

Abstract
Clinical and pathophysiological evidences connect migraine and the cerebellum. Literature on documented cerebellar abnormalities in migraine, however, is relatively sparse. Cerebellar involvement may be observed in 4 types of migraines: in the widespread migraine with aura (MWA) and migraine without aura (MWoA) forms; in particular subtypes of migraine such as basilar-type migraine (BTM); and in the genetically driven autosomal dominant familial hemiplegic migraine (FHM) forms. Cerebellar dysfunction in migraineurs varies largely in severity, and may be subclinical. Purkinje cells express calcium channels that are related to the pathophysiology of both inherited forms of migraine and primary ataxias, mostly spinal cerebellar ataxia type 6 (SCA-6) and episodic ataxia type 2 (EA-2). Genetically driven ion channels dysfunction leads to hyperexcitability in the brain and cerebellum, possibly facilitating spreading depression waves in both locations. This review focuses on the cerebellar involvement in migraine, the relevant ataxias and their association with this primary headache, and discusses some of the pathophysiological processes putatively underlying these diseases.
Keywords: migraine, familial hemiplegic migraine, cerebellum, progressive ataxia, episodic ataxia
Migraine is a common disease that affects 10 to 12% of the population and is considered by the World Health Organization as one of the most disabling neurological disorders.1 Migraine attacks typically occur in varying intervals, each lasting 4 to 72 hours by definition. The unilateral, mostly side-shifting throbbing pain, located predominantly to the frontal parts of the cranium, may be intense enough to interrupt daily activity and worsens with physical activity. Nausea, vomiting, photo and phonophobia frequently accompany the annoying moderate to severe pain. A series of different neurological focal abnormalities named aura (from the Greek “breath,” gentle breeze), mostly visual in nature, but also sometimes sensory, motor, or dysphasic, may occur in close association with the pain, typically before the headache onset.2 The International Headache Society (IHS) classifies migraine headaches, among other less frequent subtypes, as migraine with aura (MWA), or migraine without aura (MWoA), according to the presence of aura symptoms.3
Pathophysiological Theories in Migraine
The mechanisms underlying migraine attacks remain fairly unknown, although accumulating data have demonstrated that this ailment is a primary brain disorder.4 A dispute whether migraine had either a nervous or a vascular origin, polarizing the 2 so-called “vascular” and “neuronal” theories, has been present for many years,5 but the central nervous system more probably seems be the ultimate source of migraine. The hitherto suitable vascular theory, which popularized the expression “vascular headache,” has been challenged by the information that aura and headache did not parallel changes in the vasculature.6 The possibility that abnormal brain hyperexcitability primarily originates migraine attacks is now widely accepted,7 and the disease threshold, at least partially, seems to be determined by genetic predisposition.8 The hyperexcitability has been confirmed by the relatively higher susceptibility of the migrainous cortex to phosphene induction secondary to transcranial magnetic stimulation.9 It seems, therefore, that the vascular responses take place because of primarily triggered events in the nervous system intimacy.
Spreading Depression
Spreading depression (SD) consists of a spreading wave of depolarization associated with a reduction of the cortical activity that lasts for minutes with a propagation speed of around 3 mm/min. The expression “cortical spreading depression” (CSD) is widespread, but since this phenomenon is not exclusively cortical—it has been recorded in various tissues including the basal ganglia and thalamus,10,11 cerebellum,12-15 tectum and olfactory bulb,12 retina,16-22 and spinal cord23—we believe that “spreading depression” is a better denomination.
In 1945, Leão and Morrison suggested for the first time that SD could be related to the pathophysiology of migraine24 and Leão postulated that circulatory changes were in close connection with SD waves.25 SD compatible circulatory changes were subsequently found in migraineurs, making the possibility of SD being an important phenomenon in this disease even more attractive.6 SD is accompanied by an initial hyperperfusion, followed by prolonged and pronounced spreading hypoperfusion.26 The genetically hyperexcitable brain in migraine probably facilitates paroxysms of SD-like phenomena initiating each of them the cascade of events ultimately leading to the attacks. Functional imaging studies support the possibility of SD underlying migraine episodes.27 The trigeminovascular system comprised of the trigeminal fibers innervating meningeal and brain vessels is activated by SD,28 leading to plasma extravasation and vasodilatation (neurogenic inflammation) in the dura mater.29 The ability of triptans, a class of 5-HT1 agonists, to block neurogenic inflammation and neuropeptide release centrally, has supported the defense of its use as effective antimigraine agents.30-32
The Cerebellum
Although Herophilus (335 to 280 B.C.) is usually cited for firstly recognizing the cerebellum (from Latin, “small brain”) as distinct from the brain, Aristotle did so before (“The history of animals” book I, part XVI, 350 B.C.). Galen (131 to 200 A.D.) called the vermis “the worm-like outgrowth,” Luigi Rolando (1773 to 1831) concluded the cerebellum was a motor structure, and Marie-Jean-Pierre Flourens (1794 to 1867) finally linked the cerebellum to coordination.33,34 The relatively simpler structure of the cerebellum is highly specific and uniform, with cells arranged in layers in the cerebellar cortex connected each other by a repetitive microcircuitry.35 The Purkinje cells are the source of cerebellar output. Therefore, malfunction in Purkinje cells severely impairs motor planning and coordination.
CEREBELLAR DISORDERS IN COMMON MWA AND MWoA FORMS OF MIGRAINE
In spite of the fact that balance changes and vertigo have been recognized in migraine, only a few studies have specifically assessed cerebellar function between or during attacks. In migraine, stabilometry studies have revealed ictal and interictal balance abnormalities in treatment-free patients.36,37 Vestibulocerebellar function also seems compromised in migraineurs, with abnormal nystagmus in calorimetric testing and decrease in saccadic eye-movement accuracy.38 In addition, subclinical cerebellar impairment expressed as a lack of fine coordination has been shown interictally in migraineurs.39 Altogether, these findings indicate that migraine affects cerebellar function.39
It is not surprising that vestibular abnormalities may be detected in migraine patients, as about 2/3 of migraineurs are sensitive to motion and 1/4 may present with paroxysmal vertigo.40,41 Although a positive family history and previous motion sickness in childhood do not contribute to the diagnosis of MWoA, vestibular abnormalities are associated with this type of headache.42,43 Visual dysfunction may also impair coordination and probably impacts balance in migraine.44 Spatiotemporal function and motion processing are reportedly abnormal in migraineurs interictally45,46 and visual fields and contrast functions differ from controls.47
BASILAR-TYPE MIGRAINE
Cerebellar dysfunction has been recognized in relation to special forms of migraine for many years. The expression “cerebellar migraine” was used in some German48,49 and Czech50 early publications. In 1961, Bickerstaff described what he called “basilar artery migraine,”51 making the expression “basilar migraine” popular in neurology. According to the IHS, BTM is characterized by aura symptoms clearly originating from the brainstem and/or both hemispheres, without motor deficits.3 Symptoms may include dysarthria, vertigo, tinnitus, hypacusia, diplopia, visual symptoms, ataxia, decreased level of consciousness, and bilateral paresthesias.52 BTM has been considered more prevalent in adolescent girls with very positive family histories, but a recent analysis does not support BTM, which presents with ataxia in 5% of the cases, as a distinct migraine subform.53 The pathophysiology of BTM is not known. Circulatory changes and episodes of stroke putatively related to basilar-type migraine have been reported.54 Such infarcts have also been reported in the thalamus55 and the occipital areas.56-58 Knowing the genetic mechanisms behind certain forms of migraine, scrutiny indicates that many migraine patients previously described according to their clinical pictures as “cerebellar migraine” or “basilar migraine,” probably carried one of the known ion channel related mutations. A mutation at the FHM2 locus at the ATP1A2 gene has been described in familial BTM without hemiplegia, suggesting a connection between BTM and hemiplegic migraine.59 BTM most probably represents a variation of MWA rather than another migraine subtype, as 95% of the BTM patients experience typical aura as in MWA.53
FAMILIAL HEMIPLEGIC MIGRAINE AND THE CEREBELLUM-RELATED DISORDERS
FHM is an autosomal dominant disorder characterized by migraine attacks with hemiplegic aura. The diagnosis is based on the presence of aura including motor weakness and at least one first- or second-degree relative suffering from migraine with aura that presents with motor deficits.3 A multitude of associated symptoms may be present, including ataxia, seen in one-third of the families.60 Three types of FHM have been described so far: FHM-1 is consequent to mutations of the CACNA1A gene coding for a P/Q calcium channel;61 FHM-2 is due to the mutation of the ATP1A2 gene coding for the alpha2 subunit of the Na/K astrocytic ATPase;62,63 and FHM-3 follows a mutation of the SCN1A gene coding for a neuron voltage-gated sodium channel.64 The FHM phenotype includes hemiplegic migraine, seizure, prolonged coma, hyperthermia, sensory deficit, and transient or permanent cerebellar signs, such as ataxia, nystagmus, and dysarthria.65
In FHM-1, the CACNA1A gene encodes the α1A (CAV2.1) subunit of the high voltage-gated P/Q type of calcium channel. This channel is expressed throughout the central nervous system, particularly in the cerebellar Purkinje cells, where it mediates depolarization-induced Ca2+ influx into presynaptic terminals and glutamate release.66,67 P/Q calcium channels play a pivotal role in neurotransmitter release68 and influence neuronal excitability.69 The consequences of different missense mutations in the CACNA1A gene may lead to gain-of-function of human P/Q-type calcium channels, although not all studies agree in this respect.70 New animal models may provide important insights in this field. A knockin mouse expressing the human R192Q pure FHM-1 mutation was genetically engineered and recently studied. This mouse shows gain-of-function P/Q Ca2+ channel function as evidenced by opening of calcium channels at lower levels of depolarization, lower threshold for SD and faster propagation speed.71 These findings open the possibility of SD-like phenomena in the cerebellum as a justification for cerebellar dysfunction in migraine patients. Human evidence confirming this hypothesis is however not yet available.
