

Abstract
In response to NMDA receptor stimulation, CaMKIIα moves rapidly from a diffuse distribution within the shafts of neuronal dendrites to a clustered postsynaptic distribution. However, less is known about CaMKIIα localization and trafficking within neuronal somata. Here we use a novel recombinant probe capable of labeling endogenous CaMKIIα in living rat neurons to examine its localization and trafficking within the somata of cortical neurons. This probe, which was generated using an mRNA display selection, binds to endogenous CaMKIIα at high affinity and specificity following expression in rat cortical neurons in culture. In ∼45% of quiescent cortical neurons, labeled clusters of CaMKIIα 1–4 μm in diameter were present. Upon exposure to glutamate and glycine, CaMKIIα clusters disappeared in a Ca2+-dependent manner within seconds. Moreover, minutes after the removal of glutamate and glycine, the clusters returned to their original configuration. The clusters, which also appear in cortical neurons in sections taken from mouse brains, contain actin and disperse upon exposure to cytochalasin D, an actin depolymerizer. In conclusion, within the soma, CaMKII localizes and traffics in a manner that is distinct from its localization and trafficking within the dendrites.
Introduction
CaMKIIα is a Ca2+-dependent seronine/threonine kinase that plays a critical role in synaptic plasticity (Soderling, 2000; Coultrap and Bayer, 2012; Lisman et al., 2012) and demonstrates remarkable trafficking behavior in response to increases in cytosolic Ca2+. GFP-tagged CaMKIIα translocates rapidly and reversibly to postsynaptic sites within dendritic spines upon robust activation of NMDA receptors with glutamate and glycine (Shen and Meyer, 1999; Shen et al., 2000; Rose et al., 2009). Moderate NMDA receptor activation causes GFP-CaMKIIα to cluster at gephyrin-positive inhibitory synaptic sites along the dendrites (Marsden et al., 2010). In addition to its distinct trafficking at postsynaptic sites, CaMKII also traffics to presynaptic sites upon activation with Ca2+ from intracellular stores in Drosophila (Shakiryanova et al., 2011).
Information about the trafficking of CaMKIIα has come from experiments where the movements of exogenously expressed, GFP-tagged proteins were visualized (Shen and Meyer, 1999). Although GFP tagging enables proteins to be visualized in living cells (Marshall et al., 1995), many overexpressed proteins do not localize or traffic in the same manner as their endogenous counterparts, and can cause gain-of-function phenotypes (El-Husseini et al., 2000; Lardi-Studler et al., 2007). Recently, we have developed an alternative approach to visualizing proteins in living cells that avoids these drawbacks. cDNAs encoding antibody-like proteins, known as FingRs (Fibronectin intrabodies generated with mRNA display; Gross et al., 2013) are transfected into cells, which produce GFP-tagged FingRs that bind specifically to endogenous proteins, allowing them to be visualized. Probes that bind target molecules with high affinity and specificity are selected from a large library of mRNA-protein fusions encoding a scaffold based on the 10th type III fibronectin domain from human fibronectin with random amino acid substitutions (Roberts and Szostak, 1997; Koide et al., 1998; Takahashi et al., 2003; Olson and Roberts, 2007). Subsequently, an intracellular localization assay is used to select for FingRs that fold correctly and work well in the reduced environment of the cytoplasm. We have shown that these probes label the postsynaptic markers gephyrin and PSD95 in living cells in an accurate manner without perturbing neuronal function (Gross et al., 2013).
We generated a FingR that accurately labels endogenous CaMKIIα and allows visualization of its localization and trafficking in living neurons. We found that endogenous CaMKIIα localized and trafficked within dendrites in a manner very similar to that found in previous experiments with exogenous, tagged CaMKIIα. In contrast, within the soma, CaMKIIα labeling revealed large, stable clusters 1–4 μm in diameter in ∼45% of cortical neurons. These CaMKIIα clusters rapidly and reversibly dispersed in a Ca2+-dependent manner within seconds after stimulation with glutamate and glycine. Withdrawal of glutamate caused the clusters to reappear in exactly the same locations and with exactly the same configurations as before stimulation. Thus, our CaMKIIα-FingR probe demonstrates that CaMKIIα trafficking behavior in the cell body is dramatically different from its behavior in the dendrites, possibly reflecting distinct roles in the two compartments.
Materials and Methods
Target protein induction/purification/biotinylation.
DNA encoding the rat CaMKIIα C-terminal association domain (amino acids 336–478) was inserted into the pET28 plasmid (Novagen) downstream of a biotin acceptor peptide (AviTag, Avidity) and a 6X-His tag was transformed into BL21 (DE3) pLysS Escherichia coli (Stratagene). A transformed colony was inoculated into 2 ml of LB overnight and 1.5 ml of the overnight culture was grown in 2 L of broth at 37°C until OD600 = 0.8. AviTag-6XHis-CaMKIIα protein expression was induced with 0.4 mm isopropyl β-d-thiogalactoside for 3 h. The cells were centrifuged and the cell pellets were frozen at −20°C overnight. The following day, the cell pellets were resuspended in 100 ml of ice-cold Buffer A (20 mm Tris, pH 8.0, 100 mm NaCl, 5 mm β-mercaptoethanol) plus 1 μg/ml aprotinin plus 2 μg/ml leupeptin plus 2 μm sodium orthovanadate plus 2 mm PMSF plus 50 μg/ml DNase. The resuspended cells were sonicated and centrifuged at 6000 rpm for 15 min to pellet insoluble material. The soluble lysate was loaded onto a HisPur Cobalt resin column (Pierce) equilibrated with Buffer A and washed in Buffer B (20 mm Tris, pH 8.0, 100 mm NaCl, 5 mm β-mercaptoethanol, 20 mm imidazole) using a BioLogic DuoFlow system (Bio-Rad). The protein was eluted from the column by a gradient of increasing imidazole concentration with Buffer C (20 mm Tris, pH 8.0, 100 mm NaCl, 5 mm β-mercaptoethanol, 500 mm imidazole). Fractions containing protein were combined and then concentrated and exchanged into biotin ligation buffer (20 mm Tris, pH 8.0, 25 mm NaCl, 2.5 mm β-mercaptoethanol) using a Centricon-10 filter (Amicon). An in vitro biotin ligase reaction was performed to covalently link biotin to the acceptor peptide with BirA (Avidity) according to the manufacturer's instructions. Size exclusion chromatography was performed with a Superdex 200 column (GE Healthcare) equilibrated with storage buffer (20 mm Tris, pH 8.0, 100 mm NaCl, 5 mm DTT) to purify the protein and confirm the tetradecameric state of the complex. Fractions containing biotin-AviTag-6XHis-CaMKIIα oligomer were combined and concentrated to ∼0.69 mg/ml. To confirm the protein was biotinylated, a sample was incubated with streptavidin-conjugated agarose (Pierce) in a pull-down assay. The purified protein was aliquoted and stored at −20°C.
mRNA display selection.
