

Abstract
Cytochrome c oxidase (CcO), the terminal enzyme in the mitochondrial respiratory chain, catalyzes the four-electron reduction of dioxygen to water in a binuclear center comprised of a high-spin heme (heme a3) and a copper atom (CuB) coordinated by three histidine residues. As a minimum model for CcO, a mutant of sperm whale myoglobin, named CuBMb, has been engineered, in which a copper atom is held in the distal heme pocket by the native E7 histidine and two nonnative histidine residues. In this work, the role of the copper in regulating ligand binding in CuBMb was investigated. Resonance Raman studies show that the presence of copper in CO-bound CuBMb leads to a CcO-like distal heme pocket. Stopped-flow data show that, upon the initiation of the CO binding reaction, the ligand first binds to the Cu+; it subsequently transfers from Cu+ to Fe2+ in an intramolecular process, similar to that reported for CcO. The high CO affinity toward Cu+ and the slow intramolecular CO transfer rate between Cu+ and Fe2+ in the CuBMb/Cu+ complex are analogous to those in Thermus thermophilus CcO (TtCcO) but distinct from those in bovine CcO (bCcO). Additional kinetic studies show that, upon photolysis of the NO-bound CuBMb/Cu+ complex, the photolyzed ligand transiently binds to Cu+ and subsequently rebinds to Fe2+, accounting for the 100% geminate recombination yield, similar to that found in TtCcO. The data demonstrate that the CuBMb/Cu+ complex reproduces essential structural and kinetic features of CcO and that the complex is more akin to TtCcO than to bCcO.
Cytochrome c oxidase (CcO) is the terminal enzyme in the mitochondrial respiratory chain. It translocates protons across the inner membrane of mitochondria by utilizing the chemical energy harnessed from the four-electron reduction reaction of dioxygen to water.1–3 The oxygen reduction chemistry takes place in a binuclear center, comprised of a high-spin heme group (heme a3) and a CuB center ~5 Å from the heme iron atom (Figure 1).4,5 The presence of the CuB in the binuclear center introduces an intriguing twist to the ligand binding properties of the heme iron as compared to other hemeproteins, due to the high affinity of CuB toward diatomic heme iron ligands. As such, it is well-established that ligand binding in CcO can be described as a two-step reaction:
| (1) |
Figure 1.
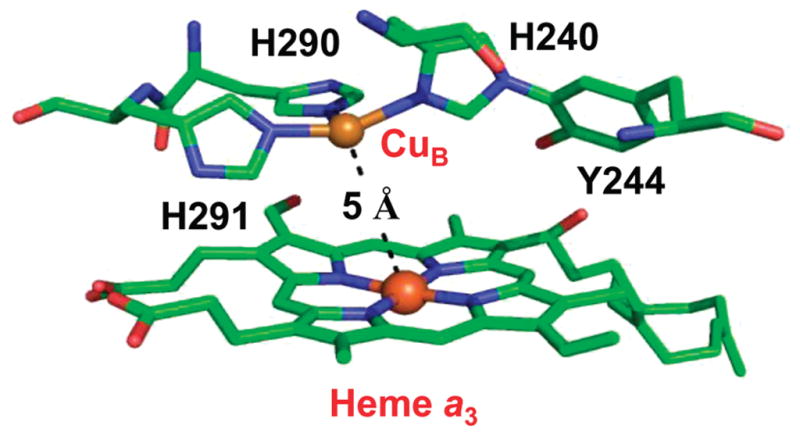
Structure of the binuclear center of bovine cytochrome c oxidase. The Fe–CuB distance is 5 Å. CuB is coordinated by H291, H290, and H240. H240 forms a covalent bond with Y244 due to posttranslational modification. The PDB code is 2OCC.
Here, L represents a diatomic heme ligand, CO, NO, or O2. In the first step of the reaction, L migrates into the heme pocket and binds to CuB+ in a bimolecular binding process. In the subsequent step of the reaction, L dissociates from CuB+ and transfers to Fe2+ in an intramolecular process.
Intriguingly, the relative affinity of the heme ligands toward Fe2+ and CuB+ was found to vary significantly among the various members of the heme-copper oxidase family,6,7 despite the fact that they possess similar catalytic sites. In particular, the CO affinity of ba3 oxidases (such as TtCcO from Thermus thermophilus) toward CuB is higher than that of aa3 oxidases (such as bCcO from bovine), owing to a very rapid on-rate and a 100-fold slower transfer rate from CuB to the heme iron, implying that ba3 oxidases could be an efficient trap for O2 under conditions of low oxygen tension.8,9 In addition to the distinct ligand binding properties, the optical change associated with the P → F transition during single turnover in aa3 oxidases is not observed in ba3 oxidases, indicating that there is no proton uptake at the binuclear center in ba3 during this transition, manifesting its unique proton translocation mechanism.10,11 Furthermore, TtCcO exhibits NO reductase activity, which is not present in aa3 oxidases, suggesting that in addition to binding to the heme iron, NO may bind to CuB via its oxygen atom in TtCcO, but not in aa3 oxidases.12 Taken together, it is clear that the ba3 center in TtCcO has very different ligand interactions as compared to the aa3 oxidases; however, the structural basis accounting for the differences among differing members of the oxidase family of enzymes has not been determined. In addition, despite its importance, the exact mechanism by which CuB modulates ligand binding and oxygen chemistry in CcO enzymes remains unclear.13,14
One useful approach to unravel functional mechanisms of CcO is through the studies of model porphyrin complexes mimicking the binuclear center of the enzyme.15 These model complexes, as summarized in several excellent reviews,16–19 typically include a minimum scaffold of the binuclear center, a heme, and a nearby copper atom chelated by three or four nitrogen ligands. In addition, in some of the models, one of the copper ligands, a histidine, was covalently bonded to a tyrosine, resembling the posttranslational modification of a histidine ligand of CuB in CcO.20–22 These models have been demonstrated to catalyze the reduction of O2 to water, and they offer valuable information regarding how the architecture of the binuclear center may affect the ligand binding properties and reactivity of the heme iron.23–26
In contrast to the wide variety of synthetic model complexes, there is only one protein-based model for CcO reported in the literature.27 In this sperm whale myoglobin (swMb)-based model, named CuBMb, the B10 Leu 29 and CD1 Phe residues are mutated to histidine; along with the native histidine at the E7 position, the three histidine residues constitute a copper binding site in the distal pocket of the heme, mimicking the binuclear center in CcO.27 Electron paramagnetic resonance data of the cyanide adduct of copper-bound CuBMb demonstrated that the Fe3+ and Cu2+ in the active site are antiferromagnetically coupled, as a result of a bridged heme Fe3+–CN−–Cu2+ structure, confirming the proximity of the two metal ions.27,28 Biochemical studies show that CuBMb binds copper tightly, with an equilibrium dissociation constant (Kd) of 9 μM.27 In addition, upon exposure to atmospheric oxygen, CuBMb forms a stable O2 adduct, albeit with a lower affinity as compared to the wild-type protein27,28 Addition of Cu+ to CuBMb facilitates O2 binding and reduction (by receiving an electron from Cu+ like that found in CcO); however, instead of producing the ferryl-heme intermediate as observed in the CcO reaction, it promotes the degradation of the prosthetic heme to verdoheme,28 presumably a result of the lack of protons required for heterolytic cleavage of the O-O bond.29 In addition to the oxygen reaction, CuBMb also exhibits NO reductase activity30 in the presence of Cu+, but not in its absence, with an efficiency similar to that of TtCcO,9 manifesting the important role of the copper ion in the catalytic properties of CuBMb.
