

Abstract
AIM: To examine transforming growth factor-β1 (TGF-β1) promoter methylation in gastric cancer and to determine if Helicobacter pylori (H. pylori) or interleukin (IL)-1β could induce TGF-β1 hypermethylation in vitro.
METHODS: We examined the frequency and extent of TGF-β1 promoter methylation using methylation-specific PCR in the gastric tissues from 47 gastric cancer patients and 39 non-gastric cancer subjects. H. pylori infection was confirmed by a positive result from either a serological test, histological analysis or C13 urea breath test. GES-1 and MKN-45 cells co-cultured with H. pylori or treated with IL-1β for 12, 24 and 48 h in vitro tested the effects of H. pylori or IL-1β on TGF-β1.
RESULTS: Twenty-four/forty-seven (51%) cases of gastric cancer (GC) tissues showed TGF-β1 promoter methylation, 15/47 (31.9%) cases of matched non-cancerous gastric mucosa tissues from the GC patients, and 11/39 (28%) case of the normal gastric mucosa tissues from non-GC subjects showed TGF-β1 promoter methylation (51% vs 28%, P < 0.05). Significantly higher levels of methylation of TGF-β1 were found in the tumor tissues than in non-tumor tissues from GC patients (0.24 ± 0.06 vs 0.17 ± 0.04, P < 0.05) and normal gastric tissues from non-GC subjects (0.24 ± 0.06 vs 0.15 ± 0.03, P < 0.05). TGF-β1 methylation was found in 48.3% of H. pylori-positive gastric mucosal tissues whereas only 23.1% of H. pylori-negative gastric mucosal tissues showed TGF-β1 methylation (48.3% vs 23.1%, P < 0.05). IL-1β appeared to induce a dose-dependent methylation of TGF-β1 and the strongest methylation was observed in GES-1 cells treated with 2.5 ng/mL of IL-1β for 48 h. Further studies showed that pre-treatment of GES-1 cells with 20 ng/mL IL-1RA for 1 h could partially abolish the effect of IL-1β on TGF-β1 methylation. Infection of GES-1 cells by H. pylori was not found to induce significant TGF-β1 promoter methylation.
CONCLUSION: Our data revealed that TGF-β1 promoter is methylated in GC patients. IL-1β may be an important mediator for H. pylori induced gene methylation during GC development.
Keywords: Transforming growth factor-β1, Interleukin-1β, Methylation, Helicobacter pylori, Gastric cancer
Core tip: In vitro studies showed that GES-1 cells exposed to Helicobacter pylori (H. pylori) did not show significant transforming growth factor-β1 (TGF-β1) methylation. However, treatment of the GES-1 cells with interleukin (IL)-1β led to a dose-dependent methylation of TGF-β1, which was partially abolished by IL-1RA. The high levels of TGF-β1 promoter methylation in H. pylori positive patients was likely the result of H. pylori-induced inflammation rather than H. pylori itself. IL-1β may be an important mediator for H. pylori-induced gene methylation during gastric cancer development.
INTRODUCTION
Genetic and epigenetic alterations in tumor suppressor genes or oncogenes are implicated in cancer formation. DNA methylation is the major form of epigenetic change in eukaryotic genomes. It involves the addition of a methyl group to the carbon 5 position of the cytosine ring within the CpG dinucleotide. CpG islands (CGIs) are regions of the genome that contain a large number of CpG dinucleotide repeats. In mammalian genomes, CGIs usually extend for 300-3000 base pairs. They are located within, and close to, sites of about 40% of gene promoters. It is estimated that in mammalian genomes, about 80% of CpG dinucleotides are methylated. However, CpG dinucleotides in regions abundant in GC pairs, such as CGIs, are normally protected from DNA methylation, and this is an important controlling mechanism for gene promoters and gene expression[1]. Although most CGIs linked to promoters are non-methylated, recent studies have revealed that promoter CpG hypermethylation, associated with transcriptional inactivation, may play a pivotal role in tumorigenesis[2].
Gastric cancer (GC) remains a major health threat because of its high incidence, poor prognosis and limited treatment options. Multiple epigenetic and genetic alterations have been identified in GC patients. High levels of aberrant CpG island methylation and DNA methylation in the gastric mucosae correlate with increased GC risk, and as such, they are increasingly recognized as candidate markers for GC[3].
Clinical and epidemiological studies have demonstrated that Helicobacter pylori (H. pylori) infection is correlates strongly with aberrant methylation in GC, whereas eradication of H. Pylori significantly reduces gene methylation[4,5]. Clearly, H. Pylori-induced aberrant methylation plays a role in GC formation. However, the precise molecular mechanisms of how H. Pylori might induce aberrant CpG island methylation remain elusive.
Chronic inflammation is a well-known promoting factor for many cancers. Approximately 15%-20% of all human cancers are related to chronic inflammation[6]. GC is a typical inflammation-related malignancy, being closely linked to H. Pylori-induced chronic inflammation in gastric mucosa. Chronic inflammation in the esophagus and colon may precipitate aberrant methylation, but whether H. pylori itself or the chronic inflammation caused by H. pylori infection induces methylation in CGIs remains controversial[7,8].
