

Abstract
AIM: To investigate the effect of propofol on human pancreatic cells and the molecular mechanism of propofol action.
METHODS: We used the human pancreatic cancer cell line MIAPaCa-2 for in vitro studies measuring growth inhibition and degree of apoptotic cell death induced by propofol alone, gemcitabine alone, or propofol followed by gemcitabine. All experiments were conducted in triplicate and carried out on three or more separate occasions. Data were means of the three or more independent experiments ± SE. Statistically significant differences were determined by two-tailed unpaired Student’s t test and defined as P < 0.05.
RESULTS: Pretreatment of cells with propofol for 24 h followed by gemcitabine resulted in 24%-75% growth inhibition compared with 6%-18% when gemcitabine was used alone. Overall growth inhibition was directly correlated with apoptotic cell death. We also showed that propofol potentiated gemcitabine-induced killing by downregulation of nuclear factor-κB (NF-κB). In contrast, NF-κB was upregulated when pancreatic cancer cells were exposed to gemcitabine alone, suggesting a potential mechanism of acquired chemoresistance.
CONCLUSION: Inactivation of the NF-κB signaling pathway by propofol might abrogate gemcitabine-induced activation of NF-κB, resulting in chemosensitization of pancreatic tumors to gemcitabine.
Keywords: Pancreatic cancer, Propofol, Gemcitabine, Nuclear factor-κB, Apoptosis
Core tip: Pretreatment of cells with propofol for 24 h followed by gemcitabine resulted in significant growth inhibition compared with gemcitabine alone. Overall growth inhibition correlated directly with apoptotic cell death. Propofol potentiated gemcitabine-induced killing by downregulation of nuclear factor-κB (NF-κB). In contrast, NF-κB was upregulated when pancreatic cancer cells were exposed to gemcitabine alone. These results suggested that inactivation of the NF-κB signaling pathway by propofol abrogated gemcitabine-induced activation of NF-κB resulting in the chemosensitization of pancreatic tumors to gemcitabine.
INTRODUCTION
Pancreatic cancer has the poorest prognosis of all major cancers, with an overall 5-year survival rate of around 5%[1]. The current clinical standard of care for advanced pancreatic cancer is gemcitabine, a cytotoxic nucleoside analog. Treatment with gemcitabine results in a tumor response rate of 12% and a median survival time of 5 mo[2].
Drug resistance (both intrinsic and acquired) is thought to be a major reason for the limited benefit of most pancreatic cancer therapies[3]. Recent studies have indicated that targeted therapies in combination with gemcitabine can have statistically significant benefits[4]. However, the results to date remain insufficient, and new approaches to improving the effectiveness of gemcitabine are needed. One of the targets considered for combination therapy that has received wide attention is the transcription factor nuclear factor-κB (NF-κB)[5]. Pan et al[6] and Kong et al[7] reported that inhibition of NF-ĸB might be useful for pancreatic cancer therapy, as it increases gemcitabine sensitivity in pancreatic cancer cells. Recent studies also indicate that combination therapy with a targeted medicine that inhibits NF-κB activity potentiated the antitumor effects of gemcitabine in pancreatic cancer cells[8-10].
Propofol is an intravenous anesthetic that is used to induce and maintain anesthesia, and to sedate and calm patients in intensive care. Increasing evidence suggests that propofol might be neuroprotective against ischemic neuronal injury in animal models of cerebral ischemia[11-13]. Xi et al[14] and Li et al[15] found that the neuroprotective effects of propofol against neuronal apoptosis might be a consequence of regulation of Bcl-2, caspase-3 and Bax. Propofol has protective effects against digestive injury. It inhibits HMGB1 expression and TLR4/MyD88/NF-κB-mediated inflammatory responses, and hampers apoptosis, which might contribute to its protective action against ethanol-induced gastric mucosal injury[16]. Propofol also has anticancer properties. Siddiqui et al[17] found that combinations of propofol and docosahexaenoate or propofol and eicosapentaenoate significantly induced apoptosis and inhibited cell adhesion and migration in breast cancer cells. Propofol inhibits MMP-2 and -9 expression, suppressing lung cancer cell invasion and migration[18]. Propofol induces proliferation and promotes invasion of gallbladder cancer cells through activation of Nrf2[19]. Li et al[20] showed that propofol reduced the level of MMP in breast cancer cells by inhibition of NF-κB pathways, significantly restraining migration and invasion of breast cancer cells. Propofol extensively counteracts the oxidative/nitrative and multiple apoptotic effects of doxorubicin in rat hearts[21].
Most human pancreatic tumors show high levels of activated NF-κB, which mediates survival signaling and confers resistance to conventional therapeutics. Therefore, targeting NF-κB could be an effective therapeutic approach. The mechanism by which NF-κB stimulates cell survival is not fully understood; however, recent studies showed that activation of NF-κB leads to the activation of a series of survival factors, including bcl-2. This allows cancer cells to resist induction of apoptosis[7]. Many conventional cancer chemotherapeutic agents such as vinblastine, vincristine, daunomycin, doxorubicin, campothecin, cisplatin, and etoposide activate NF-κB. This activation results in resistance to apoptosis, which results in poor clinical outcomes for pancreatic cancer patients. Inactivating NF-κB activity induces apoptosis and abrogates de novo or acquired chemoresistance[22]. Based on these results, we hypothesized that propofol might block multiple intracellular signaling pathways that are known to confer a high degree of chemoresistance by pancreatic cancer cells, abrogating either de novo or acquired chemoresistance. Although the development of alternative gemcitabine schedules and chemotherapy combinations continues, we report our observations in support of our hypothesis that better pancreatic cancer cell killing is feasible by using propofol with gemcitabine. Our results are primarily due to inactivation of NF-κB signaling in vitro.