The mechanisms behind the neurological symptom complex linked to CACNA1A, ATP1A2, and SCN1A genes, respectively involved with FHM 1, 2, and 3, remain partially unclear. Noteworthy is the fact that, despite the type of ion channel involved, all mutations result in hyperexcitability and may be related to hemiplegic migraine, epilepsy, and/or ataxic disorders.
Cerebellar symptoms in FHM have been recognized in many families (Table). Such symptoms may be produced by lesion in the cerebellum itself or in structures with afferent or efferent cerebellar connections, such as the brainstem. Thus, the exact origin of symptoms such as nystagmus and ataxia in migraine patients cannot be definitely related to the cerebellum. On the other hand, the atrophy found in FMH and the calcium channel abnormalities in the cerebellum indicate that symptoms are probably cerebellar in nature.
Table.
Cerebellar Symptoms in Earlier FHM Descriptions
| Author (Reference) | Year | Gender | Age | Possible Cerebellar/Vestibular Sings and Symptoms |
|---|---|---|---|---|
| Ohta et al (151) | 1967 | Male | 59 | Unsteady gait, mild dysarthria, incoordination of the limbs. |
| Male | 30 | Apparent horizontal nystagmus on lateral gaze | ||
| Female | 19 | – | ||
| Male | 13 | – | ||
| Male | 59 | Slow speech and unsteady gait | ||
| Young et al (152) | 1970 | Male | 33 | Mild ataxia, worse during migraine attacks, nystagmus |
| Male | 32 | Nystagmus | ||
| Male | 4 | Nystagmus | ||
| Male | 4 | Nystagmus | ||
| Codina et al† (153) | 1971 | Female | 44 | Nystagmus |
| Male | 14 | Nystagmus | ||
| Male | 49 | Nystagmus | ||
| Zifkin et al‡ (154) | 1980 | Male | 22 | Nystagmus |
Further cases described without cerebellar or vestibular abnormalities:
Four cases;
one case.
Around 20% of the hemiplegic migraine patients show permanent mild cerebellar deficits.72 Unconsciousness, fever, and confusion may occur associated with the hemiplegic attacks and ataxia, usually accompanied by cerebellar atrophy.73,74
SPINOCEREBELAR AND EPISODIC ATAXIAS
The CACNA1A mutations are also involved with cerebellar diseases, namely episodic ataxia type 2 (EA-2) and spinocerebellar ataxia type 6 (SCA-6). Hereditary EAs are genetic conditions typically characterized by recurrent clumsiness triggered by exertion, stress, or fatigue with a favorable response to acetazolamide.75,76 Spinocerebellar ataxias (SCA) are genetic non-paroxysmal, moderate to severe ataxias of late onset characterized by progressive cerebellar degeneration leading to incoordination. Other cerebellar symptoms associated with spinal cord signs, such as motor deficit, as well as vibratory and proprioceptive sensory loss.75 The myriad of cerebellar symptoms include dysarthria, dysmetria, tremor, and nystagmus of various types.77
A series of EA mutations have been found so far,76,78-80 and a complete loss of the P/Q function has been suggested to underlie the pathophysiology of EA-2.81 Different nomenclature in successive descriptions have confused the understanding of non-progressive ataxias.82-84 SCA-6 has been associated with small expansions of a CAG repeat at the 3′ end of the CACNA1A gene, and point mutations are responsible for the allelic disorders related to EA-2.60,79,85-87 The genetics behind these phenotypes, however, may vary.88 Regardless of the mutation type, hyperexcitability seem to stand behind all the different phenotypes. Interestingly, a mutation in the glutamate transporter excitatory aminoacid transporter 1 (EAAT1) is also related to episodic ataxia (EA), seizures, migraine, and alternating hemiplegia.89 EAAT1 is expressed particularly in the cerebellum and brain stem. The mutation in EAAT1 may lead to a reduced capacity for glutamate reuptake, increasing hyperexcitability. This reproduces the pathophysiological conditions present in channelopaties leading to FHM, episodic/progressive ataxias and coma after minor head trauma.
SCA-6 represents the form of progressive ataxia with closest relation to FHM pathophysiology, as this form of SCA is also linked to the CACNA1A gene.90,91 Different mutations have been linked to the phenotype of SCA-6, sometimes associated with FHM.92 There may be marked cerebellar atrophy on MR examination in these patients.93 Not only mutations occur at the same gene, but in 20% of FHM patients permanent cerebellar symptoms are present.94,95
The phenotypes of such disorders may vary between and within families.91,96 EA-2 patients may sometimes have non-hemiplegic migraine, which presents after the onset of the ataxic symptoms.97 Interictally, EA patients may present constant cerebellar symptoms and signs such as nystagmus and cerebellar atrophy. The migraine-progressive episodic ataxias symptoms interchange indicate that the cerebellar disorders related to channelopathies intermingle and may represent different aspects from the same abnormality. Mechanisms behind ataxias in migraine disorders most probably involve membrane dysfunction. Purkinje cells, where P/Q-type calcium channels are mostly expressed, fire according to intrinsic regular spontaneous pacemaking.98 This intrinsic pacemaking activity is irregular in P/Q-mutant Purkinje cells as well as in w-agatoxin IVA-blocked P/Q-type calcium channel in wild Purkinje cells. The defective P/Q calcium current decreases the function of calcium-activated potassium (KCa) channels, which are fundamental for the precision of the Purkinje cells intrinsic firing. EBIO, a channel activator that increases the affinity of KCa channels for calcium, recovers the regular firing in affected Purkinje cells.99 This makes the KCa channel a potential therapeutic target not only for EA-2, but also for related symptoms in migraine disorders.