In this work, we sought to use optical absorption and resonance Raman spectroscopy to systematically investigate how the presence of the copper ion in the active site of CuBMb modulates the equilibrium and dynamic ligand binding properties of the heme prosthetic group embedded in its protein matrix.
Experimental Procedures
Materials
CO/NO and the 13C18O/15N16O isotopes were obtained from Tech Air (White Plains, NY) and Icon Isotopes (Summit, NJ), respectively; all other chemicals were from Sigma-Aldrich Corp. (St. Louis, MO). All solutions were prepared with deionized water (Millipore). The CuBMb protein samples were prepared as described elsewhere.27,28 The protein concentration used for the equilibrium optical and Raman measurements was 2–40 μM; that for the stopped-flow and photolysis measurement was 2–10 μM. All the protein samples were prepared in 50 mM Tris buffer, pH 7.4. To produce the copper-bound CuBMb, 60–100 μM CuSO4 was added into the protein solution. To generate the CO− and NO− adducts, the protein samples, purged with argon gas and reduced with a minimal amount of sodium dithionite or ascorbate, were injected with CO or NO gas in gastight syringes.
Optical Absorption and Raman Measurements
The optical absorption spectra were taken on a UV2100 spectrophotometer from Shimadzu Scientific Instruments, Inc. (Columbia, MD) with a spectral slit width of 1 nm. To measure the resonance Raman spectra, the 413.1 nm excitation from a Kr ion laser (Spectra Physics, Mountain View, CA) was focused to a ~30 μm spot on the spinning quartz cell rotating at ~6000 rpm. The scattered light, collected at right angles to the incident laser beam, was focused on the 100 μm wide entrance slit of a 1.25 m Spex spectrometer equipped with a 1200 grooves/mm grating (Horiba Jobin Yvon, Edison, NJ), where it was dispersed and then detected by a liquid nitrogen cooled CCD detector (Princeton Instruments, Trenton, NJ). A holographic notch filter (Kaiser, Ann Arbor, MI) was used to remove the laser line. The Raman shift was calibrated by using indene and an acetone/ferricyanide mixture for the 200–1700 and 1700–2300 cm−1 spectral windows, respectively. The laser power was kept <25 μW for the CO complex to avoid photodissociation of the heme-bound CO. The spectral acquisition times for the νFe-CO and νC-O measurements were ~20 h. For other measurements, the laser power was kept <1 mW, and the spectral acquisition times were ~2 h.
Flash Photolysis Measurements
The CO association kinetics was measured at room temperature with a nanosecond laser flash photolysis system (LKS.60 from Applied Photophysics Ltd., Leatherhead, UK).31,32 In this system, the 532 nm output (~5 ns, 110 mJ) from a Nd:YAG laser (Brilliant B from Big Sky Laser Technologies, Inc.) was employed as the photolysis beam. The output from a 150 W xenon arc lamp, at right angles to the photolysis beam, was used as the probe beam. The probe beam passed through a monochromator prior to reaching the quartz cuvette (4 × 10 mm with a 10 mm optical path) containing the sample. The light transmitted through the sample entered a second monochromator, which was synchronized with the first monochromator, and was detected by a photomultiplier tube (PMT; model 1P28, Hamamatsu Corp.). The signal from the PMT was transferred to a digital oscilloscope (Infinium, Agilent Technologies) and then to a personal computer for subsequent analysis. Typically, five or six kinetic traces were averaged to obtain a satisfactory signal-to-noise ratio.
Stopped-Flow Measurements
The stopped-flow kinetic measurements were performed at 20 °C with a π*180 stopped-flow instrument from Applied Photophysics Ltd., equipped with a PMT as described elsewhere.33 To ensure absolute anaerobic conditions, prior to each experiment, the entire solution flow-path in the stopped-flow system was flushed with an anaerobic sodium dithionite solution several times with gastight syringes. At the last run, the anaerobic sodium dithionite solution was allowed to sit for 30 min to quench any residual oxygen. The dithionite solution was subsequently washed away with Ar-purged anaerobic buffer prior to sample loading. The ferrous deoxy derivative of CuBMb was prepared by reducing the ferric enzyme, prepurged with argon gas, with a stoichiometric amount of sodium dithionite in an anaerobic buffer, as guided by optical absorption measurements. All the deoxy samples were transferred with gastight syringes to the stopped-flow system. The data were analyzed using rapid-kinetics software developed and provided by Applied Photophysics.
Results and Discussion
The ferric form of CuBMb exhibits absorption maxima at 408, 500, and 628 nm, similar to those of the wild-type swMb. They are consistent with a mixture of six-coordinate high- and low-spin hemes with a water molecule as the distal ligand, as reported previously.27 Likewise, the reduced deoxy and CO-derivative of CuBMb display absorption maxima at 433/557 and 422/541/575 nm, respectively (Figure 2a), analogous to those of the wild-type swMb.34 The binding of Cu+ in the reduced deoxy CuBMb only slightly perturbs the spectrum, whereas that in the CO derivative introduces a pronounced shoulder on the high-wavelength side of the Soret band, indicating incomplete binding of CO.
Figure 2.