MATERIALS AND METHODS
Cell culture
The MIA-PaCa-2 human pancreatic cancer cell line was obtained from the American Type Culture Collection and cultured in Dulbecco modified Eagle’s medium supplemented with 10% fetal calf serum, sodium pyruvate, nonessential amino acids, L-glutamine, penicillin/streptomycin antibiotics, and vitamins. Cells were maintained in a humidified incubator containing 10% carbon dioxide at 37 °C. Cells underwent serum starvation for 24 h before treatment with propofol or/and gemcitabine.
Drug treatment
For single-agent treatment, MIA-PaCa-2 cells were treated with 10-100 μmol/L propofol or 0.5 mmol/L Na2CO3 (vehicle control) for 72 h; 100 μmol/L per milliliter propofol for 24, 48, 72 h; or 10, 25, 50, 100 μmol/L gemcitabine for 72 h. For combined treatment, MIA-PaCa-2 cells were treated with 50 or 100 μmol/L per milliliter propofol for 24 h, then exposed to 10-100 μmol/L gemcitabine for an additional 72 h.
Growth inhibition by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
MIA-PaCa-2 cells were seeded at 3 × 103 cells per well in 96-well microtiter culture plates. After overnight incubation, medium was replaced with fresh medium containing propofol 0-100 μmol/L diluted from a 10 mmol/L stock. After 24-72 h incubation, 20 μL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/mL in PBS) was added to each well and incubated for 2 h. Supernatant was aspirated and the MTT formazan formed by metabolically viable cells was dissolved in 100 μL isopropanol. Plates were mixed for 30 min on a shaker and absorbance was measured at 595 nm using a plate reader (TECAN, Durham, NC, United States). MIA-PaCa-2 cells were also treated with 25 μmol/L propofol for 24 h and exposed to 0-100 μmol/L gemcitabine for an additional 72 h before MTT assay.
DNA ladder analysis for apoptosis
Cytoplasmic DNA was extracted from MIA-PaCa-2 cells treated with 100 μmol/L propofol or 0.5 mmol/L Na2CO3 (vehicle control) for 24-72 h; or 10-100 μmol/L propofol for 72 h using 10 mmol/L Tris (pH 8.0), 1 mmol/L EDTA, and 0.2% Triton X-100. MIA-PaCa-2 cells were also treated with 25 μmol/L propofol for 24 h and exposed to 0-100 μmol/L of gemcitabine for an additional 72 h before cytoplasmic DNA extraction. Lysate was centrifuged for 15 min at 13000 g to separate fragmented DNA (soluble) from intact chromatin (nuclear pellet). Supernatant from lysates was treated with RNase followed by SDS-Proteinase K digestion, phenol chloroform extraction, and isopropanol precipitation. DNA was separated by 1.5% agarose gels stained with ethidium bromide for DNA visualization by UV light.
Terminal transferase dUTP nick-end labeling assay for apoptosis
Apoptosis was evaluated by terminal transferase dUTP nick-end labeling (TUNEL) assay according to the manufacturer’s instructions for MIA-PaCa-2 cells treated with 100 μmol/L propofol or 0.5 mmol/L Na2CO3 (vehicle control) for 24-72 h; or 10-100 μmol/L propofol for 72 h; or 25 μmol/L propofol for 24 h followed by 0-100 μmol/L of gemcitabine for 72 h. TUNEL-positive cells were colored using diaminobenzidine as chromogen and counterstained with hematoxylin. The percentage of TUNEL-positive cells was assessed in five randomly selected fields per section. All assays were performed in quadruplicate.
Quantification of apoptosis by enzyme-linked immunosorbent assay
The Cell Apoptosis enzyme-linked immunosorbent assay (ELISA) Detection Kit (Chemicon International, Temecula, CA, United States) was used to detect apoptosis in MIA-PaCa-2 cells according to the manufacturer’s protocol. MIA-PaCa-2 cells were treated with 10-100 μmol/L propofol for 72 h or with 50 μmol/L propofol for 24-72 h; or 50 μmol/L propofol for 24 h followed by 0-100 μmol/L gemcitabine for 72 h. After treatment, cytoplasmic histone DNA fragments from MIA-PaCa-2 cells were extracted and bound to immobilized anti-histone. Peroxidase-conjugated anti-DNA was used to detect immobilized histone DNA fragments. After addition of peroxidase substrate, spectrophotometric absorbance of samples was determined using an ULTRA Multifunctional Microplate Reader (TECAN) at 405 nm.