COMA, CEREBELLUM, AND MIGRAINE
One of the conditions associated with cerebellar dysfunction, FHM and the CACNA1A gene is fatal coma after mild head trauma.100-102 Some mutations have been related to this phenotype. Patients carrying the T666M mutation in CACNA1A gene,103 but not exclusively as the chromosome 1 has also been implicated in this kind of abnormality104—may present coma following relatively mild head trauma, with brain edema and sometimes long-lasting coma.101-103,105-107 The S218L mutation was shown to produce particularly severe brain edema after trauma.108
As a hypothesis, the mechanisms leading to coma can be understood as follows: minor trauma, a relatively irrelevant depolarizing stimulus in healthy subjects, may elicit SD in patients with a particularly marked Cav2.1 channel gain of function, both in the brain and cerebellum. Further activation may then take place through a positive feedback leading to Cav2.1-dependent glutamate release, activation of NMDA receptors, de novo increase of extracelullar K+, glutamate release, and more NMDA receptor activation.109 SD may disrupt the blood–brain barrier by activating MMP-9, one of the proteases implicated in BBB opening,110 leading to brain edema and coma. Interestingly, the long-lasting edema and coma take place after a time interval following the trauma. This indicates that the process is not dependent on immediate neuronal impulses and neurotransmitters release, but on time consuming progressive changes. Moreover, the resulting pathophysiological state is a self-perpetuating process with a relatively slow recovery rate. Positive SD and calcium waves (see below) feedbacks in particularly excitable subjects would fit with these requirements. Transient global amnesia (TGA), a potentially SD related disorder,111 may also be induced by minor head trauma, just as coma in some patients with genetic forms of migraine where cerebellar abnormalities may be present.112
THE ACETAZOLAMIDE EFFECT
Acetazolamide, a reversible inhibitor of the enzyme carbonic anhydrase, is a drug known for its benefit in EA-2.79,113,114 Acetazolamide-responsive episodic symptoms, typical of EA-2, have also been shown in SCA-6.115 The effect of acetazolamide in EAs was found in 1978 by chance, when patients received this drug after being erroneously diagnosed as periodic paralysis.114 Acetazolamide response has been described in FHM with associated ataxia74 and in migraineurs without cerebellar symptoms.116
Acetazolamide does not usually diminish the frequency or intensity of FHM, being mostly indicated for use in EA-2. However, there are 2 FHM reports with clear acetazolamide response.74,116 Formal trials using acetazolamide in migraine are few. In an open uncontrolled pilot study, the efficacy and tolerability of acetazolamide were addressed in 22 MWA patients. 68.2% reported a reduction of MA episodes higher than 50%.117 A randomized clinical trial was performed comparing 500 mg oral acetazolamide versus placebo in 53 IHS migraine patients (27 in the placebo group). This study had to be interrupted prematurely due to many side effects related withdrawals. So far, the authors did not find a difference between the active drug and placebo.118 Acetazolamide was also shown to interrupt aura status in 3 patients.119
The acetazolamide mechanism of action in episodic ataxia type 2 (EA-2) is still mysterious. It is interesting that topiramate, an effective antimigraine prophylactic agent, shares with acetazolamide the property of carbonic anhydrase inhibition.120 Besides, it was recently reported to suppress the susceptibility to cortical spreading depression in experimental animals.121 Acetazolamide induces metabolic acidosis. It is possible that this drug increases the extracelullar concentration of free protons in the brain tissue including the cerebellum.113 Since calcium channels are sensitive to pH changes, acetazolamide could restore normal function in mutant calcium channels through acidification. However, acetazolamide does not modify the channel properties through either pH-dependent or pH-independent mechanisms.122 Alternatively, since acetazolamide activate largeconductance KCa channels, which are in normal conditions exclusively activated in Purkinje cells by P/Q-type calcium channels, it is possible that this drug acts by restoring Purkinje cells pacemaking properties.99
CEREBELLAR CIRCULATORY CHANGES
Circulatory changes may take place in the cerebellum during migraine attacks. Following sumatriptan administration, a vasoconstricting antimigraine agent, infarction has been described in the cerebellum, showing that this area was probably predisposed to ischemia as compared to other regions.123 Decreased perfusion and cerebellar symptoms, including dysarthria, ataxia, and dizziness have been described in migraine.124,125 Such circulatory changes can outlast the symptoms.125 Stroke in the posterior circulation has been reported in migraine54,123 including in children,126 mostly diagnosed as “basilar migraine.” The posterior circulation territory, particularly the cerebellum, shows significantly increased risk for infarct-like MRI findings compared to the remaining of the nervous system. The highest risk is in MWA with at least 1 attack per month, in the absence of stroke history.127 According to the CAMERA study, the percent of all these small, infarct-like lesions in the posterior circulation in MWA, MWoA, and controls were 81, 47, and 44%, respectively; the majority was in vascular border zones; and multiple posterior circulation lesions were identified exclusively among the migraine patients.128
The nature and pathophysiology of such infratentorial lesions are not known. Since the cerebellar circulation has relatively few anastomoses, it is prone to watershed infarcts.129 SD related reduction in rCBF could, theoretically, induce more infarcts in this territory as compared to areas where collateral circulation is available. Although subjects do not present overt stroke symptoms, it is possible the subclinical cerebellar signs and symptoms in migraine36,38,39 are secondary to small infarcts in the posterior circulation.
SPREADING DEPRESSION AND THE CEREBELLUM
Leão and Martins-Ferreira first published a 24 line note on SD in the cerebellum, quadrigeminal plate, and olfactory bulb12 and mentioned that the cerebellum is naturally resistant to SD. Fifková et al described SD in the rat cerebellum13 and Young wrote on the SD in the elasmobranch fish (Raja erinacea, Raja ocellata).14 As also pointed by Nicholson in 1984, reviewing cerebellar SD in different species,15 the cerebellum does not easily supports this phenomenon, unless some “conditioning” takes place. This may happen by raising the extracelullar K+, removing most of the NaCl, or replacing the chloride with another anion. During SD, extracellular calcium concentration falls, reflecting Ca2+ influx with consequent intracellular Ca2+ overload, that may, if sufficiently high, promote cell death.130 Just as in the isolated retina and hippocampus, also in the turtle cerebellum SD occurs in the absence of blood flow, meaning that SD is not dependent on vascular or blood influence.15 If cerebellar SD is related to EA-2, pH changes alone may be not sufficient for explaining the acetazolamide effect. Alternatively, SD could occur in the cerebellum through facilitating mechanisms not involving pH reduction.
Other cortical self-propagating waves with potential implications in cerebellar diseases and migraine have been demonstrated. Spreading acidification and depression (SAD) has been observed in the rat cerebellar cortex following suprathreshold electrical stimulation.131 Substantial differences show that SAD and SD are not the same phenomenon. SAD spreads at a greater rate of 50 to 110 m/s, continues for 1 to 2 minutes, is accompanied by a powerful suppression of the pre and postsynaptic responses, with a refractory period of 90 seconds. Differently from SD, SAD induces no extracellular DC shift, do not change blood vessels and has a shorter recovery period. Besides, the conditioning required for SD in the cerebellum is not required to elicit SAD. While SD propagates radially outwards from the initiating point, SAD spreads perpendicularly to an activated beam of parallel fibers, which makes its spreading pattern dependent on the cerebellar cortex neuronal architecture. Pharmacologically, AMPA receptor blocking, which has little effect on SD, affects SAD, the opposite occurring with NMDA receptor blocking. SAD depends on extracellular Ca2+, while SD does not depend that strictly.132 SAD has been implicated in the pathophysiology of EA-1, where pathology is related to a Kv1.1 voltage-gated potassium channel abnormality,133 and is not likely to be involved with the cerebellar symptoms in migraine.
Astrocytes respond to glutamate with rapid calcium influx that propagate as waves from one cell to its neighbors.134 The so-called calcium waves (CW) constitute a signaling system that allows astrocytes to rapidly activate adjacent astrocytes and neurons, through gap junctions, and extracellular messengers,135,136 modulating synaptic transmission and neuronal activity.137 CWs are also triggered by neuronal activity138 and may be involved in blood flow regulation. CWs have been implicated in cortical spreading depression. They were demonstrated in cell cultures and tissue preparations in different cell populations,139, 140 and precede SD waves in hippocampal cultures.141,142 Although these 2 forms of waves are related, SD does occur in calcium-free incubated hippocampal slices where CWs are abolished, demonstrating that the latter is not an obligatory requirement for the former.142 Since FHM and the related CACNA1A mutations diseases directly involve calcium fluxing, it is tempting to consider that CWs associated with SD might have a pathophysiological role in this context.143 The glutamate release induced by abnormal Cav2.1 channels in migraine could theoretically lead to not only SD, but also CW activation and further vasodilatation, contributing particularly to the phenotype of brain edema and coma following head trauma. The astrocytes’ role in brain water homeostasis regulation144 also supports this possibility.
STRUCTURAL CHANGES IN THE CEREBELLUM AND MIGRAINE
Few studies have specifically addressed cerebellar structural changes in migraine. Dichgans et al found Magnetic Resonance Spectroscopy (1H-MRS) abnormalities in FHM-1 with reduced N-acetyl-aspartate (NAA), glutamate and elevated myo-inositol (mI) in the cerebellum, compatible with neuronal damage. Increased pH in the cerebrum and cerebellum, which normalized following acetazolamide treatment, as well as high lactate peak in half of the subjects has been reported in EA-2 patients.145 Autopsy studies have shown pathological abnormalities in SCA including mild atrophy of the cerebellar folia, reduced number of Purkinje cells especially in the vermis, swelling of the Purkinje cell axons, decrease in granular cells, reduced number of dendrites in the molecular layers of Purkinje cells, and cerebellar cortical degeneration with reduced thickness of the molecular layer.100, 146 In FHM, cerebellar vermis atrophy and cortical cerebellar degeneration accompanied with Bergman glia proliferation have been described.147
FINAL REMARKS
Taken together, the data suggest that the cerebellum is implicated not only with FHM, but also with more typical migraine forms such as MWA and MWoA. The ionic and signaling changes present in migraine may affect also the cerebellum potentially leading to cerebellar dysfunction (Fig.). Cerebellar symptomatology, which does not depend on the presence of headache, may be episodic, suggesting an underlying transitory neuropathological change in the cerebellum such as SD; or present as a constant-progressive disorders. In this case, an increase in Ca2+ influx secondary to defective Ca2+ channels expressed by Purkinje cells would favor apoptosis, possibly in a cumulative, slowly progressive pattern. Alternatively, cumulative microvascular ischemia in watershed cerebellar areas secondary to successive migraine attacks could also impair cerebellar function with time in some cases. The pain may be produced by CGRP-containing sensory nerves activated by SD in the anterior circulation (trigeminal fibers) and/or posterior circulation (C2 fibers). Trigeminal fibers may also be partially activated by SD in some parts of the cerebellum as the rostral third of the basilar artery as well as the superior cerebellar artery are innervated by the trigeminal nerve.148,149
Fig.