Selection of CaMKIIα-specific fibronectin binders was performed according to an established protocol (Olson et al., 2008) with slight modification. In the first round of selection, PCR amplification was performed on a library consisting of ∼1012 molecules of the human fibronectin 10th type III domain with residues in the BC (7 aa) and the FG (10 aa) loops randomized (Olson and Roberts, 2007). The DNA library was transcribed in vitro with T7 RNA polymerase to produce mRNA. A DNA oligonucleotide containing puromycin on its 3′ end (pF30P, Oligo Synthesis Resource, Yale School of Medicine) was ligated to the 3′ end of the mRNA with T4 DNA ligase (New England Biolabs). The puromycin-conjugated mRNA was purified with Urea PAGE (Owl Separation Systems) and electroeluted with an Elutrap (Schleicher & Schuell BioScience). The library was then desalted with a Centrisep column (Princeton Separations). An in vitro translation reaction was performed in rabbit reticulocyte lysate (Promega) and fusion of puromycin-conjugated mRNA to library protein was facilitated by the addition of potassium and magnesium (Liu et al., 2000). The reaction was stored overnight at −80°C. The mRNA-protein fusion library was purified with oligo(dT) cellulose type 7 (GE Healthcare) at 4°C in oligo(dT) buffer (100 mm Tris, pH 8.0, 1 m NaCl, 0.2% Triton X-100), eluted in distilled water at room temperature, desalted with a Centrisep column (Princeton Separations) and reverse transcribed with Superscript II (Invitrogen). The purified mRNA-protein fusion library was incubated with AviTag-6XHis-CaMKIIα immobilized on Neutravidin-conjugated acrylamide beads (Pierce) at 4°C with rotation for 1 h in selection buffer [20 mm Tris, pH 8.0, 150 mm NaCl, 0.02% Tween 20, 1 mm DTT (Sigma-Aldrich), 0.5 mg/ml bovine serum albumin (Sigma-Aldrich), 0.1 mg/ml sheared salmon sperm DNA (Ambion), 10% fetal bovine serum (Invitrogen), 2 μg/ml streptavidin (Pierce), and 0.1% saturated biotin (Sigma-Aldrich)]. After washing the beads 5 times with selection buffer, the bound fibronectins served as a template for PCR amplification. The amplified DNA was subjected to another round of selection. While in Rounds 1 and 2 the target protein was immobilized on Neutravidin acrylamide, in Rounds 3–8 the target was immobilized on streptavidin agarose. The selection took place at 4°C in Rounds 1–4, room temperature in Rounds 5 and 6, and 37°C in Rounds 7 and 8. In vitro binding of fibronectins isolated from Rounds 4–8 was measured by scintillation counting of mRNA-fused fibronectins labeled with 35S-methionine (MP Biomedical) that were oligo(dT) purified and incubated with bead-immobilized CaMKIIα or beads alone (background) in selection buffer at 4°C.
cDNA constructs.
The genes encoding fibronectins selected from Round 6 of the CaMKIIα selection were inserted upstream of eGFP in the plasmid pGW (CaMKIIα FingR 6.X-GFP). GTS-SA-CaMKIIα was constructed by inserting the Golgi-targeting sequence of the Uukuniemi virus G1 protein (amino acids 398–459; Andersson et al., 1997) upstream of the streptavidin gene, which was upstream of the rat CaMKIIα association domain gene (amino acids 336–478). GFP-CaMKIIα was constructed by inserting the eGFP gene upstream of the full-length rat CaMKIIα gene. mKate2-β actin was constructed by inserting the mKate2 gene (Evrogen) upstream of the rat β actin gene.
COS7 cell Golgi-localizing FingR screen.
COS7 cells (ATCC) were cultured in DMEM (ATCC) supplemented with 10% fetal bovine serum (Invitrogen) and 25 μg/ml gentamicin (Invitrogen) in a 5% CO2 incubator. COS7 cells were transfected via Effectene (Qiagen) with the Golgi-localized GTS-SA-CaMKIIα construct and a construct encoding a single fibronectin isolated from Round 6 of the CaMKIIα selection fused to eGFP (CaMKIIα FingR 6.X-GFP). After 16 h of expression, the cells were fixed, permeabilized, and stained according to the immunocytochemistry protocol in this section. The 11 fibronectin proteins containing unique sequences isolated from Round 6 of the selection (Table 1) were screened for their ability to colocalize significantly with the Golgi-localized CaMKIIα protein.
Table 1.
Sequence of the 10FNIII domain of human fibronectin from clones amplified from Round 6 of the CaMKIIα mRNA display selection

aLoops of the 10FNIII domain containing randomized residues.
Dissociated cultures.
Dissociated cultures were made as previously described (Lewis et al., 2009). All cells were transfected using CalPhos (Clontech) at 12–19 d in vitro (DIV) using the manufacturer's suggested protocol. Experimental protocols were conducted according to the National Institutes of Health guidelines for animal research and were approved by the Institutional Animal Care and Use Committee at the University of Southern California.
Coimmunoprecipitation of CaMKIIα.
A 100 mm dish containing rat dissociated cortical neurons, 12 DIV, was transduced with 4 × 105 infectious units of lentivirus encoding CaMKIIα FingR-GFP and allowed to express for 5 d. The lysate was centrifuged at 14,000 rpm at 4°C for 15 min to remove insoluble material. The cells were then lysed in 1 ml of ice-cold lysis buffer (150 mm NaCl, 1 mm EDTA, 10 mm Tris, pH 8.0, 1% NP40, 0.12 mg/ml PMSF, 2 μg/ml leupeptin, 1 μg/ml aprotinin, 10 mm NaF, and 1 μg/ml pepstatin; all Sigma-Aldrich). The soluble lysate was cleared with protein A Sepharose beads (Pierce) for 1 h at 4°C with rotation. Ten microliters of anti-GFP agarose (MBL International) was added to the cleared lysate and incubated overnight at 4°C with rotation. The following day, the agarose beads were separated from the lysate and washed three times with 500 μl of wash buffer (same as lysis buffer but with 0.1% NP40). Five microliter samples of the original lysate (input), the anti-GFP cleared lysate (flow through), and the anti-GFP agarose beads (immunoprecipitation) were separated by 12% SDS-PAGE and transferred to nitrocellulose membranes (Biorad). The blots were probed with mouse anti-CaMKIIα (Thermo Fisher Scientific) at 1:2000, mouse anti-gephyrin (Synaptic Systems) at 1:1000, or mouse anti-GFP (Invitrogen) at 1:2000. Probes were detected with IRDye 800CW goat anti-mouse (LI-COR) or IRDye 680CW goat anti-mouse (LI-COR), both at 1:10,000. Images were captured with the Odyssey Infrared Imaging System (LI-COR).
Immunocytochemistry.