Electrophoretic mobility shift assay
Cell extracts were prepared using a commercially available nuclear extraction kit according to the manufacturer’s protocol (Pierce, Rockford, IL, United States). Electrophoretic mobility shift assay (EMSA) was performed according to the provided protocol (Promega). Briefly, cells were washed with cold PBS and suspended in 0.15 mL lysis buffer (10 mmol/L HEPES pH 7.9, 10 mmol/L KCl, 0.1 mmol/L EDTA, 0.1 mmol/L EGTA, 1 mmol/L DTT, 1 mmol/L PMSF, 2 μg/mL leupeptin, 2 μg/mL aprotinin, and 0.5 mg/mL benzamidine). Cells were swelled on ice for 20 min and 4.8 μL 10% NP40 was added. Tubes were vigorously mixed for a few seconds and microcentrifuged. The nuclear pellet was resuspended in 30 μL ice-cold nuclear extraction buffer (20 mmol/L HEPES pH 7.9, 0.4 mol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L DTT, 0.5 mmol/L PMSF, 2 μg/mL leupeptin, 2 μg/mL aprotinin, and 0.5 mg/mL benzamidine) and incubated on ice with intermittent mixing. Tubes were microcentrifuged for 5 min at 4 °C, and supernatant (nuclear extract) was collected in cold Eppendorf tubes and stored at -70 °C. Protein content was measured by bicinchoninic acid method. EMSA used 5 μg of nuclear proteins incubated with IRDye-700-labeled NF-κB oligonucleotide. Incubation mixture was 2 μg of poly (deoxyinosinic - deoxycytidylic acid) in binding buffer. DNA-protein complexes were separated from free oligonucleotides on 8.0% native polyacrylamide gels using buffer containing 50 mmol/L Tris, 200 mmol/L glycine (pH 8.5), and 1 mmol/L EDTA and visualized by an Odyssey Infrared Imaging System using Odyssey Software Release 1.1. Equal protein loading was ensured by immunoblotting 10 μg of nuclear protein with anti-retinoblastoma.
Statistical analysis
All experiments were conducted in triplicate and carried out on three or more separate occasions. Data were means of the three or more independent experiments ± SE. Statistically significant differences were determined by two-tailed unpaired Student’s t test and defined as P < 0.05.
RESULTS
Effect of propofol on cell proliferation
MIA-PaCa-2 cells were treated with 0-100 μmol/L propofol over 72 h, and cell viability was determined by MTT assay. Treatment with 10, 25, 50, or 100 μmol/mL of propofol for 72 h resulted in 95%, 87%, 64%, and 51% of cell growth relative to control, respectively (Figure 1A). Similar results were found with exposure to 100 μmol/mL propofol for 24, 48, and 72 h (Figure 1B). Treatment of MIA-PaCa-2 cells with propofol resulted in dose- and time-dependent inhibition of cell proliferation, demonstrating that propofol applied as a single agent was an effective inhibitor of pancreatic cancer cell growth.
Figure 1.
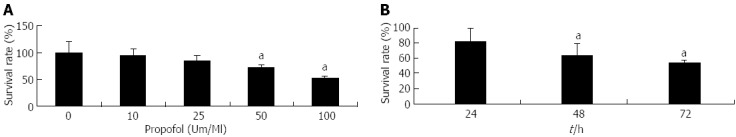
Evaluation by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay of pancreatic cancer MIA-PaCa-2 cell viability after propofol pretreatment. A: Cells were either untreated or treated with 10-100 μmol/mL propofol for 72 h; B: Cells treated with 100 μmol/mL propofol for 24, 48, or 72 h. aP < 0.05 vs control group.
Effect of propofol on apoptosis
MIA-PaCa-2 cells were treated with 10-100 μmol/L propofol for 72 h, or 100 μmol/L propofol for 24-72 h. Apoptosis was determined by TUNEL, DNA ladder and ELISA assays. Treatment with 10, 25, 50, or 100 μmol/mL propofol for 72 h resulted in 1.6%, 4.1%, 9.7%, or 13.8% apoptosis relative to controls (Figure 2A). Similar results were found with 100 μmol/mL propofol for 24, 48, or 72 h (data not shown). Treatment of MIA-PaCa-2 cells with propofol resulted in dose- and time-dependent promotion of apoptosis, demonstrating that propofol used as a single agent was an effective promoter of pancreatic cancer cell death. DNA ladder (Figure 2B) and ELISA (Figure 2C) assays gave the same results as TUNEL assays.
Figure 2.
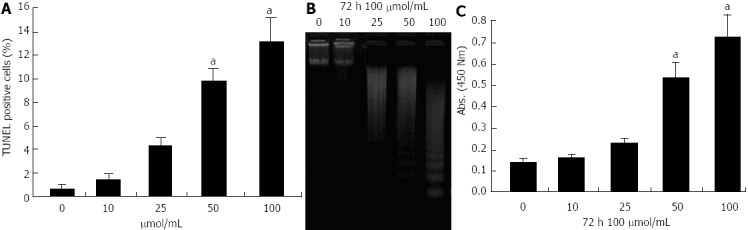
Evaluation of apoptosis of pancreatic cancer MIA-PaCa-2 cells after propofol treatment using terminal transferase dUTP nick-end labeling, DNA ladder and enzyme-linked immunosorbent assays. A: Propofol-induced apoptotic cell death by terminal transferase dUTP nick-end labeling (TUNEL) after 72 h of 10-100 μmol/L propofol; B: DNA ladder indicative of apoptosis in pancreatic cancer cells treated with 10-100 μmol/mL propofol for 72 h. C: Propofol-induced apoptosis measured by enzyme-linked immunosorbent assay after 72 h of 10-100 μmol/L propofol. aP < 0.05 vs control group.