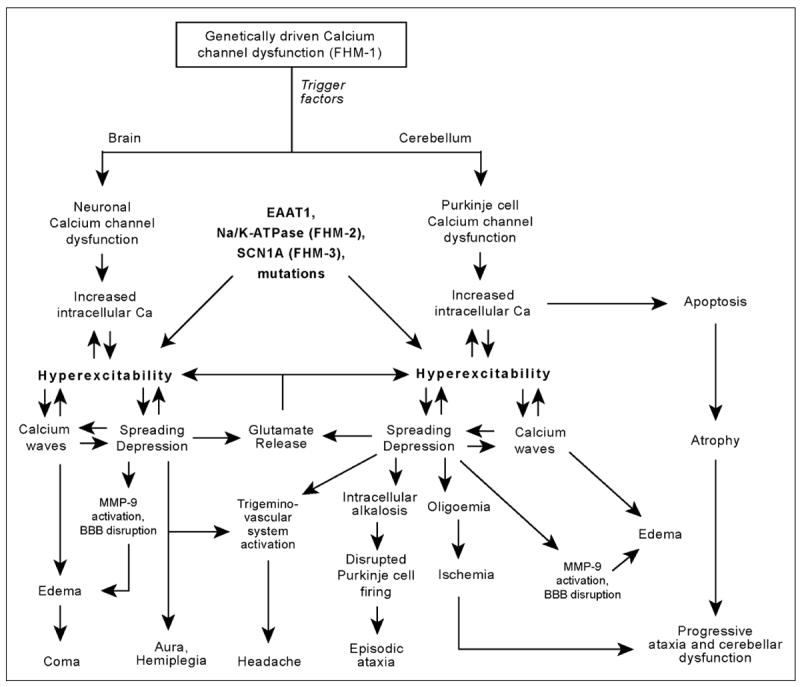
The brain and the cerebellum may share common pathophysiological mechanisms leading to different clinical pictures, which combine in diverse ways, largely varying in severity. Hyperexcitability, the pivotal abnormality in migraine, may be due to inherited calcium channel malfunction, as in CANCA1A mutations, or other mutations, such as excitatory aminoacid transporter 1 (EAAT1), Na/k-ATPase, or SCN1A. Trigger factors in susceptible individuals, such as trauma or angiography, would lead to paroxysmal spreading depression and related calcium waves, leading to temporary dysfunction in both cerebrum and cerebellum. Headache may be explained by SD activation of trigeminal (anterior circulation, the rostral third of the basilar artery and the superior cerebellar artery) and/or C2 sensory fibers (remaining vessels of the vertebrobasilar system). Progressive symptoms could be the result of cumulative ischaemic lesions and progressive atrophy provoked by exaggerated calcium influx. BBB: blood–brain barrier and MMP-9: matrix metalloprotease.
Knowledge on the genetic mechanisms leading to dysfunction in ion channels, ion pumps, and transporters has improved our understanding of migraine and related cerebellar disorders, although puzzling questions still remain. It is unclear how a multitude of phenotypes including minor trauma with edema and coma, fever, pleocytosis, hemiplegic migraine, and cerebellar ataxias, is related to a single mutation. The clinical picture in EA, for example, may vary to a great extent, such as from isolated mild ataxia to a constellation of symptoms suggestive of cerebellum, brainstem, and cortex dysfunction.150 This may indicate that phenotypic pleomorphism is a rule rather than an exception in these ailments. If an SD-like phenomenon underlies this group of diseases, it is likely that it may sometimes either not be clinically expressed, or manifest in different forms or degrees.
Cases reported as “basilar migraine,” “footballer’s migraine” or “cerebellar migraine” do not seem to constitute distinct entities. They may actually correspond to mere variations within the migraine channelopaty spectrum. As the molecular mechanisms implicated in migraine, ataxia, coma after minor trauma, and related disorders are better understood, it seems probable that clinical terms will be reviewed, and classifications will be established on a genetic-biochemical basis.
Acknowledgments
The study was supported by a NIH grant 5PO1 NS 35611-09. MV is indebted to CAPES–Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Ministry of Education, Brazil; and Fulbright, USA, for a visiting professor scholarship. The authors acknowledge Professor Michael Moskowitz for his reviewing of this manuscript. Suggestions and comments by Dr. Alexandre Façanha daSilva and Cristina Granziera are appreciated.
Abbreviations
- IHS
International Headache Society
- MWA
migraine with aura
- MWoA
migraine without aura
- SD
spreading depression
- GABA
gamma-aminobutyric acid
- SCA
spinal cerebellar ataxia
- BTM
basilar-type migraine
- FHM
familial hemiplegic migraine
- EAAT1
excitatory aminoacid transporter 1
- EA
episodic ataxia
- TGA
transient global amnesia
- SPECT
single photon emission computed tomography
- CW
calcium waves
Footnotes
Conflict of Interest: None
References
- 1.Leonardi M, Steiner TJ, Scher AT, Lipton RB. The global burden of migraine: Measuring disability in headache disorders with WHO’s Classification of Functioning, Disability and Health (ICF) J Headache Pain. 2005;6:429–440. doi: 10.1007/s10194-005-0252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goadsby PJ. Migraine: Diagnosis and management. Intern Med J. 2003;33:436–442. doi: 10.1046/j.1445-5994.2003.00453.x. [DOI] [PubMed] [Google Scholar]
- 3.Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders. Cephalalgia. 2004;24(suppl 1) doi: 10.1111/j.1468-2982.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 4.Goadsby PJ, Lipton RB, Ferrari MD. Migraine–current understanding and treatment. N Engl J Med. 2002;346:257–270. doi: 10.1056/NEJMra010917. [DOI] [PubMed] [Google Scholar]
- 5.Parsons AA, Strijbos PJ. The neuronal versus vascular hypothesis of migraine and cortical spreading depression. Curr Opin Pharmacol. 2003;3:73–77. doi: 10.1016/s1471-4892(02)00016-4. [DOI] [PubMed] [Google Scholar]
- 6.Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann Neurol. 1981;9:344–352. doi: 10.1002/ana.410090406. [DOI] [PubMed] [Google Scholar]
- 7.Welch KM. Brain hyperexcitability: The basis for antiepileptic drugs in migraine prevention. Headache. 2005;45(suppl 1):S25–S32. doi: 10.1111/j.1526-4610.2005.4501008.x. [DOI] [PubMed] [Google Scholar]
- 8.Haan J, Kors EE, Vanmolkot KR, van den Maagden-berg AM, Frants RR, Ferrari MD. Migraine genetics: An update. Curr Pain Headache Rep. 2005;9:213–220. doi: 10.1007/s11916-005-0065-9. [DOI] [PubMed] [Google Scholar]
- 9.Young WB, Oshinsky ML, Shechter AL, Gebeline-Myers C, Bradley KC, Wassermann EM. Consecutive transcranial magnetic stimulation: Phosphene thresholds in migraineurs and controls. Headache. 2004;44:131–135. doi: 10.1111/j.1526-4610.2004.04028.x. [DOI] [PubMed] [Google Scholar]
- 10.Trachtenberg MC, Hull CD, Buchwald NA. Electro-physiological concomitants of spreading depression in caudate and thalamic nuclei of the cat. Brain Res. 1970;20:219–231. doi: 10.1016/0006-8993(70)90290-8. [DOI] [PubMed] [Google Scholar]
- 11.Vinogradova LV, Koroleva VI, Bures J. Re-entry waves of Leao’s spreading depression between neocortex and caudate nucleus. Brain Res. 1991;538:161–164. doi: 10.1016/0006-8993(91)90392-9. [DOI] [PubMed] [Google Scholar]
- 12.Leão AAP, Martins-Ferreira H. Nota acerca da depressão alastrante no cerebelo, tubérculo quadrigêmino anterior e bulbo olfativo. An Acad Bras Cienc. 1961;22:34–40. [Google Scholar]
- 13.Fifková E, Bures J, Koshtoyants OK, Krivánek J. T.W. Leão’s spreading depression in the cerebellum of the rat. Experientia. 1961;17:572–573. doi: 10.1007/BF02156433. [DOI] [PubMed] [Google Scholar]
- 14.Young W. Spreading depression in elasmobranch cerebellum. Brain Res. 1980;199:113–126. doi: 10.1016/0006-8993(80)90234-6. [DOI] [PubMed] [Google Scholar]
- 15.Nicholson C. Comparative neurophysiology of spreading depression in the cerebellum. An Acad Bras Cienc. 1984;56:481–494. [PubMed] [Google Scholar]
- 16.Martins-Ferreira H, de Oliveira Castro G, Struchiner CJ, Rodrigues PS. Circling spreading depression in isolated chick retina. J Neurophysiol. 1974;37:773–784. doi: 10.1152/jn.1974.37.4.773. [DOI] [PubMed] [Google Scholar]
- 17.Martins-Ferreira H, de Oliveira Castro G. Spreading depression in isolated chick retina. Vision Res. 1971;11(suppl 3):71–84. doi: 10.1016/0042-6989(71)90038-1. [DOI] [PubMed] [Google Scholar]