Cells were fixed with 4% paraformaldehyde (Elecron Microscopy Sciences) in PBS for 5 min and washed three times with PBS for 5 min each. Cells were then permeabilized and blocked for 30 min in blocking solution (1% bovine serum albumin (Sigma-Aldrich), 5% normal goat serum (MP Biomedicals), and 0.1% Triton X-100 (Sigma-Aldrich) in PBS. Primary antibodies were diluted in blocking solution and incubated with cells for 30 min. Cells were washed three times with PBS for 5 min and then incubated with secondary antibodies diluted in blocking solution for 30 min in the dark. Cells were washed 3 times with PBS for 5 min and were mounted with Fluoromount-G (Elecron Microscopy Sciences). Primary antibodies used were chicken anti-GFP (Aves Labs) at 1:1000, mouse anti-CaMKIIα (Invitrogen) at 1:1000, and mouse anti-CaMKIIβ (Invitrogen) at 1:2000. Secondary antibodies used were goat anti-chicken Alexa Fluor 488 (Invitrogen) at 1:1000, goat anti-mouse Alex Fluor 488 (Invitrogen) at 1:1000, goat anti-mouse IgG2bκ 488 (Invitrogen) at 1:1000, goat anti-mouse Alex Fluor 594 (Invitrogen) at 1:1000, goat anti-mouse IgG2a Alex Fluor 647 (Invitrogen) at 1:1000, phalloidin-Alexa Fluor 594 (Invitrogen) at 13.2 nm, and biotin-rhodamine (American Qualex) at 1:1000. Cells were imaged on a MRC-1024 confocal microscope (Biorad) or an IX-81 inverted fluorescence microscope (Olympus).
CaMKIIα cluster characterization/quantitation.
Somatic CaMKIIα clusters were defined as CaMKIIα-positive or CaMKIIα.FingR-GFP-positive spheroids in the cell body of neurons. Clusters were 1–4 μm in diameter and had an average fluorescence intensity of >20% of surrounding cell body regions. To quantitate the frequency of cluster occurrence in vitro, rat dissociated cortical neurons 12–13 DIV were transfected with either CaMKIIα.FingR-GFP, GFP alone, or GFP-CaMKIIα. After 16 h, expression cells were fixed, permeabilized, and stained for GFP and CaMKIIα (see Immunocytochemistry). GFP-positive cells were imaged and analyzed for cluster occurrence (n = 60 for CaMKIIα.FingR.GFP-expressing cells, n = 60 for GFP-expressing cells, and n = 50 for GFP-CaMKIIα-expressing cells). To quantitate the frequency of cluster occurrence in vivo, 5 μm-thick sections of postnatal day 90 adult mouse cortex were fixed, permeabilized, and stained for CaMKIIα and DAPI (to identify nuclei and thus cell bodies). DAPI-positive cell bodies of CaMKIIα-positive cells were analyzed for cluster occurrence (n = 190 cells). Analysis was performed by a blind observer and significance of cluster occurrence between conditions was determined by Fisher's exact test. Images were taken on an IX-81 inverted fluorescence microscope (Olympus) and analyzed with ImageJ software (National Institutes of Health).
Live imaging.
Rat dissociated cortical neurons made from brains from animals of both sexes were cultured on 35 mm Delta T dishes (Bioptechs) and transfected 12–19 DIV with CaMKIIα.FingR-GFP. After 16 h, expression culture media was replaced with 10 mm HEPES plus 1× HBSS (Invitrogen) and imaged at 37° on an IX-81 inverted fluorescence microscope (Olympus). Cells were stimulated with addition of 50 μm glutamate plus 5 μm glycine to the bath and images were captured every 0.5 s for 30–60 s during stimulation. In experiments exploring the Ca2+ dependence of cluster disaggregation, cells were incubated first in 10 mm HEPES plus 1× HBSS plus 5 mm EGTA for 5 min before imaging. In experiments exploring the reaggregation of clusters, cells were stimulated with 50 μm glutamate plus 5 μm glycine for 75 s and the imaging buffer was replaced with 10 mm HEPES plus low-Ca2+ 1× HBSS (0.1× Ca2+, i.e., 0.126 mm) for 10 min before reimaging. In experiments exploring the effects of F-actin disruption on CaMKIIα clusters, cells were imaged once before addition of 2 μm cytochalasin D (Tocris Bioscience) or DMSO (vehicle) for 1.5–2 h incubation. Cells were then fixed, permeabilized, and stained for GFP and phalloidin-Alexa Fluor 594 before reimaging. In experiments exploring the localization of F-actin immediately after cluster disaggregation, neurons 12–14 DIV were cotransfected with CaMKII.FingR-GFP and mKate2-β actin. After 16 h, the cells were stimulated with glutamate/glycine and imaged as described above.
To plot the dynamics of CaMKIIα cluster disaggregation in each neuron examined, a region of interest (ROI #1) was drawn around a single CaMKIIα.FingR-GFP cluster using ImageJ software. Another similarly shaped ROI was drawn in an adjacent (<1 μm away) region of the cell body that contained no cluster (ROI #2). The ratio of fluorescence intensity (Icluster/background) was plotted for each image captured every 0.5 s as a function of time following glutamate/glycine stimulation (t = 0–21.5 s). Each line in a plot indicates one cluster/noncluster ratio in a single neuron. In experiments where CaMKIIα.FingR-GFP was coexpressed with mKate2-β actin, ROI #1 indicates both the CaMKIIα and actin cluster region while ROI #2 indicate both the CaMKIIα and actin adjacent noncluster region. In these experiments only two time points (t = 0 s and t = 21.5 s) were plotted.
Results
Generation of a recombinant antibody-like protein that recognizes CaMKIIα
To visualize endogenous CaMKIIα in living neurons, we generated a recombinant antibody-like protein, or FingR, that binds CaMKIIα with high affinity and specificity and reports its localization and trafficking. We used mRNA display, an in vitro selection method capable of generating protein aptamers such that both the aptamer and the gene that encodes it are copurified (Roberts and Szostak, 1997). The selection was performed using a library of 1012 distinct mRNA-protein fusions composed of the 10th type III fibronectin (10FNIII) domain from human fibronectin with 17 aa residues randomized within the BC and FG loops (Fig. 1; Olson and Roberts, 2007). This scaffold has a β sheet loop structure similar to that of the Fv region of antibodies, yet it has no disulfide bonds, making it stable even in reduced environments (Koide et al., 1998). As a target epitope, we chose the C-terminal association domain of CaMKIIα (Hoelz et al., 2003; Rosenberg et al., 2006). We used the multimerized form of this domain to select for proteins that bind to the exterior of the complex in a region with no known binding partners.
Figure 1.

mRNA display selection on CaMKIIα association domain. A, A round of selection consisting of (1) in vitro transcription of the fibronectin DNA library, (2) ligation of the resulting mRNA (red) to a DNA linker (black) fused to puromycin (P), (3) in vitro translation of mRNA and covalent linkage of fibronectin polypeptide (blue) to puromycin (randomized BC and FG loops in yellow), (4) oligo(dT) purification of mRNA-polypeptide fusions, (5) reverse transcription of mRNA to produce cDNA (black), (6) exposure of the mRNA/DNA-puromycin-polypeptide library to CaMKIIα association domain oligomers immobilized on agarose beads, (7) purification of mRNA/DNA-puromycin-polypeptides that bind at high affinity to CaMKIIα through affinity chromatography, and (8) PCR amplification of purified mRNA/DNA-puromycin-polypeptides to reconstitute a new DNA library of CaMKIIα-specific fibronectins. B, Percentage of mRNA/DNA-puromycin-polypeptides in Rounds 4–7 that bind to either CaMKIIα immobilized on agarose beads (+ target) or to beads alone (− target) in a hot binding assay. The percentage binding to target increases with later rounds, with negligible nonspecific binding.