Propofol potentiates growth inhibition by gemcitabine in MIA-PaCa-2 cells
MIAPaCa-2 cells are resistant to gemcitabine treatment. We found that gemcitabine treatment did not result in obvious MIAPaCa-2 growth inhibition (Figure 3). We assessed the effect on cell viability of pretreatment and cotreatment of propofol and gemcitabine by MTT assay. Cells were pretreated with propofol (50 μmol/mL) alone or in combination with a single dose of gemcitabine (10, 25, 50 and 100 μmol/L), and viable cells were evaluated by MTT assay 72 h after treatment. Doses were chosen based upon a preliminary dose escalation study (data not shown). Treatment of MIA-PaCa-2 cells with a single dose of gemcitabine (10, 25, 50 and 100 μmol/L) for 72 h resulted in only 6% to 18% loss of viability. However, pretreatment with propofol for 24 h followed by treatment with gemcitabine resulted in 24% to 75% loss of viable MIA-PaCa-2 cells (Figure 3). These results suggested that the combination of propofol with low therapeutic doses of gemcitabine elicited significantly greater inhibition of cancer cell growth compared with either agent alone. This suggested that lower toxic side effects are likely to occur in normal cells. Inhibition of cell growth and viability as assessed by MTT assay might be due to the induction of apoptosis by propofol and gemcitabine. We therefore investigated whether gemcitabine in combination with propofol induced more apoptosis than either agent alone.
Figure 3.
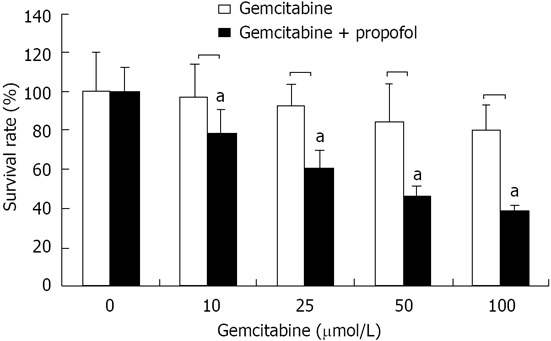
Cell viability by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay of pancreatic cancer MIA-PaCa-2 cells after propofol pretreatment. MIA-PaCa-2 cells pretreated with propofol (50 μmol/mL) for 24 h followed by coincubation with gemcitabine (10, 25, 50 and 100 μmol/L) for 72 h were analyzed for viable cells by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. Viable cells were evaluated relative to gemcitabine-treated controls and interpreted as % viable cells. Data are averages of four to five independent experiments. aP < 0.05 vs control group.
Propofol sensitizes MIA-PaCa-2 cells to apoptosis induced by gemcitabine
By TUNEL analysis, treatment of MIA-PaCa-2 cells with propofol (25 μmol/mL) for 72 h resulted in only 4.6% apoptosis (Figure 2 A), and treatment with 10, 25, 50 and 100 μmol/L gemcitabine) for 72 h resulted in 1.2% to 4.6% apoptosis (Figure 4A). Only low levels of apoptosis were detected with single-agent treatment. Figure 4B has representative TUNEL data of MIA-PaCa-2 cells pretreated with propofol (50 μmol/mL) for 24 h followed by coincubation with gemcitabine (25 μmol/L) for 72 h. Apoptosis measured by ELISA and DNA ladder assays gave the same results (data not shown). Figure 4C has Representative DNA ladder assay data of MIA-PaCa-2 cells pretreated with propofol (50 μmol/mL) for 24 h followed by coincubation with gemcitabine (25 μmol/L) for 72 h.
Figure 4.
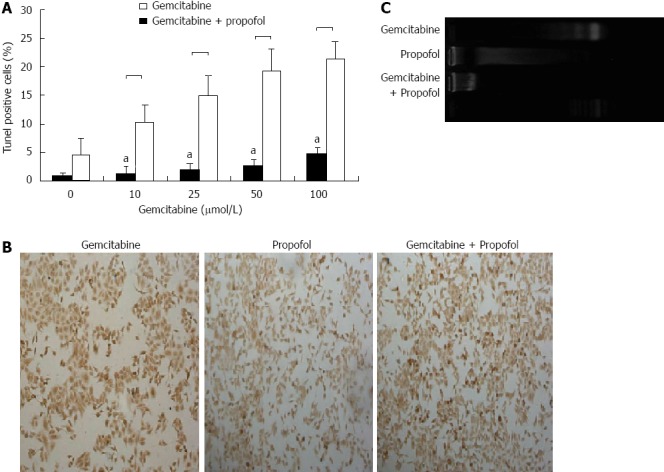
Evaluation of apoptosis by terminal transferase dUTP nick-end labeling and DNA ladder assays in pancreatic cancer MIA-PaCa-2 cells after propofol pretreatment. A: Sensitization of pancreatic tumor MIA-PaCa-2 cells to propofol- and/or gemcitabine-induced apoptosis measured by terminal transferase dUTP nick-end labeling (TUNEL) assay after 24 h of pretreatment with propofol (50 μmol/mL), gemcitabine (0-100 μmol/L), or propofol and gemcitabine combined for 72 h. Increased apoptosis was evident in the combination treatment group relative to individual treatment groups. aP < 0.05 vs propofol and gemcitabine combined group; B: Representative TUNEL image of MIA-PaCa-2 cells pretreated with propofol (50 μmol/mL) for 24 h followed by coincubation with gemcitabine (50 μmol/L) for 72 h; C: Representative DNA ladder image of MIA-PaCa-2 cells pretreated with propofol (50 μmol/mL) for 24 h followed by coincubation with gemcitabine (50 μmol/L) for 72 h.