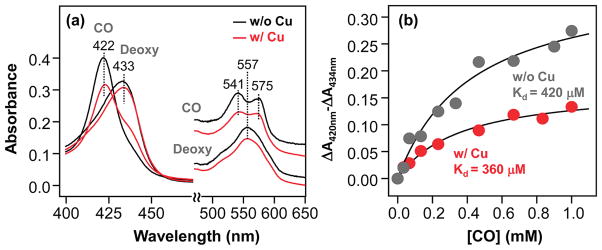
Optical absorption spectra of the deoxy and CO-bound derivatives of CuBMb (a) and the CO titration curves of CuBMb (b), in the presence and absence of 64 μM Cu+. The solid lines in (b) are the best-fit curves based on a single CO binding site in CuBMb. The Kd of CO was determined to be 420 and 360 μM for the Cu+-free and Cu+-bound CuBMb, respectively. In both cases, full saturation was not achieved even at a CO concentration of 1 mM. The protein concentration was 3.1 μM.
Equilibrium Structural Properties of the CO-Bound CuBMb
To examine how Cu+ binding affects the CO binding properties, we titrated the enzyme with CO in the presence and in the absence of copper. As shown in Figure 2b, in the absence of Cu+, the Kd for CO was found to be ~420 μM, which is ~10000-fold higher than that of the wild-type swMb (Kd ≈ 0.037 μM).35 In the presence of Cu+, the Kd slightly decreases to ~360 μM. As a result of the low affinity, in both copper-free and copper-bound CuBMb, the heme iron is not fully saturated with CO even at CO-saturated conditions (1 mM). It is noteworthy that although the CO affinity of the CuBMb/Cu+ complex is significantly lower than those of bCcO (Kd ≈ 0.3μM) and TtCcO (Kd ≈ 9 μM) (see Table 1),8 the structural and kinetic properties the CuBMb/Cu+ complex exhibit surprising resemblance to those of the CcO enzymes (vide infra).
Table 1.
CO Binding Constants of the CuBMb/Cu+ Complex As Compared to Those of the CcO from Thermus thermophilus (TtCcO) and Bovine (bCcO)
To examine possible interaction between the copper and heme-bound CO in the CuBMb/Cu+ complex, such as that found in CcO enzymes, resonance Raman studies were conducted in the presence and in the absence of Cu+. As shown in Figure 3a, in the copper-free derivative, the νFe-CO, δFe-C-O, and νC-O modes were identified at 520, 582, and 1947 cm−1, respectively, on the basis of 12C16O-13C18O isotope substitution experiments. Binding of Cu+ to CuBMb causes the shift of the νFe-CO and νC-O modes to 513 and 1967 cm−1, respectively, as well as the reduction in intensity of the δFe-C-O mode.
Figure 3.
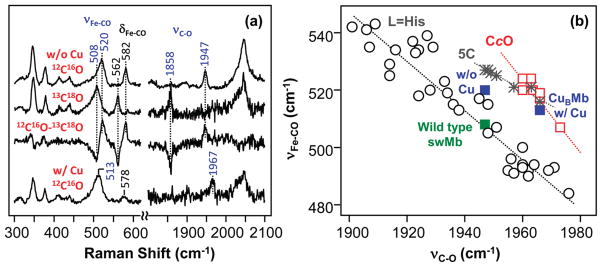
Resonance Raman spectra of CO-bound CuBMb in the presence and in the absence of 100 μM Cu+ (a) and the νFe-CO versus νC-O inverse correlation plot of the data (b). The top two spectra in (a) were obtained from Cu+-free CuBMb; the bottom spectrum was obtained from Cu+-bound CuBMb, in the presence of either 12C16O or 13C18O. The 12C16O - 13C18O difference spectrum was obtained from the Cu+-free sample. In the difference spectrum all the heme modes cancel out, while the remaining positive and negative bands are associated with 12C16O- and 13C18O-related modes, respectively. The data shown in the νFe-CO versus νC-O inverse correlation plot in (b) are taken either from this work or from refs 40, 52, and 53.
The νFe-CO and νC-O frequencies typically follow an inverse correlation, due to the resonance between the following two extreme structures of the L-Fe-CO moiety (where L is the proximal heme iron ligand):
| (2) |
In a positive polar distal environment, form I is destabilized, leading to higher νFe-CO and lower νC-O frequencies; consequently, the associated data point is located in the upper-left corner of the correlation plot (Figure 3b). As the positive polarity is reduced, the data moves along the correlation line toward the lower-right corner. The offset of a given line in the correlation plot is determined by the electronic properties of the proximal heme ligand (L). As a result, the data associated with heme proteins with histidine as a proximal ligand stay on a line (the L ) His line) distinct from the P450 line, with thiolate as a proximal ligand, and from the line associated with five-coordinate CO-bound heme complexes (the 5C line). Although the CcO family of enzymes possesses a heme group with histidine as the proximal ligand, their associated data points lie on a correlation line distinct from the histidine correlation line. The shift of the CcO data away from the typical histidine line has been attributed to a highly bent Fe-C-O moiety36 and/or a compressed Fe-CO bond37 due to the proximity of the Cu+.
In the absence of Cu+, the CuBMb data point (with νFe-CO/ νC-O at 520/1947 cm−1) lies within the body of points that are associated with the histidine correlation line, similar to the observation for the wild-type swMb. The slightly higher frequency of νFe-CO and lower frequency of νC-O in the CuBMb complex with respect to those of the wild-type swMb indicate that it possesses a distal heme pocket with a somewhat higher electrostatic potential-stabilizing structure, II in eq 2 with respect to structure I. Upon Cu+ binding, the νFe-CO/ νC-O data point shifts up to 513/1967 cm−1, which is at the intersection of the 5C and CcO lines. To determine if the CuBMb/Cu+ data point is associated with the 5C, with a broken or weakened Fe-His bond, or with the CcO line, we measured the Fe-His stretching mode (νFe-His) in the reduced deoxy form of the protein. Interestingly, the νFe-His mode of the CuBMb/Cu+ complex is detected at 220 cm−1 (Supporting Information, Figure S1), the same as that of the wild-type swMb, indicating that the proximal Fe-His bond remains intact and that the CuBMb/Cu+ data point is associated with the CcO line, instead of the 5C line. Consistent with this assignment, the high-frequency region of the CO-bound complex is characteristic of that of a six-coordinate CO-bound derivative (data not shown). The localization of the CuBMb/Cu+ data point on the CcO line supports the proposals that Cu+ is located in close proximity to the heme iron in the CuBMb/Cu+ complex and that the Fe-C-O moiety is forced to be bent and/or compressed by the nearby Cu+. The data confirm the validity of using CuBMb/Cu+ as a model for CcO; they also demonstrate that the distinct location of the CcO line with respect to the histidine line in the νFe-CO- νC-O inverse correlation plot is a result of a distal effect, instead of a proximal effect.