H. pylori infection is characterized by infiltration of inflammatory cells, such as neutrophils and lymphocytes, into the gastric mucosa, as well as increased production of inflammatory cytokines[9,10]. Interleukin (IL)-1β is a pro-inflammatory cytokine primarily secreted by activated monocytes/macrophages in response to bacterial infection. IL-1β mediates many pathophysiological events during host-environment interactions. Recent studies have demonstrated that the levels of several inflammatory cytokines including IL-1β, IL-6, IL-8 and tumor necrosis factor α (TNF-α) are significantly higher in the gastric mucosal tissues from H. pylori-positive patients than those from H. pylori-negative patients[11-13]. It was further demonstrated that IL-1β could directly induce promoter methylation of E-cadherin, an important extracellular matrix component involved in the maintenance of epithelial stability: H. pylori-induced methylation of E-cadherin promoter was mediated through IL-1β[14].
Transforming growth factor-β1 (TGF-β1) is an anti-inflammatory cytokine with multiple, and perhaps even opposite, biological effects in many tissues. TGF-β1 was shown to inhibit the growth of epithelial cells, but stimulates the proliferation of mesenchymal cells[15,16]. Many studies have shown that TGF-β1 is overexpressed in epithelial cancers and exerts its transforming potential through several mechanisms, such as stimulating the progression of stromal cells, promoting angiogenesis and suppressing immune surveillance[17]. However, TGF-β1 was reported to function as a tumor suppressor, because it could inhibit potently the proliferation of many types of cancer cells derived from breast, prostate, lung, colon and liver[18,19]. Furthermore, methylation-induced silencing of TGF-β1 has been implicated in the development of several solid tumors, and silencing of TGF-β1 signaling through methylation of the gene encoding its receptor have been reported[20,21].
It has been reported that the expression level of host TGF-β1 in gastric mucosa was an important determinant for the pathogenesis of H. pylori-associated gastric diseases[22-24]. The gastric mucosa of TGF-β1 null mice exhibit similar changes to those observed in H. pylori-associated gastritis, and these mice were found to progressively develop inflammatory diseases and die within 3-4 wk of birth[25]. However, the role of TGF-β1 methylation in the development of H. pylori-related GC remains largely unknown.
In this study, we aimed investigate if H. pylori could induce promoter methylation of TGF-β1 in GC and whether IL-1β plays a role in this process.
MATERIALS AND METHODS
Patients and specimens
This study involved 47 consecutive GC patients (35 males and 12 females, mean age 56.2 years) who underwent gastrectomy and 39 consecutive non-GC subjects (28 males and 11 females, mean age 52.1 years) who underwent upper gastroduodenoscopy. H. pylori infection was confirmed by a positive result from either one of the following diagnostic approaches: serological test, histological analysis or C13 urea breath test. No patients received prior H. pylori eradication therapy. Ninety-four gastric specimens (two from each patient, from tumor and non-tumor gastric mucosa) from GC patients and 39 gastric mucosa tissues from non-GC subjects were collected. The tissues were snap-frozen in liquid nitrogen and subsequently stored at -80 °C for the studies described below. Patients who received eradication therapy for H. pylori before the study and those with severe systemic diseases (such as major organ failure, server infection, autoimmune disease, organ transplantation and immunosuppressive therapy) were excluded.
Cell culture
Human gastric epithelium cell line GES-1 was kindly provided by Professor Bingdong Zhu (School of Basic Medical Sciences, Lanzhou University). Cells were cultured in DMEM medium containing 10% FBS supplemented with 100 IU/mL penicillin, 100 IU/mL streptomycin and maintained at 37 °C in a humidified atmosphere with 5% CO2.
Bacterial strain and conditions
NCTC11637, a CagA-positive strain of H. pylori, was purchased from the American Type Cell Culture (ATCC) (Rockville, MI, United States). H. pylori were cultured on 4.2% Columbia Blood A gar (Youkang Foundation of Biological Science and Technology Beijing Co. Ltd. Beijing, China) containing 7.5% normal sheep blood and 0.5% antibiotics (vancomycin, 10 mg/mL; polymyxin, 0.025 mg/mL; and amphotericin B, 10 mg/mL) under microaerophilic conditions for 72 h. Bacteria were harvested and re-suspended in sterile phosphate buffered saline (PBS) and counted by absorbance at 660 nm (1 OD660 = 1 × 108 colony forming units/mL).