MIA-PaCa-2 cells pretreated with propofol (25 μmol/mL) for 24 h, followed by coincubation with gemcitabine (10, 25, 50 and 100 μmol/L) for 72 h, showed significant cell apoptosis by DNA ladder, TUNEL assay and ELISA assay. These results were consistent with the growth inhibition MTT assays, and suggested that loss of viable cells by propofol and gemcitabine was partly due to the induction of apoptosis.
Propofol inhibits NF-κB DNA-binding activity
Consistent with earlier reports, constitutively active NF-κB DNA-binding activity was found in nuclear extracts from MIA-PaCa-2 cells. Band specificity was confirmed by supershift. Based on reports indicating the potential of propofol to abrogate constitutive and inducible NF-κB in halothane-induced rat liver[23], we analyzed whether propofol abrogated basal constitutive activation of NF-κB in MIA-PaCa-2 cells. To evaluate the effect of propofol in MIA-PaCa-2 cells, semiconfluent cells were treated with 0, 10, 25, 50 or 100 μmol/mL propofol for 72 h. As shown in Figure 5A, incubation with 50 μmol/mL propofol for 72 h resulted in a significant decrease in NF-κB DNA-binding activity in the MIA-PaCa-2 cells, and incubation with 100 μmol/mL propofol for 72 h resulted in complete disappearance of the activity. MIA-PaCa-2 cells treated with 100 μmol/L propofol for 24-72 h resulted in gradually reduced NF-κB DNA-binding activity (Figure 5B). These results clearly suggested that propofol was effective at downregulating NF-κB DNA-binding activity. We found no alterations in the nuclear protein content of retinoblastoma, which was used as protein loading control.
Figure 5.
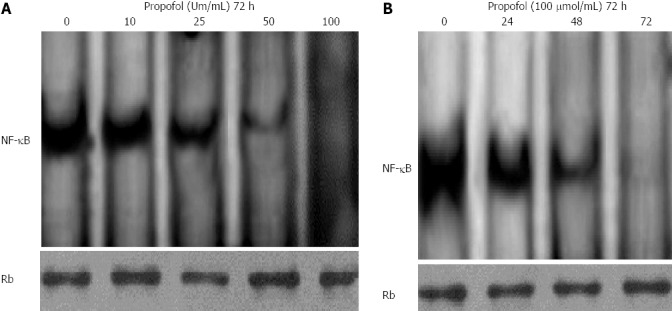
Propofol inhibits constitutively active nuclear factor-κB in MIA-PaCa-2 cells. A: MIA-PaCa-2 cells were treated with 0, 10, 25, 50, or 100 μmol/mL propofol for 72 h and nuclear extracts were probed for nuclear factor-κB (NF-κB) binding to a DNA consensus sequence. Results of NF-κB DNA-binding activity by electrophoretic mobility shift assay (EMSA); B: MIA-PaCa-2 cells were treated with 100 μmol/mL propofol for 24, 48, or 72 h and nuclear extracts were probed for NF-κB binding to a DNA consensus sequence. Results of NF-κB DNA-binding activity by EMSA. Retinoblastoma protein in the nuclear extract was used as a loading control.
Propofol abrogates NF-κB activation induced by gemcitabine
Next, we analyzed whether gemcitabine induced NF-κB DNA-binding activity and whether inactivation of NF-κB by propofol abrogated the chemoresistant phenotype of MIA-PaCa-2 cells, resulting in more pronounced gemcitabine-induced apoptosis. First, we analyzed dose and time responses to gemcitabine by induction of NF-κB in MIA-PaCa-2 cells. Nuclear extracts were prepared from MIA-PaCa-2 cells treated with 10, 25, 50 or 100 μmol/L gemcitabine for up to 72 h and analyzed for NF-κB DNA-binding activity by EMSA. Our results showed dose escalation of gemcitabine, with significant upregulation of constitutive NF-κB DNA-binding activity after gemcitabine treatment (Figure 6A). Pretreatment of cells with 100 μmol/mL propofol for 24 h abrogated gemcitabine-induced activation of NF-κB DNA-binding activity (Figure 6B). These results showed that propofol downregulates NF-κB DNA-binding activity in unstimulated conditions and inhibits gemcitabine- induced NF-κB activity. This might be the molecular mechanism of gemcitabine-induced cell death in propofol-pretreated cells.