Kinetic Properties of CO-Bound CuBMb
To determine the impact of the copper on the ligand binding kinetics of CuBMb, laser flash photolysis experiments were carried out. When CO is photolyzed from the heme iron with a short laser pulse, it can either rapidly rebind to the iron (i.e. geminate recombination) or migrate away from the distal pocket into the protein matrix, where it can subsequently return to the distal pocket via a CO-concentration independent process (i.e. also geminate recombination), or escape into the bulk solution followed by a bimolecular CO rebinding process in a CO concentration dependent fashion (i.e. bimolecular recombination). The branching ratio of the processes depends on the dynamic properties of the protein. For most hemeproteins, CO recombination following photolysis is dominated by the bimolecular processes,35 especially in CcO.38
As shown in Figure 4a, in the absence of copper, the transient absorption difference spectrum of CuBMb obtained at 0.5 μs following the photolysis exhibits positive and negative peaks at 437 and 420 nm, respectively, in good agreement with the difference spectrum calculated from the equilibrium spectra shown in Figure 2a, confirming that the bleaching of the CO-bound species is concurrent with the production of the deoxy species. The CO-rebinding kinetics of CuBMb (Figure 4b) follows a single-exponential process, with a rate constant linearly dependent on the CO concentration (see the inset), indicating that the observed kinetics reflects the bimolecular rebinding process. The kon and koff determined from the slope and intercept of the linear fit of the data are 4.8 × 102 M−1 s−1 and 0.18 s−1, respectively, leading to a Kd value of 370 μM, which is consistent with that determined by equilibrium measurements (420 μM), confirming the reliability of the kinetic measurements. As compared to those of the wild-type swMb, the kon of CuBMb is ~1000-fold slower, while the koff is ~10-fold faster,35 accounting for its ~10000-fold lower CO affinity. It is important to note that the unusually slow CO on-rate of CuBMb is the slowest among the swMb mutants reported so far.39,40 The quantum yield for the geminate recombination phase, which is too fast to be resolved by our instrumentation, is estimated to be ~12% on the basis of the amplitude of the observed kinetic trace as compared to the absorbance value predicted from the equilibrium spectra shown in Figure 2a. The quantum yield of CuBMb is significantly higher than that observed in the wild-type swMb (~4%),35 indicating that the mutation modulates the swMb structure, thereby retarding ligand escape into free solution.
Figure 4.
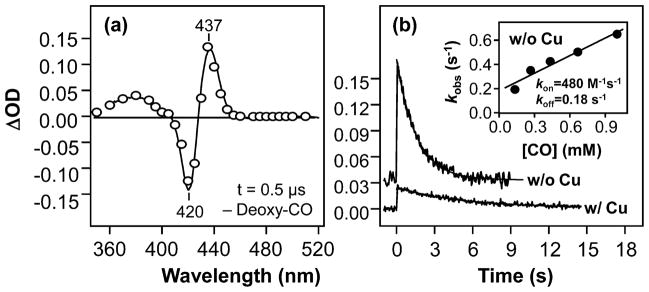
Transient absorption difference spectrum (t = 0.5 μs) (a) and the kinetic traces (b) obtained following photodissociation of CO from CuBMb. The protein and CO concentrations were 5.2 μM and 1 mM, respectively. The spectrum in (a) was obtained in the absence of Cu+; the solid spectrum represents the difference spectrum calculated from the equilibrium deoxy and CO-bound spectra shown in Figure 2a. The kinetic traces in (b) were obtained in the presence and in the absence of 64 μM Cu+, as indicated; the solid curves are the single-exponential fits of the data. In the presence of Cu, the observed rate constant for CO rebinding is 0.19 s−1. The inset in (b) shows the plot of the observed rate constant of the Cu+-free CuBMb as a function of CO concentration; the kon and koff values were determined from the slope and intercept of the best-fit line (solid line) to be 4.8 × 102 M−1 s−1 and 0.18 s−1, respectively.
In the presence of copper, a single-exponential kinetic phase with an apparent rate constant of 0.19 s−1 (Figure 4b) was observed following the photolysis in the presence of 1 mM CO. The missing CO geminate recombination yield was estimated to be ~31%, much higher than that observed in the copper-free CuBMb. The significantly higher geminate recombination yield is plausibly due to the fact that the photolyzed CO can transiently bind to Cu+, thereby accelerating CO rebinding to the heme iron. With the flash photolysis method, bimolecular rebinding kinetics of CuBMb/Cu+ could not be reliably measured as a function of CO concentration owing to the weak absorption changes due to the high geminate recombination yield and the low population of CO-bound protein at low CO concentration (see Figure 2b). As an alternative approach, we carried out stopped-flow measurements by directly mixing the deoxy protein with various concentrations of CO.
As shown in Figure 5a, the binding of CO (50 μM) to the CuBMb/Cu+ complex follows biexponential kinetics. In the first phase, a decrease in the absorbance at 435 nm was observed, concurrent with an increase in the absorbance at 420 nm, suggesting that it is associated with the coordination of CO to the deoxy heme. In contrast, in the second phase, an increase in the absorbance at 435 nm and a decrease in the absorbance at 420 nm were observed. Additional studies show that the rate constant of the first kinetic phase is dependent on the [CO] in a hyperbolic fashion (Figure 5b), while that of the second phase is [CO]-independent (Supporting Information, Figure S2). Hyperbolic [CO]-dependent kinetics has been widely observed in the CcO family of enzymes.6,8 In CcO reactions, CO binds to Cu+, prior to its intramolecular transfer to Fe2+, as illustrated by the first two steps of the reaction illustrated below:
| (3) |
Figure 5.
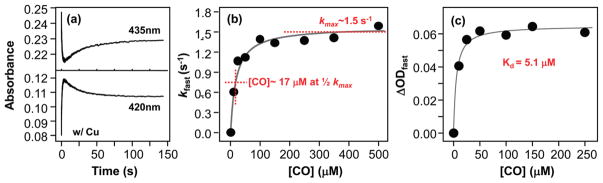
CO binding kinetic traces of CuBMb in the presence of Cu+ (a) and plots of the rate constant (b) and amplitude (c) of the fast kinetic phase as a function of the CO concentration. The biexponential kinetic traces in (a) were obtained by following the changes in the deoxy band (435 nm) and in the CO-bound band (420 nm) by mixing 4 μM CuBMb with 50 μM CO in the presence of 100 μM Cu+ (final concentrations) in pH 7.4 buffer in a stopped-flow system. The rate constants in (b) were obtained from a single-exponential fit of the fast phase of the kinetic traces. The amplitudes in (c) were calculated as ΔOD435 nm – ΔOD420 nm of the fast phase of the kinetic traces.