- 18.Martins-Ferreira H. Depressão alastrante na retina. An Acad Bras Cienc. 1962;34:44. [Google Scholar]
- 19.Martins-Ferreira H. Spreading depression in the chicken retina. In: Ookawa T, editor. The Brain and Behavior of the Fowl. 1. Tokyo: Japan Scientific Societies Press; 1983. pp. 317–333. [Google Scholar]
- 20.Maranhão-Filho PA, Martins-Ferreira H, Vincent MB, Ribeiro LJ, Novis SA. Sumatriptan blocks spreading depression in isolated chick retina. Cephalalgia. 1997;17:822–825. doi: 10.1046/j.1468-2982.1997.1708822.x. [DOI] [PubMed] [Google Scholar]
- 21.Martins-Ferreira H. Propagation of spreading depression in isolated chick retina. In: Lehmenkühler A, Grotemaier KH, Tegtmeier F, editors. Migraine: Basic Mechanisms and Treatment. Monchen: Wien, Baltimore: Urban and Schwarzenberg; 1993. pp. 533–546. [Google Scholar]
- 22.do Carmo RJ, Martins-Ferreira H. Spreading depression of Leao probed with ion-selective micro-electrodes in isolated chick retina. An Acad Bras Cienc. 1984;56:401–421. [PubMed] [Google Scholar]
- 23.Streit DS, Ferreira Filho CR, Martins-Ferreira H. Spreading depression in isolated spinal cord. J Neurophysiol. 1995;74:888–890. doi: 10.1152/jn.1995.74.2.888. [DOI] [PubMed] [Google Scholar]
- 24.Leão AAP, Morison RS. Propagation of spreading cortical depression. J Neurophysiol. 1945;8:33–45. [Google Scholar]
- 25.Leão AAP. Pial circulation and spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:391–396. [Google Scholar]
- 26.Edvinsson L, Uddman R. Neurobiology in primary headaches. Brain Res Brain Res Rev. 2005;48:438–456. doi: 10.1016/j.brainresrev.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 27.Hadjikhani N, Sanchez Del Rio M, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687–4692. doi: 10.1073/pnas.071582498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moskowitz MA, Nozaki K, Kraig RP. Neocortical spreading depression provokes the expression of c-fos protein-like immunoreactivity within trigeminal nucleus caudalis via trigeminovascular mechanisms. J Neurosci. 1993;13:1167–1177. doi: 10.1523/JNEUROSCI.13-03-01167.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moskowitz MA. The neurobiology of vascular head pain. Ann Neurol. 1984;16:157–168. doi: 10.1002/ana.410160202. [DOI] [PubMed] [Google Scholar]
- 30.Buzzi MG, Moskowitz MA. The antimigraine drug, sumatriptan (GR43175), selectively blocks neurogenic plasma extravasation from blood vessels in dura mater. Br J Pharmacol. 1990;99:202–209. doi: 10.1111/j.1476-5381.1990.tb14679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buzzi MG, Moskowitz MA. Evidence for 5-HT1B/1D receptors mediating the antimigraine effect of sumatriptan and dihydroergotamine. Cephalalgia. 1991;11:165–168. doi: 10.1046/j.1468-2982.1991.1104165.x. [DOI] [PubMed] [Google Scholar]
- 32.Jansen I, Edvinsson L, Mortensen A, Olesen J. Sumatriptan is a potent vasoconstrictor of human dural arteries via a 5-HT1-like receptor. Cephalalgia. 1992;12:202–205. doi: 10.1046/j.1468-2982.1992.1204202.x. [DOI] [PubMed] [Google Scholar]
- 33.Barlow JS. The Cerebellum and Adaptative Control. New York: Cambridge University Press; 2002. [Google Scholar]
- 34.Schmahmann JD. Disorders of the cerebellum: Ataxia, dysmetria of thought, and the cerebellar cognitive affective syndrome. J Neuropsychiatry Clin Neurosci. 2004;16:367–378. doi: 10.1176/jnp.16.3.367. [DOI] [PubMed] [Google Scholar]
- 35.Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998;21:375–382. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- 36.Ishizaki K, Mori N, Takeshima T, et al. Static stabilometry in patients with migraine and tension-type headache during a headache-free period. Psychiatry Clin Neurosci. 2002;56:85–90. doi: 10.1046/j.1440-1819.2002.00933.x. [DOI] [PubMed] [Google Scholar]
- 37.Cho AA, Clark JB, Rupert AH. Visually triggered migraine headaches affect spatial orientation and balance in a helicopter pilot. Aviat Space Environ Med. 1995;66:353–358. [PubMed] [Google Scholar]
- 38.Harno H, Hirvonen T, Kaunisto MA, et al. Subclinical vestibulocerebellar dysfunction in migraine with and without aura. Neurology. 2003;61:1748–1752. doi: 10.1212/01.wnl.0000098882.82690.65. [DOI] [PubMed] [Google Scholar]
- 39.Sandor PS, Mascia A, Seidel L, de Pasqua V, Schoenen J. Subclinical cerebellar impairment in the common types of migraine: A three-dimensional analysis of reaching movements. Ann Neurol. 2001;49:668–672. [PubMed] [Google Scholar]
- 40.Baloh RW. Neurotology of migraine. Headache. 1997;37:615–621. doi: 10.1046/j.1526-4610.1997.3710615.x. [DOI] [PubMed] [Google Scholar]
- 41.Cutrer FM, Baloh RW. Migraine-associated dizziness. Headache. 1992;32:300–304. doi: 10.1111/j.1526-4610.1992.hed3206300.x. [DOI] [PubMed] [Google Scholar]
- 42.Toglia JU, Thomas D, Kuritzky A. Common migraine and vestibular function. Electronystagmographic study and pathogenesis. Ann Otol Rhinol Laryngol. 1981;90(3pt1):267–271. doi: 10.1177/000348948109000315. [DOI] [PubMed] [Google Scholar]
- 43.Kayan A, Hood JD. Neuro-otological manifestations of migraine. Brain. 1984;107(pt4):1123–1142. doi: 10.1093/brain/107.4.1123. [DOI] [PubMed] [Google Scholar]
- 44.Guerraz M, Yardley L, Bertholon P, et al. Visual vertigo: Symptom assessment, spatial orientation and postural control. Brain. 2001;124(pt8):1646–1656. doi: 10.1093/brain/124.8.1646. [DOI] [PubMed] [Google Scholar]
- 45.McKendrick AM, Vingrys AJ, Badcock DR, Heywood JT. Visual dysfunction between migraine events. Invest Ophthalmol Vis Sci. 2001;42:626–633. [PubMed] [Google Scholar]
- 46.McKendrick AM, Badcock DR. Motion processing deficits in migraine. Cephalalgia. 2004;24:363–372. doi: 10.1111/j.1468-2982.2004.00679.x. [DOI] [PubMed] [Google Scholar]
- 47.Yenice O, Onal S, Incili B, Temel A, Afsar N, Tanrida Gcaron T. Assessment of spatial-contrast function and short-wavelength sensitivity deficits in patients with migraine. Eye. 2006 doi: 10.1038/sj.eye.6702251. [DOI] [PubMed] [Google Scholar]
- 48.Polak O, Grof P. Kleinhirnmigräne bei basilarer impression. Psychiatr Neurol (Basel) 1966;152:246–257. [PubMed] [Google Scholar]
- 49.Heidrich R. Cerebellar migraine. Psychiatr Neurol Med Psychol (Leipz) 1961;13:42–46. [PubMed] [Google Scholar]
- 50.Chrást B. Migraena cerebellaris. Bratisl Lékarskék Listy. 1954;9:271–276. [PubMed] [Google Scholar]
- 51.Bickerstaff ER. Basilar artery migraine. Lancet. 1961;277:15–17. [Google Scholar]
- 52.Golden GS, French JH. Basilar artery migraine in young children. Pediatrics. 1975;56:722–726. [PubMed] [Google Scholar]
- 53.Kirchmann M, Thomsen LL, Olesen J. Basilar-type migraine. Clinical, epidemiologic and genetic features. Neurology. 2006;66:880–886. doi: 10.1212/01.wnl.0000203647.48422.dd. [DOI] [PubMed] [Google Scholar]
- 54.Bernsen HJ, Van de Vlasakker C, Verhagen WI, Prick MJ. Basilar artery migraine stroke. Headache. 1990;30:142–144. doi: 10.1111/j.1526-4610.1990.hed3003142.x. [DOI] [PubMed] [Google Scholar]