Figure 6.
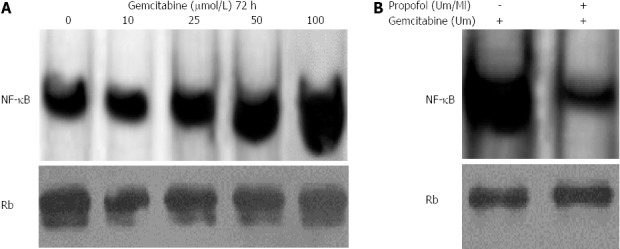
Propofol abrogates gemcitabine-induced nuclear factor-κB in MIA-PaCa-2 cells. A: MIA-PaCa-2 cells were exposed to 10-100 μmol/L gemcitabine for 72 h. Nuclear extracts were analyzed by electrophoretic mobility shift assay (EMSA); B: Propofol abrogated gemcitabine induced nuclear factor-κB (NF-κB) in MIA-PaCa-2 cells exposed to 100 μmol/L/mL propofol for 24 h followed by incubation with 100 μmol/L gemcitabine. Nuclear extracts were harvested at 72 h and analyzed by EMSA. Propofol pretreatment downregulated gemcitabine-induced NF-κB with 100 μmol/L gemcitabine.
DISCUSSION
Despite rapid advances in diagnostic and operative techniques, pancreatic cancer remains one of the most difficult human malignancies to treat. This is partly due to the advanced stage of the disease and de novo chemoresistant behavior towards cytotoxic chemotherapeutic agents and/or radiotherapy. In recent years, this problem has been addressed by a combinatorial approach. Several randomized studies have shown a significant increase in patient response rates when different classes of chemotherapeutic agents are used in combination. However, a major problem is high treatment-associated toxicity with no added benefit in significant overall survival[24-26]. The FOLFIRINOX regimen (bolus and infusional 5-fluorouracil, leucovorin, irinotecan, and oxaliplatin) is a new option for patients with metastatic pancreatic cancer and has good performance. Compared with gemcitabine, FOLFIRINOX was associated with a survival advantage, but had increased toxicity[27]. These limitations could be overcome by rational chemotherapeutic combinations in which toxic agents are used in lower doses, and treatment efficacy is increased by use of a nontoxic agent with a different mechanism of action.
Propofol is a fairly new induction agent introduced in the 1980s. Animal studies have demonstrated the neuroprotective ability of propofol. Recent studies of propofol effects during global cerebral ischemia-reperfusion injury in rats (4-vessel occlusion) concluded that propofol inhibited neuronal death induced by brain ischemia[10,14,28]. Propofol is also a glutamate antagonist at the NMDA-receptor level and a calcium-channel antagonist[29], has GABAergic activity and antioxidant properties[29], and reduces excitotoxicity[30]. Increasing evidence suggests that propofol has anticancer properties[17-20].
In this study, we used propofol in combination with a commonly used chemotherapeutic agent, gemcitabine, and tested its efficacy against the pancreatic cancer cell line MIA-PaCa-2. This preclinical study documented that sensitization of cancer cells was achieved by propofol during gemcitabine-induced killing, as shown by more pronounced cell death compared with single-agent treatment. We found that propofol pretreatment significantly enhanced tumor cell killing compared with either agent alone. This observation is important because 75% growth inhibition could be achieved using the same doses of gemcitabine that produced only 18% growth inhibition when gemcitabine was used alone. Consistent with the previously observed apoptotic effect of propofol, we showed that propofol alone not only significantly promoted pancreatic cancer MIA-PaCa-2 cell death and apoptosis, but also promoted gemcitabine-induced apoptosis as determined by MTT, ELISA, TUNEL staining and DNA ladder assays. In addition, we found for the first time that propofol inhibited the NF-κB activity in the MIA-PaCa-2 cells as demonstrated by EMSA assay. Together, these observations suggested that propofol strongly sensitized pancreatic cancer cells to gemcitabine-induced apoptosis.
Our results also showed that gemcitabine alone activated NF-κB, resulting in reduced apoptosis. This supported the model that NF-κB activation inhibits apoptosis. In addition, our in vitro results showed that propofol alone or propofol pretreatment followed by gemcitabine treatment abrogated NF-κB activation and increased the apoptotic index, suggesting that inhibition of NF-κB is mechanistically associated with sensitization of pancreatic cancer cells to apoptosis.
In conclusion, our findings are consistent with the hypothesis that chemosensitivity of pancreatic cancer cells is enhanced by pretreatment with propofol and that this effect is mediated by inactivation of NF-κB DNA-binding activity leading to apoptosis. Although in clinical practice the use of propanolol in combination with gemcitabine might not result in this enhancement, our findings provide a new insight into propofol in cancer treatment. Our results suggest that propofol can be an anesthetic agent that reduces pain and might also be important in inhibiting the growth of pancreatic cancer cells in pancreatic cancer therapy.
COMMENTS
Background
Propofol is a popular agent for anesthesia and long-term sedation. Recently, propofol was found to be effective at inducing apoptosis and possibly contributing to anti-tumor activity.