At low [CO], the observed kinetics is rate-limited by the bimolecular binding of CO to Cu+ in the protein; it increases monotonically in a linear fashion as [CO] increases. At high [CO], the observed kinetics is rate-limited by the intramolecular CO transfer from Cu+ to Fe2+, which is independent of [CO], accounting for the saturation behavior shown in Figure 5b. On this basis, the intramolecular CO transfer rate from Cu+ to Fe2+, k2 + k−2, can be estimated to be 1.5 s−1, the maximum rate constant observed at saturating conditions. Intriguingly, the k2 + k−2 value of the CuBMb/Cu+ complex is similar to that of TtCcO (8.8 s−1)8 but much smaller than those of bCcO (1030 s−1)38 and RsCcO (800 s−1).6 On the other hand, the [CO]1/2, defined as the [CO] required to reach the half-maximum rate (i.e., 0.75 s−1 in this case), reflects the pre-equilibrium constant, k−1/k1, associated with the CO binding reaction to Cu+.6 It is estimated to be ~17 μM for the CuBMb/Cu+ complex (Figure 5b), which is again similar to that of TtCcO (<100 μM) and much smaller than those of bCcO (11 mM) and RsCcO (16 mM, see Table 1).6,8,38
The slow kinetic phase, with a [CO]-independent rate constant of ~0.037 s−1 (Figure 5 and Supporting Information, Figure S2), unlike the fast phase, is unprecedented. To investigate the origin of this kinetic phase, we plotted the amplitude of the fast phase as a function of [CO] and found that the product of the fast phase, [Fe2+-CO Cu+]* in eq 3, exhibits a Kd of ~5.1 μM (Figure 5c), which is ~70-fold smaller than that of the equilibrium state (360 μM, see Figure 2b). The data indicate that the [Fe2+-CO Cu+]* state produced immediately following intramolecular CO transfer from Cu+ to Fe2+ is a metastable state with a higher CO affinity as compared to that of the equilibrium state (Fe2+-CO Cu+). We hypothesize that the intramolecular CO transfer causes the reorganization of the three histidine residues coordinated to the Cu+, due to the change in its coordination state, and that the resulting metastable state relaxes to its equilibrium state with a rate constant of ~0.037 s−1, as illustrated by the last step of the reaction in eq 3. As the equilibrium state has a much higher Kd for CO, the structural relaxation is associated with partial dissociation of CO from the heme iron, accounting for the increase in the absorbance at 435 nm and the concurrent decrease in the absorbance at 420 nm.
It is noteworthy that, at the saturation condition (1 mM CO, see Figure 5b), the rate of intramolecular CO transfer reaction in the stopped-flow system, ca. 1.5 s−1, is ~8-fold faster than that observed in the flash photolysis experiment (0.19 s−1 in Figure 4b). We propose that, in the flash photolysis measurements, the nonequilibrium conformation associated with the deoxy photoproduct, indicated by ** in eq 4, has not yet relaxed to the equilibrium conformation (Fe2+ Cu+) prior to CO binding to Cu+ and its subsequent transfer to Fe2+; as a result, the intramolecular CO transfer reaction experiences a higher activation barrier, thereby accounting for the slower reaction rate.
| (4) |
In CcO, a similar nonequilibrium deoxy state has been identified following CO photolysis, due to slow relaxation of the proximal iron-histidine bond.41 It is plausible that a similar slow proximal iron-histidine bond relaxation occurs in the CuBMb/Cu+ complex following CO photolysis, accounting for the [Fe2+ Cu+]** state, although it remains to be confirmed by time-resolved measurements on the iron-histidine stretching mode.
Analysis of the kinetic results for the CO-bound CuBMb/Cu+ complex revealed several important features: (1) the presence of Cu+ in CuBMb promotes ligand rebinding following photo-dissociation (as reflected by a higher geminate rebinding yield); (2) CuB modulates the bimolecular CO association kinetics by providing a temporary docking site for CO prior to its binding to heme iron, similar to that observed in CcO; (3) the CO transfer reaction from Cu+ to Fe2+ and the photodissociation reaction of CO from the heme iron in the CuBMb/Cu+ complex introduce conformational changes to its protein matrix, again analogous to the observations in CcO; and (4) the replacement of copper with Ag+ or Zn2+ ions in the CuBMb/Cu+ abolished these effects (Supporting Information, Figure S3), demonstrating that the perturbations in CO binding properties of CuBMb/Cu+ are specific to copper.
Equilibrium Structural Properties of NO-Bound CuBMb
CuBMb has been shown to be able to convert NO to N2O in the presence of Cu+, but not in its absence.30 In an effort to understand how Cu+ binding in CuBMb modulates the reactivity of the heme to promote the reaction, we studied the equilibrium and kinetic properties of the NO adducts of CuBMb.
As shown in Figure 6a, the NO adduct of CuBMb exhibits a Soret maximum at 420 nm and visible maxima at 546 and 580 nm, similar to those reported for the wild-type swMb.42 As in the CO adduct, the optical spectrum of the NO adduct is not significantly sensitive to the Cu+ binding. In the resonance Raman spectra of copper-free CuBMb-NO, the νFe-NO, δFe-N-O, and νN-O modes were identified at 566, 457, and 1598 cm−1, respectively, on the basis of isotope substitution experiments (Figure 6b). The δFe-N-O mode is similar to that of the wild-type protein, whereas the νFe-NO and νN-O modes are up- and down-shifted, respectively, as compared to those of the wild-type protein (bottom trace). The data indicate that the three histidine residues in the distal heme pocket of CuBMb cause the changes in the environment and conformation of the Fe-N-O moiety.43 It is noted that the split of the νN-O mode with 15N18O is a result of its coupling to the porphyrin mode at 1583 cm−1, as reported for other heme proteins.43
Figure 6.
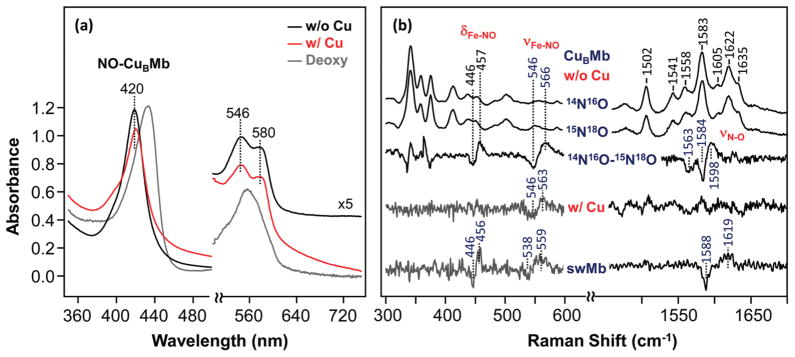
Optical absorption (a) and resonance Raman (b) spectra of the NO adducts of ferrous CuBMb in the presence or absence Cu+. The spectra in (a) and (b) were obtained in the presence or absence of 79 and 100 μM Cu+, respectively. In (a), the spectrum of the Cu+-free deoxy CuBMb is shown as a reference. In (b), the spectrum of the NO-bound wild-type swMb is shown as a reference. The protein concentrations in (a) and (b) were 10 and 40 μM, respectively, prepared by adding NO to dithionite-reduced protein under anaerobic conditions.