In each round of an mRNA display selection, single molecules consisting of 10FNIII polypeptide domains fused to the mRNA that encodes them are exposed to the target (Fig. 1A). The mRNA-protein fusions that bind to the target are purified using affinity chromatography and then amplified to reconstitute a library of molecules that preferentially bind to the target. To monitor the ability of library members to bind to the CaMKIIα target, we performed a radioactive pull-down assay with the fibronectins isolated after Rounds 4–8. In Round 5, library members bound to target significantly above background. In Round 7, target binding peaked at 22.5% (Fig. 1B). Since the diversity of target-specific fibronectin sequences was likely large in the round just before peak binding, we amplified and sequenced members of the library from Round 6. Of the 13 clones sequenced, two were present in multiple copies, indicating that the selection was converging (Table 1). To test whether any of the 11 unique CaMKIIα FingRs from this round bound to the target in a cytoplasmic environment, we engineered a GFP tag downstream of the gene encoding them and coexpressed the resulting proteins (CaMKIIα.FingR.6.X-GFP) in COS cells coexpressing the target protein fused to a Golgi targeting sequence (GTS-SA-CaMKIIα; Fig. 2A; Andersson et al., 1997). Tight colocalization of CaMKIIα.FingR.6.1-GFP with GTS-SA-CaMKIIα at the Golgi membrane revealed that the FingR bound to target with high affinity and specificity in the reducing environment of the cytoplasm (Fig. 2B–D). In contrast, another clone from Round 6, CaMKIIα.FingR.6.2-GFP, failed to colocalize significantly with target (Fig. 2E–H).
Figure 2.
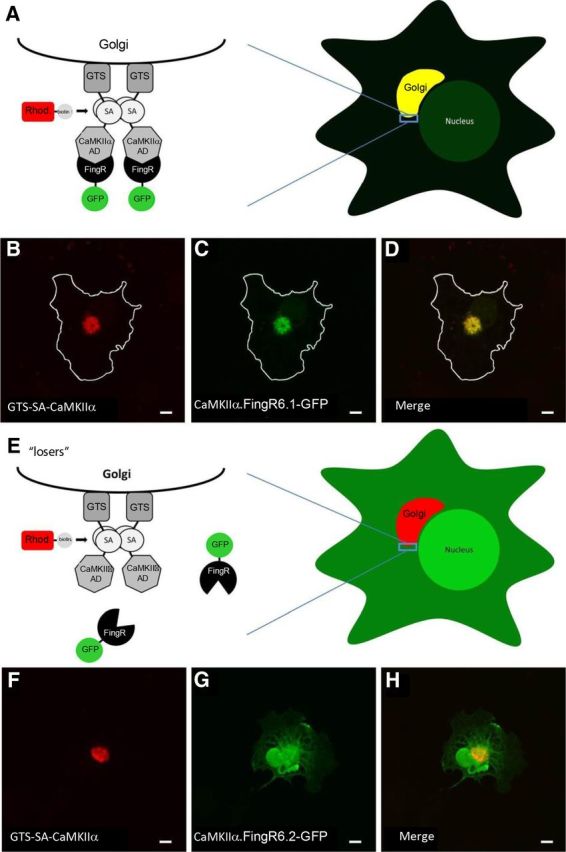
Intracellular Golgi localization assay for FingR/target binding. A, Schematic shows CaMKIIα target targeted to the surface of the Golgi through a Golgi targeting signal (GTS) following expression in COS-7 cells. Coexpressed FingRs that bind to the CaMKIIα target are colocalized with the target at the Golgi. B, C, COS-7 cell coexpressing a Golgi-targeted CaMKIIα association domain (B, GTS-SA-CaMKIIα, red) and a FingR that binds to CaMKIIα (C, CaMKIIα.FingR6.1-GFP, green). D, Both GTS-SA-CaMKIIα and CaMKIIα.FingR6.1-GFP colocalize at the Golgi. E, Schematic of a FingR that does not bind to target following expression in COS-7 cells. F, GTS-SA-CaMKIIα (red) targeted to the Golgi. G, CaMKIIα.FingR6.2-GFP (green) is localized nonspecifically and does not colocalize with GTS-SA-CaMKIIα. H, Merged image shows that CaMKIIα.FingR6.2-GFP and GTS-SA-CaMKIIα do not overlap, indicating that the FingR does not bind at high affinity to the target. Scale bars, 5 μm.
To determine whether CaMKIIα.FingR 6.1-GFP (henceforth referred to as CaMKIIα.FingR-GFP) binds to endogenous CaMKIIα, we expressed CaMKIIα.FingR-GFP in 12 DIV rat cortical neurons in dissociated culture. Following expression for 16 h, the neurons were fixed and immunostained for endogenous CaMKIIα so that its distribution could be compared with that of CaMKIIα.FingR-GFP. We found that CaMKIIα.FingR-GFP showed a striking overlap with endogenous CaMKIIα, which is consistent with the two proteins binding with high affinity (Fig. 3A–C). To further test for interaction between CaMKIIα.FingR-GFP and endogenous CaMKIIα, we assessed whether the FingR could coimmunoprecipitate endogenous CaMKIIα following expression in cortical neurons. We infected dissociated neurons (12 d in culture) with a lentivirus encoding CaMKIIα.FingR-GFP and allowed it to express for 5 d. Following cell lysis and probing of the lysate with anti-GFP conjugated beads, CaMKIIα.FingR-GFP precipitated nearly all of the endogenous CaMKIIα protein, whereas the unrelated protein gephyrin remained exclusively in the lysate (Fig. 3D). This result indicates CaMKIIα.FingR-GFP forms a complex with CaMKIIα protein in a highly efficient manner. In addition, in a hot binding assay, 46% of CaMKIIα.FingR-GFP bound to CaMKIIα, whereas only 3% bound to CaMKIIβ, indicating that binding to CaMKIIα is highly specific (data not shown).
Figure 3.

CaMKIIα.FingR-GFP colocalizes with endogenous CaMKIIα in neurons. A, B, CaMKIIα.FingR-GFP (A, green) expressed in a dissociated cortical neuron colocalizes similarly to CaMKIIα (B, red). C, Yellow color of merged image of CaMKIIα and CaMKIIα.FingR-GFP indicates colocalization of the two proteins. D, Immunoprecipitation of CaMKIIα.FingR-GFP expressed in cortical neurons in dissociated culture results in coprecipitation of virtually 100% of endogenous CaMKIIα, indicated by its absence in flow through (FT). In contrast, endogenous gephyrin is not coprecipitated. Scale bars, 5 μm.
Previously, it has been shown that GFP-CaMKIIα clusters specifically at postsynaptic excitatory sites in cultured neurons following stimulation of NMDA receptors by glutamate and glycine (Shen and Meyer, 1999; Shen et al., 2000; Rose et al., 2009). To determine whether CaMKIIα.FingR-GFP exhibits similar trafficking, we expressed it in 19 DIV cortical neurons in culture for 16 h and then exposed the cultures to 50 μm glutamate and 5 μm glycine. Within 1 min after adding glutamate and glycine, CaMKIIα.FingR-GFP changed from being diffusely localized to forming puncta located on dendritic spines (Fig. 4). Thus, CaMKIIα.FingR-GFP appears to target to postsynaptic sites in the same manner as GFP-CaMKIIα following stimulation with glutamate and glycine, suggesting that binding of CaMKIIα.FingR-GFP does not disrupt the localization or trafficking of endogenous CaMKIIα. These results also suggest that trafficking of CaMKIIα in cortical neurons is similar to its trafficking in hippocampal neurons (Shen and Meyer, 1999).