Research frontiers
Propofol was found to effectively induce apoptosis and possibly contribute to anti-tumor activity. Research is needed on whether propofol might inhibit growth, promote apoptosis, and increase gemcitabine sensitivity in pancreatic cancer cells.
Innovations and breakthroughs
Propofol has anticancer properties. The combination of propofol and docosahexaenoate or propofol and eicosapentaenoate might significantly induce apoptosis and inhibit cell adhesion and migration in breast cancer cells. The results of this study show that pretreatment of cells with propofol for 24 h followed by gemcitabine resulted in 24%-75% growth inhibition compared with 6%-18% for gemcitabine used alone. Overall growth inhibition was directly correlated with apoptosis. The authors also showed that propofol potentiated gemcitabine-induced killing by downregulation of nuclear factor-κB (NF-κB). In contrast, NF-κB was upregulated when pancreatic cancer cells were exposed to gemcitabine alone, suggesting a potential mechanism of acquired chemoresistance.
Applications
This study suggests that inactivation of the NF-κB signaling pathway by propofol could abrogate gemcitabine-induced activation of NF-κB, resulting in the chemosensitization of pancreatic tumors to gemcitabine.
Terminology
Propofol is an intravenous anesthetic used to induce and maintain anesthesia, and to sedate and calm patients in intensive care units.
Peer review
In this experimental study, the authors show that propofol induces apoptosis and increases in vitro gemcitabine sensitivity in pancreatic cancer cells by inhibition of NF-κB activity. The manuscript is interesting.
Footnotes
P- Reviewers Manfredi S, Sperti C S- Editor Gou SX L- Editor Logan S E- Editor Ma S
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.O’Reilly EM, Abou-Alfa GK. Cytotoxic therapy for advanced pancreatic adenocarcinoma. Semin Oncol. 2007;34:347–353. doi: 10.1053/j.seminoncol.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Long J, Zhang Y, Yu X, Yang J, LeBrun DG, Chen C, Yao Q, Li M. Overcoming drug resistance in pancreatic cancer. Expert Opin Ther Targets. 2011;15:817–828. doi: 10.1517/14728222.2011.566216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 5.Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- 6.Pan X, Arumugam T, Yamamoto T, Levin PA, Ramachandran V, Ji B, Lopez-Berestein G, Vivas-Mejia PE, Sood AK, McConkey DJ, et al. Nuclear factor-kappaB p65/relA silencing induces apoptosis and increases gemcitabine effectiveness in a subset of pancreatic cancer cells. Clin Cancer Res. 2008;14:8143–8151. doi: 10.1158/1078-0432.CCR-08-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kong R, Sun B, Jiang H, Pan S, Chen H, Wang S, Krissansen GW, Sun X. Downregulation of nuclear factor-kappaB p65 subunit by small interfering RNA synergizes with gemcitabine to inhibit the growth of pancreatic cancer. Cancer Lett. 2010;291:90–98. doi: 10.1016/j.canlet.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Ahn DW, Seo JK, Lee SH, Hwang JH, Lee JK, Ryu JK, Kim YT, Yoon YB. Enhanced antitumor effect of combination therapy with gemcitabine and guggulsterone in pancreatic cancer. Pancreas. 2012;41:1048–1057. doi: 10.1097/MPA.0b013e318249d62e. [DOI] [PubMed] [Google Scholar]
- 9.Guo HC, Bu HQ, Luo J, Wei WT, Liu DL, Chen H, Tong HF, Wang ZH, Wu HY, Li HH, et al. Emodin potentiates the antitumor effects of gemcitabine in PANC-1 pancreatic cancer xenograft model in vivo via inhibition of inhibitors of apoptosis. Int J Oncol. 2012;40:1849–1857. doi: 10.3892/ijo.2012.1389. [DOI] [PubMed] [Google Scholar]
- 10.Wang YW, Wang SJ, Zhou YN, Pan SH, Sun B. Escin augments the efficacy of gemcitabine through down-regulation of nuclear factor-κB and nuclear factor-κB-regulated gene products in pancreatic cancer both in vitro and in vivo. J Cancer Res Clin Oncol. 2012;138:785–797. doi: 10.1007/s00432-012-1152-z. [DOI] [PubMed] [Google Scholar]
- 11.Velly LJ, Guillet BA, Masmejean FM, Nieoullon AL, Bruder NJ, Gouin FM, Pisano PM. Neuroprotective effects of propofol in a model of ischemic cortical cell cultures: role of glutamate and its transporters. Anesthesiology. 2003;99:368–375. doi: 10.1097/00000542-200308000-00018. [DOI] [PubMed] [Google Scholar]
- 12.Zheng YY, Lan YP, Tang HF, Zhu SM. Propofol pretreatment attenuates aquaporin-4 over-expression and alleviates cerebral edema after transient focal brain ischemia reperfusion in rats. Anesth Analg. 2008;107:2009–2016. doi: 10.1213/ane.0b013e318187c313. [DOI] [PubMed] [Google Scholar]