Although copper binding does not affect the electronic transition bands of CuBMb, it leads to the total loss of intensity of the δFe-N-O and νN-O modes (Figure 6b), as well as a 3 cm−1 down-shift of the νFe-NO mode. The spectral pattern is not only different from that of the wild-type swMb but also distinct from those of other CcOs (Supporting Information, Table S1), as highlighted by its significantly higher νFe-NO frequency. On the basis of the studies conducted by Pinakoulaki et al.,44 the stronger Fe-NO bond (as indicated by a higher νFe-NO frequency) in CcO is associated with a stronger proximal iron-histidine bond; the higher νFe-NO of the CuBMb/Cu+ complex is hence consistent with its higher νFe-His frequency as compared to those of CcO enzymes (220 vs 193–214 cm−1, see Supporting Information, Table S1).41
Kinetic Properties of NO-Bound CuBMb
To determine the kinetic properties of the NO adduct of ferrous CuBMb, flash photolysis studies were carried out. The transient absorption difference spectrum obtained at 1.0 μs following NO photolysis exhibits a positive and a negative peak at 436 and 417 nm, respectively (Supporting Information, Figure S4), reflecting the bleaching of the NO-bound species and the concomitant production of the deoxy species. The difference spectrum is in good agreement with that calculated from the equilibrium spectra shown in Figure 6a, indicating that there is no intermediate populated other than the deoxy protein. As shown in Figure 7a, a single-exponential kinetic phase was observed at 440 nm. Additional studies show that the rate constant depends linearly on [NO], indicating that the observed kinetics is associated with a bimolecular NO rebinding reaction. The bimolecular rate constant derived from the slope of the linear fit of the data (see the inset in Figure 7a) is 1.6 × 105 M−1 s−1, which is ~100 times slower than that of the wild-type protein (1.7 × 107 M−1 s−1),45 indicating that the mutation blocks ligand access to the heme binding site, as found in the CO binding reaction.
Figure 7.
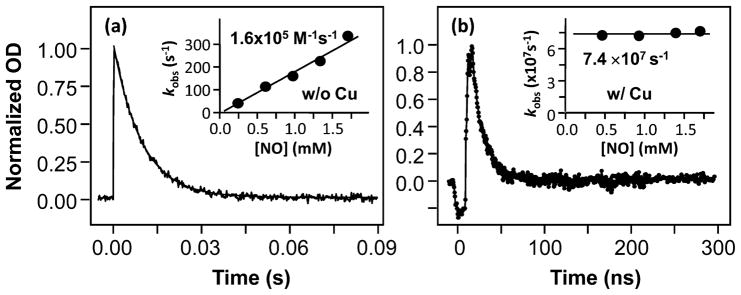
Kinetic traces obtained following photodissociation of NO from CuBMb in the absence (a) or presence (b) of 65 μM Cu+. The protein and NO concentrations were 10 μM and 1.7 mM, respectively. The reduced protein solution was prepared with 9.0 mM ascorbate. The solid curves are the single-exponential fits of the data. The insets show plots of the observed rate constants as a function of NO concentration. In the absence of Cu+, the bimolecular rate constant was determined to be 1.6 × 105 M−1 s−1 on the basis of the slope of the best-fit solid line. In the presence of Cu+, the rate (7.4 × 107 s−1) was independent of the NO concentration.
In the presence of Cu+, a much faster single-exponential kinetic phase with a rate constant of 7.4 × 107 s−1 (t ≈ 15 ns) was observed (Figure 7b). The observed kinetic phase was found to be independent of [NO]. In addition, the missing geminate recombination yield estimated from the amplitude of the kinetic phase was significantly higher in the presence of Cu+ than in its absence (although the values cannot be qualitatively determined with our instrumentation). The kinetic behavior of the CuBMb/Cu+ complex is similar to that of TtCcO, which exhibits a geminate phase with extremely high yield (~80%) and a lifetime of ~15 ns.46 It is believed that, like the CO reaction, the photodissociated NO from TtCcO first binds to the Cu+, followed by intramolecular NO transfer from Cu+ to Fe2+, as illustrated in eq 5.
| (5) |
Here, [Fe2+ Cu+]* stands for the photodissociated deoxy species. The 15 ns geminate phase of TtCcO has been attributed to intramolecular NO transfer from Cu+ to Fe2+.46 Likewise, we assigned the 15 ns kinetic phase of the CuBMb/Cu+ complex to the same process, while the missing geminate phase was attributed to conformational changes associated with NO binding to the Cu+.
Taken together, our data show that the presence of Cu+ drastically alters the NO binding dynamics. Instead of escaping into free solution, all the photodissociated NO completely rebinds to the heme iron following a transient stop at the Cu+ center. Although the NO reductase activity of CuBMb/Cu+ requires simultaneous binding of two NO molecules in the binuclear center to form the hypothesized hyponitrite,47 under the equilibrium conditions employed in this work, there is no evidence that a second NO molecule is present, implying that, when the first NO is already present in the heme iron site, the NO affinity toward Cu+ is low. This scenario is consistent with the low NO reductase activity of the enzyme (~2 NO per enzyme per minute),30 similar to that observed in TtCcO.9 It suggests that the NO reductase reaction occurs through a concerted mechanism with NO only transiently bound to CuB.12 This behavior of CuBMb/Cu+ is in sharp contrast to those of bovine and bacterial aa3 oxidases, in which NO binds to both the heme iron and CuB but there is no NO reductase activity observed.48 It has been suggested that the binding of a second NO in the binuclear center of the oxidase family of enzymes is sensitive to the Fe-CuB distance, which has been estimated to be ~5.3 Å for CuBMb/Cu+ on the basis of computational modeling studies.27 This distance is similar to that of TtCcO (4.4 Å)49 as well as that of the bo3 CcO from Escherichia coli (5.3 Å),50 both of which exhibit NO reductase activity.12,47 Nonetheless, the Fe-CuB distances in bCcO and PdCcO (a CcO from Paracoccus denitrificans) are also in a similar range, but they do not display any NO reductase activity, suggesting that, in addition to the relative distance, a favorable region orientation of the CuB center with respect to Fe in the binuclear center is important for the NO reductase activity.
