 carriers. Data are shown as mean ± SEM.
B. M-IV metabolite plasma concentration-time curves by CYP2C8 genotype. Closed circles represent the CYP2C8*1/*1 genotype. Open circles represent CYP2C8*2 carriers. Data are shown as mean ± SEM.
Table 3.
M-III and M-IV Pharmacokinetic Parameters by CYP2C8 Genotype Group
| Parameter |
CYP2C8*1/*1 (n = 9) |
CYP2C8*2 carriers (n = 8) |
P value |
|---|---|---|---|
| M-III AUC0-48 (ng*h/ml) | 7602 ± 2221 | 6639 ± 2643 | 0.11 |
| M-III Cmax (ng/ml) | 256 ± 70 | 200 ± 65 | 0.38 |
| M-III to pioglitazone AUC0-48 ratio | 1.20 ± 0.37 | 0.70 ± 0.15 | 0.006 |
| M-IV AUC0-48 (ng*h/ml) | 6414 ± 2000 | 8466 ± 3683 | 0.23 |
| M-IV Cmax (ng/ml) | 81 ± 29 | 123 ± 77 | 0.37 |
| M-IV to pioglitazone AUC0-48 ratio | 0.97 ± 0.35 | 0.89 ± 0.23 | 0.59 |
| M-III to M-IV AUC0-48 ratio | 1.22 ± 0.26 | 0.82 ± 0.26 | 0.006 |
Data are expressed as mean ± SD
Discussion
Few studies have systematically investigated the impact of CYP2C8*2 on drug disposition in populations of African descent. In order to address this knowledge gap, we prospectively evaluated the association between CYP2C8*2 and pioglitazone pharmacokinetics in healthy African-American volunteers. We found that pioglitazone plasma exposure and half-life did not differ significantly between CYP2C8*2 carriers and wild-type homozygotes. However, the ratios of M-III to pioglitazone AUC0-48 and M-III to M-IV AUC0-48 were significantly lower in CYP2C8*2 carriers compared with the CYP2C8*1/*1 genotype group. Taken together, these results suggest that CYP2C8*2 is associated with decreased pioglitazone metabolism in healthy African-American volunteers.
CYP2C8 plays a major role in the metabolism of pioglitazone to its primary M-IV metabolite and the conversion of M-IV to M-III (a secondary metabolite). 8, 9 To date, most clinical investigations have focused on the impact of the CYP2C8*3 allele on pioglitazone disposition. 14, 16 Specifically, CYP2C8*3 has been associated with increased metabolism, decreased pioglitazone plasma exposure, and higher M-IV/pioglitazone and M-III/pioglitazone AUC ratios compared with wild-type homozygotes. 14, 16 However, CYP2C8*3 rarely occurs in individuals of African descent and is therefore not a likely contributor to interindividual variability in pioglitazone pharmacokinetics or the disposition of other CYP2C8 substrates in African populations.
CYP2C8*2 is common in African-Americans and appears to have functional consequences in vitro.3 In recombinant cell expression systems, CYP2C8*2 was associated with decreased intrinsic clearance of paclitaxel and amodiaquine and lower total CYP2C8 protein content, than was wild-type enzyme.3-5 In our study, the M-III/pioglitazone AUC ratio was significantly lower in CYP2C8*2 carriers than in wild-type homozygotes, which supports the hypothesis that CYP2C8*2 contributes to decreased CYP2C8 metabolism in vivo. We did not observe significant differences in plasma exposures of the M-III or M-IV metabolites between genotype groups. This supports data from previous studies showing that while gemfibrozil, a potent CYP2C8 inhibitor, did not significantly alter M-III or M-IV AUCs compared with placebo, the metabolite-to-parent ratios were significantly decreased following gemfibrozil administration. 9, 17 We also observed that the M-III/M-IV AUC0-48 ratiodiffered significantly between genotype groups, but the M-IV/pioglitazone AUC0-48 ratio did not. It is possible that genetic alterations in CYP2C8 may have a larger impact on the secondary step in pioglitazone metabolism, i.e., M-IV to M-III, rather than parent to M-IV. Alternatively, African-Americans may have a unique pioglitazone metabolism profile compared with other race or ethnic groups. Both of these hypotheses merit further study in additional nonCaucasian cohorts.
In terms of clinical relevance, both M-IV and M-III are active metabolites and possess 40% to 60% of the glucose-lowering potency of the parent drug. 8, 10 At steady-state, the active metabolites make up 50% to 70% of total pioglitazone concentrations (i.e., parent drug plus active metabolites).10 Interindividual variability in parent pioglitazone and/or metabolite exposure due to genetic polymorphisms may impact drug efficacy and the risk of concentration-dependent adverse effects, such as edema. To the best of our knowledge, however, the extent to which the parent drug versus its metabolites mediate adverse effects has not been fully elucidated in humans.