Infection of gastric epithelial cells by H. pylori
To establish an in vitro model of H. pylori infected gastric mucosa, GES-1 cells were grown to 80% confluence under the above-mentioned conditions. Cells were infected with live H. pylori at H. pylori/cell ratios of 5:1, 10:1, 50:1, and 100:1. To determine the involvement of IL-1β in H. pylori-induced pathology, GES-1 cells were pre-treated with human interleukin-1 receptor antagonist (IL-1RA) (Peprotech, Rocky Hill, NJ, United States) for 1 h before H. pylori infection. IL-1RA was used at various concentrations (10, 20, 50 and 100 ng/mL) to select the concentration at which it can effectively block the IL-1β signaling. 20 ng/mL for 48 h was found to be an effective dose. All cells were cultured in 6-well plates at 37 °C in a humidified atmosphere for 12, 24 and 48 h.
Treatment of GES-1 cells with IL-1
GES-1cells were pre-treated with or without various concentrations of IL-1RA (10, 20, 50 and 100 ng/mL) for 1 h, followed by treatment with different concentrations IL-1β (Peprotech, Rocky Hill, NJ, United States) (0.1, 0.25, 1.0 and 2.5 ng/mL) for 12, 24, and 48 h in fresh serum-free DMEM medium.
Methylation-specific polymerase chain reaction
Genomic DNA was extracted from each sample using the TIANamp Genomic DNA Kit (Tiangen Biotech Beijing Co. Ltd, Beijing, China), according to the manufacturer’s instructions. The extracted DNA was treated with sodium bisulfite using the EZ DNA Methylation-GoldTM Kit (Zymo Research, Los Angeles, CA, United States), according to the manufacturer’s instructions. After bisulfite treatment, DNA was purified using a Zymo-SpinTM IC Column (Zymo Research, Los Angeles, CA, United States) and resuspended in 10 μL of dilution buffer (M-Elution Buffer, Zymo Research, Los Angeles, CA, United States). DNA methylation of the TGF-β1 promoter was analyzed by methylation-specific polymerase chain reaction (MSPCR), using Zymo TaqTM PreMix (Zymo Research), according to the manufacturer’s instructions. Briefly, the bisulfite-modified DNA (2 μL) was amplified using specific primers for methylated and unmethylated sequences of TGF-β1. The primer sequences used in the study were are as follows. For methylated TGF-β1, forward: TATATCGTTCGTAAAGTTATAGCGT, reverse: AACATAAAAAAACTAAACCACCGTC. For unmethylated TGF-β1, forward: ATTTATATTGTTTGTAAAGTTATAGTGT, and reverse: AACATAAAAAAACTAAACCACCATC. PCR was performed in a 50-μL reaction system, which contains Zymo TaqTM PreMix (25 μL), forward primer (10 μmol/L) 4 μL, reverse primer (10 μmol/L) 4 μL, DNA template (2 μL), and double distilled water (15 μL). The reactions were hot-started at 97 °C for 10 min, followed by 40 cycles of reactions (15 s at 95 °C, 35 s for annealing, and 30 s at 72 °C) and a final 7-min extension in a Thermal Cycler (Veriti, ABI Co., Foster, CA, United States). For positive controls, we used CpGenome Universal Methylated DNA (Intergen, New York, NY, United States). Five microliters of PCR products were separated by electrophoresis on 2% agarose gel stained with ethidium bromide, and imaged using a VersaDoc Imaging System (Bio-Rad Laboratories Co., Ltd. Hercules, CA, United States).
Densitometric analysis of TGF-β1 methylation Levels
Quantity One software v4.62 (Bio-Rad Laboratories Co., Ltd. Hercules, CA, United States) was used to perform densitometric analyses of methylated and un-methylated bands of TGF-β1, and the results were presented as the mean of three independent experiments. The methylation levels were calculated as the ratio of the value of methylated band to methylated plus unmethylated bands.
Statistical analysis
The frequencies of promoter methylation were compared between the two groups by two-sided Fisher’s exact test or Pearson χ2 test. Differences in methylation levels of TGF-β1 between two groups were examined by Student’s t test. A P value of < 0.05 was considered statistically significant.
RESULTS
SMethylation frequency and levels of TGF-β1 in GC and non-GC subjects
As shown in Table 1, 24/47 (51%) cases of GC tissues showed TGF-β1 promoter methylation, 15/47 (31.9%) cases of matched non-cancerous gastric mucosa tissues from the GC patients, and 11/39 (28%) cases of the normal gastric mucosa tissues from non-GC subjects showed TGF-β1 promoter methylation (51% vs 28%, P = 0.032).
Table 1.