- 55.Sabharwal RK, Mehndiratta MM, Gupta M, Anjaneyulu A, Malhotra LK, Khwaja G. Cerebellar and thalamic infarctions in basilar artery migraine. J Assoc Physicians India. 1990;38:237–238. [PubMed] [Google Scholar]
- 56.Ganji S, Williams W, Furlow J. Bilateral occipital lobe infarction in acute migraine: Clinical, neurophysiological, and neuroradiological study. Headache. 1992;32:360–365. doi: 10.1111/j.1526-4610.1992.hed3207360.x. [DOI] [PubMed] [Google Scholar]
- 57.Seto H, Shimizu M, Futatsuya R, et al. Basilar artery migraine. Reversible ischemia demonstrated by Tc-99m HMPAO brain SPECT. Clin Nucl Med. 1994;19:215–218. [PubMed] [Google Scholar]
- 58.Muellbacher W, Mamoli B. Prolonged impaired consciousness in basilar artery migraine. Headache. 1994;34:282–285. doi: 10.1111/j.1526-4610.1994.hed3405282.x. [DOI] [PubMed] [Google Scholar]
- 59.Ambrosini A, D’Onofrio M, Grieco GS, et al. Familial basilar migraine associated with a new mutation in the ATP1A 2 gene. Neurology. 2005;65:1826–1828. doi: 10.1212/01.wnl.0000187072.71931.c0. [DOI] [PubMed] [Google Scholar]
- 60.Kors EE, Haan J, Giffin NJ, et al. Expanding the phenotypic spectrum of the CACNA1A gene T666M mutation: A description of 5 families with familial hemiplegic migraine. Arch Neurol. 2003;60:684–688. doi: 10.1001/archneur.60.5.684. [DOI] [PubMed] [Google Scholar]
- 61.Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 62.Ducros A, Joutel A, Vahedi K, et al. Mapping of a second locus for familial hemiplegic migraine to 1q21-q23 and evidence of further heterogeneity. Ann Neurol. 1997;42:885–890. doi: 10.1002/ana.410420610. [DOI] [PubMed] [Google Scholar]
- 63.Vanmolkot KR, Kors EE, Hottenga JJ, et al. Novel mutations in the Na+, K+−ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol. 2003;54:360–366. doi: 10.1002/ana.10674. [DOI] [PubMed] [Google Scholar]
- 64.Dichgans M, Freilinger T, Eckstein G, et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371–377. doi: 10.1016/S0140-6736(05)66786-4. [DOI] [PubMed] [Google Scholar]
- 65.Spadaro M, Ursu S, Lehmann-Horn F, et al. A G301R Na+/K+ -ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics. 2004;5:177–185. doi: 10.1007/s10048-004-0183-2. [DOI] [PubMed] [Google Scholar]
- 66.Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, Adams ME. P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature. 1992;355:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- 67.Mori Y, Friedrich T, Kim MS, et al. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- 68.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 69.Pineda JC, Waters RS, Foehring RC. Specificity in the interaction of HVA Ca2+ channel types with Ca2+-dependent AHPs and firing behavior in neocortical pyramidal neurons. J Neurophysiol. 1998;79:2522–2534. doi: 10.1152/jn.1998.79.5.2522. [DOI] [PubMed] [Google Scholar]
- 70.Hans M, Luvisetto S, Williams ME, et al. Functional consequences of mutations in the human alpha1A calcium channel subunit linked to familial hemiplegic migraine. J Neurosci. 1999;19:1610–1619. doi: 10.1523/JNEUROSCI.19-05-01610.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van den Maagdenberg AM, Pietrobon D, Pizzorusso T, et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41:701–710. doi: 10.1016/s0896-6273(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 72.Ducros A, Denier C, Joutel A, et al. Recurrence of the T666M calcium channel CACNA1A gene mutation in familial hemiplegic migraine with progressive cerebellar ataxia. Am J Hum Genet. 1999;64:89–98. doi: 10.1086/302192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vahedi K, Denier C, Ducros A, et al. CACNA1A gene de novo mutation causing hemiplegic migraine, coma, and cerebellar atrophy. Neurology. 2000;55:1040–1042. doi: 10.1212/wnl.55.7.1040. [DOI] [PubMed] [Google Scholar]
- 74.Battistini S, Stenirri S, Piatti M, et al. A new CACNA1A gene mutation in acetazolamide-responsive familial hemiplegic migraine and ataxia. Neurology. 1999;53:38–43. doi: 10.1212/wnl.53.1.38. [DOI] [PubMed] [Google Scholar]
- 75.Margolis RL. The spinocerebellar ataxias: Order emerges from chaos. Curr Neurol Neurosci Rep. 2002;2:447–456. doi: 10.1007/s11910-002-0072-8. [DOI] [PubMed] [Google Scholar]
- 76.Hawkes CH. Familial paroxysmal ataxia: Report of a family. J Neurol Neurosurg Psychiatry. 1992;55:212–213. doi: 10.1136/jnnp.55.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harada H, Tamaoka A, Watanabe M, Ishikawa K, Shoji S. Downbeat nystagmus in two siblings with spinocerebellar ataxia type 6 (SCA 6) J Neurol Sci. 1998;160:161–163. doi: 10.1016/s0022-510x(98)00250-0. [DOI] [PubMed] [Google Scholar]
- 78.Friend KL, Crimmins D, Phan TG, et al. Detection of a novel missense mutation and second recurrent mutation in the CACNA1A gene in individuals with EA-2 and FHM. Hum Genet. 1999;105:261–265. doi: 10.1007/s004390051099. [DOI] [PubMed] [Google Scholar]
- 79.Jen J, Kim GW, Baloh RW. Clinical spectrum of episodic ataxia type 2. Neurology. 2004;62:17–22. doi: 10.1212/01.wnl.0000101675.61074.50. [DOI] [PubMed] [Google Scholar]
- 80.Denier C, Ducros A, Durr A, Eymard B, Chassande B, Tournier-Lasserve E. Missense CACNA1A mutation causing episodic ataxia type 2. Arch Neurol. 2001;58:292–295. doi: 10.1001/archneur.58.2.292. [DOI] [PubMed] [Google Scholar]
- 81.Guida S, Trettel F, Pagnutti S, et al. Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am J Hum Genet. 2001;68:759–764. doi: 10.1086/318804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kvistad PH, Dahl A, Skre H. Autosomal recessive non-progressive ataxia with an early childhood debut. Acta Neurol Scand. 1985;71:295–302. doi: 10.1111/j.1600-0404.1985.tb03203.x. [DOI] [PubMed] [Google Scholar]
- 83.Tranebjaerg L, Teslovich TM, Jones M, et al. Genome-wide homozygosity mapping localizes a gene for autosomal recessive non-progressive infantile ataxia to 20q11-q13. Hum Genet. 2003;113:293–295. doi: 10.1007/s00439-003-0967-8. [DOI] [PubMed] [Google Scholar]
- 84.Steckley JL, Ebers GC, Cader MZ, McLachlan RS. An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology. 2001;57:1499–1502. doi: 10.1212/wnl.57.8.1499. [DOI] [PubMed] [Google Scholar]
- 85.Mantuano E, Veneziano L, Jodice C, Frontali M. Spinocerebellar ataxia type 6 and episodic ataxia type 2: Differences and similarities between two allelic disorders. Cytogenet Genome Res. 2003;100:147–153. doi: 10.1159/000072849. [DOI] [PubMed] [Google Scholar]
- 86.Frontali M. Spinocerebellar ataxia type 6: Channelopathy or glutamine repeat disorder? Brain Res Bull. 2001;56:227–231. doi: 10.1016/s0361-9230(01)00574-3. [DOI] [PubMed] [Google Scholar]
- 87.Jen J. Familial episodic ataxias and related ion channel disorders. Curr Treat Options Neurol. 2000;2:429–431. doi: 10.1007/s11940-000-0041-y. [DOI] [PubMed] [Google Scholar]