CYP2C8*2 may also have important implications for other CYP2C8 substrates, particularly those that are used often in African populations. 18-20 One such agent is amodiaquine, which is used to treat malaria, and relies heavily on CYP2C8 for metabolism. Given the high frequency of the CYP2C8*2 allele in African individuals, it would be prudent to more intensively evaluate the influence of CYP2C8*2 on the pharmacokinetics, pharmacodynamics, and toxicity of relevant CYP2C8 substrates in African populations.
Limitations
There are several limitations of our study that deserve to be acknowledged. First, the CYP2C8*2 carrier group included only one CYP2C8*2 homozygote. This individual’s pharmacokinetic data were not remarkably different than the heterozygotes. Nonetheless, additional studies are needed to assess whether a gene-dose relationship exists for CYP2C8*2 and pioglitazone metabolism. Second, we intentionally used a prospective CYP2C8*2 genotype enrichment study design. As such, we did not evaluate other polymorphisms in CYP2C8 or variants in other CYP enzymes that play a minor role in pioglitazone metabolism (i.e., CYP3A4, CYP1A2, CYP2D6). Third, a post hoc sample size calculation revealed that nine subjects in the CYP2C8*1/*1 genotype group and eight subjects in the CYP2C8*2 carrier group would have 80% power to detect an 82% difference in mean pioglitazone AUC0-∞. Thus, our study was underpowered to detect smaller differences in pioglitazone AUC between genotype groups. This was likely driven by higher variability in pioglitazone plasma concentrations than originally anticipated, and the presence of covariates (e.g., smoking) in the population. Additional studies with larger sample sizes are needed to further explore the relationship between CYP2C8*2 genotype and pioglitazone plasma exposure in African-American individuals. Lastly, our assessment of M-III and M-IV plasma concentrations was only 48 hours postpioglitazone dose, with limited time points in the terminal elimination phase. Therefore, we could not adequately assess the half-lives of these metabolites, which have ranged from 22-39 hours in previous reports. 8, 9, 17, 21, 22
Conclusion
CYP2C8*2 influences pioglitazone pharmacokinetics in vivo, which was most evident in the 48-hour plasma exposure ratios of M-III/pioglitazone and M-III/M-IV. Given the common frequency of CYP2C8*2 and its potential functional significance, the influence of CYP2C8*2 on the pharmacokinetics of pioglitazone and other CYP2C8 substrates merits further study in individuals of African descent.
Supplementary Material
Acknowledgments
We would like to thank the study volunteers for their participation, and the nursing and administrative staff at the University of Colorado Hospital Clinical and Translational Research Center for assisting with the conduct of the study. The study was funded by National Institutes of Health (NIH) grants K23 DK073197 (to CLA) and UL1 TR000154 (to University of Colorado). The research utilized the services of the Medicinal Chemistry Core facility (MFW) housed within the Department of Pharmaceutical Sciences at the University of Colorado Skaggs School of Pharmacy and Pharmaceutical Sciences. The Medicinal Chemistry Core facility receives funding via the Colorado Clinical and Translational Sciences Institute (CCTSI), which is supported in part by CTSA grant UL1TR000154 from NIH/NCRR. Contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
Footnotes
The study results were presented as a poster presentation at the American Society of Clinical Pharmacology and Therapeutics Annual Meeting, Indianapolis, Indiana, March 6-9, 2013.