Figure 4.

CaMKIIα.FingR-GFP translocates to dendritic spines upon excitatory stimulation. A, CaMKIIα.FingR-GFP expressed in cortical neurons in dissociated culture localizes in a diffuse fashion. B, 54 s after addition of 50 μm glutamate and 5 μm glycine to the bath CaMKIIα.FingR-GFP localizes specifically to puncta corresponding to the tips of dendritic spines. Scale bars, 5 μm.
CaMKIIα aggregates appear in neuronal somata
Although the trafficking of CaMKIIα in dendrites has been well characterized, much less is known about its trafficking within the soma. Following expression of CaMKIIα.FingR-GFP in cortical neurons in dissociated culture, we found that in many cells it had a somatic distribution that was distinctly different from its distribution within dendrites. CaMKIIα.FingR-GFP was present in prominent clusters ∼1–4 μm in diameter within the soma of ∼45% of living cells in culture (n = 60 cells; Fig. 5A). To determine whether expression of CaMKIIα.FingR-GFP was responsible for the formation of the clusters, we examined staining of endogenous CaMKIIα in cells expressing GFP alone (Fig. 5C). We found that the percentage of GFP-expressing cells containing CaMKIIα clusters (43%; Fig. 5F; n = 60 cells) was not significantly different from that found in cells expressing CaMKIIα.FingR-GFP, indicating that expression of the FingR likely did not promote formation and/or retention of the clusters (p > 0.99, Fisher's exact test). Note that the appearance of clusters in live cells (Fig. 5A) and in fixed cells (Fig. 5C) was quite similar, suggesting that the clusters were not artifacts of fixation. In cells expressing GFP-CaMKIIα, the percentage of cells containing clusters within the cell body increased with increasing amounts of transfected DNA encoding GFP-CaMKIIα (34, 50, and 62% for 0.1, 1, and 8 μg, respectively; Fig. 5B,F; n = 50 cells each). Furthermore, increasing the amount of GFP-CaMKIIα expressed in the cell significantly increases the number of clusters (7.01 ± 1 clusters for 0.1 μg of GFP-CaMKIIα vs 16 ± 4 clusters for 1 μg of GFP-CaMKIIα; n = 20, p < 0.001, t test). These results indicate that GFP-CaMKIIα expression influences CaMKIIα cluster formation within the cell body, a significant drawback to using tagged, exogenous CaMKIIα for studying endogenous CaMKIIα clusters in somata. It should be noted, however, that it is possible that this clustering could be a result of GFP–GFP interactions, which might be reduced using a version of GFP that is less prone to multimerization (Bayer et al., 2006). To determine whether the presence of clusters was an artifact of dissociated culture, we examined the distribution of CaMKIIα in fixed sections from adult mice immunostained with an anti-CaMKIIα antibody (Fig. 5D,E). Approximately 40% of cortical neurons had clusters of CaMKIIα in the somata (n = 190 cells), a number that is not statistically distinct from the percentage of neurons with similar clusters in culture (Fig. 5F; p > 0.77, Fisher's exact test). Thus, clusters appear in approximately the same proportion of cortical neurons, both in culture and in vivo, and their formation is not induced by expression of CaMKIIα.FingR-GFP. Note that the difference in the appearance of clusters in intact tissue versus those in dissociated neurons could be due to actual differences in the clusters in the two different conditions or merely due to differences in fixation between dissociated cultures and intact brain sections.
Figure 5.

CaMKIIα clusters in the cell bodies of cortical neurons in vitro and in vivo. A, CaMKIIα.FingR-GFP localizes in clusters ∼1–4 μm in diameter in the cell bodies of live cortical neurons. B, GFP-CaMKIIα also clusters in the cell body, but the number of clusters was larger and the intensity more variable than clusters of CaMKIIα.FingR-GFP in live cortical neurons. C, Endogenous CaMKIIα, stained with an antibody is present in clusters in the cell bodies of fixed cortical neurons in culture. D, E, Sections of postnatal day (P) 90 mouse cortex stained with anti-CaMKIIα antibody show clusters within neuronal somata. F, Approximately 45% of cells in dissociated culture showed clusters of CaMKIIα when marked with CaMKIIα.FingR-GFP or with anti-CaMKIIα antibody staining. A similar percentage was found in sections cut from mouse cortex and stained with a CaMKIIα antibody. Although clusters were found in ∼35% of cells transfected with 0.1 μg of CaMKIIα-GFP, increasing the amount of CaMKIIα-GFP caused an increase in the percentage of cells exhibiting clusters, suggesting that expression of the exogenous protein facilitated cluster formation. Approximately 40% of cells in cortices cut from P90 mouse cortex are positive for CaMKIIα clusters. Scale bars, 10 μm.
Dynamics of CaMKIIα clusters
To study the dynamics of clusters of CaMKIIα in living neurons, we exposed dissociated cortical neurons to 50 μm glutamate and 5 μm glycine, which causes CaMKIIα to cluster at postsynaptic sites. Strikingly, we found that within 10 s after addition of glutamate and glycine the clusters dispersed in 100% of cells (Fig. 6A–C). In particular, the ratio of the intensity of CaMKIIα.FingR-GFP inside a cluster, versus background intensity within the cell body was significantly higher than 1, Icluster/background = 1.39 +/− 0.04 at t = 0 (n = 13 cells), whereas Icluster/background = 1.06 +/− 0.01 at t = 10 s, a significant difference (p < 0.0001, Wilcoxon). Because Ca2+ plays a prominent role in both the activation of CaMKIIα and its aggregation at excitatory synaptic sites in dendrites (Shen and Meyer, 1999), we tested whether Ca2+ influx is necessary for the disaggregation of cell body clusters. To do so, we incubated the cells first in the Ca2+ chelator EGTA followed by addition of glutamate and glycine. Under these circumstances, there was no difference in the appearance of clusters before and after addition of glutamate and glycine (Fig. 6D–F). In addition, quantitation confirmed these qualitative results [Icluster/background = 1.50 ± 0.05 at t = 0 s (n = 10), whereas Icluster/background = 1.49 ± 0.05 at t = 10 s, a difference that is not significant (p > 0.8, Wilcoxon)]. Thus, Ca2+ influx is necessary for the disaggregation of CaMKIIα clusters. We also tested whether CaMKIIα clusters would reaggregate following dispersal if glutamate and glycine were removed from the bath and replaced with a low-Ca2+ buffer. The clusters reappeared in exactly the same places and with the same shapes as the originals, suggesting the existence of spatially fixed loci that could mediate reaggregation (Fig. 7A,C,E,G). Note that CaMKIIα clusters in the dendrites went from diffuse, to clustered, and then to diffuse again during this time (Fig. 7B,D,F). A time course of the relative intensity of CaMKIIα in clusters versus background CaMKIIα (Icluster/background) confirms the qualitative results (Fig. 7H). In addition, Icluster/background at time points 0, 0.5, and 15 min (Icluster/background, 0 = 1.37 ± 0.03; Icluster/background, 0.5 = 1.05 ± 0.02; Icluster/background, 15 = 1.21 ± 0.04, n = 5) are all significantly different from one another (p < 0.001, ANOVA).