Methylation frequency for transforming growth facto-β1 in gastric tissues
| Case ID |
GC patients (n = 47) |
Non-GC subjects (n = 39) |
|||
| H. pylori |
TGF-β1 methylation |
||||
| Tumor | Non-tumor | H. pylori | TGF-β1 methylation | ||
| 1 | + | Methylated | Methylated | + | |
| 2 | + | + | Methylated | ||
| 3 | + | Methylated | Methylated | + | Methylated |
| 4 | + | Methylated | + | ||
| 5 | + | Methylated | + | ||
| 6 | + | + | |||
| 7 | + | + | |||
| 8 | + | + | |||
| 9 | + | Methylated | + | ||
| 10 | + | Methylated | Methylated | + | |
| 11 | + | + | Methylated | ||
| 12 | + | + | |||
| 13 | + | Methylated | + | Methylated | |
| 14 | + | + | |||
| 15 | + | Methylated | Methylated | + | |
| 16 | + | + | Methylated | ||
| 17 | + | + | |||
| 18 | + | Methylated | + | Methylated | |
| 19 | + | Methylated | Methylated | + | |
| 20 | + | + | Methylated | ||
| 21 | + | Methylated | Methylated | + | |
| 22 | + | + | Methylated | ||
| 23 | + | - | Methylated | ||
| 24 | + | - | |||
| 25 | + | Methylated | - | ||
| 26 | + | Methylated | Methylated | - | |
| 27 | + | - | |||
| 28 | + | - | |||
| 29 | + | Methylated | Methylated | - | |
| 30 | + | Methylated | Methylated | - | Methylated |
| 31 | + | Methylated | - | ||
| 32 | + | - | |||
| 33 | + | - | |||
| 34 | + | Methylated | Methylated | - | Methylated |
| 35 | + | - | |||
| 36 | + | Methylated | - | ||
| 37 | + | Methylated | Methylated | - | |
| 38 | + | Methylated | - | ||
| 39 | - | - | |||
| 40 | - | Methylated | Methylated | ||
| 41 | - | Methylated | Methylated | ||
| 42 | - | ||||
| 43 | - | ||||
| 44 | - | ||||
| 45 | - | ||||
| 46 | - | Methylated | Methylated | ||
| 47 | - | ||||
GC: Gastric cancer; TGF-β1: Transforming growth factor-β1; H. pylori: Helicobacter pylori.
To evaluate the levels of TGF-β1 methylation, we analyzed quantitatively the MSPCR product bands by their fluorescence intensities. Typical MSPCR bands are shown in Figure 1A, and the quantitative data for the methylation levels of TGF-β1 in gastric tissues are shown in Figure 1B. Significantly higher levels of methylation of TGF-β1 were found in the tumor tissues (0.24 ± 0.06) than in non-tumor tissues from GC patients (0.17 ± 0.04) (P = 0.001) and normal gastric tissues from non-GC subjects (0.15 ± 0.03) (P = 0.001).
Figure 1.
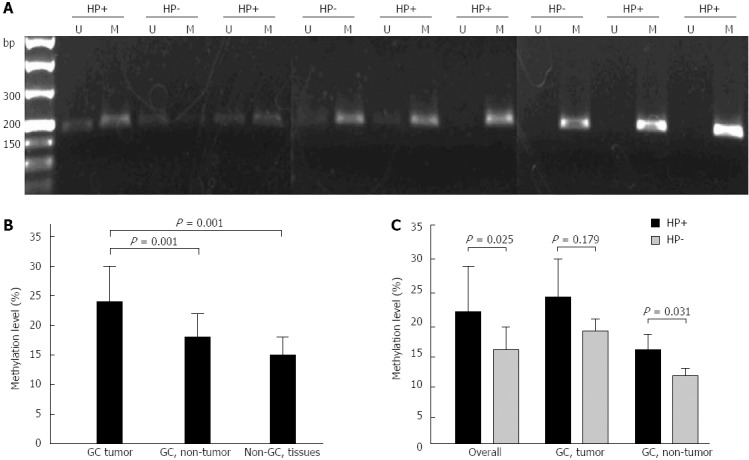
Detection of transforming growth factor-β1 promoter methylation by methylation-specific polymerase chain reaction. Genomic DNA was extracted from human gastric tissues. A: Representative methylation-specific polymerase chain reaction results from gastric cancer (GC) tissues with or without Helicobacter pylori (H. pylori) infection are shown; B: Levels of transforming growth factor-β1 (TGF-β1) promoter methylation in GC tissues, non-cancerous gastric mucosa from the GC patients (GC, non-tumor) and normal gastric mucosa from non-GC subjects (Non-GC tissues); C: Impact of H. pylori status on the levels of TGF-β1 promoter methylation in GC tissues (GC, tumor), non-cancerous gastric mucosa from the GC patients (GC, non-tumor) and combined samples (overall). HP+: H. pylori-positive; HP-: H. pylori-negative; U: Unmethylated; M: Methylated.
Methylation frequency and levels of TGF-β1 in H. pylori-positive and H. pylori-negative subjects
To examine the impact of H. pylori on the methylation of TGF-β1, we compared the methylation frequency of TGF-β1 in H. pylori-positive and H. pylori-negative GC tissues and non-cancerous gastric tissues. As shown in Table 1 and best shown in Table 2, 21/38 (55.3%) cases of H. pylori-positive GC tissues showed TGF-β1 methylation, whereas only 3/9 (33.3%) cases of H. pylori-negative GC tissues showed TGF-β1 methylation. In non-cancerous gastric tissues obtained from GC patients, the frequency of TGF-β1 methylation appeared to be similar between H. pylori-positive and H. pylori-negative gastric tissues (31.6% vs 33%, P = 0.919). In normal gastric mucosa from non-GC patients, H. pylori-positive tissues exhibited more frequent TGF-β1 methylation than the H. pylori-negative gastric tissues (36.4% vs 17.6%, P = 0.288). Overall, TGF-β1 methylation was found in 48.3% of H. pylori-positive gastric mucosal tissues, whereas only 23.1% of H. pylori-negative gastric mucosal tissues showed TGF-β1 methylation (48.3% vs 23.1%, P = 0.029).