References
- 1.Totah RA, Rettie AE. Cytochrome P450 2C8: substrates, inhibitors, pharmacogenetics, and clinical relevance. Clin Pharmacol Ther. 2005;5:341–52. doi: 10.1016/j.clpt.2004.12.267. [DOI] [PubMed] [Google Scholar]
- 2.Daily EB, Aquilante CL. Cytochrome P450 2C8 pharmacogenetics: a review of clinical studies. Pharmacogenomics. 2009;9:1489–510. doi: 10.2217/pgs.09.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dai D, Zeldin DC, Blaisdell JA, et al. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics. 2001;7:597–607. doi: 10.1097/00008571-200110000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Parikh S, Ouedraogo JB, Goldstein JA, Rosenthal PJ, Kroetz DL. Amodiaquine metabolism is impaired by common polymorphisms in CYP2C8: implications for malaria treatment in Africa. Clin Pharmacol Ther. 2007;2:197–203. doi: 10.1038/sj.clpt.6100122. [DOI] [PubMed] [Google Scholar]
- 5.Gao Y, Liu D, Wang H, Zhu J, Chen C. Functional characterization of five CYP2C8 variants and prediction of CYP2C8 genotype-dependent effects on in vitro and in vivo drug-drug interactions. Xenobiotica. 2010;7:467–75. doi: 10.3109/00498254.2010.487163. [DOI] [PubMed] [Google Scholar]
- 6.Jaakkola T, Laitila J, Neuvonen PJ, Backman JT. Pioglitazone is metabolised by CYP2C8 and CYP3A4 in vitro: potential for interactions with CYP2C8 inhibitors. Basic Clin Pharmacol Toxicol. 2006;1:44–51. doi: 10.1111/j.1742-7843.2006.pto_437.x. [DOI] [PubMed] [Google Scholar]
- 7.Muschler E, Lal J, Jetter A, et al. The role of human CYP2C8 and CYP2C9 variants in pioglitazone metabolism in vitro. Basic Clin Pharmacol Toxicol. 2009;6:374–9. doi: 10.1111/j.1742-7843.2009.00457.x. [DOI] [PubMed] [Google Scholar]
- 8.Eckland DA, Danhof M. Clinical pharmacokinetics of pioglitazone. Exp Clin Endocrinol Diabetes. 2000;(Suppl 2):S234–42. [Google Scholar]
- 9.Deng LJ, Wang F, Li HD. Effect of gemfibrozil on the pharmacokinetics of pioglitazone. Eur J Clin Pharmacol. 2005;11:831–6. doi: 10.1007/s00228-005-0042-6. [DOI] [PubMed] [Google Scholar]
- 10.Takeda Pharmaceuticals America, Inc. Actos (pioglitazone) package insert. Deerfield, IL: 2012. [Google Scholar]
- 11.Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events. 2004 Dec; http://www.niaid.nih.gov/labsandresources/resources/daidsclinrsrch/documents/daidsaegradingtable.pdf.
- 12.Andrisin TE, Humma LM, Johnson JA. Collection of genomic DNA by the noninvasive mouthwash method for use in pharmacogenetic studies. Pharmacotherapy. 2002;8:954–60. doi: 10.1592/phco.22.12.954.33598. [DOI] [PubMed] [Google Scholar]
- 13.Little JL, Wempe MF, Buchanan CM. Liquid chromatography-mass spectrometry/mass spectrometry method development for drug metabolism studies: Examining lipid matrix ionization effects in plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;2:219–30. doi: 10.1016/j.jchromb.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 14.Tornio A, Niemi M, Neuvonen PJ, Backman JT. Trimethoprim and the CYP2C8*3 allele have opposite effects on the pharmacokinetics of pioglitazone. Drug Metab Dispos. 2008;1:73–80. doi: 10.1124/dmd.107.018010. [DOI] [PubMed] [Google Scholar]
- 15.Kalliokoski A, Neuvonen M, Neuvonen PJ, Niemi M. No significant effect of SLCO1B1 polymorphism on the pharmacokinetics of rosiglitazone and pioglitazone. Br J Clin Pharmacol. 2008;1:78–86. doi: 10.1111/j.1365-2125.2007.02986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aquilante CL, Kosmiski LA, Bourne DW, et al. Impact of the CYP2C8 *3 polymorphism on the drug-drug interaction between gemfibrozil and pioglitazone. Br J Clin Pharmacol. 2013;1:217–26. doi: 10.1111/j.1365-2125.2012.04343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaakkola T, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics of pioglitazone. Clin Pharmacol Ther. 2005;5:404–14. doi: 10.1016/j.clpt.2004.12.266. [DOI] [PubMed] [Google Scholar]
- 18.Gil JP, Gil Berglund E. CYP2C8 and antimalaria drug efficacy. Pharmacogenomics. 2007;2:187–98. doi: 10.2217/14622416.8.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adjei GO, Kristensen K, Goka BQ, et al. Effect of concomitant artesunate administration and cytochrome P4502C8 polymorphisms on the pharmacokinetics of amodiaquine in Ghanaian children with uncomplicated malaria. Antimicrob Agents Chemother. 2008;12:4400–6. doi: 10.1128/AAC.00673-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paganotti GM, Gallo BC, Verra F, et al. Human genetic variation is associated with Plasmodium falciparum drug resistance. J Infect Dis. 2011;11:1772–8. doi: 10.1093/infdis/jir629. [DOI] [PubMed] [Google Scholar]
- 21.Jaakkola T, Backman JT, Neuvonen M, Laitila J, Neuvonen PJ. Effect of rifampicin on the pharmacokinetics of pioglitazone. Br J Clin Pharmacol. 2006;1:70–8. doi: 10.1111/j.1365-2125.2005.02515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaakkola T, Backman JT, Neuvonen M, Niemi M, Neuvonen PJ. Montelukast and zafirlukast do not affect the pharmacokinetics of the CYP2C8 substrate pioglitazone. Eur J Clin Pharmacol. 2006;7:503–9. doi: 10.1007/s00228-006-0136-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.