Figure 6.

CaMKIIα clusters rapidly disperse upon exposure to high concentrations of Ca2+. A, CaMKIIα.FingR-GFP labels clusters in the cell bodies of cortical neurons in culture. B, Ten seconds following the addition of 50 μm glutamate and 5 μm glycine, the clusters in A have disappeared. C, Time courses of the ratio of fluorescence of CaMKIIα.FingR-GFP at a point within a cluster versus at a point outside of the clusters (Icluster/background) for 10 different cells show that the clusters disappear at similar rates following exposure of cells to 50 μm glutamate and 5 μm glycine. D, Clusters labeled by CaMKIIα.FingR-GFP in cortical neurons in culture in the presence of 5 mm EGTA. E, Following exposure to 50 μm glutamate and 5 μm glycine for 46 s in the presence of 5 mm EGTA, the clusters in D do not change in shape or intensity. F, Time courses of Icluster/background for CaMKIIα.FingR-GFP in 10 different cells shows that clusters do not disperse when exposed to 50 μm glutamate and 5 μm glycine in the presence of 5 mm EGTA. Scale bars, 10 μm.
Figure 7.

Following dispersal, clusters labeled with CaMKIIα.FingR-GFP re-form in their original positions and shapes. A, Clusters labeled with CaMKIIα.FingR-GFP in a cortical neuron in culture. B, Dendrite from the same neuron as in A shows faint, diffuse staining by CaMKIIα.FingR-GFP. C, Clusters in A dispersed following addition of 50 μm glutamate and 5 μm glycine. D, CaMKIIα.FingR-GFP labeled tight clusters in same dendrite as in B following addition of 50 μm glutamate and 5 μm glycine. E, Following washout of glutamate and glycine and incubation for 10 min, the clusters re-formed in their original positions and shapes. F, Same dendrite as in B and E showed diffuse labeling by CaMKIIα.FingR-GFP following washout of glutamate and glycine. G, Superimposition of the images of clusters before and after dispersal and re-formation confirms that they re-formed in the same locations and configurations as the original clusters. H, Time course of Icluster/background for CaMKIIα.FingR-GFP in the presence (t = 0 to t = 75 s) and absence (t > 75 s) of glutamate and glycine. Error bars represent SEM. Scale bars, 10 μm.
CaMKIIα clusters associate with F-actin
Since our previous experiments suggest CaMKIIα clusters associate with and dissociate from fixed structures within the soma, we tested whether they colocalize with actin, which has been shown to associate with CaMKIIβ (Shen et al., 1998). Immunostaining of cultured cortical neurons with antibodies detecting CaMKIIα, CaMKIIβ, and phalloidin, a toxin that labels the F-actin cytoskeleton, revealed that CaMKIIα and phalloidin colocalized precisely in somatic clusters (Fig. 8A,C,D), suggesting the clusters of CaMKIIα associate with F-actin structures. Although there was some overlap of phalloidin staining with CaMKIIβ, it was not as pronounced (Fig. 8B–D). To determine whether disruption of F-actin could disperse CaMKIIα clusters, we imaged CaMKIIα.FingR-GFP-expressing cells before and after addition of cytochalasin D, an inhibitor of actin polymerization, or DMSO (vehicle). Addition of cytochalasin D to the bath, which eliminated phalloidin staining (Fig. 9C), caused clusters of CaMKIIα.FingR-GFP to disperse [Fig. 9A,B,G; Icluster/background = 1.28 ± 0.04 at t = 0; Icluster/background = 0.98 ± 0.02 at t = 1.75 h; a significant difference (p < 0.01, Wilcoxon)]. In contrast, DMSO had no effect on either phalloidin staining (Fig. 9F), or CaMKIIα.FingR-GFP clusters [Fig. 9D,E,G; Icluster/background = 1.36 ± 0.06 at t = 0, n = 5 cells; Icluster/background = 1.34 ± 0.06 at t = 1.75 h; a difference that is not significant (p > 0.8, Wilcoxon)]. Thus, the presence of intact actin filaments is necessary for clustering of CaMKIIα.
Figure 8.

CaMKIIα and CaMKIIβ colocalize with F-actin in clusters within the cell body. A, Cluster in the cell body of a cortical neuron in culture stained with anti-CaMKIIβ antibody. B, C, Same cell as in A colabeled with anti-CaMKIIα (B) or with phalloidin (C). D, Merge confirms that expression patterns of CaMKIIα (blue), CaMKIIβ, (green), and actin (red) overlap. Scale bars, 10 μm.
Figure 9.

Cytochalasin D disruption of F-actin causes CaMKII clusters to disperse. A, CaMKIIα.FingR-GFP-labeled clusters in cell body of a cortical neuron in culture. B, Addition of cytochalasin D (2 μm) for 98 min results in dispersal of the clusters in A. C, Phalloidin staining of cell in B and C verifies that addition of cytochalasin D mediated dispersal of actin clusters. D–F, Clusters in a cortical pyramidal neuron labeled with CaMKIIα.FingR-GFP (D) are not dispersed by DMSO (E), which also leaves clusters of actin filaments intact (F). G, Icluster/background at t = 0 s and t = 21.5 s for CaMKIIα.FingR-GFP following addition of either cytochalasin D or DMSO. Scale bars, 10 μm.
To determine whether F-actin clusters disappear simultaneously with clusters of CaMKIIα.FingR-GFP upon stimulation of NMDA receptors, we coexpressed CaMKIIα.FingR-GFP along with the far-red fluorescent protein mKate2 fused to β-actin (mKate2-β actin) in cultured neurons and stimulated the cells with glutamate and glycine. CaMKIIα.FingR-GFP and mKate2-β-actin colocalized precisely in soma clusters and CaMKIIα.FingR-GFP clusters dispersed rapidly (<10 s) upon addition of glutamate and glycine (Fig. 10A–C). For CaMKIIα.FingR-GFP, Icluster/background = 1.46 ± 0.03 at t = 0 (n = 5 cells), whereas Icluster/background = 1.01 ± 0.02 at t = 20 s, a difference that is significant (p < 0.01, Wilcoxon). In contrast, mKate2-β-actin clusters were still clearly visible beyond 20 s, a point at which all of the CaMKIIα.FingR-GFP clusters had completely dispersed (Fig. 10D–F). However, for mKate2-β-actin, Icluster/background = 2.7 ± 0.3 at t = 0 (n = 5 cells), whereas Icluster/background = 1.8 ± 0.1 at t = 20 s, a difference that is still significant (p < 0.02, Wilcoxon). This result suggests that F-actin clusters remain in the cell body upon stimulation with glutamate and glycine, although the amount of F-actin within them is significantly reduced. Thus, our data are consistent with the idea that clusters of F-actin act as platforms that sequester CaMKIIα within the soma under basal Ca2+ levels. Following a rise in Ca2+, CaMKIIα clusters are rapidly dispersed, causing an abrupt increase in the concentration of free CaMKIIα within the soma.