- 13.Cui D, Wang L, Qi A, Zhou Q, Zhang X, Jiang W. Propofol prevents autophagic cell death following oxygen and glucose deprivation in PC12 cells and cerebral ischemia-reperfusion injury in rats. PLoS One. 2012;7:e35324. doi: 10.1371/journal.pone.0035324. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Xi HJ, Zhang TH, Tao T, Song CY, Lu SJ, Cui XG, Yue ZY. Propofol improved neurobehavioral outcome of cerebral ischemia-reperfusion rats by regulating Bcl-2 and Bax expression. Brain Res. 2011;1410:24–32. doi: 10.1016/j.brainres.2011.06.060. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Han B, Ma X, Qi S. The effects of propofol on hippocampal caspase-3 and Bcl-2 expression following forebrain ischemia-reperfusion in rats. Brain Res. 2010;1356:11–23. doi: 10.1016/j.brainres.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Ye HH, Wu KJ, Fei SJ, Zhang XW, Liu HX, Zhang JL, Zhang YM. Propofol participates in gastric mucosal protection through inhibiting the toll-like receptor-4/nuclear factor kappa-B signaling pathway. Clin Res Hepatol Gastroenterol. 2013;37:e3–15. doi: 10.1016/j.clinre.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 17.Siddiqui RA, Zerouga M, Wu M, Castillo A, Harvey K, Zaloga GP, Stillwell W. Anticancer properties of propofol-docosahexaenoate and propofol-eicosapentaenoate on breast cancer cells. Breast Cancer Res. 2005;7:R645–R654. doi: 10.1186/bcr1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu KC, Yang ST, Hsia TC, Yang JS, Chiou SM, Lu CC, Wu RS, Chung JG. Suppression of cell invasion and migration by propofol are involved in down-regulating matrix metalloproteinase-2 and p38 MAPK signaling in A549 human lung adenocarcinoma epithelial cells. Anticancer Res. 2012;32:4833–4842. [PubMed] [Google Scholar]
- 19.Zhang L, Wang N, Zhou S, Ye W, Jing G, Zhang M. Propofol induces proliferation and invasion of gallbladder cancer cells through activation of Nrf2. J Exp Clin Cancer Res. 2012;31:66. doi: 10.1186/1756-9966-31-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Q, Zhang L, Han Y, Jiang Z, Wang Q. Propofol reduces MMPs expression by inhibiting NF-κB activity in human MDA-MB-231 cells. Biomed Pharmacother. 2012;66:52–56. doi: 10.1016/j.biopha.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Lai HC, Yeh YC, Wang LC, Ting CT, Lee WL, Lee HW, Wang KY, Wu A, Su CS, Liu TJ. Propofol ameliorates doxorubicin-induced oxidative stress and cellular apoptosis in rat cardiomyocytes. Toxicol Appl Pharmacol. 2011;257:437–448. doi: 10.1016/j.taap.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 22.Arlt A, Gehrz A, Müerköster S, Vorndamm J, Kruse ML, Fölsch UR, Schäfer H. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene. 2003;22:3243–3251. doi: 10.1038/sj.onc.1206390. [DOI] [PubMed] [Google Scholar]
- 23.Brasil LJ, San-Miguel B, Kretzmann NA, Amaral JL, Zettler CG, Marroni N, González-Gallego J, Tuñón MJ. Halothane induces oxidative stress and NF-kappaB activation in rat liver: protective effect of propofol. Toxicology. 2006;227:53–61. doi: 10.1016/j.tox.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 24.Berlin JD, Rothenberg ML. Chemotherapeutic advances in pancreatic cancer. Curr Oncol Rep. 2003;5:219–226. doi: 10.1007/s11912-003-0113-8. [DOI] [PubMed] [Google Scholar]
- 25.Diaz-Rubio E. New chemotherapeutic advances in pancreatic, colorectal, and gastric cancers. Oncologist. 2004;9:282–294. doi: 10.1634/theoncologist.9-3-282. [DOI] [PubMed] [Google Scholar]
- 26.Arends JJ, Sleeboom HP, Leys MB, Ten Bokkel Huinink D, de Jong RS, Smit JM, Nortier JW, Tesselaar ME. A phase II study of raltitrexed and gemcitabine in patients with advanced pancreatic carcinoma. Br J Cancer. 2005;92:445–448. doi: 10.1038/sj.bjc.6602368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 28.Ergün R, Akdemir G, Sen S, Taşçi A, Ergüngör F. Neuroprotective effects of propofol following global cerebral ischemia in rats. Neurosurg Rev. 2002;25:95–98. doi: 10.1007/s101430100171. [DOI] [PubMed] [Google Scholar]
- 29.Fu L, Tang R, Bao N, Wang J, Ma H. Ketamine and propofol in combination induce neuroapoptosis and down-regulate the expression of N-methyl-D-aspartate glutamate receptor NR2B subunit in rat forebrain culture. Pharmazie. 2011;66:771–776. [PubMed] [Google Scholar]
- 30.Huang Y, Zitta K, Bein B, Scholz J, Steinfath M, Albrecht M. Effect of propofol on hypoxia re-oxygenation induced neuronal cell damage in vitro*. Anaesthesia. 2013;68:31–39. doi: 10.1111/j.1365-2044.2012.07336.x. [DOI] [PubMed] [Google Scholar]