- 88.Jodice C, Mantuano E, Veneziano L, et al. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum Mol Genet. 1997;6:1973–1978. doi: 10.1093/hmg/6.11.1973. [DOI] [PubMed] [Google Scholar]
- 89.Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65:529–534. doi: 10.1212/01.wnl.0000172638.58172.5a. [DOI] [PubMed] [Google Scholar]
- 90.Zhuchenko O, Bailey J, Bonnen P, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 91.Alonso I, Barros J, Tuna A, et al. Phenotypes of spinocerebellar ataxia type 6 and familial hemiplegic migraine caused by a unique CACNA1A missense mutation in patients from a large family. Arch Neurol. 2003;60:610–614. doi: 10.1001/archneur.60.4.610. [DOI] [PubMed] [Google Scholar]
- 92.Alonso I, Barros J, Tuna A, et al. A novel R1347Q mutation in the predicted voltage sensor segment of the P/Q-type calcium-channel alpha-subunit in a family with progressive cerebellar ataxia and hemiplegic migraine. Clin Genet. 2004;65:70–72. doi: 10.1111/j..2004.00187.x. [DOI] [PubMed] [Google Scholar]
- 93.Richter S, Dimitrova A, Maschke M, et al. Degree of cerebellar ataxia correlates with three-dimensional MRI-based cerebellar volume in pure cerebellar degeneration. Eur Neurol. 2005;54:23–27. doi: 10.1159/000087241. [DOI] [PubMed] [Google Scholar]
- 94.Ducros A, Denier C, Joutel A, et al. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med. 2001;345:17–24. doi: 10.1056/NEJM200107053450103. [DOI] [PubMed] [Google Scholar]
- 95.Elliott MA, Peroutka SJ, Welch S, May EF. Familial hemiplegic migraine, nystagmus, and cerebellar atrophy. Ann Neurol. 1996;39:100–106. doi: 10.1002/ana.410390115. [DOI] [PubMed] [Google Scholar]
- 96.Kors EE, Melberg A, Vanmolkot KR, et al. Childhood epilepsy, familial hemiplegic migraine, cerebellar ataxia, and a new CACNA1A mutation. Neurology. 2004;63:1136–1137. doi: 10.1212/01.wnl.0000138571.48593.fc. [DOI] [PubMed] [Google Scholar]
- 97.Jen J, Yue Q, Nelson SF, et al. A novel nonsense mutation in CACNA1A causes episodic ataxia and hemiplegia. Neurology. 1999;53:34–37. doi: 10.1212/wnl.53.1.34. [DOI] [PubMed] [Google Scholar]
- 98.Nam SC, Hockberger PE. Analysis of spontaneous electrical activity in cerebellar Purkinje cells acutely isolated from postnatal rats. J Neurobiol. 1997;33:18–32. doi: 10.1002/(sici)1097-4695(199707)33:1<18::aid-neu3>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 99.Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- 100.Kors EE, Terwindt GM, Vermeulen FL, et al. Delayed cerebral edema and fatal coma after minor head trauma: Role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol. 2001;49:753–760. doi: 10.1002/ana.1031. [DOI] [PubMed] [Google Scholar]
- 101.Fitzsimons RB, Wolfenden WH. Migraine coma. Meningitic migraine with cerebral oedema associated with a new form of autosomal dominant cerebellar ataxia. Brain. 1985;108(pt3):555–577. doi: 10.1093/brain/108.3.555-a. [DOI] [PubMed] [Google Scholar]
- 102.Munte TF, Muller-Vahl H. Familial migraine coma: A case study. J Neurol. 1990;237:59–61. doi: 10.1007/BF00319672. [DOI] [PubMed] [Google Scholar]
- 103.Wada T, Kobayashi N, Takahashi Y, Aoki T, Watanabe T, Saitoh S. Wide clinical variability in a family with a CACNA1A T666m mutation: Hemiplegic migraine, coma, and progressive ataxia. Pediatr Neurol. 2002;26:47–50. doi: 10.1016/s0887-8994(01)00371-x. [DOI] [PubMed] [Google Scholar]
- 104.Tan BB. Migraine versus glaucoma–A diagnostic dilemma. Ann Acad Med Singapore. 1990;19:856–858. [PubMed] [Google Scholar]
- 105.Corbin D, Martyr T, Graham AC. Migraine coma. J Neurol Neurosurg Psychiatry. 1991;54:744. doi: 10.1136/jnnp.54.8.744-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Echenne B, Ducros A, Rivier F, et al. Recurrent episodes of coma: An unusual phenotype of familial hemiplegic migraine with linkage to chromosome 1. Neuropediatrics. 1999;30:214–217. doi: 10.1055/s-2007-973493. [DOI] [PubMed] [Google Scholar]
- 107.Sareen D. Interesting case of migraine presenting with recurrent episodes of migraine coma. J Assoc Physicians India. 2000;48:1031. [PubMed] [Google Scholar]
- 108.Tottene A, Pivotto F, Fellin T, Cesetti T, van den Maagdenberg AM, Pietrobon D. Specific kinetic alterations of human CaV2. 1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J Biol Chem. 2005;280:17678–17686. doi: 10.1074/jbc.M501110200. [DOI] [PubMed] [Google Scholar]
- 109.Pietrobon D. Migraine: New molecular mechanisms. Neuroscientist. 2005;11:373–386. doi: 10.1177/1073858405275554. [DOI] [PubMed] [Google Scholar]
- 110.Gursoy-Ozdemir Y, Qiu J, Matsvaka N, et al. Cortical spreading depression activates and upregulates MMP-9. J Clin Invest. 2004;113:1447–1455. doi: 10.1172/JCI21227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Olesen J, Jorgensen MB. Leao’s spreading depression in the hippocampus explains transient global amnesia: A hypothesis. Acta Neurol Scand. 1986;73:219–220. doi: 10.1111/j.1600-0404.1986.tb03267.x. [DOI] [PubMed] [Google Scholar]
- 112.Haas DC, Ross GS. Transient global amnesia triggered by mild head trauma. Brain. 1986;109(pt2):251–257. doi: 10.1093/brain/109.2.251. [DOI] [PubMed] [Google Scholar]
- 113.Bain PG, O’Brien MD, Keevil SF, Porter DA. Familial periodic cerebellar ataxia: A problem of cerebellar intracellular pH homeostasis. Ann Neurol. 1992;31:147–154. doi: 10.1002/ana.410310205. [DOI] [PubMed] [Google Scholar]
- 114.Griggs RC, Moxley RT, 3rd, Lafrance RA, Mc-Quillen J. Hereditary paroxysmal ataxia: Response to acetazolamide. Neurology. 1978;28:1259–1264. doi: 10.1212/wnl.28.12.1259. [DOI] [PubMed] [Google Scholar]
- 115.Jen JC, Yue Q, Karrim J, Nelson SF, Baloh RW. Spinocerebellar ataxia type 6 with positional vertigo and acetazolamide responsive episodic ataxia. J Neurol Neurosurg Psychiatry. 1998;65:565–568. doi: 10.1136/jnnp.65.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Athwal BS, Lennox GG. Acetazolamide responsiveness in familial hemiplegic migraine. Ann Neurol. 1996;40:820–821. doi: 10.1002/ana.410400526. [DOI] [PubMed] [Google Scholar]
- 117.de Simone R, Marano E, Di Stasio E, Bonuso S, Fiorillo C, Bonavita V. Acetazolamide efficacy and tolerability in migraine with aura: A pilot study. Headache. 2005;45:385–386. doi: 10.1111/j.1526-4610.2005.05077_3.x. [DOI] [PubMed] [Google Scholar]
- 118.Vahedi K, Taupin P, Djomby R, et al. Efficacy and tolerability of acetazolamide in migraine prophylaxis: A randomised placebo-controlled trial. J Neurol. 2002;249:206–211. doi: 10.1007/pl00007866. [DOI] [PubMed] [Google Scholar]
- 119.Haan J, Sluis P, Sluis LH, Ferrari MD. Acetazolamide treatment for migraine aura status. Neurology. 2000;55:1588–1589. doi: 10.1212/wnl.55.10.1588. [DOI] [PubMed] [Google Scholar]