Table 2.
Impact of Helicobacter pylori on the frequency of transforming growth factor-β1 methylation in gastric tissues
|
TGF-β1 methylation in GC patients |
TGF-β1 methylation in Non-GC patients |
Total1 | ||
| Tumor | Non-tumor | Normal gastric mucosa | ||
| H. pylori (+) | 21/38 (55.3%) | 12/38 (31.6%) | 8/22 (36.4%) | 41/98 (41.8%) |
| H. pylori (-) | 3/9 (33%) | 3/9 (33%) | 3/17 (17.6%) | 9/35 (25.7%) |
Combined tumor and non-tumor tissue. GC: Gastric cancer; TGF-β1: Transforming growth factor-β1; H. pylori: Helicobacter pylori.
Further densitometric analysis showed that H. pylori-positive GC tumor tissues exhibited much higher levels of TGF-β1 methylation than H. pylori-negative GC tumor tissues (Figure 1A and C). Furthermore, higher levels of TGF-β1 methylation were also found in H. pylori positive non-GC mucosal tissues (Figure 1C). Overall, more TGF-β1 methylation was present in H. pylori positive gastric mucosa than in H. pylori-negative gastric tissues (0.23 ± 0.06 vs 0.16 ± 0.03, P = 0.025) (Figure 1C).
Effect of IL-1β signaling on TGF-β1 promoter methylation
To examine the mechanisms of TGF-β1 methylation, GES-1 cells were incubated in the presence or absence of different concentrations of IL-1β (0.1, 0.25, 1.0 and 2.5 ng/mL) for 12, 24 and 48 h, and TGF-β1 promoter methylation was then measured by MSPCR. As shown in Figure 2A, IL-1β appeared to induce a dose-dependent methylation of TGF-β1, with the strongest methylation being observed in GES-1 cells treated with 2.5 ng/mL of IL-1β for 48 h. Further studies showed that pre-treatment of GES-1 cells with 20 ng/mL of IL-1RA for 1 h could partially abolish the effect of IL-1β on TGF-β1 methylation (Figure 2B).
Figure 2.
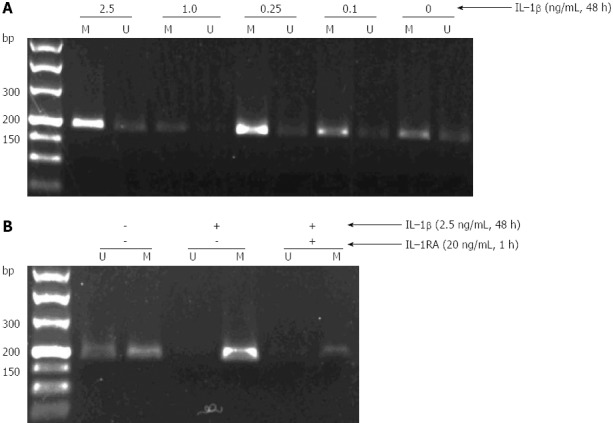
Induction of transforming growth factor-β1 methylation by interleukin-1β in GES-1 cells. A: Treatment of GES-1 cells by interleukin (IL)-1β led to a dose-dependent methylation of transforming growth factor (TGF)-β1; B: IL-1β-induced TGF-β1 methylation in GES-1cells was partially abolished by IL-1RA. U: Unmethylated; M: Methylated; H. pylori: Helicobacter pylori.
H. pylori alone dose not induce TGF-β1 methylation in vitro
Infection of GES-1 cells by H. pylori did not induce significant TGF-β1 promoter methylation (data not shown).
DISCUSSION
Promoter hypermethylation leading to epigenetic inactivation of tumor suppressor genes plays a pivotal role in tumorigenesis. Aging, chronic inflammation, and viral and bacterial infections promote methylation of promoter CpG islands and may represent the “environmental” triggers of carcinogenesis. The stomach is one of the organs that constantly undergoes DNA methylation of CpG islands in its epithelial cells. The oncogenic role of H. pylori for gastric malignancies, mainly gastric carcinoma and MALT lymphoma, has been well documented, and as such, H. pylori has been designated a Class I carcinogen. However, the mechanism by which H. pylori induces GC remained poorly defined.
In the present study, we have revealed, for the first time, that significantly more frequent and higher levels of TGF-β1 promoter methylation are present in GC patients than in non-cancerous controls. Although aging is a recognized risk factor for DNA methylation, from the current study, the impact of age (and sex) on TGF-β1 DNA methylation could be excluded, because our study populations in each group were well-balanced in their distribution of age and sex.