Conclusions
The data reported here substantiate the value of the CuBMb/Cu+ complex as a model for CcO enzymes. The resonance Raman studies of the CO-bound CuBMb/Cu+ complex show that the presence of copper, held in position by three histidine residues in the heme distal pocket, leads to a CcO-like heme environment, with the ligand situated in close proximity to the copper. Kinetic studies show that the Cu+ in the CuBMb/Cu+ complex plays an important role in regulating ligand entry and exit from the binuclear center, similar to that observed in CcO,6,8,38 by providing a temporary ligand docking site. They also show that, during the CO binding reaction, the coordination of the ligand to Cu+ and its subsequent transfer to Fe2+ induce conformational changes to the protein matrix, possibly due to the change in the coordination state of the Cu+, as proposed for similar conformational changes observed in the CcO family of enzymes.38 It is noteworthy that, in CcO enzymes, the conformational changes have been proposed to play critical roles in controlling ligand escape and in coupling oxygen chemistry to proton pumping.1,36,51
The CuBMb/Cu+ complex exhibits several ligand binding parameters similar to those of TtCcO, including (1) high geminate CO/NO recombination yields following its photodissociation and (2) high CO affinity toward Cu+ and slow intramolecular CO transfer rate between Cu+ and Fe2+ (as reflected by k−1/k1 and k2 + k−2 listed in Table 1, respectively). Intriguingly, these ligand binding properties are distinct from those of bCcO, highlighting the subtle structural differences between bCcO and the CuBMb/Cu+ complex, as well as the TtCcO. Along the same lines, previous studies showed that the CuBMb/Cu+ complex exhibits NO reductase activities, like TtCcO, but distinct from bCcO, which does not exhibit any NO reductase activity. These data indicate that the CuBMb/Cu+ complex is a better model for TtCcO than bCcO. Further studies of the CuBMb/Cu+ complexes, for example with the incorporation of an engineered His-Tyr moiety, would be beneficial for better understanding of the structural basis for the functional similarities and differences between these protein systems.
Supplementary Material
Acknowledgments
The project is supported by GM074982 to D.L.R. and GM06221 to Y.L. We thank Dr. Tsuyoshi Egawa for valuable discussions.
Footnotes
Supporting Information Available: Figure S1, Raman spectra of the Fe-His stretching mode; Figure S2, rate constant for the slow kinetic phase of the reaction of CO with CuBMb/Cu+; Figure S3, pseudo-first-order CO rebinding constant of CuBMb in the presence of various metals; Figure S4, transient absorption difference spectrum following photodissociation of NO from ferrous CuBMb; Table S1, vibrational mode frequencies of the model complexes compared to the swMb and the CcO frequencies. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Denis L. Rousseau, Email: rousseau@aecom.yu.edu.
Syun-Ru Yeh, Email: syeh@aecom.yu.edu.
References
- 1.Musser SM, Stowell MH, Chan SI. Adv Enzymol Relat Areas Mol Biol. 1995;71:79–208. doi: 10.1002/9780470123171.ch3. [DOI] [PubMed] [Google Scholar]
- 2.Brzezinski P. Trends Biochem Sci. 2004;29:380–387. doi: 10.1016/j.tibs.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Gennis RB. Front Biosci. 2004;9:581–591. doi: 10.2741/1237. [DOI] [PubMed] [Google Scholar]
- 4.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 5.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 6.Aagaard A, Gilderson G, Gomes CM, Teixeira M, Brzezinski P. Biochemistry. 1999;38:10032–10041. doi: 10.1021/bi990473m. [DOI] [PubMed] [Google Scholar]
- 7.Smirnova IA, Zaslavsky D, Fee JA, Gennis RB, Brzezinski P. J Bioenerg Biomembr. 2008;40:281–287. doi: 10.1007/s10863-008-9157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giuffre A, Forte E, Antonini G, D’Itri E, Brunori M, Soulimane T, Buse G. Biochemistry. 1999;38:1057–1065. doi: 10.1021/bi9815389. [DOI] [PubMed] [Google Scholar]
- 9.Giuffre A, Stubauer G, Sarti P, Brunori M, Zumft WG, Buse G, Soulimane T. Proc Natl Acad Sci USA. 1999;96:14718–14723. doi: 10.1073/pnas.96.26.14718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fee JA, Case DA, Noodleman L. J Am Chem Soc. 2008;130:15002–15021. doi: 10.1021/ja803112w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siletsky SA, Belevich I, Jasaitis A, Konstantinov AA, Wikstrom M, Soulimane T, Verkhovsky MI. Biochim Biophys Acta. 2007;1767:1383–1392. doi: 10.1016/j.bbabio.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi T, Lin IJ, Chen Y, Fee JA, Moenne-Loccoz P. J Am Chem Soc. 2007;129:14952–14958. doi: 10.1021/ja074600a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belevich I, Verkhovsky MI. Antioxid Redox Signal. 2008;10:1–29. doi: 10.1089/ars.2007.1705. [DOI] [PubMed] [Google Scholar]
- 14.Brzezinski P, Gennis RB. J Bioenerg Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ibers JA, Holm RH. Science. 1980;209:223–235. doi: 10.1126/science.7384796. [DOI] [PubMed] [Google Scholar]
- 16.Chufan EE, Puiu SC, Karlin KD. Acc Chem Res. 2007;40:563–572. doi: 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]
- 17.Collman JP, Boulatov R, Sunderland CJ, Fu L. Chem Rev. 2004;104:561–588. doi: 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]
- 18.Collman JP, Decreau RA. Chem Commun (Camb) 2008:5065–5076. doi: 10.1039/b808070b. [DOI] [PubMed] [Google Scholar]