- 120.Shank RP, Doose DR, Streeter AJ, Bialer M. Plasma and whole blood pharmacokinetics of top-iramate: The role of carbonic anhydrase. Epilepsy Res. 2005;63:103–112. doi: 10.1016/j.eplepsyres.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 121.Ayata C, Jin H, Kudo C, Dalkara T, Moskowitz MA. Suppression of cortical spreading depression in migraine prophylaxis. Ann Neurol. 2006;59:652–661. doi: 10.1002/ana.20778. [DOI] [PubMed] [Google Scholar]
- 122.Spacey SD, Hildebrand ME, Materek LA, Bird TD, Snutch TP. Functional implications of a novel EA2 mutation in the P/Q-type calcium channel. Ann Neurol. 2004;56:213–220. doi: 10.1002/ana.20169. [DOI] [PubMed] [Google Scholar]
- 123.Jayamaha JE, Street MK. Fatal cerebellar infarction in a migraine sufferer whilst receiving sumatriptan. Intensive Care Med. 1995;21:82–83. doi: 10.1007/BF02425161. [DOI] [PubMed] [Google Scholar]
- 124.Crawford JS, Konkol RJ. Familial hemiplegic migraine with crossed cerebellar diaschisis and unilateral meningeal enhancement. Headache. 1997;37:590–593. doi: 10.1046/j.1526-4610.1997.3709590.x. [DOI] [PubMed] [Google Scholar]
- 125.Lee TG, Solomon GD, Kunkel RS, Raja S. Reversible cerebellar perfusion in familial hemiplegic migraine. Lancet. 1996;348:1383. doi: 10.1016/S0140-6736(05)65447-5. [DOI] [PubMed] [Google Scholar]
- 126.Harbaugh RE, Saunders RL, Reeves AG. Pediatric cerebellar infarction: Case report and review of the literature. Neurosurgery. 1982;10:593–596. doi: 10.1227/00006123-198205000-00008. [DOI] [PubMed] [Google Scholar]
- 127.Kruit MC, van Buchem MA, Hofman PA, Bakkers JT, Terwindt GM, Ferrari MD, et al. Migraine as a risk factor for subclinical brain lesions. JAMA. 2004;291:427–434. doi: 10.1001/jama.291.4.427. [DOI] [PubMed] [Google Scholar]
- 128.Kruit MC, Launer LJ, Ferrari MD, van Buchem MA. Infarcts in the posterior circulation territory in migraine. The population-based MRI CAMERA study. Brain. 2005;128:2068–2077. doi: 10.1093/brain/awh542. [DOI] [PubMed] [Google Scholar]
- 129.Duvernoy H, Delon S, Vannson JL. The vascularization of the human cerebellar cortex. Brain Res Bull. 1983;11:419–480. doi: 10.1016/0361-9230(83)90116-8. [DOI] [PubMed] [Google Scholar]
- 130.Guerini D, Coletto L, Carafoli E. Exporting calcium from cells. Cell Calcium. 2005;38:281–289. doi: 10.1016/j.ceca.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 131.Chen G, Hanson CL, Dunbar RL, Ebner TJ. Novel form of spreading acidification and depression in the cerebellar cortex demonstrated by neutral red optical imaging. J Neurophysiol. 1999;81:1992–1998. doi: 10.1152/jn.1999.81.4.1992. [DOI] [PubMed] [Google Scholar]
- 132.Ebner TJ, Chen G. Spreading acidification and depression in the cerebellar cortex. Neuroscientist. 2003;9:37–45. doi: 10.1177/1073858402239589. [DOI] [PubMed] [Google Scholar]
- 133.Comu S, Giuliani M, Narayanan V. Episodic ataxia and myokymia syndrome: A new mutation of potassium channel gene Kv1.1. Ann Neurol. 1996;40:684–687. doi: 10.1002/ana.410400422. [DOI] [PubMed] [Google Scholar]
- 134.Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: Long-range glial signaling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- 135.Charles A. Intercellular calcium waves in glia. Glia. 1998;24:39–49. doi: 10.1002/(sici)1098-1136(199809)24:1<39::aid-glia5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 136.Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254–9262. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- 138.Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- 139.Newman EA, Zahs KR. Calcium waves in retinal glial cells. Science. 1997;275:844–847. doi: 10.1126/science.275.5301.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Guthrie PB, Knappenberger J, Segal M, Bennett MV, Charles AC, Kater SB. ATP released from astrocytes mediates glial calcium waves. J Neurosci. 1999;19:520–528. doi: 10.1523/JNEUROSCI.19-02-00520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kunkler PE, Kraig RP. Calcium waves precede electrophysiological changes of spreading depression in hippocampal organ cultures. J Neurosci. 1998;18:3416–3425. doi: 10.1523/JNEUROSCI.18-09-03416.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Basarsky TA, Duffy SN, Andrew RD, MacVicar BA. Imaging spreading depression and associated intracellular calcium waves in brain slices. J Neurosci. 1998;18:7189–7199. doi: 10.1523/JNEUROSCI.18-18-07189.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pietrobon D. Calcium channels and channelopathies of the central nervous system. Mol Neurobiol. 2002;25:31–50. doi: 10.1385/MN:25:1:031. [DOI] [PubMed] [Google Scholar]
- 144.del Zoppo GJ, Hallenbeck JM. Advances in the vascular pathophysiology of ischemic stroke. Thromb Res. 2000;98:73–81. doi: 10.1016/s0049-3848(00)00218-8. [DOI] [PubMed] [Google Scholar]
- 145.Sappey-Marinier D, Vighetto A, Peyron R, Broussolle E, Bonmartin A. Phosphorus and proton magnetic resonance spectroscopy in episodic ataxia type 2. Ann Neurol. 1999;46:256–259. doi: 10.1002/1531-8249(199908)46:2<256::aid-ana17>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 146.Sasaki H, Kojima H, Yabe I, et al. Neuropathological and molecular studies of spinocerebellar ataxia type 6 (SCA6) Acta Neuropathol (Berl) 1998;95:199–204. doi: 10.1007/s004010050787. [DOI] [PubMed] [Google Scholar]
- 147.Takahashi T, Arai N, Shimamura M, et al. Autopsy case of acute encephalopathy linked to familial hemiplegic migraine with cerebellar atrophy and mental retardation. Neuropathology. 2005;25:228–234. doi: 10.1111/j.1440-1789.2005.00604.x. [DOI] [PubMed] [Google Scholar]
- 148.Ruskell GL, Simons T. Trigeminal nerve pathways to the cerebral arteries in monkeys. J Anat. 1987;155:23–37. [PMC free article] [PubMed] [Google Scholar]
- 149.Saito K, Moskowitz MA. Contributions from the upper cervical dorsal roots and trigeminal ganglia to the feline circle of Willis. Stroke. 1989;20:524–526. doi: 10.1161/01.str.20.4.524. [DOI] [PubMed] [Google Scholar]
- 150.Baloh RW, Yue Q, Furman JM, Nelson SF. Familial episodic ataxia: Clinical heterogeneity in four families linked to chromosome 19p. Ann Neurol. 1997;41:8–16. doi: 10.1002/ana.410410105. [DOI] [PubMed] [Google Scholar]
- 151.Ohta M, Araki S, Kuroiwa Y. Familial occurrence of migraine with a hemiplegic syndrome and cerebellar manifestations. Neurology. 1967;17(8pt1):813–817. doi: 10.1212/wnl.17.8.813. [DOI] [PubMed] [Google Scholar]
- 152.Young GF, Leon-Barth CA, Green J. Familial hemiplegic migraine, retinal degeneration, deafness, and nystagmus. Arch Neurol. 1970;23:201–209. doi: 10.1001/archneur.1970.00480270011002. [DOI] [PubMed] [Google Scholar]
- 153.Codina A, Acarin PN, Miquel F, Noguera M. Migraine Hémiplégique associée à un nystagmus. Rev Neurol (Paris) 1971;124:526–530. [PubMed] [Google Scholar]
- 154.Zifkin B, Andermann E, Andermann F, Kirkham T. An autosomal dominant syndrome of hemiplegic migraine, nystagmus, and tremor. Ann Neurol. 1980;8:329–332. doi: 10.1002/ana.410080319. [DOI] [PubMed] [Google Scholar]