TGF-β1 shows biphasic effects in tumorigenesis[26]. In the initial stage, it may function as a tumor suppressor by inhibiting cell growth. This was demonstrated in breast cancer in which constitutive activation of the TGF-β1 pathway prolonged the latency of tumorigenesis or resulted in smaller tumor formation in mice[27,28]. However, in the later stage of tumorigenesis (i.e., when the tumors are well established), activation of the TGF-β1 signaling can strongly promote tumor progression[29].
Chronic inflammation-induced methylation of promoter CGIs has been closely linked to the development of human cancers[30,31]. In gastric cancer, whether the gene hypermethylation observed in H. pylori-infected individuals is caused directly by H. pylori, or the gene hypermethylation results from H. pylori-induced gastric inflammation remains a topic of debate. In our study, we first examined if H. pylori could directly cause methylation of TGF-β1. We made use of a gastric epithelial cell line GES-1, and co-cultured this cell line with H. pylori. Contrary to our initial hypothesis, H. pylori did not induce apparent promoter methylation of TGF-β1. However, treatment of GES-1 cells with IL-1β led to a marked methylation of the TGF-β1 promoter, and this was partially reversed by antagonizing IL-1β signaling using its receptor blocker IL-1RA. IL-1β is an important pro-inflammatory cytokine that initiates and amplifies the inflammatory responses to H. pylori infection. IL-1β is closely linked to DNA methylation of gastric epithelial cells, particularly in H. pylori infected individuals. We think that IL-1β may be an important mediator in H. pylori-induced TGF-β1 methylation[32,33].
Our in vitro data did not correlate with the in vivo data, which showed that H. pylori infected individuals, particularly H. pylori-positive GC tissues, showed more frequent and higher levels of TGF-β1 methylation. This inconsistency may be explained in several ways. Firstly, the acute infection of GES-1 cells by H. pylori may not be a valid model for studying the impact of H. pylori on gastric mucosa. It was reported that chronic, rather than acute, H. pylori infection was responsible for methylation induction. Secondly, H. pylori may not directly induce hypermethylation, but rather it induces inflammation and the production of pro-inflammatory cytokines, such IL-1β, is likely a contributing factor for TGF-β1 methylation. Our data support this view. Lastly, inflammatory cytokines or molecules may also be involved in H. pylori-induced methylation. For example, nitric oxide may be involved in H. pylori infection-related DNA methylation[34].
COMMENTS
Background
Hypermethylation of promoter CpG islands (CGIs) leads to functional silencing of some tumor suppressor genes and is thus involved in carcinogenesis. Chronic inflammation is closely associated with cancer formation. Inflammation-induced gene methylation is an important mechanism for inflammation-associated cancers, of which gastric cancer is a classical example. During gastric cancer formation, frequent aberrant CGI methylation has been reported and the role of Helicobacter pylori (H. pylori) infection during this process is controversial. Inactivation of transforming growth factor-β1 (TGF-β1) by promoter methylation has been implicated as an important mechanism for the development of several malignancies, such lung and prostate cancers; however, the role of TGF-β1 methylation in gastric cancer remains unknown.
Research frontiers
Multiple epigenetic and genetic alterations have been identified in gastric cancer (GC) patients. High level of aberrant CpG island methylation in the gastric mucosae is correlated with increased GC risk, and as such, they are increasingly recognized as candidate markers for GC. Clinical and epidemiological studies have demonstrated that H. pylori infection is strongly correlated with aberrant methylation in GC. However, the precise molecular mechanism by which H. pylori might induce aberrant CpG island methylation remain elusive. GC is a typical inflammation-related malignancy, being closely linked to H. pylori induced chronic inflammation in the gastric mucosa. Whether H. pylori itself or the chronic inflammation caused by H. pylori infection can induce methylation in CGIs remains controversial.
Innovations and breakthroughs
TGF-β1 is an anti-inflammatory cytokine with multiple effects in many tissues. Many studies have shown that TGF-β1 is overexpressed in epithelial cancers and exerts its transforming potential through several mechanisms, such as stimulating the progression of stromal cells, promoting angiogenesis and suppressing immune surveillance. In the present study, the authors have revealed, for the first time, that significantly more frequent and higher levels of TGF-β1 promoter methylation is present in GC patients with H. pylori infection than in non-cancerous controls. In vitro studies showed that normal gastric epithelial cell line GES-1 cells exposed to H. pylori did not show significant TGF-β1 methylation. However, treatment of the GES-1 cells with IL-1β led to a dose-dependent methylation of TGF-β1. The data show that H. pylori may not directly induce hypermethylation, but rather this bacteria induced inflammation and the production of pro-inflammatory cytokines, such IL-1β, is likely a contributing factor for TGF-β1 methylation.