- 19.Kim E, Chufan EE, Kamaraj K, Karlin KD. Chem Rev. 2004;104:1077–1133. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 20.Collman JP, Decreau RA, Zhang C. J Org Chem. 2004;69:3546–3549. doi: 10.1021/jo0499625. [DOI] [PubMed] [Google Scholar]
- 21.Kim E, Kamaraj K, Galliker B, Rubie ND, Moenne-Loccoz P, Kaderli S, Zuberbuhler AD, Karlin KD. Inorg Chem. 2005;44:1238–1247. doi: 10.1021/ic048907b. [DOI] [PubMed] [Google Scholar]
- 22.Liu JG, Naruta Y, Tani F, Chishiro T, Tachi Y. Chem Commun (Camb) 2004:120–121. doi: 10.1039/b311538k. [DOI] [PubMed] [Google Scholar]
- 23.Chufan EE, Mondal B, Gandhi T, Kim E, Rubie ND, Moenne-Loccoz P, Karlin KD. Inorg Chem. 2007;46:6382–6394. doi: 10.1021/ic700363k. [DOI] [PubMed] [Google Scholar]
- 24.Collman JP, Devaraj NK, Decreau RA, Yang Y, Yan YL, Ebina W, Eberspacher TA, Chidsey CE. Science. 2007;315:1565–1568. doi: 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collman JP, Ghosh S, Dey A, Decreau RA, Yang Y. J Am Chem Soc. 2009;131:5034–5035. doi: 10.1021/ja9001579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lucas HR, Meyer GJ, Karlin KD. J Am Chem Soc. 2009;131:13924–13925. doi: 10.1021/ja906172c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sigman JA, Kwok BC, Lu Y. J Am Chem Soc. 2000;122:8192–8196. [Google Scholar]
- 28.Sigman JA, Kim HK, Zhao X, Carey JR, Lu Y. Proc Natl Acad Sci USA. 2003;100:3629–3634. doi: 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang N, Zhao X, Lu Y. J Am Chem Soc. 2005;127:16541–16547. doi: 10.1021/ja052659g. [DOI] [PubMed] [Google Scholar]
- 30.Zhao X, Yeung N, Russell BS, Garner DK, Lu Y. J Am Chem Soc. 2006;128:6766–6767. doi: 10.1021/ja058822p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu C, Egawa T, Wainwright LM, Poole RK, Yeh SR. J Biol Chem. 2007;282:13627–13636. doi: 10.1074/jbc.M609397200. [DOI] [PubMed] [Google Scholar]
- 32.Guallar V, Lu C, Borrelli K, Egawa T, Yeh SR. J Biol Chem. 2009;284:3106–3116. doi: 10.1074/jbc.M806183200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu C, Mukai M, Lin Y, Wu G, Poole RK, Yeh SR. J Biol Chem. 2007;282:25917–25928. doi: 10.1074/jbc.M704415200. [DOI] [PubMed] [Google Scholar]
- 34.Springer BA, Sligar SG. Proc Natl Acad Sci USA. 1987;84:8961–8965. doi: 10.1073/pnas.84.24.8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carver TE, Rohlfs RJ, Olson JS, Gibson QH, Blackmore RS, Springer BA, Sligar SG. J Biol Chem. 1990;265:20007–20020. [PubMed] [Google Scholar]
- 36.Das TK, Tomson FL, Gennis RB, Gordon M, Rousseau DL. Biophys J. 2001;80:2039–2045. doi: 10.1016/S0006-3495(01)76177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ibrahim M, Xu C, Spiro TG. J Am Chem Soc. 2006;128:16834–16845. doi: 10.1021/ja064859d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Einarsdottir O, Dyer RB, Lemon DD, Killough PM, Hubig SM, Atherton SJ, Lopez-Garriga JJ, Palmer G, Woodruff WH. Biochemistry. 1993;32:12013–12024. doi: 10.1021/bi00096a011. [DOI] [PubMed] [Google Scholar]
- 39.Phillips GN, Jr, Teodoro ML, Li T, Smith B, Olson JS. J Phys Chem B. 1999;103:8817–8829. [Google Scholar]
- 40.Lu C, Egawa T, Mukai M, Poole RK, Yeh SR. Methods Enzymol. 2008;437:255–286. doi: 10.1016/S0076-6879(07)37014-6. [DOI] [PubMed] [Google Scholar]
- 41.Oertling WA, Surerus KK, Einarsdottir O, Fee JA, Dyer RB, Woodruff WH. Biochemistry. 1994;33:3128–3141. doi: 10.1021/bi00176a048. [DOI] [PubMed] [Google Scholar]
- 42.Ionascu D, Gruia F, Ye X, Yu A, Rosca F, Beck C, Demidov A, Olson JS, Champion PM. J Am Chem Soc. 2005;127:16921–16934. doi: 10.1021/ja054249y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coyle CM, Vogel KM, Rush TS, Kozlowski PM, Williams R, Spiro TG, Dou Y, Ikeda-Saito M, Olson JS, Zgierski MZ. Biochemistry. 2003;42:4896–4903. doi: 10.1021/bi026395b. [DOI] [PubMed] [Google Scholar]
- 44.Pinakoulaki E, Ohta T, Soulimane T, Kitagawa T, Varotsis C. J Am Chem Soc. 2005;127:15161–15167. doi: 10.1021/ja0539490. [DOI] [PubMed] [Google Scholar]
- 45.Olson JS, Phillips GN., Jr J Biol Chem. 1996;271:17593–17596. doi: 10.1074/jbc.271.30.17593. [DOI] [PubMed] [Google Scholar]
- 46.Pilet E, Nitschke W, Rappaport F, Soulimane T, Lambry JC, Liebl U, Vos MH. Biochemistry. 2004;43:14118–14127. doi: 10.1021/bi0488808. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi T, Lin MT, Ganesan K, Chen Y, Fee JA, Gennis RB, Moenne-Loccoz P. Biochemistry. 2009;48:883–890. doi: 10.1021/bi801915r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stubauer G, Giuffre A, Brunori M, Sarti P. Biochem Biophys Res Commun. 1998;245:459–465. doi: 10.1006/bbrc.1998.8457. [DOI] [PubMed] [Google Scholar]
- 49.Soulimane T, Buse G, Bourenkov GP, Bartunik HD, Huber R, Than ME. EMBO J. 2000;19:1766–1776. doi: 10.1093/emboj/19.8.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abramson J, Riistama S, Larsson G, Jasaitis A, Svensson-Ek M, Laakkonen L, Puustinen A, Iwata S, Wikstrom M. Nat Struct Biol. 2000;7:910–917. doi: 10.1038/82824. [DOI] [PubMed] [Google Scholar]
- 51.Qin L, Liu J, Mills DA, Proshlyakov DA, Hiser C, Ferguson-Miller S. Biochemistry. 2009;48:5121–5130. doi: 10.1021/bi9001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spiro TG, Wasbotten IH. J Inorg Biochem. 2005;99:34–44. doi: 10.1016/j.jinorgbio.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 53.Pinakoulaki E, Ohta T, Soulimane T, Kitagawa T, Varotsis C. J Biol Chem. 2004;279:22791–22794. doi: 10.1074/jbc.C400124200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.