Figure 10.

CaMKIIα clusters dissociate from F-actin clusters upon stimulation. A, Clusters labeled with CaMKIIα.FingR-GFP (green) in cortical neurons. B, Addition of 50 μm glutamate and 5 μm glycine for 7.5 s results in dispersal of the clusters. C, Icluster/background for CaMKIIα.FingR-GFP shows that the clusters have completely dispersed within 21.5 s. D, Clusters of β actin fused to mKate2 in the same cell as in A. E, Addition of 50 μm glutamate and 5 μm glycine for 7.5 s does not disperse the clusters. F, Time course of Icluster/background for mKate2-β actin during exposure to 50 μm glutamate and 5 μm glycine. Scale bars, 10 μm.
Discussion
CaMKIIα clusters in synaptic sites in dendritic spines upon activation of NMDA receptors, a phenomenon that plays an important role in both the induction and maintenance of LTP and spatial learning (Sanhueza et al., 2011). In addition, CaMKIIα clusters at nonsynaptic sites, including ones in the soma, under ischemic conditions of low pH (Aronowski et al., 1992), as well as following prolonged exposure to glutamate and glycine (Dosemeci et al., 2000; Hudmon et al., 2005; Vest et al., 2009). Here we describe a novel clustering behavior of CaMKIIα that occurs exclusively in the cell bodies of almost half of cortical neurons both in culture and in vivo. Unlike the clustering of CaMKIIα in dendrites, the clustering observed in this article occurs under resting, nonischemic conditions. Exposure of cells to glutamate and glycine leads to dispersal of clusters in <10 s in a Ca2+-dependent manner. If glutamate and glycine are removed following dispersal of clusters, they re-form within 10 min in exactly the same configurations that they had been in before dispersal. Clusters of CaMKIIα are colocalized with clusters of CaMKIIβ and with F-actin. The fact that F-actin clusters do not disappear upon stimulation with glutamate and glycine suggests that F-actin might be part of a cytoskeletal structure that provides a platform for initial formation and re-formation of CaMKIIα clusters.
The observations of CaMKIIα clustering in this study were enabled by its labeling with a novel recombinant probe, CaMKIIα.FingR, that binds to CaMKIIα with high affinity and specificity. CaMKIIα.FingR was selected from a library whose members consist of the 10FNIII scaffold of human fibronectin with 17 random residues within the BC and FG loops using mRNA display, an in vitro selection procedure, and an intracellular screen based on colocalization. CaMKIIα.FingR displayed remarkable specificity, binding to the association domain of CaMKIIα at a rate >20-fold higher than to CaMKIIβ, despite >76% identity between the target regions of the two CaMKII isoforms. Localization of CaMKIIα.FingR-GFP was identical to that of endogenous CaMKIIα, and the trafficking exhibited by CaMKIIα.FingR-GFP in spines in response to stimulation by glutamate and glycine was similar to that demonstrated by GFP-CaMKIIα. A crucial advantage of the CaMKIIα.FingR-GFP over GFP-CaMKIIα is that it does not increase the amount of CaMKIIα in the cells or induce aberrant clustering.
The effect of Ca2+ on clustering of CaMKIIα in the cell body is, paradoxically, the opposite of its effect on clustering in spines. The recruitment of CaMKIIα to the actin clusters in the cell body likely occurs through interaction with CaMKIIβ, which has an actin-binding domain that is adjacent to a Ca2+/calmodulin-binding domain (Shen et al., 1998). Binding of Ca2+/calmodulin to CaMKIIβ blocks binding of CaMKIIβ to F-actin (O'Leary et al., 2006; Lin and Redmond, 2008), which suggests a mechanism by which influx of Ca2+ could lead to dissociation of CaMKIIα from actin. In spines, clustering is mediated by the binding of CaMKIIα to the NMDA receptor GluN2B (Bayer et al., 2001, 2006; O'Leary et al., 2011). The fact that the cell body has relatively little GluN2B (Rao et al., 1998) could partially explain the difference between CaMKIIα trafficking there and in dendritic spines.
The experiments described in this article raise a number of questions regarding the CaMKIIα function. For instance, what is the significance of CaMKIIα clusters appearing in 40–45% of cortical neurons both in vivo and in culture? There was no obvious morphological distinction between neurons with and without CaMKIIα clusters, and clusters were found in both excitatory and inhibitory neurons. Although cells containing clusters in cortical sections appeared to be particularly prevalent in layer IV, they were present in all layers. It is also not known what functional significance clustering CaMKIIα in the cell body and releasing it following Ca2+ influx might have. CaMKIIβ was shown to associate with spike-like actin protrusions from the cell body of immature (5 DIV) hippocampal neurons (Lin and Redmond, 2008). The appearance of actin “spikes,” which surround the perimeter of the cell body and resemble stress fibers found in fibroblasts, suggests that they might be involved in cell adhesion. In contrast, the actin clusters observed in the present article tend to be present in the center of the cell body and, thus, likely do not have functions similar to those of stress fibers.
Another possibility is that the CaMKIIα associated with somatic actin platforms might be involved in regulating transcription, as CaMKII has been implicated in transcriptional control in a number of different contexts. Along with ΔFosB, it participates in a transcriptional feedback loop in the nucleus accumbens shell that is involved in the response to chronic cocaine exposure (Robison et al., 2013). In fact, the chronic addictive behavior can be blocked by even transient expression of a dominant-negative version of CaMKIIα (Loweth et al., 2013). CaMKII has also been implicated in activation of CREB in response to electrical activity and Ca2+ influx (Wheeler et al., 2008). In addition, CaMKII phosphorylates the transcription factor neuroD, which stimulates neuronal growth (Gaudillière et al., 2004). In the context of such regulatory mechanisms, it is possible that graded release of CaMKIIα from actin platforms in the cell body in response to changes in Ca2+ concentration could modulate transcription in a Ca2+-dependent and activity-dependent fashion.
Footnotes
This work was supported by National Institutes of Health Grants NS041963 and MH086381 (D.B.A.), and Grants GM083898, GM060416, and OD006117 (R.W.R.). We thank Garrett Gross, Jason Junge, Terry Takahashi, and C. Anders Olson for helpful discussions about mRNA display; Samantha Ancona for help analyzing data; and Liana Asatryan in the University of Southern California Lentivirus Core Facility.
The authors declare no competing financial interests.