Applications
High levels of TGF-β1 methylation in the gastric cancer tissue suggest that TGF-β1 functions as a tumor suppressor in H. pylori related gastric cancer.
Terminology
CpG island: CpG islands are DNA segments, at least 0.5 kb in size, that are rich in G:C and CpG content, and are often located in the promoter or 50-exon sequences of genes. Promoter CpG islands have traditionally been thought to be unmethylated in normal cells, with the exception of those on the inactive X chromosome and those associated with imprinted genes. DNA methylation: DNA methylation is the major epigenetic phenomenon of eukaryotic genomes, and involves the addition of a methyl group to the carbon 5 position of the cytosine ring within the CpG dinucleotide. DNA methylation is required for the normal development of cells, whereas aberrant methylation of CpG islands confers a selective growth advantage that results in cancerous grow.
Peer review
The manuscript by Wang et al demonstrates that high levels of TGF-β1 promoter methylation in H. pylori positive patients was the result of H. pylori-induced inflammation rather than by H. pylori itself, and that IL-1β may be an important mediator for H. pylori-induced gene methylation during gastric cancer development. The overall goal of the paper is relevant. The data presented are solid and credible. The results are interesting, clinically important and worthy of publication in World Journal of Gastroenterology.
Footnotes
Supported by The National Natural Science Foundation of China, No. 31270532; the Foundation of Key Laboratory of Digestive System Tumors of Gansu Province and the Fundamental Research Funds for the Central Universities, No. lzujbky-2011-t03-03
P- Reviewers Chi SG, Swierczynski J S- Editor Zhai HH L- Editor Stewart GJ E- Editor Zhang DN
References
- 1.Antequera F. Structure, function and evolution of CpG island promoters. Cell Mol Life Sci. 2003;60:1647–1658. doi: 10.1007/s00018-003-3088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang RM, Wang L, Chen GY, Mao B, Chang J, Zhang Y, Li TQ, Gong M, Wang YQ, Feng WJ, et al. [Randomized controlled trial on haiguiyuyang capsule in the treatment of duodenal ulcer] Sichuan Daxue Xuebao Yixueban. 2005;36:233–236. [PubMed] [Google Scholar]
- 3.Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006;12:989–995. doi: 10.1158/1078-0432.CCR-05-2096. [DOI] [PubMed] [Google Scholar]
- 4.Leung WK, Yu J, Ng EK, To KF, Ma PK, Lee TL, Go MY, Chung SC, Sung JJ. Concurrent hypermethylation of multiple tumor-related genes in gastric carcinoma and adjacent normal tissues. Cancer. 2001;91:2294–2301. [PubMed] [Google Scholar]
- 5.Chan AO, Peng JZ, Lam SK, Lai KC, Yuen MF, Cheung HK, Kwong YL, Rashid A, Chan CK, Wong BC. Eradication of Helicobacter pylori infection reverses E-cadherin promoter hypermethylation. Gut. 2006;55:463–468. doi: 10.1136/gut.2005.077776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuper H, Adami HO, Trichopoulos D. Infections as a major preventable cause of human cancer. J Intern Med. 2000;248:171–183. doi: 10.1046/j.1365-2796.2000.00742.x. [DOI] [PubMed] [Google Scholar]
- 7.Bian YS, Osterheld MC, Fontolliet C, Bosman FT, Benhattar J. p16 inactivation by methylation of the CDKN2A promoter occurs early during neoplastic progression in Barrett’s esophagus. Gastroenterology. 2002;122:1113–1121. doi: 10.1053/gast.2002.32370. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh CJ, Klump B, Holzmann K, Borchard F, Gregor M, Porschen R. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res. 1998;58:3942–3945. [PubMed] [Google Scholar]
- 9.Mattsson A, Quiding-Järbrink M, Lönroth H, Hamlet A, Ahlstedt I, Svennerholm A. Antibody-secreting cells in the stomachs of symptomatic and asymptomatic Helicobacter pylori-infected subjects. Infect Immun. 1998;66:2705–2712. doi: 10.1128/iai.66.6.2705-2712.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crabtree JE. Immune and inflammatory responses to Helicobacter pylori infection. Scand J Gastroenterol Suppl. 1996;215:3–10. [PubMed] [Google Scholar]
- 11.Kai H, Kitadai Y, Kodama M, Cho S, Kuroda T, Ito M, Tanaka S, Ohmoto Y, Chayama K. Involvement of proinflammatory cytokines IL-1beta and IL-6 in progression of human gastric carcinoma. Anticancer Res. 2005;25:709–713. [PubMed] [Google Scholar]
- 12.Moss SF, Legon S, Davies J, Calam J. Cytokine gene expression in Helicobacter pylori associated antral gastritis. Gut. 1994;35:1567–1570. doi: 10.1136/gut.35.11.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matysiak-Budnik T, Mégraud F. Helicobacter pylori infection and gastric cancer. Eur J Cancer. 2006;42:708–716. doi: 10.1016/j.ejca.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 14.Qian X, Huang C, Cho CH, Hui WM, Rashid A, Chan AO. E-cadherin promoter hypermethylation induced by interleukin-1beta treatment or H. pylori infection in human gastric cancer cell lines. Cancer Lett. 2008;263:107–113. doi: 10.1016/j.canlet.2007.12.023. [DOI] [PubMed] [Google Scholar]
- 15.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 16.Lutz M, Knaus P. Integration of the TGF-beta pathway into the cellular signalling network. Cell Signal. 2002;14:977–988. doi: 10.1016/s0898-6568(02)00058-x. [DOI] [PubMed] [Google Scholar]
- 17.Muraoka-Cook RS, Shin I, Yi JY, Easterly E, Barcellos-Hoff MH, Yingling JM, Zent R, Arteaga CL. Activated type I TGFbeta receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene. 2006;25:3408–3423. doi: 10.1038/sj.onc.1208964. [DOI] [PubMed] [Google Scholar]
- 18.Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999;59:3379–3386. [PubMed] [Google Scholar]
- 19.Fu XB, Yang YH, Sun TZ, Gu XM, Jiang LX, Sun XQ, Sheng ZY. Effect of intestinal ischemia-reperfusion on expressions of endogenous basic fibroblast growth factor and transforming growth factor betain lung and its relation with lung repair. World J Gastroenterol. 2000;6:353–355. doi: 10.3748/wjg.v6.i3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shah JN, Shao G, Hei TK, Zhao Y. Methylation screening of the TGFBI promoter in human lung and prostate cancer by methylation-specific PCR. BMC Cancer. 2008;8:284. doi: 10.1186/1471-2407-8-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo W, Dong Z, Guo Y, Kuang G, Yang Z, Shan B. Concordant repression and aberrant methylation of transforming growth factor-beta signaling pathway genes occurs early in gastric cardia adenocarcinoma. Mol Biol Rep. 2012;39:9453–9462. doi: 10.1007/s11033-012-1810-x. [DOI] [PubMed] [Google Scholar]
- 22.Li Z, Li J. Local expressions of TGF-beta1, TGF-beta1RI, CTGF, and Smad-7 in Helicobacter pylori-associated gastritis. Scand J Gastroenterol. 2006;41:1007–1012. doi: 10.1080/00365520600554477. [DOI] [PubMed] [Google Scholar]
- 23.Sun Y, Liu YQ, Feng GS, Li JY. [Role of transforming growth factor beta1 in the development of atrophic gastritis] Beijing Da Xue Xue Bao. 2009;41:635–639. [PubMed] [Google Scholar]
- 24.Jo Y, Han SU, Kim YJ, Kim JH, Kim ST, Kim SJ, Hahm KB. Suppressed Gastric Mucosal TGF-beta1 Increases Susceptibility to H. pylori-Induced Gastric Inflammation and Ulceration: A Stupid Host Defense Response. Gut Liver. 2010;4:43–53. doi: 10.5009/gnl.2010.4.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 27.Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J, Easterly E, Roebuck LR, Ryan S, Gotwals PJ, et al. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J Clin Invest. 2002;109:1551–1559. doi: 10.1172/JCI15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mourskaia AA, Dong Z, Ng S, Banville M, Zwaagstra JC, O’Connor-McCourt MD, Siegel PM. Transforming growth factor-beta1 is the predominant isoform required for breast cancer cell outgrowth in bone. Oncogene. 2009;28:1005–1015. doi: 10.1038/onc.2008.454. [DOI] [PubMed] [Google Scholar]
- 29.Hasegawa Y, Takanashi S, Kanehira Y, Tsushima T, Imai T, Okumura K. Transforming growth factor-beta1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer. 2001;91:964–971. [PubMed] [Google Scholar]
- 30.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 31.Lu H, Ouyang W, Huang C. Inflammation, a key event in cancer development. Mol Cancer Res. 2006;4:221–233. doi: 10.1158/1541-7786.MCR-05-0261. [DOI] [PubMed] [Google Scholar]
- 32.Wang M, Furuta T, Takashima M, Futami H, Shirai N, Hanai H, Kaneko E. Relation between interleukin-1beta messenger RNA in gastric fundic mucosa and gastric juice pH in patients infected with Helicobacter pylori. J Gastroenterol. 1999;34 Suppl 11:10–17. [PubMed] [Google Scholar]
- 33.Yamaoka Y, Kita M, Kodama T, Sawai N, Kashima K, Imanishi J. Induction of various cytokines and development of severe mucosal inflammation by cagA gene positive Helicobacter pylori strains. Gut. 1997;41:442–451. doi: 10.1136/gut.41.4.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katayama Y, Takahashi M, Kuwayama H. Helicobacter pylori causes runx3 gene methylation and its loss of expression in gastric epithelial cells, which is mediated by nitric oxide produced by macrophages. Biochem Biophys Res Commun. 2009;388:496–500. doi: 10.1016/j.bbrc.2009.08.003. [DOI] [PubMed] [Google Scholar]