References
- Andersson AM, Melin L, Bean A, Pettersson RF. A retention signal necessary and sufficient for Golgi localization maps to the cytoplasmic tail of a Bunyaviridae (Uukuniemi virus) membrane glycoprotein. J Virol. 1997;71:4717–4727. doi: 10.1128/jvi.71.6.4717-4727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronowski J, Grotta JC, Waxham MN. Ischemia-induced translocation of Ca2+/calmodulin-dependent protein kinase II: potential role in neuronal damage. J Neurochem. 1992;58:1743–1753. doi: 10.1111/j.1471-4159.1992.tb10049.x. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O'Leary H, Schulman H, De Koninck P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci. 2006;26:1164–1174. doi: 10.1523/JNEUROSCI.3116-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Bayer KU. CaMKII regulation in information processing and storage. Trends Neurosci. 2012;35:607–618. doi: 10.1016/j.tins.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosemeci A, Reese TS, Petersen J, Tao-Cheng JH. A novel particulate form of Ca2+/calmodulin-dependent [correction of Ca(2+)/CaMKII-dependent] protein kinase II in neurons. J Neurosci. 2000;20:3076–3084. doi: 10.1523/JNEUROSCI.20-09-03076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Gaudillière B, Konishi Y, de la Iglesia N, Yao GI, Bonni A. A CaMKII-neuroD signaling pathway specifies dendritic morphogenesis. Neuron. 2004;41:229–241. doi: 10.1016/S0896-6273(03)00841-9. [DOI] [PubMed] [Google Scholar]
- Gross GG, Junge JA, Mora RJ, Kwon HB, Olson CA, Takahashi TT, Liman ER, Ellis-Davies GC, McGee AW, Sabatini BL, Roberts RW, Arnold DB. Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron. 2013;78:971–985. doi: 10.1016/j.neuron.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelz A, Nairn AC, Kuriyan J. Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell. 2003;11:1241–1251. doi: 10.1016/S1097-2765(03)00171-0. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Lebel E, Roy H, Sik A, Schulman H, Waxham MN, De Koninck P. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J Neurosci. 2005;25:6971–6983. doi: 10.1523/JNEUROSCI.4698-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide A, Bailey CW, Huang X, Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol. 1998;284:1141–1151. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- Lardi-Studler B, Smolinsky B, Petitjean CM, Koenig F, Sidler C, Meier JC, Fritschy JM, Schwarz G. Vertebrate-specific sequences in the gephyrin E-domain regulate cytosolic aggregation and postsynaptic clustering. J Cell Sci. 2007;120:1371–1382. doi: 10.1242/jcs.003905. [DOI] [PubMed] [Google Scholar]
- Lewis TL, Jr, Mao T, Svoboda K, Arnold DB. Myosin-dependent targeting of transmembrane proteins to neuronal dendrites. Nat Neurosci. 2009;12:568–576. doi: 10.1038/nn.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Redmond L. CaMKIIbeta binding to stable F-actin in vivo regulates F-actin filament stability. Proc Natl Acad Sci U S A. 2008;105:15791–15796. doi: 10.1073/pnas.0804399105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Barrick J, Szostak JW, Roberts RW. Optimized synthesis of RNA-protein fusions for in vitro protein selection. Methods Enzymol. 2000;317:268–293. doi: 10.1016/s0076-6879(00)18058-9. [DOI] [PubMed] [Google Scholar]
- Loweth JA, Li D, Cortright JJ, Wilke G, Jeyifous O, Neve RL, Bayer KU, Vezina P. Persistent reversal of enhanced amphetamine intake by transient CaMKII inhibition. J Neurosci. 2013;33:1411–1416. doi: 10.1523/JNEUROSCI.4386-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden KC, Shemesh A, Bayer KU, Carroll RC. Selective trans location of Ca2+/calmodulin protein kinase IIalpha (CaMKIIalpha) to inhibitory synapses. Proc Natl Acad Sci U S A. 2010;107:20559–20564. doi: 10.1073/pnas.1010346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall J, Molloy R, Moss GW, Howe JR, Hughes TE. The jellyfish green fluorescent protein: a new tool for studying ion channel expression and function. Neuron. 1995;14:211–215. doi: 10.1016/0896-6273(95)90279-1. [DOI] [PubMed] [Google Scholar]
- O'Leary H, Lasda E, Bayer KU. CaMKIIbeta association with the actin cytoskeleton is regulated by alternative splicing. Mol Biol Cell. 2006;17:4656–4665. doi: 10.1091/mbc.E06-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary H, Liu WH, Rorabaugh JM, Coultrap SJ, Bayer KU. Nucleotides and phosphorylation bidirectionally modulate Ca2+/calmodulin-dependent protein kinase II (CaMKII) binding to the N-methyl-d-aspartate (NMDA) receptor subunit GluN2B. J Biol Chem. 2011;286:31272–31281. doi: 10.1074/jbc.M111.233668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson CA, Roberts RW. Design, expression, and stability of a diverse protein library based on the tenth fibronectin type III domain. Protein Sci. 2007;16:476–484. doi: 10.1110/ps.062498407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson CA, Liao HI, Sun R, Roberts RW. mRNA display selection of a high-affinity, modification-specific phospho-IkappaBalpha-binding fibronectin. ACS Chem Biol. 2008;3:480–485. doi: 10.1021/cb800069c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci. 1998;18:1217–1229. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RW, Szostak JW. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Vialou V, Mazei-Robison M, Feng J, Kourrich S, Collins M, Wee S, Koob G, Turecki G, Neve R, Thomas M, Nestler EJ. Behavioral and structural responses to chronic cocaine require a feedforward loop involving ΔFosB and calcium/calmodulin-dependent protein kinase II in the nucleus accumbens shell. J Neurosci. 2013;33:4295–4307. doi: 10.1523/JNEUROSCI.5192-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose J, Jin SX, Craig AM. Heterosynaptic molecular dynamics: locally induced propagating synaptic accumulation of CaM kinase II. Neuron. 2009;61:351–358. doi: 10.1016/j.neuron.2008.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Comolli LR, Hoelz A, Downing KH, Nairn AC, Kuriyan J. Oligomerization states of the association domain and the holoenyzme of Ca2+/CaM kinase II. Febs J. 2006;273:682–694. doi: 10.1111/j.1742-4658.2005.05088.x. [DOI] [PubMed] [Google Scholar]
- Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW, Lisman J. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci. 2011;31:9170–9178. doi: 10.1523/JNEUROSCI.1250-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiryanova D, Morimoto T, Zhou C, Chouhan AK, Sigrist SJ, Nose A, Macleod GT, Deitcher DL, Levitan ES. Differential control of presynaptic CaMKII activation and translocation to active zones. J Neurosci. 2011;31:9093–9100. doi: 10.1523/JNEUROSCI.0550-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Subramanian K, Meyer T. CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron. 1998;21:593–606. doi: 10.1016/S0896-6273(00)80569-3. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Connor JH, Shenolikar S, Meyer T. Molecular memory by reversible translocation of calcium/calmodulin-dependent protein kinase II. Nat Neurosci. 2000;3:881–886. doi: 10.1038/78783. [DOI] [PubMed] [Google Scholar]
- Soderling TR. CaM-kinases: modulators of synaptic plasticity. Curr Opin Neurobiol. 2000;10:375–380. doi: 10.1016/S0959-4388(00)00090-8. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Svoboda K, Malinow R. Experience strengthening transmission by driving AMPA receptors into synapses. Science. 2003;299:1585–1588. doi: 10.1126/science.1079886. [DOI] [PubMed] [Google Scholar]
- Vest RS, O'Leary H, Bayer KU. Differential regulation by ATP versus ADP further links CaMKII aggregation to ischemic conditions. FEBS Lett. 2009;583:3577–3581. doi: 10.1016/j.febslet.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DG, Barrett CF, Groth RD, Safa P, Tsien RW. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J Cell Biol. 2008;183:849–863. doi: 10.1083/jcb.200805048. [DOI] [PMC free article] [PubMed] [Google Scholar]